The association between sick sinus syndrome (SSS)
and atrial fibrillation (AF) has been recognized for more than 5
decades since 1968 (1) with the
first description of tachycardia-bradycardia syndrome (TBS)
reported 5 years later (2).
Tachycardia complicates approximately 50% of SSS cases (2–4). A
related condition, Bayes syndrome, involves inter-atrial block
associated with AF (5–15). Our understanding of cardiac
electrophysiology has significantly advanced with the use of
pre-clinical animal models, which are amenable to pharmacological,
physical or genetic manipulation for studying the consequences of
ion channel abnormalities (16–19), and have provided insight for
translational application (14,20–25). These studies have identified the
roles of different ion channels, such as
hyperpolarization-activated, cyclic nucleotide-gated (HCN),
Na+ and transient receptor potential (TRP) channels,
ryanodine receptors (RyR) and gap junctions (26–28), as well as tissue-level mechanisms,
in the pathogenesis of TBS. To understand the molecular basis of
how ion channel dysfunction leads to bradycardia or tachycardia,
and the causal relationship between bradycardia and tachycardia,
the mechanisms responsible for automaticity in the sinoatrial node
(SAN) and mediating action potential conduction need to be
considered.
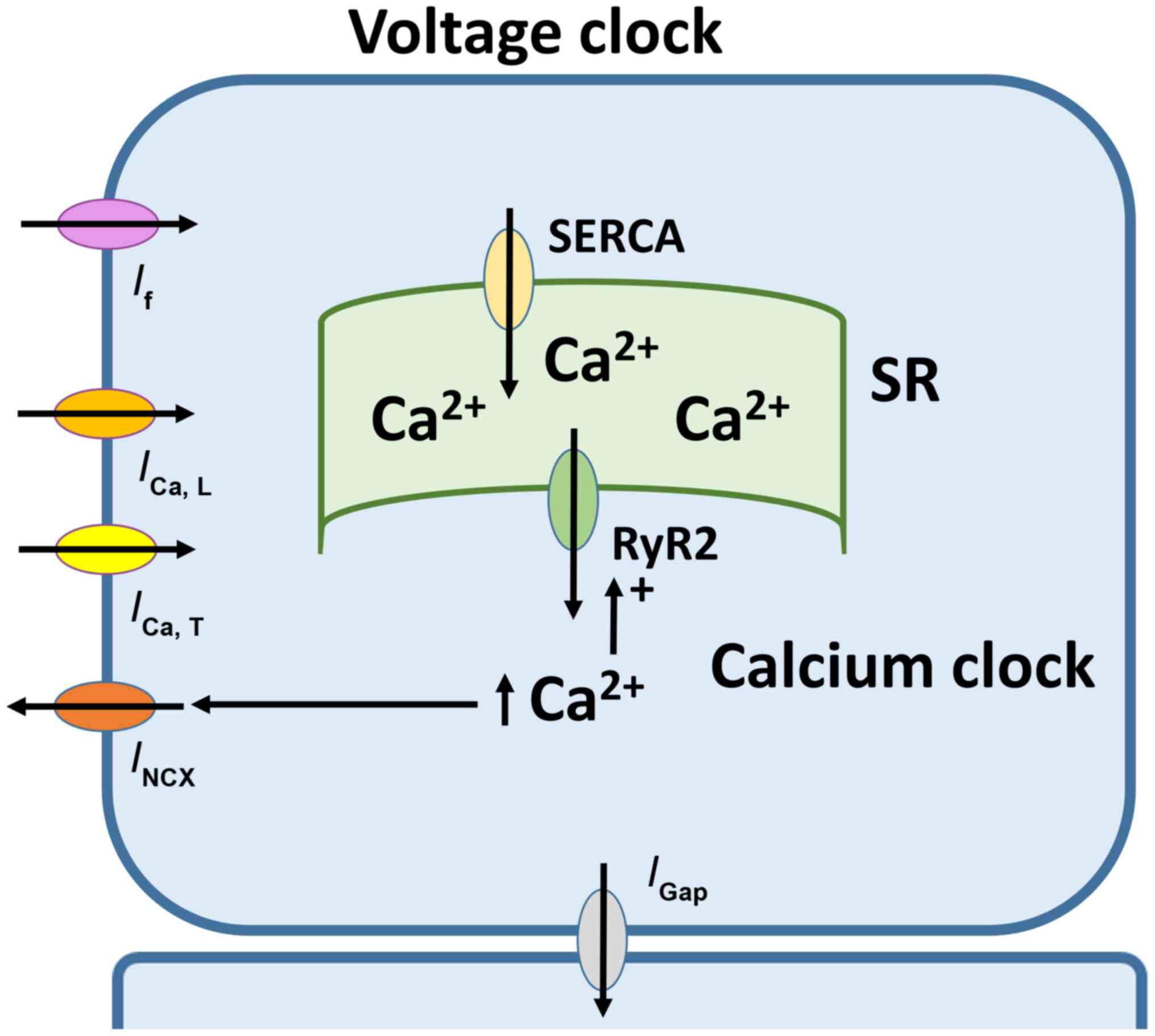
Automaticity of SAN is dependent on two closely
coupled clocks, voltage- and calcium-dependent mechanisms (Fig. 1) (29). The voltage-dependent mechanism
involves the funny current (If) mediated by HCN
channels located at the plasma membrane (30). If has several
unusual properties for a transmembrane current, including
activation by a hyperpolarized voltage, permeability to both
Na+ and K+ ions, regulation by intracellular
cAMP, and small single channel conductance (31). There are four recognized HCN
channel isoforms (1 to 4) (32).
