Introduction
Vascular smooth muscle cells (SMCs) retain
remarkable plasticity to alternate from a differentiated to a
dedifferentiated phenotype at different developmental stages or
local environmental cues (1). The
cellular switching process of SMCs from a quiescent contractile
differentiated phenotype which is associated with the high
expression of smooth muscle-specific marker genes, such as α-smooth
muscle actin (SMA), smooth muscle 22α (SM22α) and
calponin, to a synthetic dedifferentiated phenotype which is
associated with decreased levels of the marker genes plays a
critical role in many proliferative vascular diseases (1–3).
This phenotypic switch is believed to be essential for vascular
repair (3). However, for various
cardiovascular diseases, the inhibition of abnormal switching and
the control of SMC proliferation are critical therapeutic
strategies.
MicroRNAs (miRNAs or miRs) are non-coding RNAs
measuring ~22 nucleotides in length. They act as
post-transcriptional negative repressors of protein expression by
binding to the 3′-untranslated region (3′-UTR) of their target
messenger RNAs (mRNAs) (4–6).
miRNAs are known to play a critical role in cancer and
cardiovascular disorders (7).
Several miRNAs have emerged as important modulators of vascular SMC
function and phenotype (8–10).
The expression of miR-182 has been previously shown to be altered
in various types of cancer, and it inhibits the proliferation and
migration of rat Schwann cells (11–13).
Fibroblast growth factor (FGF) 9, a member of the
FGF family, is a potent mitogen secreted from bone marrow cells
(14,15). FGF9 has been reported to be one of
the direct targets of miR-182, which is associated with a variety
of vessel biological processes (13,16). Frontini et al revealed that
FGF9 stimulates smooth muscle cells (SMCs) wrapping microvessels in
implants required sonic hedgehog (SHH) and platelet-derived growth
factor (PDGF)Rβ (16). PDGFs and
their receptors (PDGFRs) are implicated in blood vessel
pathophysiology (17). PDGFRβ,
expressed in perivascular mesenchyme, particularly in vascular
mural cell (vascular SMCs and pericytes), has been established to
play important roles in SMC differentiation and dedifferentiation
(17,18).
In this study, we found that miR-182 expression in
cultured rat vascular SMCs was decreased with prolonged incubation
(number of days) or with platelet-derived growth factor (PDGF)-BB
treatment. Despite great advances in vascular SMC biology, the
molecular mechanisms responsible for SMC phenotypic modulation
remained unclear. Thus, the purpose of the study was to investigate
the role of miR-182 in the vascular SMC phenotypic switch and to
determine the underlying molecular mechanisms.
Materials and methods
Rat vascular smooth muscle cell
isolation
The animal protocols used were approved by the
Scientific Affairs Committee of Animal Research and Ethics of the
2nd Hospital of Harbin Medical University. Vascular SMCs were
isolated using the enzymatic dissociation method as previously
described (19). Abdominal and
thoracic aortic segments were isolated from Sprague-Dawley (SD)
rats (weighting about 180 g) under general anesthesia. All rats in
this study were obtained from the Model Animal Center of Harbin
Medical University. After removing the connective tissue,
adventitia and the endothelial layer, aortas were sliced into
1–2-mm-thick fragments and incubated with Dulbecco's modified
Eagle's medium (DMEM)/F12 with the addition of type 2 collagenase
(1.4 mg/ml) (Sigma, St. Louis, MO, USA) for 4–6 h in a 37°C
incubator with 95% air and 5% CO2. The fragments were
agitated to release SMCs and the cells were centrifuged at 300 × g
for 5 min. SMCs were then cultured in DMEM/F12 medium added with
10% fetal bovine serum in an incubator as described above at least
for 5 days prior to the first time trypsin digestion. The primary
cultured SMCs were then passed every 3 days, and the 4–6 passages
were used. In addition, SMCs released from the arterial fragments
(0 days), cultured SMCs at 5 day, 10 and 15 days were also
collected for examination.
Transfection of cultured SMCs with
oligonucleotides
Oligonucleotide (oligo; Bioneer Co., Ltd., Daejeon,
Korea) transfection was based on the instruction of Roche
X-tremeGENE siRNA Transfection Reagent (Roche, Mannheim, Germany).
