Introduction
Bone metabolism is strictly controlled by
osteoblastic bone formation and osteoclastic bone resorption
(1). Bone tissue is continuously
regenerated through a process called bone remodeling (2). To maintain adequate quality and
quantity of bone, formation and resorption are tightly coordinated.
This bone remodeling process begins with osteoclastic bone
resorption, followed by osteoblastic bone formation (1). Disruption of the bone remodeling
process causes metabolic bone diseases such as osteoporosis or
fracture healing distress. It is currently recognized that
osteoblasts play a crucial role also in the regulation of bone
resorption through the expression of the receptor activator of
nuclear factor-κB (RANK) ligand in response to a variety of bone
resorptive stimuli (3).
It is firmly established that tumor necrosis
factor-α (TNF-α) is a multifunctional cytokine and plays a pivotal
role in inflammation, infection and cancer (4). In bone metabolism, TNF-α reportedly
induces the activation of RANK in osteoclasts through RANK ligand
expression after binding to its specific receptors in osteoblasts,
leading to osteoclast activation (5). In addition, it has been shown that
TNF-α inhibits the differentiation of osteoblasts from precursor
cells (6). Therefore, TNF-α is
generally recognized as a potent bone resorptive agent. Regarding
the intracellular signaling mechanism of TNF-α, it is well known
that TNF-α induces activation of the inhibiting inhibitor of κB
(IκB)/nuclear factor-κB (NF-κB) pathway (7). In our previous studies (8,9),
we demonstrated that TNF-α stimulated the activation of p70 S6
kinase in osteoblast-like MC3T3-E1 cells, and that the activated
p70 S6 kinase negatively regulated TNF-α-stimulated interleukin-6
(IL-6) synthesis. IL-6, a member of the glycoprotein 130 cytokine
families (4,10), is well known to stimulate bone
resorption and induce osteoclastgenesis (4). It has been shown that IL-6 acts on
the osteoblast lineage to stimulate bone formation and maintain
strength rather than promoting osteoclast formation (11). Additionally, IL-6 reportedly has a
pivotal role in the process of bone fracture repair (12). Therefore, IL-6 secreted by
osteoblasts is currently considered as an osteotropic modulator,
leading to influencing bone formation under the condition of
increased bone remodeling (13).
However, the exact mechanism behind the TNF-α-stimulated IL-6
synthesis in osteoblasts remains to be elucidated.
Incretins are designated as small intestine-derived
hormones and are released in response to oral food intake (14). Incretins stimulate insulin
secretion from pancreatic islet β cells (14,15). Glucose-dependent insulinotropic
polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are generally
recognized as two major incretins. GIP and GLP-1 exert their
insulinotropic actions through the activation of their specific
guanine nucleotide-binding protein (G-protein)-coupled receptors
expressed on pancreatic β cells (16). It is generally established that
the binding of incretins to their receptors causes the activation
of adenylyl cyclase, resulting in the formation of cAMP from ATP
(14). Subsequent activation of
protein kinase A (PKA) leads into diverse events including altered
ion channel activity and elevation of intracellular calcium
concentrations (14). With regard
to incretin effects on bone, it has been shown that GIP stimulated
both collagen type I mRNA expression levels and alkaline
phosphatase activity in osteoblasts (17). GIP reportedly increases bone
mineral density in an ovariectomized rat model (18). As for GLP-1, it has been
demonstrated that GLP-1 receptor-deficient mice presented with an
increased number of osteoclasts and bone resorption, and suffered
from osteoporosis (19). In
addition, GLP-1 reportedly induces the differentiation of
osteoblasts (20). However, the
details underlying the effects of incretins on bone metabolism have
not yet been clarified.
In the present study, we investigated the effects of
GLP-1 or GIP on the TNF-α-induced IL-6 synthesis in osteoblast-like
MC3T3-E1 cells. We herein showed that GLP-1 and GIP enhanced the
TNF-α-stimulated IL-6 synthesis in MC3T3-E1 cells, and that the
amplifying effect was exerted via reducing the IκB/NF-κB pathway
through the adenylyl cyclase-cAMP system.
Materials and methods
Materials
GLP-1 and GIP were obtained from Peptide Institute,
Inc. (Osaka, Japan). TNF-α and mouse IL-6 enzyme-linked
immunosorbent assay (ELISA) kit were purchased from R&D System,
Inc. (Minneapolis, MN, USA). Dibutyryl cAMP (Bt2cAMP)
was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA).
Wedelolactone was purchased from Calbiochem (La Jolla, CA, USA).
N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89)
was obtained from Seikagaku Kogyo Co. (Tokyo, Japan).
Phospho-specific IκB, IκB, phospho-specific p70 S6 kinase, p70 S6
and phospho-specific cAMP response element-binding protein (CREB)
antibodies were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) antibody was obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). An ECL western blotting detection system was
obtained from GE Healthcare Life Sciences (Chalfont, UK). Other
materials and chemicals were obtained from commercial sources.
Wedelolactone and H-89 were dissolved in dimethyl sulfoxide. The
maximum concentration of dimethyl sulfoxide was 0.1%, which did not
affect either assay for IL-6 or western blot analysis.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells, which were
derived from newborn mouse calvaria (21), were maintained as previously
described (22). In brief, the
cells were cultured in α-minimum essential medium (α-MEM)
containing 10% fetal bovine serum (FBS) at 37°C in a humidified
atmosphere of 5% CO2/95% air. The cells were seeded into
35-mm diameter dishes (5×104 cells/dish) or 90-mm
diameter dishes (2×105 cells/dish) in α-MEM containing
10% FBS. After 5 days, the medium was exchanged for α-MEM
containing 0.3% FBS. The cells were then used for experiments
following a 48-h incubation period.
Assay for IL-6
The cultured cells were stimulated by 30 ng/ml of
TNF-α or vehicle in 1 ml of α-MEM containing 0.3% FBS for the
indicated periods. When indicated, the cells were pretreated with
various doses of GLP-1, GIP, wedelo-lactone or Bt2cAMP
for 60 min. Pretreatment with H-89 was performed for 60 min prior
to the stimulation by GLP-1. The conditioned medium was collected
at the end of the incubation, and the IL-6 concentrations in the
medium were then measured using the mouse IL-6 ELISA kit according
to the manufacturer's protocol.
Real-time RT-PCR
The cultured cells were pretreated with 100 nM of
GLP-1, 100 nM of GIP or vehicle for 60 min, and then stimulated by
30 ng/ml TNF-α or vehicle in α-MEM containing 0.3% FBS for 3 h.
Total RNA was isolated and transcribed into complementary DNA using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Heysham, Lancashire, UK) and the Omniscript Reverse Transcriptase
kit (Qiagen Inc., Valencia, CA, USA), respectively. Real-time
RT-PCR was performed using a LightCycler system in capillaries and
the LightCycler FastStart DNA Master SYBR-Green I provided with the
kit (Roche Diagnostics, Basel, Switzerland). Sense and antisense
primers for mouse IL-6 mRNA and GAPDH mRNA were synthesized based
on the report of Simpson et al (23). The amplified products were
determined using a melting curve analysis, according to the system
protocol. The levels of IL-6 mRNA were normalized to those of GAPDH
mRNA.
Western blot analysis
The cultured cells were pretreated with various
concentrations of GLP-1, GIP, wedelolactone or Bt2cAMP
for 60 min, and then stimulated by 30 ng/ml of TNF-α or vehicle in
α-MEM containing 0.3% FBS for the indicated periods. When
indicated, the cells were washed twice with phosphate-buffered
saline, and then lysed, homogenized and sonicated in a lysis buffer
containing 62.5 mM Tris/HCl, pH 6.8, 2% sodium dodecyl sulfate
(SDS), 50 mM dithiothreitol and 10% glycerol. SDS-polyacrylamide
gel electrophoresis (PAGE) was performed using the method of
Laemmli (24) on 10%
polyacrylamide gels. The protein was fractionated and transferred
onto an Immun-Blot PVDF membrane (Bio-Rad, Hercules, CA, USA). The
membranes were blocked with 5% fat-free dry milk in Tris-buffered
saline-Tween (TBS-T; 20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.1%
Tween-20) for 1 h before incubation with primary antibodies.