HCN4 is the predominant subtype found in the SAN (33,34). By contrast, the
Ca2+-mediated mechanism involves rhythmic release of
Ca2+ from the sarcoplasmic reticulum (SR), subsequent
reuptake by the SR Ca2+-ATPase and extrusion via the
Na+-Ca2+ exchanger (35). Together, the complex interplay of
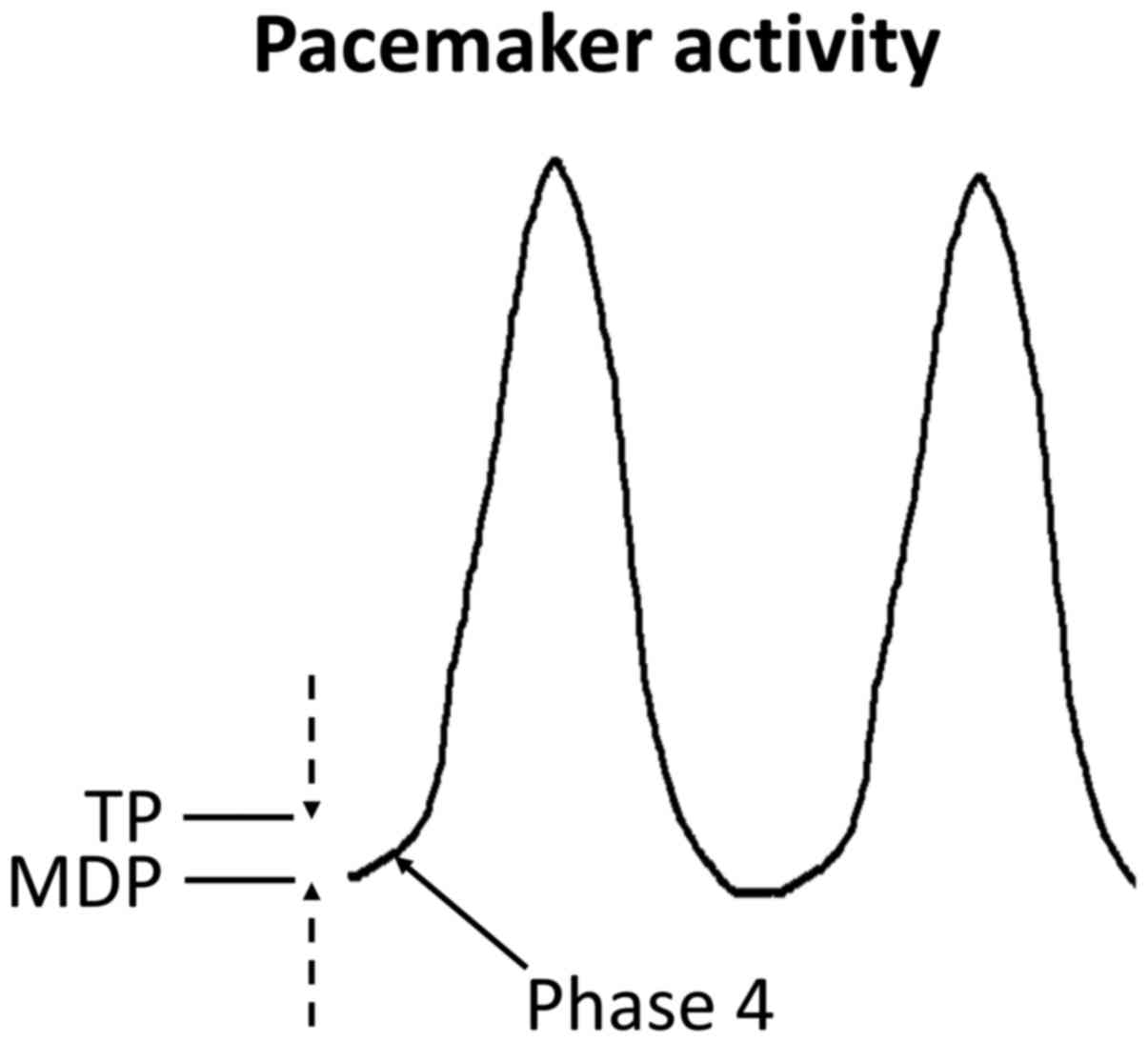
ion channels and pumps gives rise to the pacemaker action potential
(AP), which is uniquely character-ized by spontaneous
depolarization during phase 4 (Fig.
2).
Other ion channels are also involved in SAN
function, such as HCN channels, predominantly HCN4, carry the
If current which is a combination of both sodium
and potassium currents. Alterations in the highly regulated
activation and inactivation of the highly regulated cycle of ion
channels, such as an increase in late INa can
lead to arrhythmias (47). A
genetic mutation in any part of this complex pathway results in SAN
dysfunction leading to arrhythmias (50).
Conduction of APs from one myocyte to the next
occurs via gap junctions, each of which consists of two hexamers of
connexin (Cx) subunits (51–53). Cx 30.2, 40, 43 and 45 are found in
cardiac tissues (54). Cx40 is
expressed only in the atria and His-Purkinje system (55,56). Cx43 is expressed throughout the
atria and ventricles (57). Cx45
is the predominant isoform found in the core of SAN (58), whereas Cx43, Cx40 and Cx45 are
expressed in the periphery (50).
However, few gap junctions are found in the SAN core, suggesting
that intercellular coupling is not required for synchronization of
electrical activity within the node (59,60). The conventional membrane
voltage-dependent gating, transjunctional voltage-dependent gating
(61), phosphorylation (62–64), intracellular Ca2+
(65–68) and pH (69,70) as well as the surrounding lipid
environment (71–74) all regulate gap junctional
conductance.
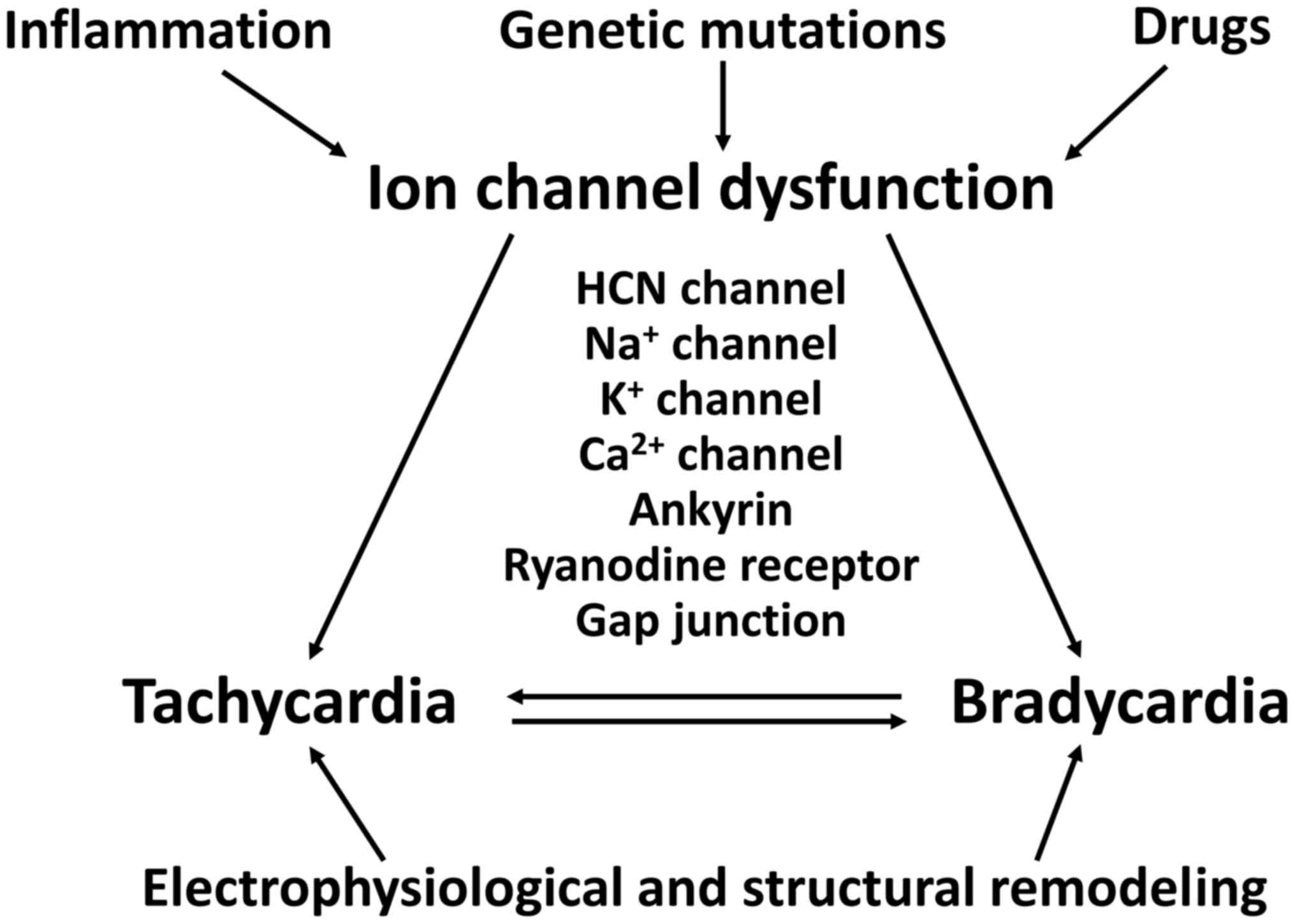
SSS can affect newborns and younger individuals, as
well as elderly individuals over 65 years of age (36,75). TBS can be caused by genetic
mutations, inflammation, ischaemia or drugs, involving both
structural and electrophysiological remodeling (Fig. 3). Broadly, TBS can involve
abnormal ion channel function, altered intercellular coupling or
tissue level mechanisms.