For miR-182 overexpression or silencing, miR-182 mimics or
inhibitors were added directly to the complexes to different final
concentrations (miR-182 mimic: sense, GCG GGU CUA GCU GCC GGA and
antisense, CGG CAG CUA GAC CCG CUU; miR-182 inhibitor: UUU GGC AAU
GGU AGA ACU CAC ACC G). A small interfering RNA (siRNA) was used to
degrade the target mRNAs for fibroblast growth factor 9 (FGF9) gene
silencing as previously described (sense, CUU CCA ACC UGU ACA AGA
and antisense, UGC UUG UAC AGG UUG GAA G) (20). Briefly, following trypsin
digestion, SMCs were inoculated in a 6-well plate. Twenty-four
hours later, the X-tremeGENE siRNA transfection reagent was used to
treat the cells in a 1:4 ratio for 20 min. Further treatment
included transfection with siRNA followed by incubation in medium
comprising 2 ml of serum-free Opti-MEMI (Invitrogen, Carlsbad, CA,
USA) using transfection reagents (vehicle), negative control,
inhibitor negative control (Bioneer Co., Ltd.), and siRNA control
(Invitrogen). The transfection efficiency of miR-182 mimic and
inhibitor was then examined by reverse transcription-quantitative
PCR (RT-qPCR) and the efficiency of FGF9 siRNA was examined by
western blot analysis, as described below. Forty-eight hours later,
the engineered SMCs were analyzed further.
miRNA and mRNA analysis by RT-qPCR
Following oligo transfection and stimulation with or
without PDGF-BB, total RNA was extracted from the SMCs using TRIzol
reagent (15596-026; Invitrogen) according to the instructions of
the manufacturer. After the abdominal and thoracic aortic segments
were isolated from the anesthetized rats, organs including aorta,
muscle, heart, lung, liver and kidney were also removed. Total RNA
was then extracted from these organs based on the instructions of
the manufaturer of TRIzol reagent. RNA was then reverse transcribed
into cDNA using the First Stand miRcute miRNA cDNA Synthesis kit
according to the instructions of the manufacturer (Tiangen Biotech,
Beijing, China). The miR-182 level was analyzed by quantitative
polymerase chain reaction (qPCR) using the Tiangen miRcute miRNA
qPCR detection system (Poly A tail addition method; miR-182 primer,
TTT GGC AAT GGT AGA ACT CAC ACC; U6 primer sequence, ACA CGC AAA
TTC GTG AAG CGT TCC). The RT-qPCR for FGF9, platelet- derived
growth factor receptor β (PDGFRβ), SMA, SM22α and calponin was
performed using the Bioneer AccuPower GreenStar qPCR PreMix system
(SMA forward, GTC AGG TCA TCA CTA TCG GCA AT and reverse, AGA GGT
CTT TAC GGA TGT CAA CGT; SM22α forward, ATG GCC AAC AAG GGT CCA TCC
and reverse, TCC ATC TGC TTG AAG ACC ATG; calponin forward, AGAGAA
GGC AGG AAC ATC ATT GGC and reverse, GTG TCA CAG TGT TCC ATG CCC
AG; FGF9 forward, ACA GGA GTG CGT GTT CAG AG and reverse, GTT CAG
GTA CTT TGT CAG GGT C; PDGFRβ forward, AGG ACA ACC GTA CCT TGG GTG
ACT and reverse, CAG TTC TGA CAC GTA CCG GGT CTC). The expression
of miR-182 relative to U6 and SMA, SM22α, calponin, FGF9, and
PDGFRβ relative to β-actin was determined by the 2−ΔΔCt
method.
Western blot analysis
Cellular proteins were extracted for western blot
analysis as previously described (20). The cell lysates were subjected to
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and run under standard conditions. Subsequently, the
proteins were removed to polyvinylidene fluoride (PVDF) membranes.
After blocking with 5% nonfat milk at room temperature for 1 h, the
PVDF membranes were incubated with diluted primary antibodies SMA
(1:400 dilution; sc-53142), SM22α (1:300 dilution; sc-51442) (both
from Santa Cruz Biotechnology, Santa Cruz, CA, USA), FGF9 (1:800
dilution; ab9743), PDGFRβ (1:1,000 dilution; ab32570) (both from
Abcam, Cambridge, MA, USA), and antibody against GAPDH (1:1,000
dilution; sc-47724; Santa Cruz Biotechnology) at 4°C overnight. The
membranes were washed, further incubated for 1 h with horseradish
peroxidase-conjugated secondary antibodies (anti-mouse and
anti-rabbit, ZB-2305 and ZB-2305; both from Zhongshan Goldenbridge
Biotechnology, Beijing, China) at 37°C. Subsequently, the membranes
were detected with BeyoECL Plus (Beyotime Institute of
Biotechnology, Haimen, China), and further analysis of protein band
densitometry was facilitated by Quantity One software (Bio-Rad,
Hercules, CA, USA).