Western blot analysis was performed as previously described
(25) using the indicated primary
antibodies with peroxidase-labeled antibodies raised in goat
against rabbit IgG being used as secondary antibodies. The primary
and secondary antibodies were diluted at 1:1,000 with 5% fat-free
dry milk in TBS-T. The peroxidase activity on the PVDF sheet was
visualized on X-ray film by means of the ECL western blotting
detection system.
Densitometric analysis
The densitometric analysis of western blotting was
performed using scanner and image analysis software program (imageJ
version 1.48; National Institutes of Health, Bethesda, MD, USA).
The phosphorylated protein levels were calculated as follows: the
background-subtracted signal intensity of each phosphorylation
signal was normalized to the respective intensity of GAPDH or p70
S6 kinase, and plotted as the fold increase in comparison to that
of the control cells treated without stimulation.
Statistical analysis
The data were analyzed by ANOVA followed by
Bonferroni method for multiple comparisons between pairs, and
P<0.05 was considered to indicate a statistically significant
difference. All data are presented as the mean ± SEM of triplicate
determinations from three independent cell preparations.
Results
Effects of GLP-1 or GIP on the
TNF-α-stimulated IL-6 release in MC3T3-E1 cells
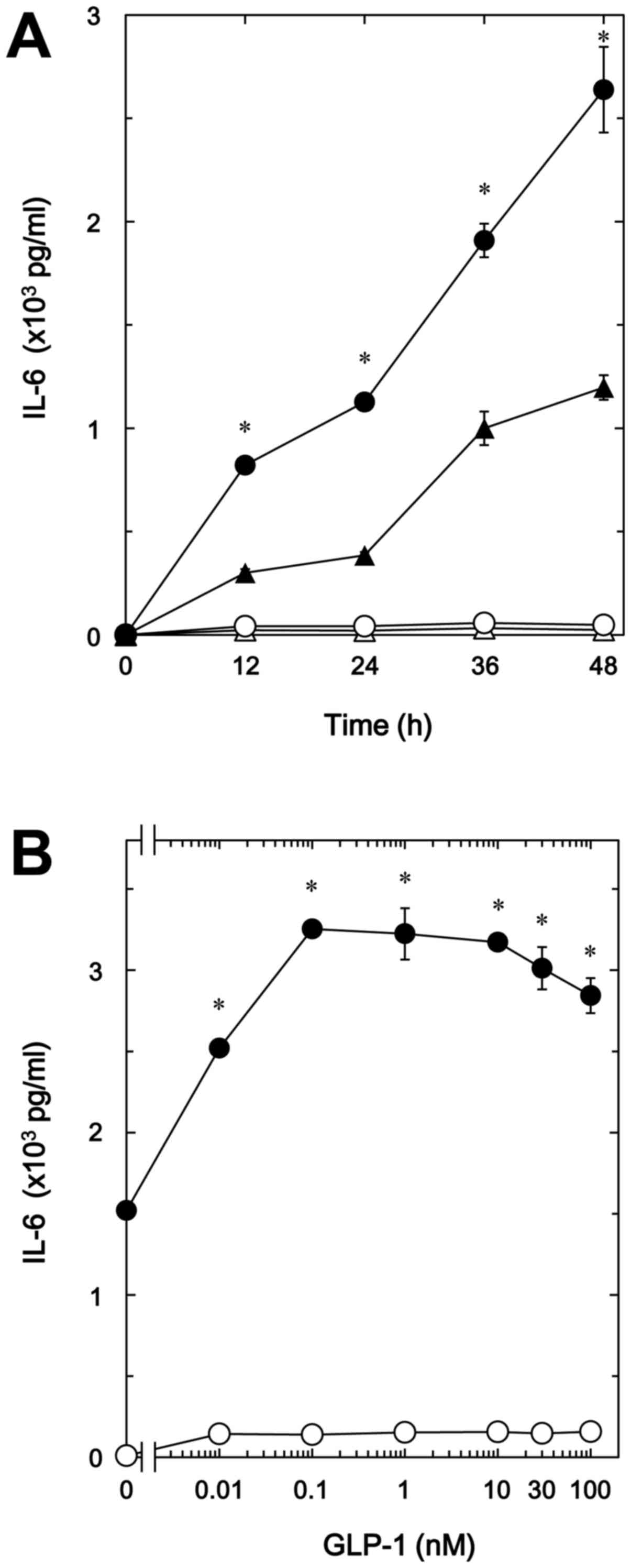
In our previous study (8), we demonstrated that TNF-α stimulates
IL-6 synthesis via sphingomyelin breakdown in osteoblast-like
MC3T3-E1 cells. We first examined the effect of GLP-1, one of the
incretins, on the TNF-α-stimulated IL-6 release in MC3T3-E1 cells.
GLP-1 significantly amplified the TNF-α-stimulated IL-6 release in
a time-dependent manner up to 48 h (Fig. 1A). The enhancing effect of GIP-1
was dose-dependent up to 100 nM (Fig.
1B). The maximum effect of GLP-1 was observed at 0.1 nM,
causing ~100% upregulation of the TNF-α effect. In addition, GIP,
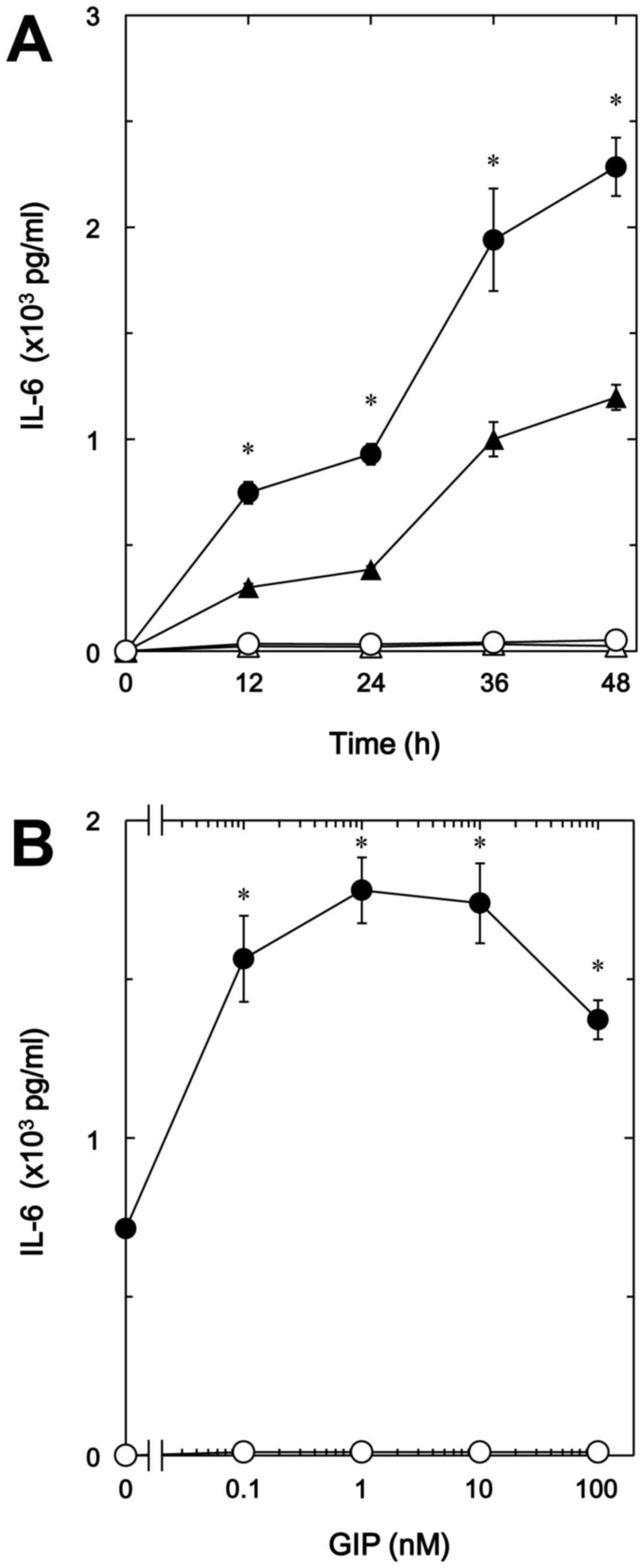
another incretin, as well as GLP-1 time-dependently enhanced the
TNF-α-stimulated IL-6 release in a dose-dependent manner up to 100
nM (Fig. 2A and B). GIP (1 nM)
caused a 150% increase in the TNF-α effect (Fig. 2B).