HCN4 is involved in mammalian cardiac pacemaking and
is predominantly expressed in the SAN (28). Loss-of-function HCN4 mutations are
known to cause atrioventricular (AV) block, long QT syndrome
(LQTS), AF, familial TBS and non-compaction cardiomyopathy in
addition to sinus bradycardia (76–80). The G1097W HCN4 mutation, which is
a loss-of-function mutation resulting in a hyperpolarizing shift of
the activation curve and reduced expression levels, demonstrates
4:1 AV block and reflex sinus tachycardia (81). A missense HCN4 mutation was found
to lead to impaired trafficking of the channel to the surface
membrane, resulting in SSS, long QT and torsade de pointes
(82). Some of these phenotypes
have been recapitulated in genetically modified mice, making them
particularly useful for modeling TBS. For example, HCN4-knockout
mice show severe sinus bradycardia complicated by AV block
(83), whereas
If-deficient mice generated by expression of a
dominant-negative, non-conductive HCN4-channel subunit exhibit
bradycardia, AV block and ventricular tachycardia (84). In this model, delayed
afterdepolarizations in SAN, AV node and Purkinje fibres were
observed, attibuted to increased SR Ca2+ load and
increased frequency of Ca2+ release from the SR
(84).
In the SAN, gap junctions contribute to automaticity
and exit conduction of APs to the myocardium surrounding nodal
tissue (111). Cx43
haploinsufficiency resulted in reduced CV in the ventricles, with
tachyarrhythmias preceding bradyarrhythmias, but little effect on
SAN function (112).
Cx40−/− mice showed intra-atrial block, ectopic rhythms
and abnormal conduction in the right atrium (113), inducible atrial tachycardia
(114), AVN and infra-Hisian
conduction delays (115).
If arrhythmia persists untreated, the structure of
the SAN can be modified and this remodeling can lead to fibrosis
and disturbance of the electrophysiology and even apoptosis of
cardiac cells. This in turn increases the risk of AF and paroxysmal
AF developing into permanent AF (28). The electrophysiological and
structure remodeling of the SAN not only lead to arrhythmias, as
discussed, but also are responsible for arrhythmias refractory to
medication and recurrence following cardioversion (28).
The causal relationship between bradycardia and
tachycardia is bidirectional. It is unclear which precipitates
which (28). Tachyarrhythmias can
promote SND, resulting in sinus bradycardia (1,2).
Patients with AF demonstrate structural abnormalities in the form
of fibrosis in their SAN (116).
Atrial tachycardia in dogs was found to lead to downregulation of
HCN2, HCN4 and KCNE1 (which modulates the α-subunit of the
K+ channel), which underlies the SND observed (27). In an atrial tachycardia pacing
model of TBS in rabbits, SND was associated with reduced HCN4
expression, both of which were reversible upon cessation of
tachycardia pacing (26). In
humans, HCN4 has been identified as a gene candidate associated
with AF from a meta-analysis of genome-wide association studies
(117). Adenosine is elevated in
the plasma of patients, and the consequent activation of adenosine
A1 receptors in the SAN is likely responsible for heart rate
reduction (118). In a canine
tachycardia-pacing model, A1 receptors were upregulated, which was
associated with prolonged SAN conduction time, conduction block
within the SAN, post-pacing pauses, shortening of atrial
repolarization durations leading to a higher propensity to AF
(119).
The current treatment options for TBS involve
removal or correction of extrinsic causes. In acute situations
where heart block is observed, the parasympathomimetic agent
atropine or beta agonist isoproterenol, or temporary pacing can be
used to overcome the conduction abnormalities. Tachyarrhythmias can
be managed by digoxin, quinidine or propranolol. Permanent pacing
using an electronic pacemaker is, at present, the only curative
option however battery life and electromagnetic interference are
often problematic.
Animal models have been extensively used for
exploring the electrophysiological basis of complex rhythm
disorders in an attempt to develop a biological pacemaker which
would be free of complications such as limited battery life
(125–129). These systems provide a platform
for elucidating the mechanisms of arrhythmogenesis in different
medical conditions (17,130–133), determining the efficacy of novel
therapeutic approaches and providing insights for translational
application (134–136). Generally, there are two
engineering biological alternatives to electronic pacemakers. The
first is a gene-based bio-artificial SAN. Ventricular
cardiomyocytes normally do not possess pacemaker activity, but they
can be induced to exhibit pacemaker function by genetic suppression
of the inward-rectifier K+ channels (137) or expression of HCN channels by
adenoviral transfer (135–145). A second approach is cell-based
bio-artificial pacemakers. This involves differentiation of human
embryonic stem cells or induced pluripotent stem cells into
cardiomyocytes (146,147). For example, human mesenchymal
stem cells pre-transfected with HCN2 channels can be used to
introduce If into surrounding cardiomyocytes that
subsequently possess pacemaker activity (148,149). Cardiomyocytes can be converted
into pacemaker cells by a cell fusion technique, where fibroblasts
engineered to express HCN1 are chemically fused to the
cardiomyocytes using chemicals such as polyethylene-glycol 1500
(150). Human embryonic stem
cells have also been differentiated into cardiomyocytes that
demonstrated intrinsic pacemaker activity, capable of pacing the
ventricular myocardium in vivo (135,151). Experimental data do not always
produce the same results when applied to animal models (152) and it would therefore be sensible
not to assume that animal models will produce the same results in a
human heart. Future research is needed to establish the safety of
these bio-artificial pacemakers, and little is known regarding
their long-term efficacy. They may provide better treatment options
for debilitating complex arrhythmias such as TBS.
In this review we summarized current literature to
understand the molecular and electrophysiological mechanisms and
discussed the current treatment and the exciting future possibility
of superior biological pacemakers which are hopefully not a too
distant possibility.
Professor Gary Tse was supported by the BBSRC and Dr
Yin Wah Fiona Chan was supported by the ESRC for their PhD studies.