SMC proliferation assay
SMCs at the 4th to 6th passages were trypsinized and
plated into 96-well culture plates (5×103 cells/well) in
DMEM/F-12 complete medium. After 24 h, the SMCs were rendered
quiescent in DMEM/F-12 supplemented with 0.5% FCS for 24 h. The
proliferation of the SMCs was examined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Solarbio, Beijing, China) or by 5-ethynyl-2′-deoxyuridine
(EdU) proliferation assay (Ribo-Bio Co., Ltd., Guangzhou, China).
MTT assays were conducted as previously described (8,9).
In brief, the SMCs in each well were incubated with 20 µl
MTT mixture (5 mg/µl). Four hours later, formazan crystals
were accumulated in live cells. Subsequently, 100 µl
dimethyl sulphoxide (DMSO) were added to each well to solubilize
the formazan crystals with 10 min shaking. The results of MTT assay
were analyzed by optical density measurement at OD490 nm. EdU
proliferation assays were conducted according to the manufacturer's
instructions, for quantitative synthesis of DNA via the
incorporation of a deoxyribonucleoside analog containing a
detectable tag. Briefly, with SMCs, with the addition of EdU at a
final concentration of 50 µM, were incubated for a further 2
h. The cells were then fixed with 4% paraformaldehyde (PFA) for 30
min, and permeabilized with PBS containing 0.5% Triton X-100 for 10
min. Subsequently, SMCs were stained with 1X Apollo®
reaction cocktail for 30 min, and with Hoechst 33342 for 30 min.
The number of Hoechst 33342-stained cells in 10 random viewing
fields were counted for analysis. The images were acquired using a
fluorescence microscope (Leica DMI4000B; Leica, Solms,
Germany).
SMC migration assay
Migration assays were performed by scratch wound
healing as previously described (10). SMCs were pre-transfected with
oligos, and plated into 6-well cultured plates (2.5×105
cells/well). SMCs, after being quiescent for 24 h, were scratched
using a 200 µl disposable pipette tip to make a single
scratch wound by streaking across a monolayer of cells. The SMCs
were then treated with or without PDGF-BB (P-4056; Sigma). Images
were captured at various times post-wounding using an Olympus IX 73
microscope equipped with a diagnostic instrument RT SPOT Digital
Camera (Olympus, Tokyo, Japan). The migrated cells of each
high-power field (×400) were counted to represent SMC
migration.
Statistical analysis
The data were analyzed using IBM SPSS software
(version 17.0; SPSS, Inc., Chicago, IL, USA). The data, averaged of
at least three repeated experiments, are expressed as mean ±
standard deviation (SD). The relative gene expression was
determined based on the mean value of the vehicle control group
defined as 100%. Data analysis was accomplished using two- tailed
unpaired Student's t-tests and ANOVA. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
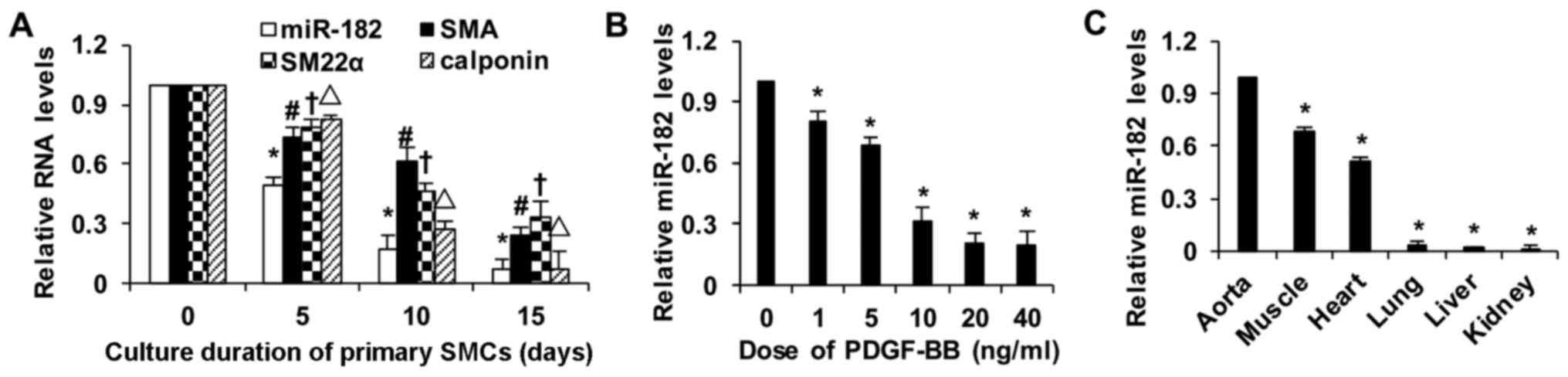
miR-182 is a novel marker for rat
vascular SMC phenotypic modulation
The levels of the differentiation marker genes,
including SMA, SM22α and calponin, declined eventually with the
prolonged incubation of rat vascular SMCs in vitro (Fig. 1A). During the phonotypic switch,
the biosynthesis of miR-182 was also gradually diminished. On the
5th day of culture, the level of miR-182 in the SMCs decreased to
almost half that of the level at 0 days. To examine the
implications of this regulation, the SMCs were treated with various
concentrations of PDGF- BB, a powerful stimulant of SMC
dedifferentiation. Treatment with PDGF-BB induced a decrease in
miR-182 expression in a dose-dependent manner; treatment with 10
ng/ml PDGF-BB markedly reduced miR-182 expression; the expression
levels of miR-182 did not differ significantly between the SMCs
stimulated with PDGF-BB 20 and 40 ng/ml (Fig. 1B). Furthermore, the miR-182
distribution levels in the aorta, heart, muscle, lung, liver and
kidney were examined by RT-qPCR. The expression level of miR-182 in
the aorta was higher than that in other tissues (Fig. 1C). Taken together, these findings
suggest that miR-182 may be a novel marker for SMC phenotypic
modulation.