Effects of GLP-1 or GIP on the
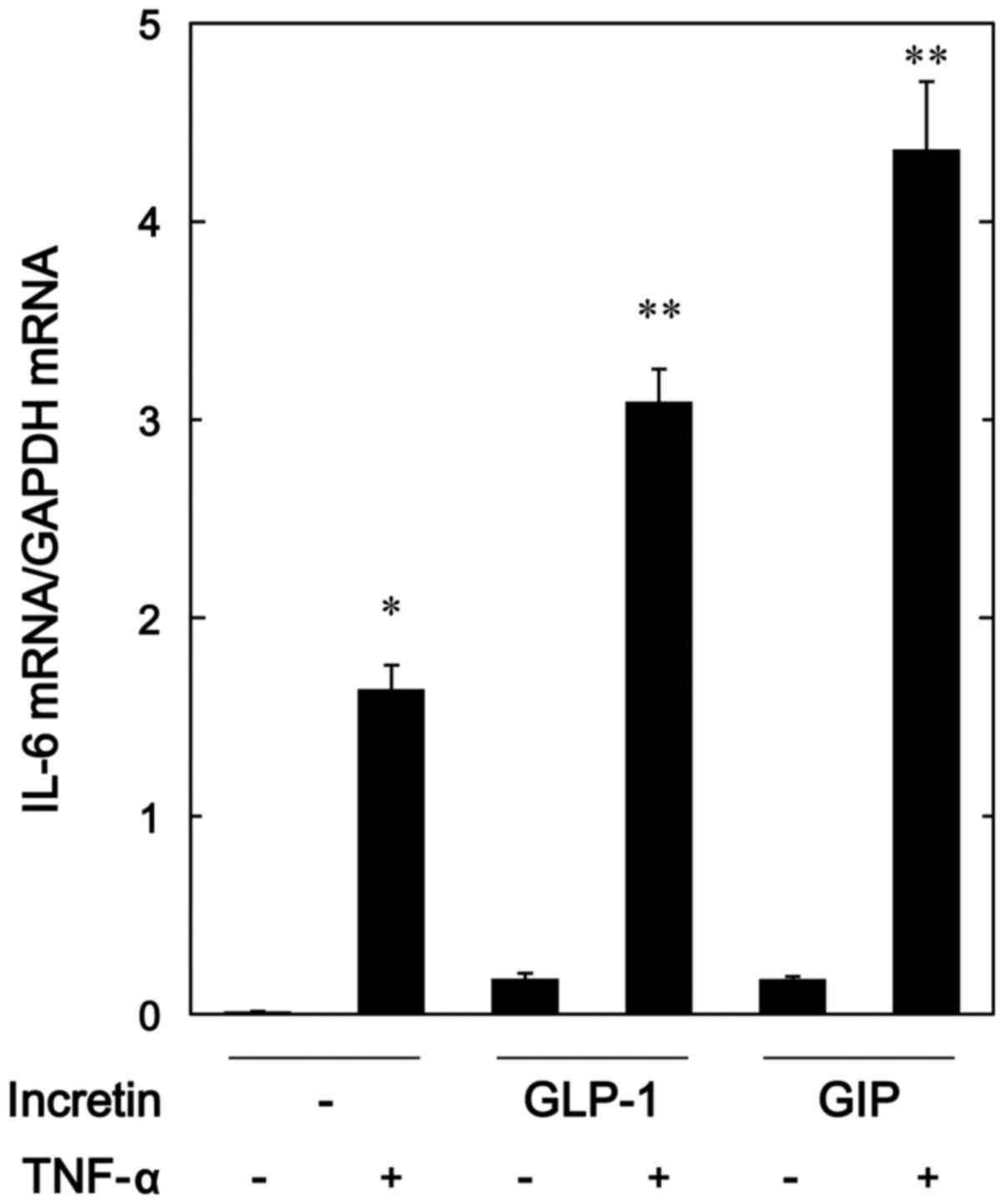
TNF-α-induced expression of IL-6 mRNA in MC3T3-E1 cells
In order to investigate whether the amplifying
effects of GLP-1 and GIP on the TNF-α-stimulated IL-6 release were
exerted through transcriptional events, we next examined the
effects of GLP-1 or GIP on the TNF-α-induced IL-6 mRNA expression
by real-time RT-PCR. Both GLP-1 and GIP, which alone had little
effect on the mRNA levels of IL-6, significantly upregulated the
expression levels of IL-6 mRNA induced by TNF-α (Fig. 3).
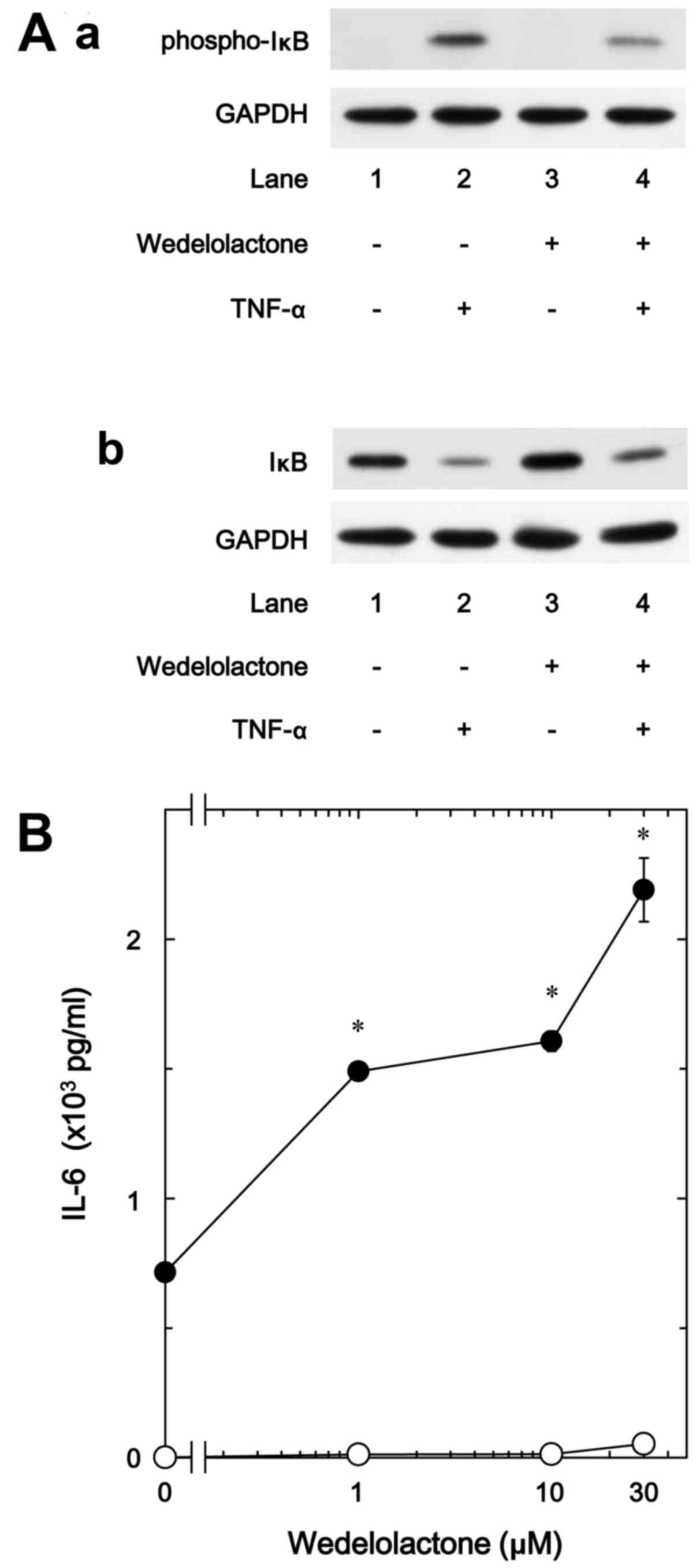
Effect of wedelolactone on the IL-6
release stimulated by TNF-α in MC3T3-E1 cells
It is generally established that cellular responses
to TNF-α are mediated through the activation of the IκB/NF-κB
pathway (7). In response to
TNF-α, IκB is phosphorylated and degraded by IκB kinase,
subsequently NF-κB is dissociated with IκB and translocated into
the nucleus, resulting in the induction of transcriptional events
(26). Therefore, in order to
clarify whether the IκB/NF-κB pathway is implicated in the
TNF-α-stimulated IL-6 synthesis in osteoblast-like MC3T3-E1 cells,
we examined the effect of wedelolactone, a selective inhibitor of
IκB kinase (27), on the IL-6
release. We found that wedelolactone truly suppressed the
TNF-α-induced phosphorylation of IκB, and restored the decrease of
total levels of IκB in the MC3T3-E1 cells (Fig. 4A). On the other hand,
wedelolactone markedly enhanced the IL-6 release stimulated by
TNF-α dose-dependently in the range between 1 and 30 µM,
suggesting that the IκB/NF-κB pathway limits the TNF-α-stimulated
IL-6 synthesis (Fig. 4B).