Professor Gary Tse is grateful to the Croucher Foundation of Hong
Kong for supporting his clinical assistant professorship.
|
1
|
Ferrer MI: The sick sinus syndrome in
atrial disease. JAMA. 206:645–646. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaplan BM, Langendorf R, Lev M and Pick A:
Tachycardia-bradycardia syndrome (so-called 'sick sinus syndrome').
Pathology, mechanisms and treatment. Am J Cardiol. 31:497–508.
1973. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rubenstein JJ, Schulman CL, Yurchak PM and
DeSanctis RW: Clinical spectrum of the sick sinus syndrome.
Circulation. 46:5–13. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gomes JA, Kang PS, Matheson M, Gough WB Jr
and El-Sherif N: Coexistence of sick sinus rhythm and atrial
flutter-fibrillation. Circulation. 63:80–86. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bayés de Luna AJ: Bloqueo a nivel
auricular. Rev Esp Cardiol. 32:5–10. 1979.
|
|
6
|
Bayes de Luna A, Fort de Ribot R, Trilla
E, Julia J, Garcia J, Sadurni J, Riba J and Sagues F:
Electrocardiographic and vector-cardiographic study of interatrial
conduction disturbances with left atrial retrograde activation. J
Electrocardiol. 18:1–13. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bayés de Luna A, Cladellas M, Oter R,
Torner P, Guindo J, Martí V, Rivera I and Iturralde P: Interatrial
conduction block and retrograde activation of the left atrium and
paroxysmal supraventricular tachyarrhythmia. Eur Heart J.
9:1112–1118. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bayés de Luna A, Oter MC and Guindo J:
Interatrial conduction block with retrograde activation of the left
atrium and paroxysmal supraventricular tachyarrhythmias: Influence
of preventive anti-arrhythmic treatment. Int J Cardiol. 22:147–150.
1989. View Article : Google Scholar
|
|
9
|
Bayés de Luna A, Guindo J, Viñolas X,
Martinez-Rubio A, Oter R and Bayés-Genís A: Third-degree
inter-atrial block and supraventricular tachyarrhythmias. Europace.
1:43–46. 1999. View Article : Google Scholar
|
|
10
|
Bayés de Luna A, Platonov P, Cosio FG,
Cygankiewicz I, Pastore C, Baranowski R, Bayés-Genis A, Guindo J,
Viñolas X, Garcia-Niebla J, et al: Interatrial blocks. A separate
entity from left atrial enlargement: A consensus report. J
Electrocardiol. 45:445–451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Conde D, Seoane L, Gysel M, Mitrione S,
Bayés de Luna A and Baranchuk A: Bayés' syndrome:The association
between interatrial block and supraventricular arrhythmias. Expert
Rev Cardiovasc Ther. 13:541–550. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baranchuk A and Bayés de Luna A: The
P-wave morphology: What does it tell us. Herzschrittmacherther
Elektrophysiol. 26:192–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baranchuk A, de Luna AB and Breithardt G:
To the Editor - The role of advanced interatrial block pattern as a
predictor of atrial fibrillation. Heart Rhythm. 13:e872016.
View Article : Google Scholar
|
|
14
|
Tse G: Both transmural dispersion of
repolarization and transmural dispersion of refractoriness are poor
predictors of arrhythmogenicity: A role for the index of Cardiac
Electrophysiological Balance (QT/QRS). J Geriatr Cardiol. In
press.
|
|
15
|
Zhao J, Liu T and Li G: Relationship
between two arrhythmias: Sinus node dysfunction and atrial
fibrillation. Arch Med Res. 45:351–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choy L, Yeo JM, Tse V, Chan SP and Tse G:
Cardiac disease and arrhythmogenesis: Mechanistic insights from
mouse models. Int J Cardiol Heart Vasc. 12:1–10. 2016.PubMed/NCBI
|
|
17
|
Tse G and Yan BP: Electrophysiological
mechanisms of long and short QT syndromes: Insights from mouse
models. IJC Heart & Vasculature. In press.
|
|
18
|
Tse G, Lai ET, Lee AP, Yan BP and Wong SH:
Electrophysiological mechanisms of gastrointestinal
arrhythmogenesis: Lessons from the heart. Front Physiol.
7:2302016.PubMed/NCBI
|
|
19
|
Tse G, Wong ST, Tse V, Lee YT, Lin HY and
Yeo JM: Cardiac dynamics: alternans and arrhythmogenesis. J
Arrhythm. In press.
|
|
20
|
Tse G: Novel conduction-repolarization
indices for the stratification of arrhythmic risk. J Geriatr
Cardiol. 13:811–812. 2016.PubMed/NCBI
|
|
21
|
Tse G: (Tpeak-Tend)/QRS and
(Tpeak-Tend)/(QT x QRS): Novel markers for predicting arrhythmic
risk in the Brugada syndrome. Europace. In press.
|
|
22
|
Tse G and Yan BP: Novel arrhythmic risk
markers incorporating QRS dispersion: QRSd × (Tpeak - Tend)/QRS and
QRSd × (Tpeak - Tend)/(QT × QRS). Ann Noninvasive Electrocardiol.
Aug 18–2016.Epub ahead of print. View Article : Google Scholar
|
|
23
|
Wong J, Tan T, Chan C, Laxton V, Chan Y,
Liu T, Wong J and Tse G: The role of connexins in wound healing and
repair: novel therapeutic approaches. Front Physiol. In press.
|
|
24
|
Tse G and Yan BP: Traditional and novel
electrocardiographic conduction and repolarization markers of
sudden cardiac death. Europace. Oct 4–2016.Epub ahead of print.
View Article : Google Scholar
|
|
25
|
Tse G, Wong ST, Tse V and Yeo JM:
Variability in local action potential durations, dispersion of
repolarization and wavelength restitution in aged wild type and
Scn5a/- mouse hearts modelling human Brugada syndrome. J Geriatr
Cardiol. In press.
|
|
26
|
Chen Z, Sun B, Tse G, Jiang J and Xu W:
Reversibility of both sinus node dysfunction and reduced HCN4 mRNA
expression level in an atrial tachycardia pacing model of
tachycardia-bradycardia syndrome in rabbit hearts. Int J Clin Exp
Pathol. 9:8526–8531. 2016.