miR-182 is a novel phenotypic modulator
of vascular SMCs
To explore the potential effect of miR-182 during
SMC phenotypic modulation, gain-of-function and loss-of-function
experiments were performed to transfect oligos into rat vascular
SMCs. For miR-182 overexpression, we added miR-182 mimics at a
final concentration of 20, 40, 60 or 80 nM to culture medium.
miR-182 expression was increased depending on the oligo
concentrations (Fig. 2A).
Conversely, miR-182 expression was markedly inhibited by
transfection with 80 nM miR-182 inhibitors. Additionally, the gene
levels of SMC-specific markers, including SMA, SM22α and calponin,
were distinctly increased by transfection with miR-182 mimics, and
the marker gene levels were significantly decreased by transfection
with miR-182 inhibitors (Fig.
2B). Similarly, as shown in Fig.
2C, the results of western blot analysis revealed that the
transfection of SMCs with miR-182 mimic or inhibitor altered the
protein levels of SMA and SM22α.
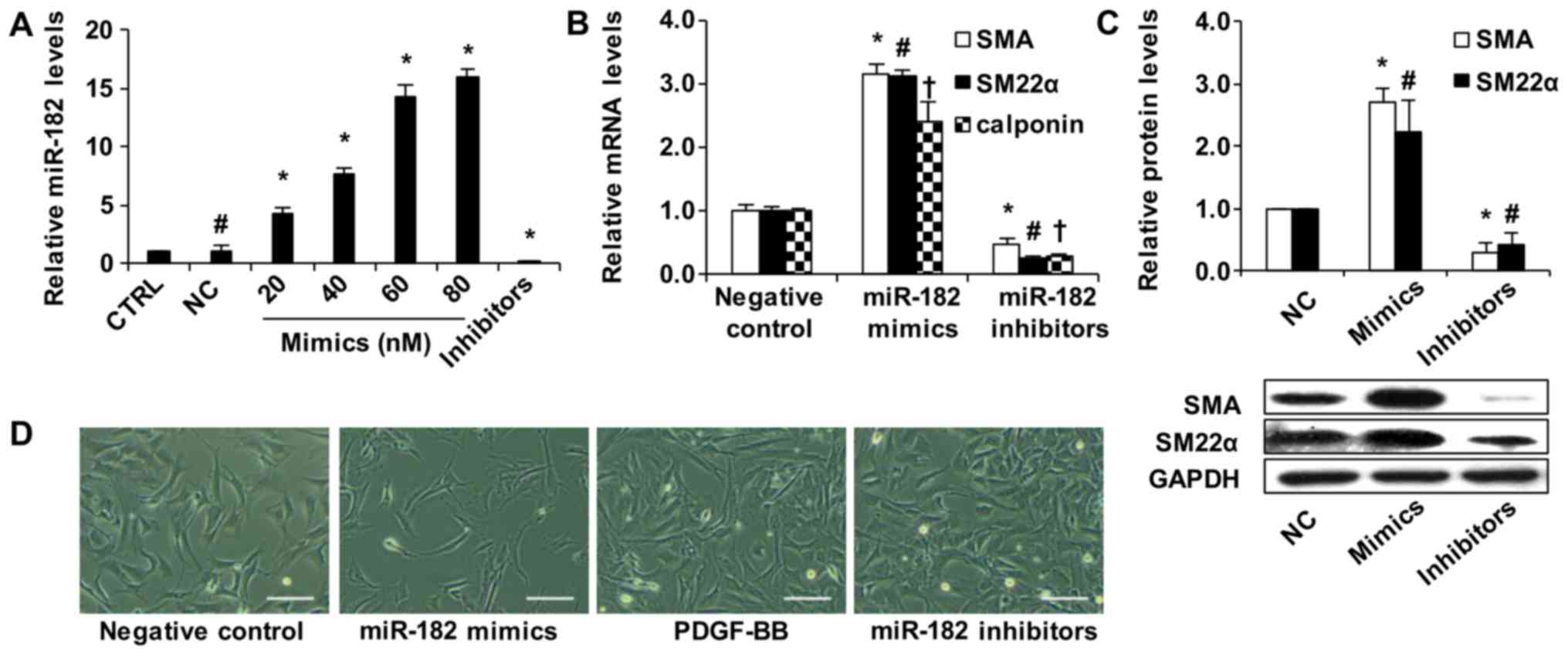 | Figure 2Role of miR-182 in the phenotypic
modulation of rat vascular smooth muscle cells (SMCs). (A)
miRNA-negative control (NC), miR-182 mimics (20, 40, 60 and 80 nM)
or inhibitors (80 nM) were transfected into rat vascular SMCs.