Wedelolactone (30 µM) caused an ~200% increase in the TNF-α
effect (Fig. 4B).
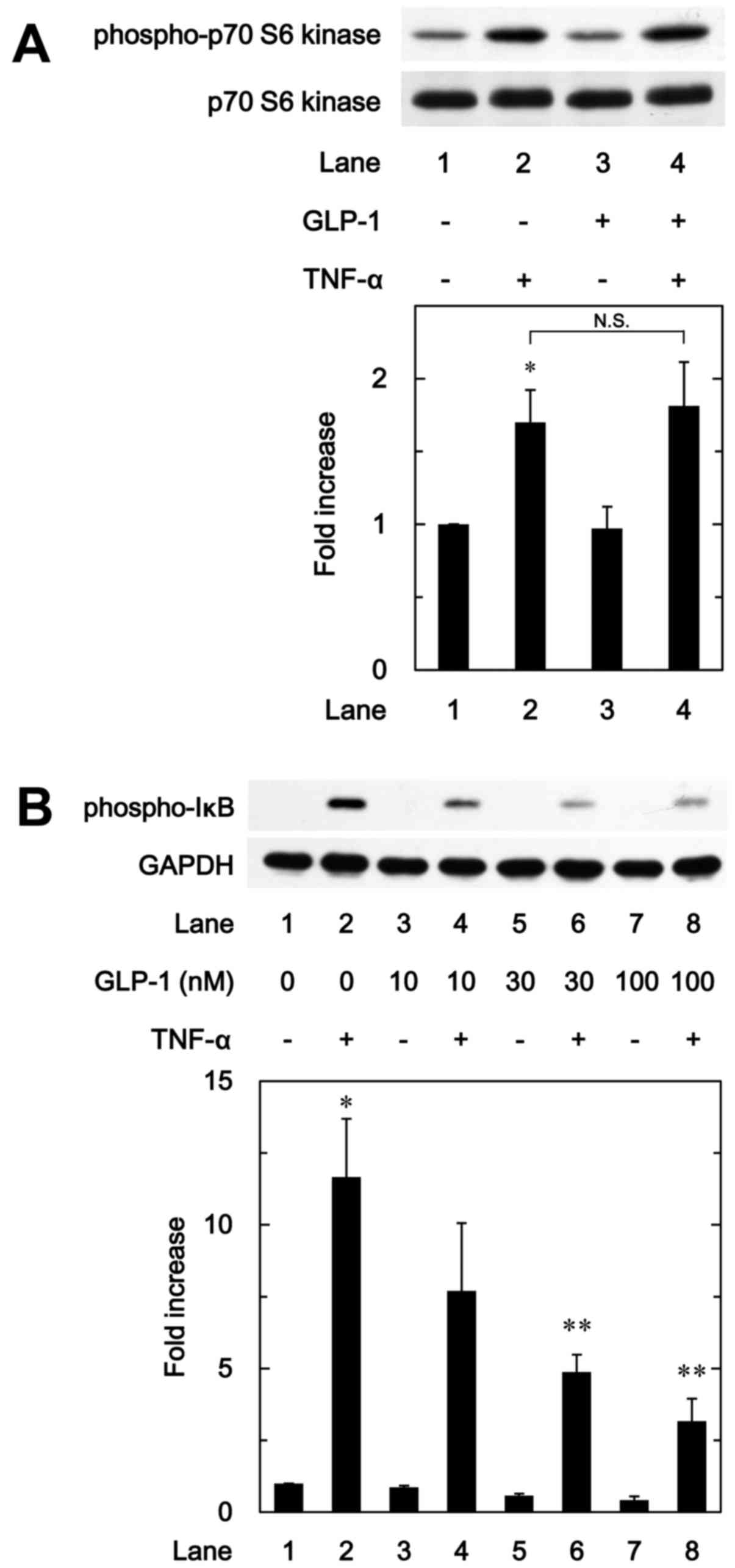
Effect of GLP-1 on the phosphorylation of
p70 S6 kinase induced by TNF-α in MC3T3-E1 cells
We have previously shown that p70 S6 kinase
activated by TNF-α negatively regulates IL-6 synthesis in
osteoblast-like MC3T3-E1 cells (9). Therefore, in order to investigate
whether the effects of incretins on the TNF-α-stimulated IL-6
synthesis are associated with the activation of p70 S6 kinase in
these cells, we further examined the effect of GLP-1 on the
TNF-α-induced phosphorylation of p70 S6 kinase. However, GLP-1
failed to affect the TNF-α-induced phosphorylation of p70 S6 kinase
(Fig. 5A).
Effect of GLP-1 on the phosphorylation of
IκB induced by TNF-α in MC3T3-E1 cells
To furthermore clarify whether the effect of GLP-1
on the TNF-α-stimulated IL-6 synthesis is associated with the
activation of the IκB/NF-κB pathway in osteoblast-like MC3T3-E1
cells, we examined the effect of GLP-1 on the TNF-α-induced
phosphorylation of IκB. GLP-1 significantly attenuated the
TNF-α-induced phosphorylation of IκB in a dose-dependent manner
between 10 and 100 nM (Fig.
5B).
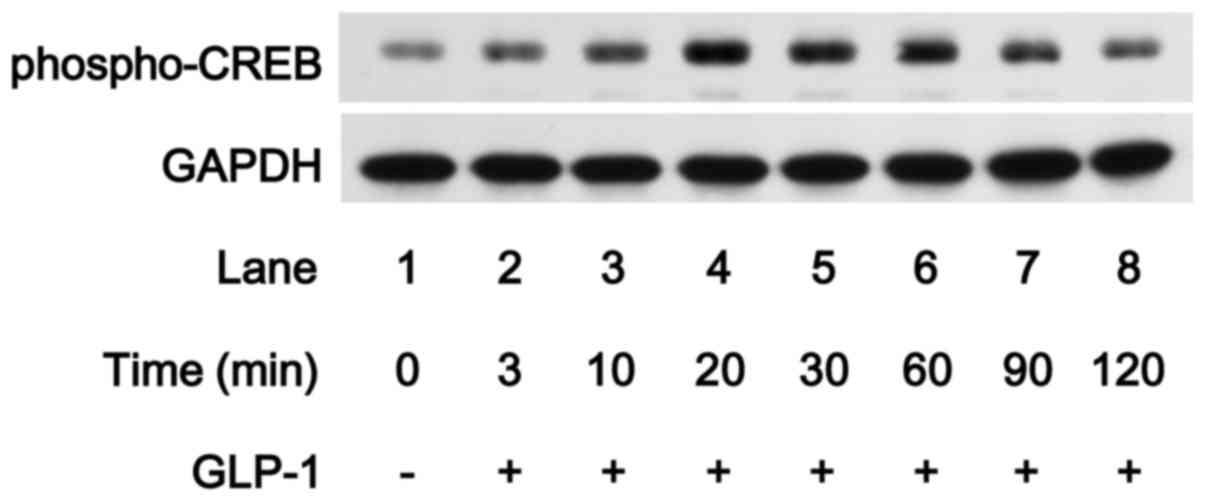
Effect of GLP-1 on the phosphorylation of
CREB in MC3T3-E1 cells
Accumulating evidence suggests that GIP and GLP-1
receptor signaling involves activation of adenylyl cyclase and
increases the production of cAMP from ATP, which leads to
cAMP-dependent activation of PKA (14). In addition, sustained elevation of
cAMP concentrations induces nuclear translocation of the catalytic
subunit of the cAMP-dependent PK, leading to CREB activation
(28). In order to clarify
whether CREB is activated by GLP-1 in osteoblast-like MC3T3-E1
cells, we examined the effect of GLP-1 on the phosphorylation of
CREB. GLP-1 markedly induced CREB phosphorylation in the MC3T3-E1
cells, and the effect of GLP-1 reached their peak 20 and 30 min
subsequent to the stimulation, respectively, and was reduced
thereafter (Fig. 6), suggesting
that GLP-1 induces the activation of adenylyl cyclase/cAMP/PKA
pathway.