|
|
27
|
Yeh YH, Burstein B, Qi XY, Sakabe M,
Chartier D, Comtois P, Wang Z, Kuo CT and Nattel S: Funny current
downregulation and sinus node dysfunction associated with atrial
tachyarrhythmia: A molecular basis for tachycardia-bradycardia
syndrome. Circulation. 119:1576–1585. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Monfredi O and Boyett MR: Sick sinus
syndrome and atrial fibrillation in older persons - A view from the
sinoatrial nodal myocyte. J Mol Cell Cardiol. 83:88–100. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lakatta EG, Vinogradova T, Lyashkov A,
Sirenko S, Zhu W, Ruknudin A and Maltsev VA: The integration of
spontaneous intracellular Ca2+ cycling and surface membrane ion
channel activation entrains normal automaticity in cells of the
heart's pacemaker. Ann N Y Acad Sci. 1080:178–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baruscotti M, Bucchi A and Difrancesco D:
Physiology and pharmacology of the cardiac pacemaker ('funny')
current. Pharmacol Ther. 107:59–79. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DiFrancesco D: Pacemaker mechanisms in
cardiac tissue. Annu Rev Physiol. 55:455–472. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ludwig A, Zong X, Jeglitsch M, Hofmann F
and Biel M: A family of hyperpolarization-activated mammalian
cation channels. Nature. 393:587–591. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi W, Wymore R, Yu H, Wu J, Wymore RT,
Pan Z, Robinson RB, Dixon JE, McKinnon D and Cohen IS: Distribution
and prevalence of hyperpolarization-activated cation channel (HCN)
mRNA expression in cardiac tissues. Circ Res. 85:e1–e6. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moroni A, Gorza L, Beltrame M, Gravante B,
Vaccari T, Bianchi ME, Altomare C, Longhi R, Heurteaux C, Vitadello
M, et al: Hyperpolarization-activated cyclic nucleotide-gated
channel 1 is a molecular determinant of the cardiac pacemaker
current I(f). J Biol Chem. 276:29233–29241. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yaniv Y, Lakatta EG and Maltsev VA: From
two competing oscillators to one coupled-clock pacemaker cell
system. Front Physiol. 6:282015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dobrzynski H, Boyett MR and Anderson RH:
New insights into pacemaker activity: Promoting understanding of
sick sinus syndrome. Circulation. 115:1921–1932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boyett MR, Honjo H and Kodama I: The
sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc
Res. 47:658–687. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gellens ME, George ALJ Jr, Chen LQ,
Chahine M, Horn R, Barchi RL and Kallen RG: Primary structure and
functional expression of the human cardiac tetrodotoxin-insensitive
voltage-dependent sodium channel. Proc Natl Acad Sci USA.
89:554–558. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stühmer W, Conti F, Suzuki H, Wang XD,
Noda M, Yahagi N, Kubo H and Numa S: Structural parts involved in
activation and inactivation of the sodium channel. Nature.
339:597–603. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kontis KJ, Rounaghi A and Goldin AL:
Sodium channel activation gating is affected by substitutions of
voltage sensor positive charges in all four domains. J Gen Physiol.
110:391–401. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Horn R, Patlak J and Stevens CF: Sodium
channels need not open before they inactivate. Nature. 291:426–427.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
West JW, Patton DE, Scheuer T, Wang Y,
Goldin AL and Catterall WA: A cluster of hydrophobic amino acid
residues required for fast Na(+)-channel inactivation. Proc Natl
Acad Sci USA. 89:10910–10914. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kellenberger S, Scheuer T and Catterall
WA: Movement of the Na+ channel inactivation gate during
inactivation. J Biol Chem. 271:30971–30979. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kellenberger S, West JW, Catterall WA and
Scheuer T: Molecular analysis of potential hinge residues in the
inactivation gate of brain type IIA Na+ channels. J Gen Physiol.
109:607–617. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kellenberger S, West JW, Scheuer T and
Catterall WA: Molecular analysis of the putative inactivation
particle in the inactivation gate of brain type IIA Na+ channels. J
Gen Physiol. 109:589–605. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Smith MR and Goldin AL: Interaction
between the sodium channel inactivation linker and domain III
S4-S5. Biophys J. 73:1885–1895. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shryock JC, Song Y, Rajamani S,
Antzelevitch C and Belardinelli L: The arrhythmogenic consequences
of increasing late INa in the cardiomyocyte. Cardiovasc Res.
99:600–611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Balser JR, Nuss HB, Chiamvimonvat N,
Pérez-García MT, Marban E and Tomaselli GF: External pore residue
mediates slow inactivation in mu 1 rat skeletal muscle sodium
channels. J Physiol. 494:431–442. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vilin YY, Makita N, George AL Jr and Ruben
PC: Structural determinants of slow inactivation in human cardiac
and skeletal muscle sodium channels. Biophys J. 77:1384–1393. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
John RM and Kumar S: Sinus Node and Atrial
Arrhythmias. Circulation. 133:1892–1900. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Koval M, Isakson BE and Gourdie RG:
Connexins, pannexins and innexins: Protein cousins with overlapping
functions. FEBS Lett. 588:11852014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Veeraraghavan R, Gourdie RG and Poelzing
S: Mechanisms of cardiac conduction: A history of revisions. Am J
Physiol Heart Circ Physiol. 306:H619–H627. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Veeraraghavan R, Poelzing S and Gourdie
RG: Intercellular electrical communication in the heart: A new,
active role for the intercalated disk. Cell Commun Adhes.
21:161–167. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Davis LM, Kanter HL, Beyer EC and Saffitz
JE: Distinct gap junction protein phenotypes in cardiac tissues
with disparate conduction properties. J Am Coll Cardiol.
24:1124–1132. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gourdie RG, Green CR, Severs NJ, Anderson
RH and Thompson RP: Evidence for a distinct gap-junctional
phenotype in ventricular conduction tissues of the developing and
mature avian heart. Circ Res. 72:278–289. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gourdie RG, Severs NJ, Green CR, Rothery
S, Germroth P and Thompson RP: The spatial distribution and
relative abundance of gap-junctional connexin40 and connexin43
correlate to functional properties of components of the cardiac
atrioventricular conduction system. J Cell Sci. 105:985–991.
1993.PubMed/NCBI
|
|
57
|
Beyer EC, Paul DL and Goodenough DA:
Connexin43: A protein from rat heart homologous to a gap junction
protein from liver. J Cell Biol. 105:2621–2629. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Davis LM, Rodefeld ME, Green K, Beyer EC
and Saffitz JE: Gap junction protein phenotypes of the human heart
and conduction system. J Cardiovasc Electrophysiol. 6:813–822.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Saffitz JE, Green KG and Schuessler RB:
Structural determinants of slow conduction in the canine sinus
node. J Cardiovasc Electrophysiol. 8:738–744. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wilders R, Verheijck EE, Kumar R, Goolsby
WN, van Ginneken AC, Joyner RW and Jongsma HJ: Model clamp and its
application to synchronization of rabbit sinoatrial node cells. Am
J Physiol. 271:H2168–H2182. 1996.PubMed/NCBI
|
|
61
|
Bukauskas FF and Verselis VK: Gap junction
channel gating. Biochim Biophys Acta. 1662:42–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Musil LS and Goodenough DA: Biochemical
analysis of connexin43 intracellular transport, phosphorylation,
and assembly into gap junctional plaques. J Cell Biol.
115:1357–1374. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sáez JC, Nairn AC, Czernik AJ, Fishman GI,
Spray DC and Hertzberg EL: Phosphorylation of connexin43 and the
regulation of neonatal rat cardiac myocyte gap junctions. J Mol
Cell Cardiol. 29:2131–2145. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kwak BR, Hermans MM, De Jonge HR, Lohmann
SM, Jongsma HJ and Chanson M: Differential regulation of distinct
types of gap junction channels by similar phosphorylating
conditions. Mol Biol Cell. 6:1707–1719. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
De Mello WC: Effect of intracellular
injection of calcium and strontium on cell communication in heart.
J Physiol. 250:231–245. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dahl G and Isenberg G: Decoupling of heart
muscle cells: Correlation with increased cytoplasmic calcium
activity and with changes of nexus ultrastructure. J Membr Biol.