Intracellular miR-182 expression was significantly increased
depending on the miR-182 mimic concentrations, as determined by
RT-qPCR (n=5). #p<0.05 compared with the control
(CTRL). *p<0.05 vs. NC. (B) The effect of miR-182
mimics (80 nM) and miR-182 inhibitors (80 nM) on the expression of
SMA, smooth muscle 22α (SM22α), and calponin, as determined by
RT-qPCR (n=4). *p<0.05 vs. NC group of the SMA
expression; #p<0.05 vs. NC group of the SM22α;
†p<0.05 vs. NC group of the calponin. (C) The effect
of miR-182 mimics (80 nM) and miR-182 inhibitors (80 nM) on the
protein expression of SMA and SM22α, as determined by western blot
analysis (n=4). *p<0.05 vs. NC group of SMA;
#p<0.05 vs. NC group of SM22α. (D) Morphology of
primary cultured rat vascular SMCs. SMCs were treated with miR-182
mimics 80 nM, inhibitors 80 nM or PDGF-BB (10 ng/ml).
Magnification, ×400. Scale bars, 50 µm. |
Subsequently, the biological role of miR-182 in
primary cultured rat vascular SMC morphology was investigated. The
primary cultured SMCs at passages 4 to 6 transfected with miRNA
negative control presented a flattened morphology as a synthetic
phenotype (Fig. 2D). The
overexpression of miR-182 altered SMC morphology from a flattened
to a spindle-like contractile state. The cells flattened
progressively when transfected with miR-182 inhibitors or treated