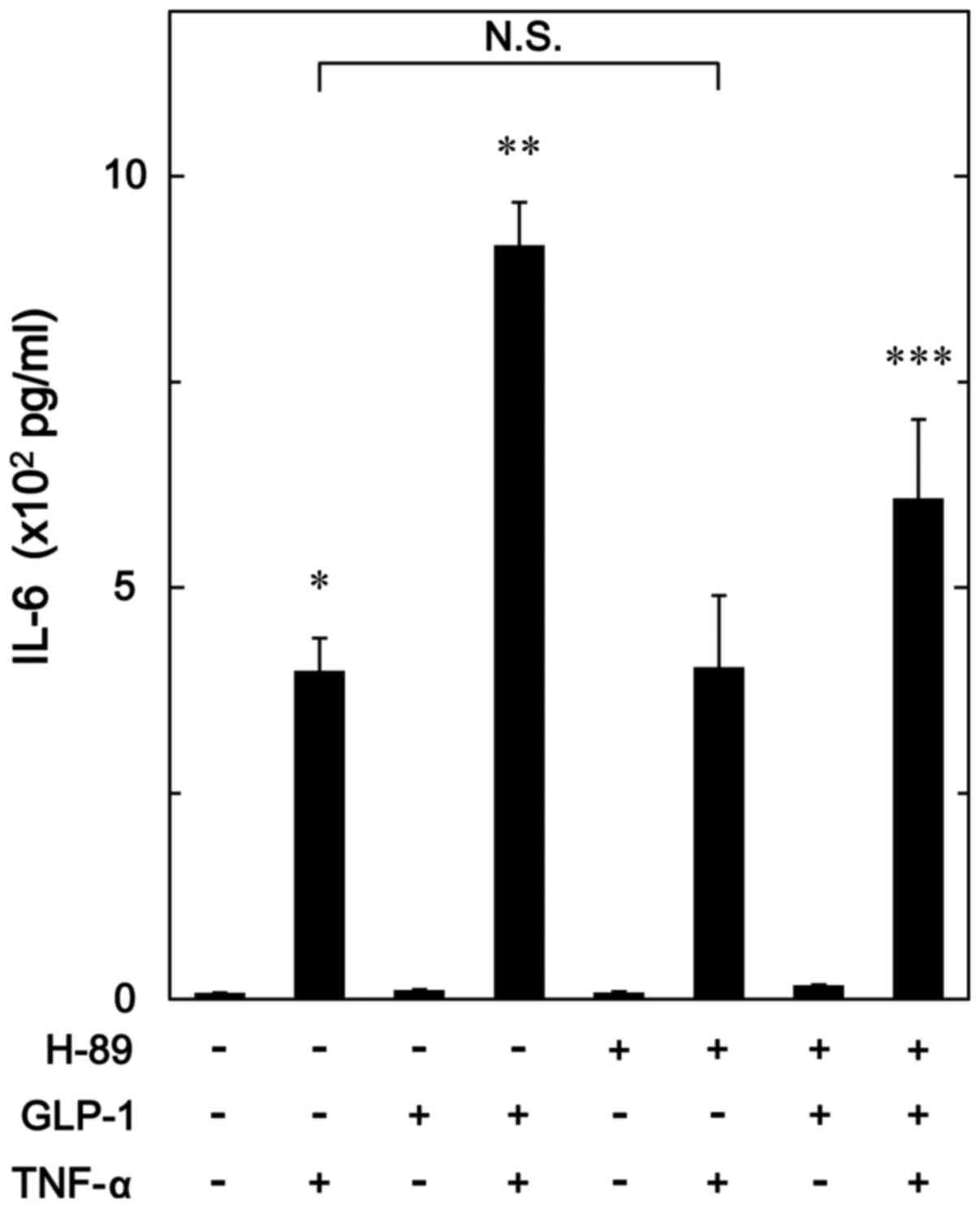
Effect of H-89 on the enhancement by
GLP-1 of TNF-α-stimulated IL-6 release in MC3T3-E1 cells
In order to investigate whether PKA activated by
GLP-1 amplifies the TNF-α-stimulated IL-6 synthesis in
osteoblast-like MC3T3-E1 cells, we examined the effect of H-89, a
potent inhibitor of PKA (29), on
the enhancement by GLP-1 of the TNF-α-stimulated IL-6 release.
H-89, which hardly affected the base line levels of IL-6 release by
itself or the levels by TNF-α alone, significantly suppressed the
amplification by GLP-1 of the IL-6 release stimulated by TNF-α
(Fig. 7).
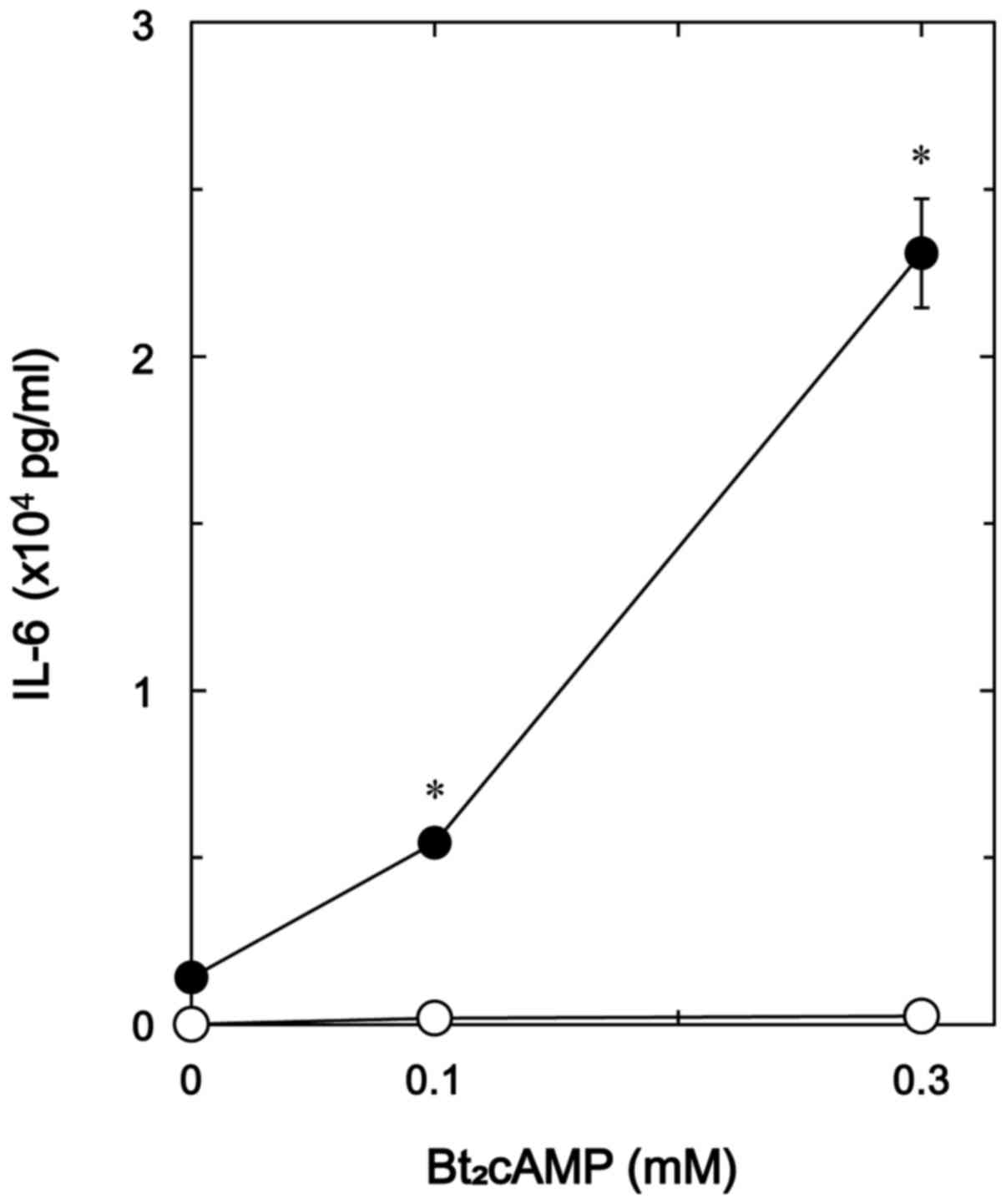
Effects of Bt2cAMP on the
TNF-α-stimulated IL-6 release and the TNF-α-induced phosphorylation
of IκB in MC3T3-E1 cells
Additionally, to elucidate whether the effect of
GLP-1 on the TNF-α-stimulated IL-6 synthesis is exerted through the
adenylyl cyclase/cAMP/PKA pathway in MC3T3-E1 cells, we examined
the effect of Bt2cAMP, a permeable analogue of cAMP, on
the TNF-α-stimulated IL-6 release. Bt2cAMP markedly
amplified the TNF-α-stimulated IL-6 release dose-dependently up to
0.3 mM (Fig. 8).