53:63–75. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Burt JM: Block of intercellular
communication: Interaction of intracellular H+ and Ca2+. Am J
Physiol. 253:C607–C612. 1987.PubMed/NCBI
|
|
68
|
Maurer P and Weingart R: Cell pairs
isolated from adult guinea pig and rat hearts: Effects of [Ca2+]i
on nexal membrane resistance. Pflugers Arch. 409:394–402. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hermans MM, Kortekaas P, Jongsma HJ and
Rook MB: pH sensitivity of the cardiac gap junction proteins,
connexin 45 and 43. Pflugers Arch. 431:138–140. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Morley GE, Taffet SM and Delmar M:
Intramolecular interactions mediate pH regulation of connexin43
channels. Biophys J. 70:1294–1302. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Meyer R, Malewicz B, Baumann WJ and
Johnson RG: Increased gap junction assembly between cultured cells
upon cholesterol supplementation. J Cell Sci. 96:231–238.
1990.PubMed/NCBI
|
|
72
|
Meyer RA, Lampe PD, Malewicz B, Baumann WJ
and Johnson RG: Enhanced gap junction formation with LDL and
apolipoprotein B. Exp Cell Res. 196:72–81. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Massey KD, Minnich BN and Burt JM:
Arachidonic acid and lipoxygenase metabolites uncouple neonatal rat
cardiac myocyte pairs. Am J Physiol. 263:C494–C501. 1992.PubMed/NCBI
|
|
74
|
Schubert AL, Schubert W, Spray DC and
Lisanti MP: Connexin family members target to lipid raft domains
and interact with caveolin-1. Biochemistry. 41:5754–5764. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yabek SM and Jarmakani JM: Sinus node
dysfunction in children, adolescents, and young adults. Pediatrics.
61:593–598. 1978.PubMed/NCBI
|
|
76
|
Schulze-Bahr E, Neu A, Friederich P, Kaupp
UB, Breithardt G, Pongs O and Isbrandt D: Pacemaker channel
dysfunction in a patient with sinus node disease. J Clin Invest.
111:1537–1545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Duhme N, Schweizer PA, Thomas D, Becker R,
Schröter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus
HA, et al: Altered HCN4 channel C-linker interaction is associated
with familial tachycardia-bradycardia syndrome and atrial
fibrillation. Eur Heart J. 34:2768–2775. 2013. View Article : Google Scholar
|
|
78
|
DiFrancesco D: HCN4, Sinus Bradycardia and
Atrial Fibrillation. Arrhythm Electrophysiol Rev. 4:9–13. 2015.
View Article : Google Scholar
|
|
79
|
Milano A, Vermeer AM, Lodder EM, Barc J,
Verkerk AO, Postma AV, van der Bilt IA, Baars MJ, van Haelst PL,
Caliskan K, et al: HCN4 mutations in multiple families with
bradycardia and left ventricular noncompaction cardiomyopathy. J Am
Coll Cardiol. 64:745–756. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Schweizer PA, Schröter J, Greiner S, Haas
J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A,
et al: The symptom complex of familial sinus node dysfunction and
myocardial noncompaction is associated with mutations in the HCN4
channel. J Am Coll Cardiol. 64:757–767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhou J, Ding WG, Makiyama T, Miyamoto A,
Matsumoto Y, Kimura H, Tarutani Y, Zhao J, Wu J, Zang WJ, et al: A
novel HCN4 mutation, G1097W, is associated with atrioventricular
block. Circ J. 78:938–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ueda K, Nakamura K, Hayashi T, Inagaki N,
Takahashi M, Arimura T, Morita H, Higashiuesato Y, Hirano Y,
Yasunami M, et al: Functional characterization of a
trafficking-defective HCN4 mutation, D553N, associated with cardiac
arrhythmia. J Biol Chem. 279:27194–27198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baruscotti M, Bucchi A, Viscomi C,
Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR,
Micheloni S, Barbuti A, et al: Deep bradycardia and heart block
caused by inducible cardiac-specific knockout of the pacemaker
channel gene Hcn4. Proc Natl Acad Sci USA. 108:1705–1710. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mesirca P, Alig J, Torrente AG, Müller JC,
Marger L, Rollin A, Marquilly C, Vincent A, Dubel S, Bidaud I, et
al: Cardiac arrhythmia induced by genetic silencing of 'funny' (f)
channels is rescued by GIRK4 inactivation. Nat Commun. 5:4664.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Makiyama T, Akao M, Shizuta S, Doi T,
Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, et al: A
novel SCN5A gain-of-function mutation M1875T associated with
familial atrial fibrillation. J Am Coll Cardiol. 52:1326–1334.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bezzina C, Veldkamp MW, van Den Berg MP,
Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G,
Bink-Boelkens MT, van Der Hout AH, et al: A single Na(+) channel
mutation causing both long-QT and Brugada syndromes. Circ Res.
85:1206–1213. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bezzina CR, Barc J, Mizusawa Y, Remme CA,
Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi
F, et al: Common variants at SCN5A–SCN10A and HEY2 are associated
with Brugada syndrome, a rare disease with high risk of sudden
cardiac death. Nat Genet. 45:1044–1049. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bezzina CR and Remme CA: Dilated
cardiomyopathy due to sodium channel dysfunction: What is the
connection. Circ Arrhythm Electrophysiol. 1:80–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bezzina CR, Rook MB, Groenewegen WA,
Herfst LJ, van der Wal AC, Lam J, Jongsma HJ, Wilde AA and Mannens
MM: Compound heterozygosity for mutations (W156X and R225W) in
SCN5A associated with severe cardiac conduction disturbances and
degenerative changes in the conduction system. Circ Res.
92:159–168. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Remme CA, Wilde AA and Bezzina CR: Cardiac
sodium channel overlap syndromes: Different faces of SCN5A
mutations. Trends Cardiovasc Med. 18:78–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tan HL, Bink-Boelkens MT, Bezzina CR,
Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, van den Berg MP,
Wilde AA and Balser JR: A sodium-channel mutation causes isolated
cardiac conduction disease. Nature. 409:1043–1047. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chang CC, Acharfi S, Wu MH, Chiang FT,
Wang JK, Sung TC and Chahine M: A novel SCN5A mutation manifests as
a malignant form of long QT syndrome with perinatal onset of
tachycardia/bradycardia. Cardiovasc Res. 64:268–278. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Letsas KP, Korantzopoulos P, Efremidis M,
Weber R, Lioni L, Bakosis G, Vassilikos VP, Deftereos S, Sideris A
and Arentz T: Sinus node disease in subjects with type 1 ECG
pattern of Brugada syndrome. J Cardiol. 61:227–231. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Girmatsion Z, Biliczki P, Bonauer A,
Wimmer-Greinecker G, Scherer M, Moritz A, Bukowska A, Goette A,
Nattel S, Hohnloser SH, et al: Changes in microRNA-1 expression and
IK1 up-regulation in human atrial fibrillation. Heart Rhythm.
6:1802–1809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Bennett V and Healy J: Organizing the
fluid membrane bilayer: Diseases linked to spectrin and ankyrin.