with PDGF-BB (10 ng/ml). miR-182 overexpression helped the SMCs
retain their contractile morphology and prevented the phenotypic
switch. On the whole, these findings suggest that miR-182 is
essential in modulating the rat vascular SMC phenotype.
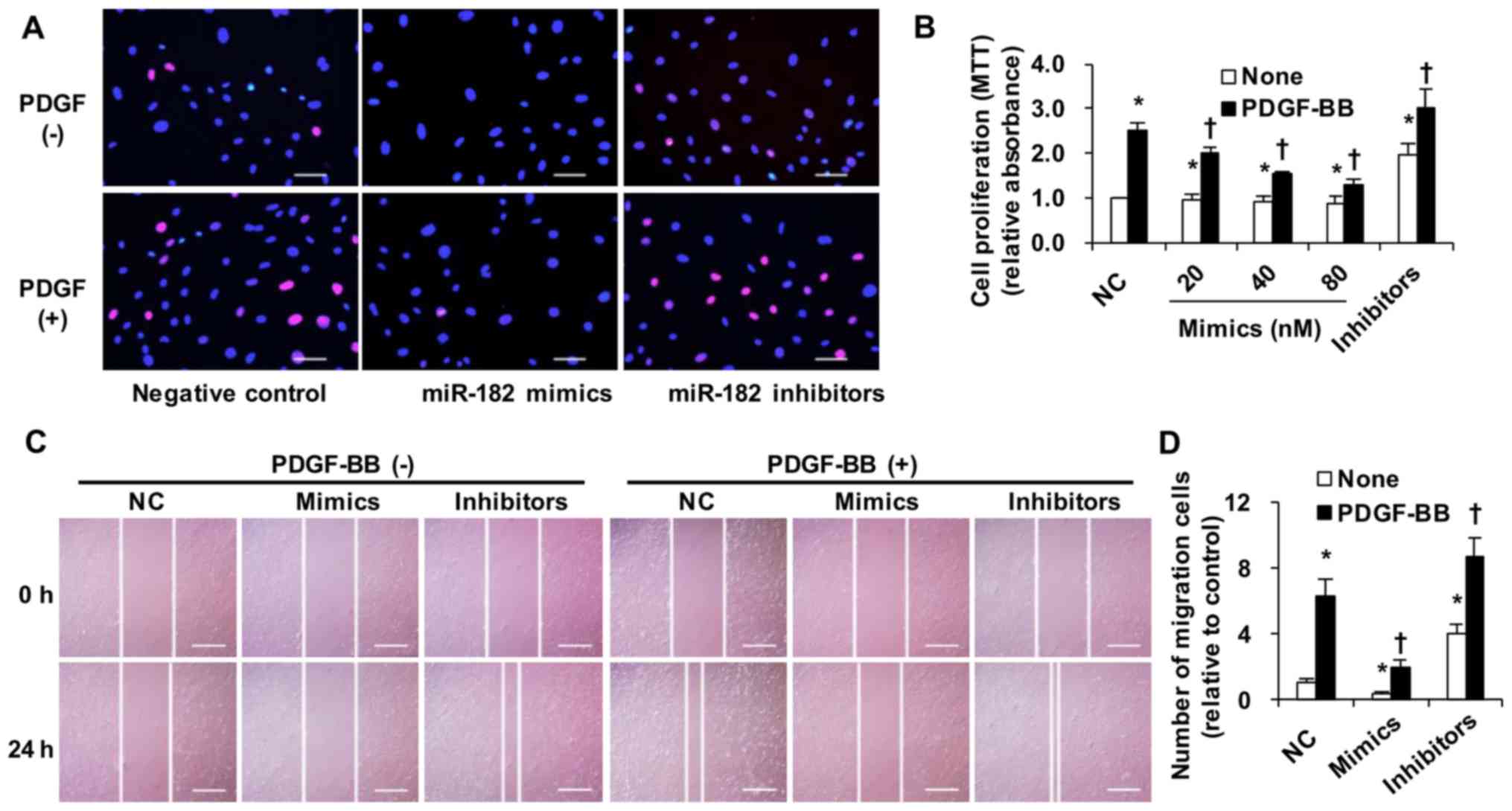
miR-182 inhibits rat vascular SMC
proliferation and migration
Typical images of EdU cell proliferation are shown
in Fig. 3A. miR-182
overexpression markedly suppressed SMC proliferation even with
PDGF-BB (5 ng/ml) treatment in vitro. In addition, PDGF-BB
treatment increased SMC proliferation, which was further enhanced
by transfection with miR-182 inhibitors. MTT assay was also
performed to determine the mitochondrial activity. Transfection
with miR-182 mimics at 80 nM significantly inhibited cell
proliferation to a greater extent than at 20 and 40 nM (Fig. 3B).
Furthermore, we examined the effect of miR-182 on
SMC migration in vitro by scratch-wound healing assay. The
migration of SMCs transfected with miR-182 mimics was markedly
inhibited even with PDGF-BB stimulation (Fig. 3C and D). However, transfection
with miR-182 inhibitors increased the PDGF-BB-induced migration of
SMCs.
To the best of our knowledge, for the first time, in
this study, we established that miR-182 effectively suppressed the
proliferation and migration of rat vascular SMCs under both
quiescent conditions and PDGF-BB stimulation.