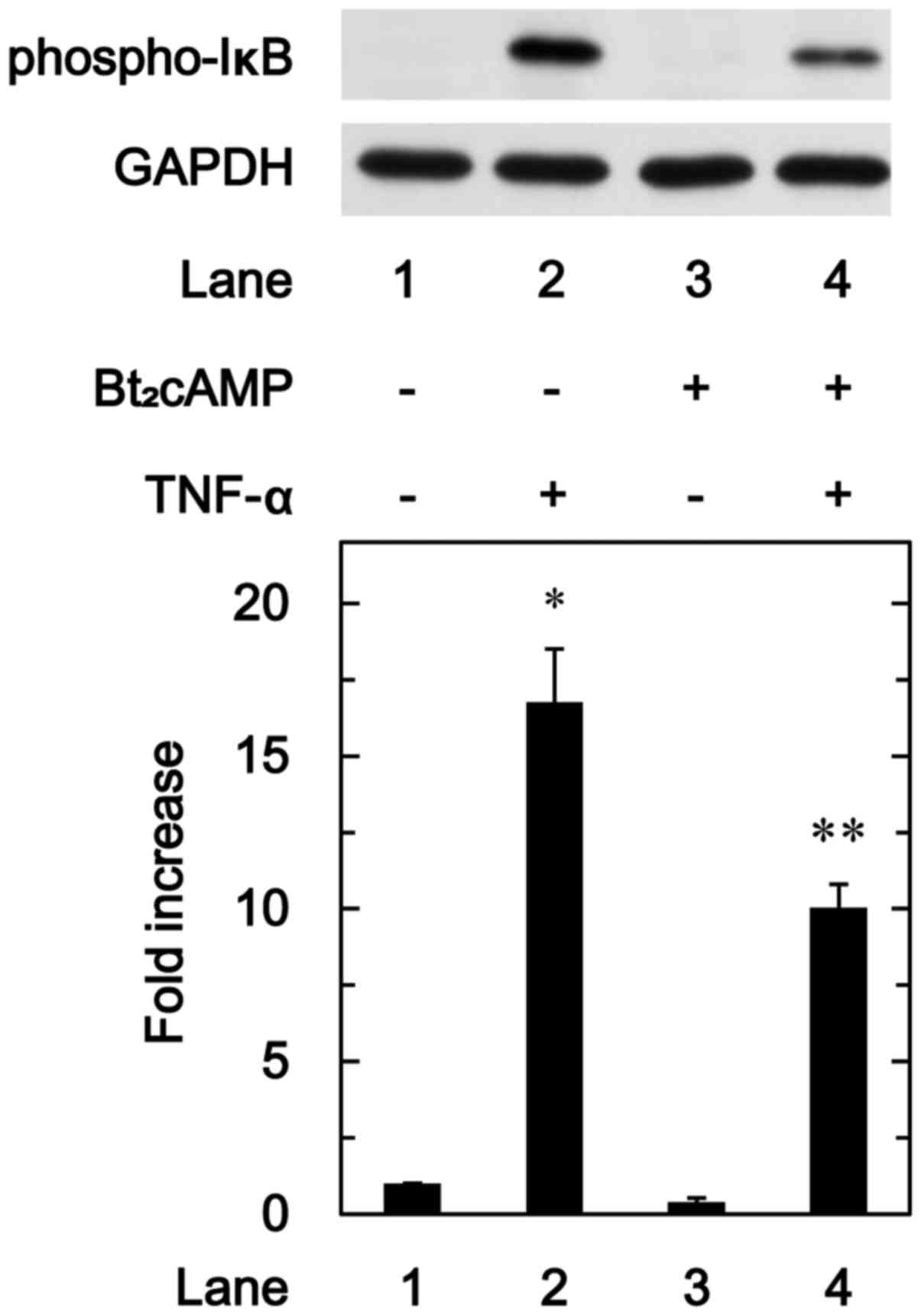
We further investigated the effect of
Bt2cAMP on the phosphorylation of IκB induced by TNF-α
in these cells. Bt2cAMP significantly attenuated the
TNF-α-induced phosphorylation of IκB (Fig. 9).
Discussion
In the present study, we demonstrated that GLP-1 and
GIP, which are well known to stimulate insulin secretion from
pancreatic β cells, significantly enhanced the TNF-α-induced
release of IL-6 in osteoblast-like MC3T3-E1 cells. Currently, GLP-1
receptor agonists are clinically used for patients with type 2
diabetes. Regarding incretins in bone metabolism, GIP receptors are
expressed in osteoblasts (30),
and GIP enhances bone formation following binding with GIP receptor
(17). On the other hand, GLP-1
reportedly suppressed osteoclastic bone resorption via calcitonin
secreted from thyroid parafollicular cells (31). In addition, we showed that the
expression levels of IL-6 mRNA induced by TNF-α were amplified by
GLP-1 and GIP. Based on our findings, it is most likely that the
amplifying effects of GLP-1 or GIP on the TNF-α-stimulated release
of IL-6 are mediated through transcriptional levels in
osteoblast-like MC3T3-E1 cells. To the best of our knowledge, this
is the first report showing that incretins, which alone had no
effect on IL-6 synthesis, upregulated the TNF-α-induced IL-6
synthesis in osteoblasts.
We next investigated the exact mechanism underlying
the TNF-α-stimulated IL-6 synthesis in osteoblast-like MC3T3-E1
cells. With regard to the intracellular signaling of TNF-α, it is
well recognized that TNF-α exerts its cellular effects through
activation of the IκB/NF-κB pathway in a variety of cell types
including osteoblasts (4). Thus,
we investigated whether the IκB/NF-κB pathway is involved in the
TNF-α-stimulated IL-6 synthesis in osteoblast-like MC3T3-E1 cells.