Trends Mol Med. 14:28–36. 2008. View Article : Google Scholar
|
|
96
|
Le Scouarnec S, Bhasin N, Vieyres C, Hund
TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, et
al: Dysfunction in ankyrin-B-dependent ion channel and transporter
targeting causes human sinus node disease. Proc Natl Acad Sci USA.
105:15617–15622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mohler PJ, Splawski I, Napolitano C,
Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT and Bennett
V: A cardiac arrhythmia syndrome caused by loss of ankyrin-B
function. Proc Natl Acad Sci USA. 101:9137–9142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mohler PJ, Schott JJ, Gramolini AO, Dilly
KW, Guatimosim S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME,
et al: Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia
and sudden cardiac death. Nature. 421:634–639. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Mohler PJ, Le Scouarnec S, Denjoy I, et
al: Defining the cellular phenotype of 'ankyrin-B syndrome'
variants: Human ANK2 variants associated with clinical phenotypes
display a spectrum of activities in cardiomyocytes. Circulation.
115:432–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mangoni ME, Couette B, Bourinet E, Platzer
J, Reimer D, Striessnig J and Nargeot J: Functional role of L-type
Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad
Sci USA. 100:5543–5548. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Trebak M, Zhang W, Ruhle B, Henkel MM,
González-Cobos JC, Motiani RK, Stolwijk JA, Newton RL and Zhang X:
What role for store-operated Ca2+ entry in muscle.
Microcirculation. 20:330–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ju YK, Lee BH, Trajanovska S, Hao G, Allen
DG, Lei M and Cannell MB: The involvement of TRPC3 channels in
sinoatrial arrhythmias. Front Physiol. 6:862015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Swaminathan PD, Purohit A, Soni S, Voigt
N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, et al:
Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J
Clin Invest. 121:3277–3288. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Luu M, Stevenson WG, Stevenson LW, Baron K
and Walden J: Diverse mechanisms of unexpected cardiac arrest in
advanced heart failure. Circulation. 80:1675–1680. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Stevenson WG, Stevenson LW, Middlekauff HR
and Saxon LA: Sudden death prevention in patients with advanced
ventricular dysfunction. Circulation. 88:2953–2961. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Faggioni M, van der Werf C and Knollmann
BC: Sinus node dysfunction in catecholaminergic polymorphic
ventricular tachycardia: Risk factor and potential therapeutic
target. Trends Cardiovasc Med. 24:273–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sumitomo N, Sakurada H, Taniguchi K, et
al: Association of atrial arrhythmia and sinus node dysfunction in
patients with catecholaminergic polymorphic ventricular
tachycardia. Circ J. 71:1606–1609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Faggioni M, Savio-Galimberti E,
Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D and Knollmann
BC: Suppression of spontaneous ca elevations prevents atrial
fibrillation in calsequestrin 2-null hearts. Circ Arrhythm
Electrophysiol. 7:313–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Glukhov AV, Kalyanasundaram A, Lou Q, Hage
LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M,
Györke S, et al: Calsequestrin 2 deletion causes sinoatrial node
dysfunction and atrial arrhythmias associated with altered
sarcoplasmic reticulum calcium cycling and degenerative fibrosis
within the mouse atrial pacemaker complex1. Eur Heart J.
36:686–697. 2015. View Article : Google Scholar
|
|
111
|
Jongsma HJ: Diversity of gap junctional
proteins: Does it play a role in cardiac excitation. J Cardiovasc
Electrophysiol. 11:228–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Eckardt D, Theis M, Degen J, Ott T, van
Rijen HV, Kirchhoff S, Kim JS, de Bakker JM and Willecke K:
Functional role of connexin43 gap junction channels in adult mouse
heart assessed by inducible gene deletion. J Mol Cell Cardiol.
36:101–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bagwe S, Berenfeld O, Vaidya D, Morley GE
and Jalife J: Altered right atrial excitation and propagation in
connexin40 knockout mice. Circulation. 112:2245–2253. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Verheule S, van Batenburg CA, Coenjaerts
FE, Kirchhoff S, Willecke K and Jongsma HJ: Cardiac conduction
abnormalities in mice lacking the gap junction protein connexin40.
J Cardiovasc Electrophysiol. 10:1380–1389. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
VanderBrink BA, Sellitto C, Saba S, Link
MS, Zhu W, Homoud MK, Estes NA III, Paul DL and Wang PJ:
Connexin40-deficient mice exhibit atrioventricular nodal and
infra-Hisian conduction abnormalities. J Cardiovasc Electrophysiol.
11:1270–1276. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Thery C, Gosselin B, Lekieffre J and
Warembourg H: Pathology of sinoatrial node. Correlations with
electrocardiographic findings in 111 patients. Am Heart J.
93:735–740. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ellinor PT, Lunetta KL, Albert CM, Glazer
L, Ritchie MD, Smith AV, Arking DE, Müller-Nurasyid M, Krijthe BP,
Lubitz SA, et al: Meta-analysis identifies six new susceptibility
loci for atrial fibrillation. Nat Genet. 44:670–675. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Funaya H, Kitakaze M, Node K, Minamino T,
Komamura K and Hori M: Plasma adenosine levels increase in patients
with chronic heart failure. Circulation. 95:1363–1365. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lou Q, Hansen BJ, Fedorenko O, Csepe TA,
Kalyanasundaram A, Li N, Hage LT, Glukhov AV, Billman GE, Weiss R,
et al: Upregulation of adenosine A1 receptors facilitates
sinoatrial node dysfunction in chronic canine heart failure by
exacerbating nodal conduction abnormalities revealed by novel
dual-sided intramural optical mapping. Circulation. 130:315–324.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li G, Liu E, Liu T, Wang J, Dai J, Xu G,
Korantzopoulos P and Yang W: Atrial electrical remodeling in a
canine model of sinus node dysfunction. Int J Cardiol. 146:32–36.
2011. View Article : Google Scholar
|
|
121
|
Herrmann S, Fabritz L, Layh B, Kirchhof P
and Ludwig A: Insights into sick sinus syndrome from an inducible
mouse model. Cardiovasc Res. 90:38–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tse G and Yeo JM: Conduction abnormalities
and ventricular arrhythmogenesis: The roles of sodium channels and
gap junctions. Int J Cardiol Heart Vasc. 9:75–82. 2015.
|
|
123
|
Pezhouman A, Cao H, Lee HH, Belardinelli
L, Weiss JN and Karagueuzian HS: Abstract 16247: Oxidative Stress
Initiates Atrial Fibrillation in Fibrotic Hearts by Early
Afterdepolarization-Mediated Triggered Activity. The Key Role of
Late INa. Circulation. 130:A162472014.
|
|
124
|
Morita N, Mandel WJ, Kobayashi Y and
Karagueuzian HS: Cardiac fibrosis as a determinant of ventricular
tachyarrhythmias. J Arrhythm. 30:389–394. 2014. View Article : Google Scholar
|
|
125
|
Tse G, Tse V and Yeo JM: Ventricular
anti-arrhythmic effects of heptanol in hypokalaemic,
Langendorff-perfused mouse hearts. Biomed Rep. 4:313–324.