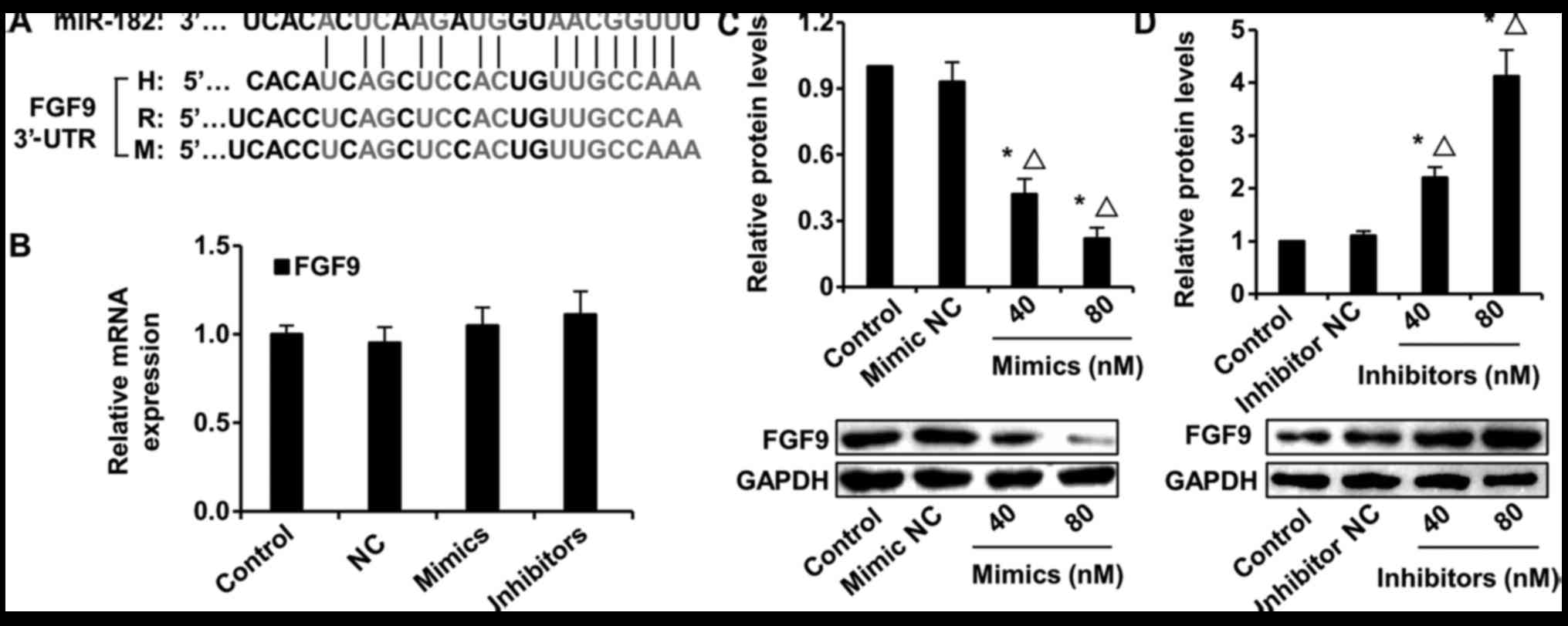
miR-182 prevents SMC phenotypic
modulation via FGF9/PDGFRβ signaling
miRNAs exert their biological functions by
inhibiting the transcription or translation of their target genes.
We used the microRNA.org database to identify the
target genes of miR-182. FGF9 is a target gene of miR-182 across
multiple species, such as human, rat and mouse (Fig. 4A). Using qPCR, we discovered that
transfection with miR-182 mimic or inhibitor slightly inhibited or
increased the FGF9 mRNA levels, respectively (Fig. 4B). However, using western blot
analysis, the protein levels of FGF9 were significantly affected by
miR-182 regulation. The FGF9 levels were decreased when the SMCs
were transfected with miR-182 mimics at 80 nM more than at 40 nM
(Fig. 4C). On the contrary, the
protein level of FGF9 was increased by transfection with miR-182
inhibitors at 80 nM more than 40 nM (Fig. 4D). These data suggest that FGF9 is
the direct target gene of miR-182 in rat vascular SMCs.
To determine the role of FGF9 in rat SMCs, we used
FGF9 siRNA transfection to achieve FGF9 downregulation. The protein
levels of the SMC-specific contractile markers, SMA and SM22α, were
significantly upregulated by FGF9 siRNA transfection (Fig. 5A). The protein levels of PDGFRβ
were decreased with the knockdown of FGF9. Additionally, as shown
in Fig. 5B and C, the knockdown
of FGF9 significantly inhibited SMC proliferation with or without
PDGF-BB stimulation, as determined by MTT and EdU assays. Moreover,
when the SMCs were transfected with FGF9 siRNA, the migration
induced by PDGF-BB was also significantly depressed (Fig. 5D and E). Therefore, the silencing
of FGF9 suppressed the differentiation, proliferation and migration
of rat SMCs.