It is firmly established that IκB binds to NF-κB and inhibits the
translocation of NF-κB into the nucleus under unstimulated
conditions (32). The
phosphorylation of IκB induces the ubiquitination of itself to
undergo proteolysis, and subsequent release of NF-κB. The released
NF-κB translocates to the nucleus and regulates the process of
transcriptional events including IL-6 synthesis (32). In the present study, we showed
that wedelolactone, a selective inhibitor of IκB kinase (27), significantly amplified the
TNF-α-stimulated IL-6 release in MC3T3-E1 cells. We found that
wedelolactone reduced the TNF-α-induced phosphorylation of IκB,
suggesting that wedelolactone actually functioned as an inhibitor
of IκB kinase. Therefore, these findings suggest that the IκB/NF-κB
pathway regulates TNF-α-stimulated IL-6 synthesis, and that the
activated pathway plays an inhibitory role in the IL-6 synthesis in
osteoblast-like MC3T3-E1 cells. In our previous study (9), we demonstrated that p70 S6 kinase
activated by TNF-α limited the IL-6 synthesis in these cells. Based
on our findings, it is most likely that the TNF-α-stimulated IL-6
synthesis is negatively regulated by the IκB/NF-κB pathway or p70
S6 kinase in osteoblast-like MC3T3-E1 cells.
We further investigated the precise mechanism
underlying the enhancement by incretins of the TNF-α-stimulated
IL-6 synthesis in osteoblast-like MC3T3-E1 cells. However, GLP-1
barely affected the phosphorylation of p70 S6 kinase in MC3T3-E1
cells. Therefore, it seems unlikely that the enhancing effect of
GLP-1 on the TNF-α-induced IL-6 synthesis is mediated through p70
S6 kinase. On the contrary, we demonstrated that GLP-1 markedly
reduced the TNF-α-stimulated phosphorylation of IκB. Therefore, our
findings suggest that GLP-1 enhances the TNF-α-stimulated IL-6
synthesis by inhibiting the IκB/NF-κB pathway but not p70 S6 kinase
in osteoblast-like MC3T3-E1 cells.
Accumulating evidence indicates that an important
intracellular signaling of incretin is the adenylyl
cyclase/cAMP/PKA pathway (14,28), and CREB is the first
transcriptional regulator targeted by the pathway. In the present
study, we showed that GLP-1 markedly induced the phosphorylation of
CREB in osteoblast-like MC3T3-E1 cells. Therefore, it seems likely
that GLP-1 activates the cAMP/PKA pathway in osteoblast-like
MC3T3-E1 cells. We next investigated the involvement of the
cAMP/PKA pathway in the enhancement by incretin of the
TNF-α-stimulated IL-6 synthesis. H-89, an inhibitor of PKA
(29), significantly suppressed
the amplification by GLP-1 of the TNF-α-stimulated IL-6 release in
MC3T3-E1 cells. Additionally, we demonstrated that
Bt2cAMP, a cell-permeable analogue of cAMP, enhanced the
TNF-α-stimulated IL-6 release whereas Bt2cAMP
significantly suppressed the TNF-α-induced phosphorylation of IκB.
Taking our present results into account as a whole, it is most
likely that the amplifying effect of GLP-1 on the TNF-α-stimulated
IL-6 synthesis is exerted via inhibition of the IκB/NFκ-B pathway
through the adenylyl cyclase-cAMP system in osteoblast-like
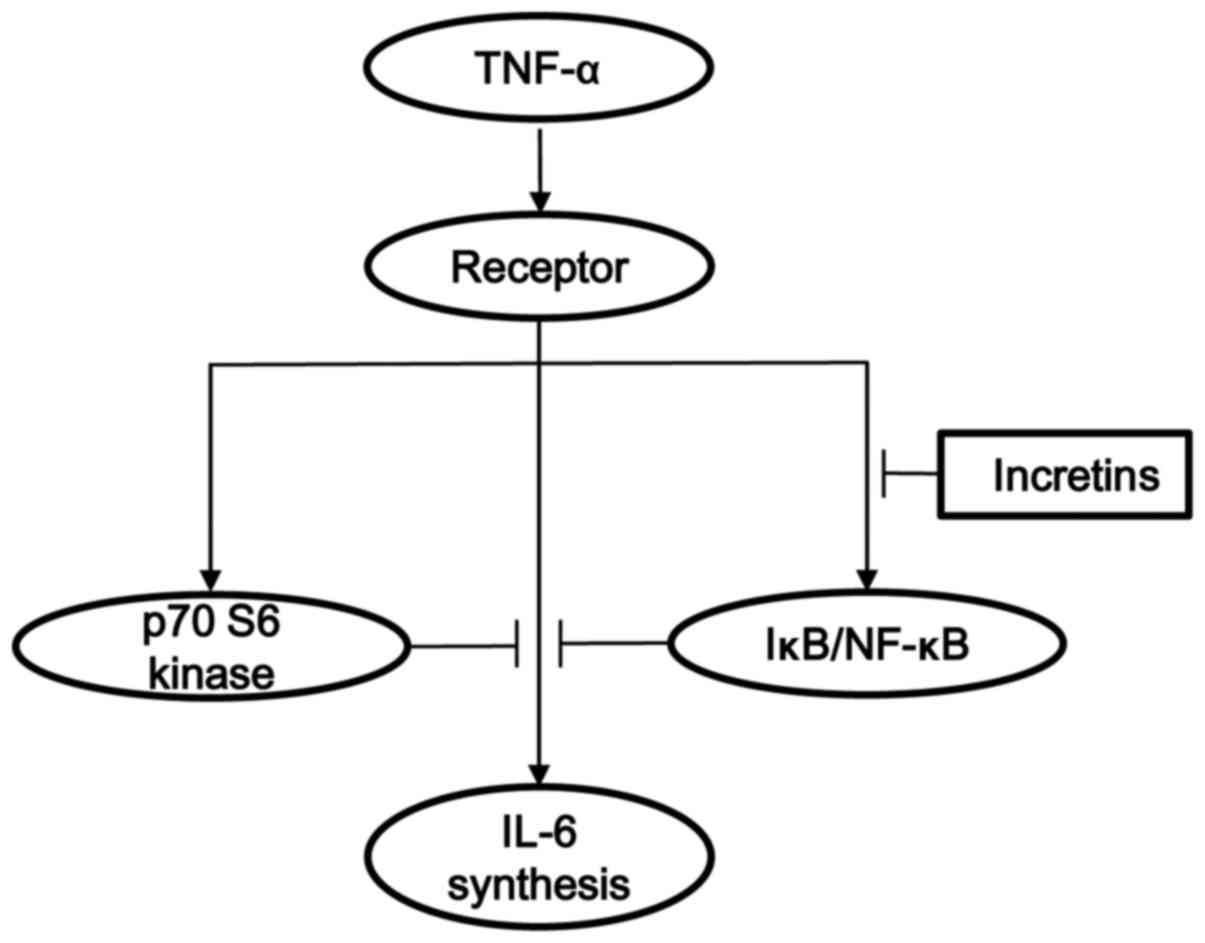
MC3T3-E1 cells. The potential mechanism of incretins in the
TNF-α-stimulated IL-6 synthesis in osteoblasts shown here is
summarized in Fig. 10.
In bone metabolism, IL-6 has been recognized to act
as a bone resorptive agent and to promote osteoclast formation
(4). However, IL-6 is currently
considered as an osteotropic factor or a bone remodeling mediator
under high turnover of bone remodeling, and subsequently stimulates
bone formation (13). In bone
remodeling, osteoclastic bone resorption is the first step, and
subsequently osteoblastic bone formation is induced (33). To maintain adequate quantity and
quality of bone, effecient bone remodeling coordinated by
osteoclasts and osteoblasts is essential. Taking our present
findings into account, it is probable that incretins secreted from
the small intestine after nutrient ingestion could affect bone
metabolism as an osteotropic modulator through the enhancement of
IL-6. On the other hand, in an early stage of bone fracture repair,
IL-6 reportedly plays a crucial role in the initiation of bone
formation (12). Additionally,
evidence is accumulating that incretins stimulate osteoblastic bone
formation and maintain bone strength (31). Therefore, our present findings may
provide a new significant aspect in the favorable effect of
incretins on fracture prevention in elderly persons.
In conclusion, our results strongly suggest that
incretins amplify the TNF-α-stimulated IL-6 synthesis in
osteoblasts, and that the enhancement by incretins is exerted via
inhibition of the IκB/NF-κB pathway through the adenylyl
cyclase-cAMP system.