2016.PubMed/NCBI
|
|
126
|
Tse G, Tse V, Yeo JM and Sun B: Atrial
anti-arrhythmic effects of heptanol in Langendorff-perfused mouse
hearts. PLoS One. 11:e01488582016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Tse G, Wong ST, Tse V and Yeo JM:
Restitution analysis of alternans using dynamic pacing and its
comparison with S1S2 restitution in heptanol-treated, hypokalaemic
Langendorff-perfused mouse hearts. Biomed Rep. 4:673–680.
2016.PubMed/NCBI
|
|
128
|
Tse G, Sun B, Wong ST, Tse V and Yeo JM:
Ventricular anti-arrhythmic effects of hypercalcaemia treatment in
hyperkalaemic, Langendorff-perfused mouse hearts. Biomed Rep.
5:301–310. 2016.PubMed/NCBI
|
|
129
|
Tse G, Yeo JM, Tse V, Kwan J and Sun B:
Gap junction inhibition by heptanol increases ventricular
arrhythmogenicity by reducing conduction velocity without affecting
repolarization properties or myocardial refractoriness in
Langendorff-perfused mouse hearts. Mol Med Rep. 14:4069–4074.
2016.PubMed/NCBI
|
|
130
|
Tse G, Lai ET, Tse V and Yeo JM: Molecular
and electrophysiological mechanisms underlying cardiac
arrhythmogenesis in diabetes mellitus. J Diabetes Res.
2016:28487592016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Tse G, Yeo JM, Chan YW, Lai ET and Yan BP:
What is the arrhythmic substrate in viral myocarditis? Insights
from clinical and animal studies. Front Physiol. 7:3082016.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tse G, Yan BP, Chan YW, Tian XY and Huang
Y: Reactive oxygen species, endoplasmic reticulum stress and
mitochondrial dysfunction: The link with cardiac arrhythmogenesis.
Front Physiol. 7:3132016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Tse G, Lai ET, Yeo JM and Yan BP:
Electrophysiological mechanisms of Bayés syndrome: Insights from
clinical and mouse studies. Front Physiol. 7:1882016.
|
|
134
|
Li RA: Gene- and cell-based bio-artificial
pacemaker: What basic and translational lessons have we learned.
Gene Ther. 19:588–595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Xue T, Cho HC, Akar FG, Tsang SY, Jones
SP, Marbán E, Tomaselli GF and Li RA: Functional integration of
electrically active cardiac derivatives from genetically engineered
human embryonic stem cells with quiescent recipient ventricular
cardiomyocytes: Insights into the development of cell-based
pacemakers. Circulation. 111:11–20. 2005. View Article : Google Scholar
|
|
136
|
Nattel S: Inward rectifier-funny current
balance and spontaneous automaticity: Cautionary notes for biologic
pacemaker development. Heart Rhythm. 5:1318–1319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Miake J, Marbán E and Nuss HB: Biological
pacemaker created by gene transfer. Nature. 419:132–133. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Azene EM, Xue T, Marbán E, Tomaselli GF
and Li RA: Non-equilibrium behavior of HCN channels: Insights into
the role of HCN channels in native and engineered pacemakers.
Cardiovasc Res. 67:263–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Qu J, Barbuti A, Protas L, Santoro B,
Cohen IS and Robinson RB: HCN2 overexpression in newborn and adult
ventricular myocytes: Distinct effects on gating and excitability.
Circ Res. 89:E8–E14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xue T, Siu CW, Lieu DK, Lau CP, Tse HF and
Li RA: Mechanistic role of I(f) revealed by induction of
ventricular automaticity by somatic gene transfer of
gating-engineered pacemaker (HCN) channels. Circulation.
115:1839–1850. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kass-Eisler A, Falck-Pedersen E, Alvira M,
Rivera J, Buttrick PM, Wittenberg BA, Cipriani L and Leinwand LA:
Quantitative determination of adenovirus-mediated gene delivery to
rat cardiac myocytes in vitro and in vivo. Proc Natl Acad Sci USA.
90:11498–11502. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Mühlhauser J, Jones M, Yamada I, Cirielli
C, Lemarchand P, Gloe TR, Bewig B, Signoretti S, Crystal RG and
Capogrossi MC: Safety and efficacy of in vivo gene transfer into
the porcine heart with replication-deficient, recombinant
adenovirus vectors. Gene Ther. 3:145–153. 1996.PubMed/NCBI
|
|
143
|
Chan YC, Siu CW, Lau YM, Lau CP, Li RA and
Tse HF: Synergistic effects of inward rectifier (I) and pacemaker
(I) currents on the induction of bioengineered cardiac
automaticity. J Cardiovasc Electrophysiol. 20:1048–1054. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lieu DK, Chan YC, Lau CP, Tse HF, Siu CW
and Li RA: Overexpression of HCN-encoded pacemaker current silences
bioartificial pacemakers. Heart Rhythm. 5:1310–1317. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Saito Y, Nakamura K, Yoshida M, Sugiyama
H, Ohe T, Kurokawa J, Furukawa T, Takano M, Nagase S, Morita H, et
al: Enhancement of Spontaneous Activity by HCN4 Overexpression in
Mouse Embryonic Stem Cell-Derived Cardiomyocytes - A Possible
Biological Pacemaker. PLoS One. 10:e01381932015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kong CW, Akar FG and Li RA: Translational
potential of human embryonic and induced pluripotent stem cells for
myocardial repair: Insights from experimental models. Thromb
Haemost. 104:30–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Weng Z, Kong CW, Ren L, Karakikes I, Geng
L, He J, Chow MZ, Mok CF, Keung W, Chow H, et al: A simple,
cost-effective but highly efficient system for deriving ventricular
cardiomyocytes from human pluripotent stem cells. Stem Cells Dev.
23:1704–1716. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Plotnikov AN, Shlapakova I, Szabolcs MJ,
Danilo P Jr, Lorell BH, Potapova IA, Lu Z, Rosen AB, Mathias RT,
Brink PR, et al: Xenografted adult human mesenchymal stem cells
provide a platform for sustained biological pacemaker function in
canine heart. Circulation. 116:706–713. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Plotnikov AN, Sosunov EA, Qu J, Shlapakova
IN, Anyukhovsky EP, Liu L, Janse MJ, Brink PR, Cohen IS, Robinson
RB, et al: Biological pacemaker implanted in canine left bundle
branch provides ventricular escape rhythms that have
physiologically acceptable rates. Circulation. 109:506–512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Cho HC, Kashiwakura Y and Marbán E:
Creation of a biological pacemaker by cell fusion. Circ Res.
100:1112–1115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kehat I, Khimovich L, Caspi O, Gepstein A,
Shofti R, Arbel G, Huber I, Satin J, Itskovitz-Eldor J and Gepstein
L: Electromechanical integration of cardiomyocytes derived from
human embryonic stem cells. Nat Biotechnol. 22:1282–1289. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Verkerk AO and Wilders R:
Hyperpolarization-activated current, If, in mathematical models of
rabbit sinoatrial node pacemaker cells. BioMed Res Int.
2013:8724542013. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Tse G: Mechanisms of cardiac arrhythmias.
J Arrhythm. 32:75–81. 2016. View Article : Google Scholar : PubMed/NCBI
|