Acknowledgments
We are very grateful to Mrs. Yumiko Kurokawa for her
skillful technical assistance. This study was supported in part by
a Grant-in-Aid for Scientific Research (grant no. 19591042) from
the Ministry of Education, Science, Sports and Culture of Japan,
and the Research Funding for Longevity Sciences (grant no. 25-4,
26-12) from the National Center for Geriatrics and Gerontology
(Obu, Japan).
References
|
1
|
Kular J, Tickner J, Chim SM and Xu J: An
overview of the regulation of bone remodelling at the cellular
level. Clin Biochem. 45:863–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chim SM, Tickner J, Chow ST, Kuek V, Guo
B, Zhang G, Rosen V, Erber W and Xu J: Angiogenic factors in bone
local environment. Cytokine Growth Factor Rev. 24:297–310. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boyce BF and Xing L: Functions of
RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem
Biophys. 473:139–146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwan Tat S, Padrines M, Théoleyre S,
Heymann D and Fortun Y: IL-6, RANKL, TNF-alpha/IL-1: Interrelations
in bone resorption pathophysiology. Cytokine Growth Factor Rev.
15:49–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zou W, Hakim I, Tschoep K, Endres S and
Bar-Shavit Z: Tumor necrosis factor-α mediates RANK ligand
stimulation of osteoclast differentiation by an autocrine
mechanism. J Cell Biochem. 83:70–83. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gilbert L, He X, Farmer P, Boden S,
Kozlowski M, Rubin J and Nanes MS: Inhibition of osteoblast
differentiation by tumor necrosis factor-α. Endocrinology.
141:3956–3964. 2000.PubMed/NCBI
|
|
7
|
Puimège L, Libert C and Van Hauwermeiren
F: Regulation and dysregulation of tumor necrosis factor
receptor-1. Cytokine Growth Factor Rev. 25:285–300. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kozawa O, Suzuki A, Kaida T, Tokuda H and
Uematsu T: Tumor necrosis factor-alpha autoregulates interleukin-6
synthesis via activation of protein kinase C. Function of
sphingosine 1-phosphate and phosphatidylcholine-specific
phospholipase C. J Biol Chem. 272:25099–25104. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Minamitani C, Tokuda H, Adachi S,
Matsushima-Nishiwaki R, Yamauchi J, Kato K, Natsume H, Mizutani J,
Kozawa O and Otsuka T: p70 S6 kinase limits tumor necrosis
factor-α-induced interleukin-6 synthesis in osteoblast-like cells.
Mol Cell Endocrinol. 315:195–200. 2010. View Article : Google Scholar
|
|
10
|
Hirano T, Yasukawa K, Harada H, Taga T,
Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K,
Iwamatsu A, et al: Complementary DNA for a novel human interleukin
(BSF-2) that induces B lymphocytes to produce immunoglobulin.
Nature. 324:73–76. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnson RW, Brennan HJ, Vrahnas C, Poulton
IJ, McGregor NE, Standal T, Walker EC, Koh TT, Nguyen H, Walsh NC,
et al: The primary function of gp130 signaling in osteoblasts is to
maintain bone formation and strength, rather than promote
osteoclast formation. J Bone Miner Res. 29:1492–1505. 2014.
View Article : Google Scholar
|
|
12
|
Fazzalari NL: Bone fracture and bone
fracture repair. Osteoporos Int. 22:2003–2006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Franchimont N, Wertz S and Malaise M:
Interleukin-6: An osteotropic factor influencing bone formation?
Bone. 37:601–606. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Holst JJ: The physiology of glucagon-like
peptide 1. Physiol Rev. 87:1409–1439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meier C, Schwartz AV, Egger A and
Lecka-Czernik B: Effects of diabetes drugs on the skeleton. Bone.
82:93–100. 2016. View Article : Google Scholar
|
|
16
|
Baggio LL and Drucker DJ: Biology of
incretins: GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bollag RJ, Zhong Q, Phillips P, Min L,
Zhong L, Cameron R, Mulloy AL, Rasmussen H, Qin F, Ding KH, et al:
Osteoblast-derived cells express functional glucose-dependent
insulinotropic peptide receptors. Endocrinology. 141:1228–1235.
2000.PubMed/NCBI
|
|
18
|
Bollag RJ, Zhong Q, Ding KH, Phillips P,
Zhong L, Qin F, Cranford J, Mulloy AL, Cameron R and Isales CM:
Glucose- dependent insulinotropic peptide is an integrative hormone
with osteotropic effects. Mol Cell Endocrinol. 177:35–41. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamada C, Yamada Y, Tsukiyama K, Yamada K,
Udagawa N, Takahashi N, Tanaka K, Drucker DJ, Seino Y and Inagaki
N: The murine glucagon-like peptide-1 receptor is essential for
control of bone resorption. Endocrinology. 149:574–579. 2008.
View Article : Google Scholar
|
|
20
|
Sanz C, Vázquez P, Blázquez C, Barrio PA,
Alvarez MM and Blázquez E: Signaling and biological effects of
glucagon-like peptide 1 on the differentiation of mesenchymal stem
cells from human bone marrow. Am J Physiol Endocrinol Metab.
298:E634–E643. 2010. View Article : Google Scholar
|
|
21
|
Sudo H, Kodama HA, Amagai Y, Yamamoto S
and Kasai S: In vitro differentiation and calcification in a new
clonal osteogenic cell line derived from newborn mouse calvaria. J
Cell Biol. 96:191–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kozawa O, Tokuda H, Miwa M, Kotoyori J and
Oiso Y: Cross-talk regulation between cyclic AMP production and
phosphoinositide hydrolysis induced by prostaglandin E2
in osteoblast-like cells. Exp Cell Res. 198:130–134. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simpson DA, Feeney S, Boyle C and Stitt
AW: Retinal VEGF mRNA measured by SYBR green I fluorescence: A
versatile approach to quantitative PCR. Mol Vis. 6:178–183.
2000.PubMed/NCBI
|
|
24
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kato K, Ito H, Hasegawa K, Inaguma Y,
Kozawa O and Asano T: Modulation of the stress-induced synthesis of
hsp27 and α B-crystallin by cyclic AMP in C6 rat glioma cells. J
Neurochem. 66:946–950. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hehlgans T and Pfeffer K: The intriguing
biology of the tumour necrosis factor/tumour necrosis factor
receptor superfamily: Players, rules and the games. Immunology.
115:1–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kobori M, Yang Z, Gong D, Heissmeyer V,
Zhu H, Jung YK, Gakidis MA, Rao A, Sekine T, Ikegami F, et al:
Wedelolactone suppresses LPS-induced caspase-11 expression by
directly inhibiting the IKK complex. Cell Death Differ. 11:123–130.
2004. View Article : Google Scholar
|
|
28
|
Yu Z and Jin T: New insights into the role
of cAMP in the production and function of the incretin hormone
glucagon-like peptide-1 (GLP-1). Cell Signal. 22:1–8. 2010.
View Article : Google Scholar
|
|
29
|
Chijiwa T, Mishima A, Hagiwara M, Sano M,
Hayashi K, Inoue T, Naito K, Toshioka T and Hidaka H: Inhibition of
forskolin-induced neurite outgrowth and protein phosphorylation by
a newly synthesized selective inhibitor of cyclic AMP-dependent
protein kinase, N-[2-(p-bromocinnamylamino)
ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma
cells. J Biol Chem. 265:5267–5272. 1990.PubMed/NCBI
|
|
30
|
Nuche-Berenguer B, Portal-Núñez S, Moreno
P, González N, Acitores A, López-Herradón A, Esbrit P, Valverde I
and Villanueva-Peñacarrillo ML: Presence of a functional receptor
for GLP-1 in osteoblastic cells, independent of the cAMP-linked
GLP-1 receptor. J Cell Physiol. 225:585–592. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seino Y and Yabe D: Glucose-dependent
insulinotropic polypeptide and glucagon-like peptide-1: Incretin
actions beyond the pancreas. J Diabetes Investig. 4:108–130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hoffmann A and Baltimore D: Circuitry of
nuclear factor-κB signaling. Immunol Rev. 210:171–186. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karsenty G and Wagner EF: Reaching a
genetic and molecular understanding of skeletal development. Dev
Cell. 2:389–406. 2002. View Article : Google Scholar : PubMed/NCBI
|