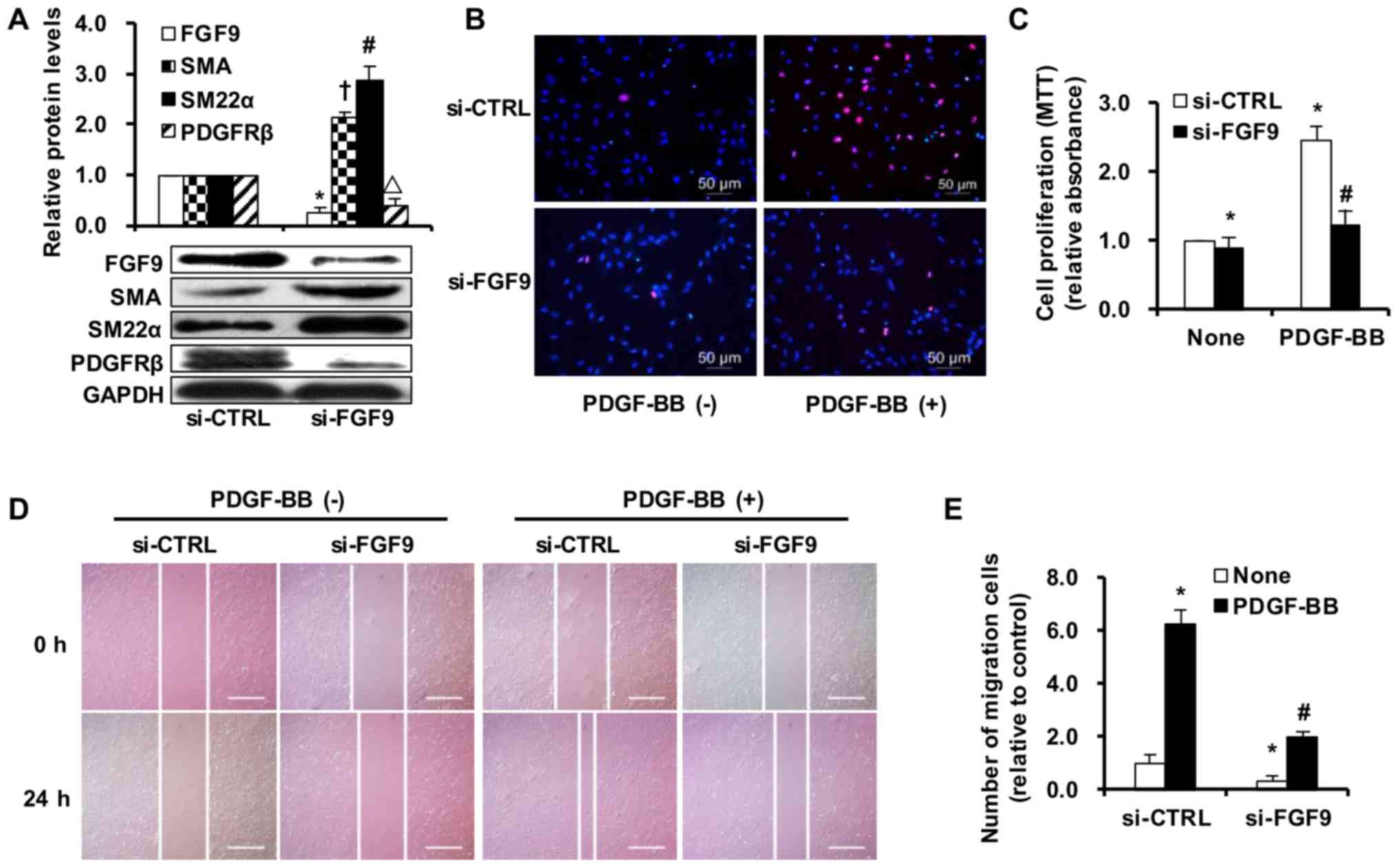 | Figure 5FGF9 plays an important role in the
differentiation, proliferation and migration of rat smooth muscle
cells (SMCs). (A) The protein levels of FGF9 in SMCs were markedly
inhibited by transfection with FGF9 siRNA (100 nM) compared with
siRNA control (si-CTRL, 100 nM) transfection (n=5;
*p<0.05). With FGF9 knockdown, the protein levels of
SMA and smooth muscle 22α (SM22α) were upregulated, but the level
of platelet-derived growth factor receptor β (PDGFRβ) was
downregulated as determined by western blot and densitometric
analyses (n=5). †p<0.05 vs. si-CTRL of SMA;
#p<0.05 vs. si-CTRL of SM22α; △p<0.05
vs. Si-CTRL of PDGFRβ; (B and C) Knockdown of FGF9 significantly
inhibited SMC proliferation with or without PDGF-BB stimulation, as
determined by EdU [(B) magnification, ×400. Scale bars, 50
µm] and MTT assays [(C) n=5. *p<0.05 vs.
si-CTRL without PDGF-BB treatment; #p<0.05 vs.
si-CRTL with PDGF-BB treatment] assays. (D) Typical images of
Scratch-wound healing assay (scale bars, 50 µm). The
migration of SMCs was significantly suppressed by si-FGF9
transfection, even with PDGF-BB stimulation. (E) Quantification of
migrated SMCs. Data of the number of migrated cells relative to
si-CTRL without PDGF-BB stimulated group are presented as the means
± SD (n=5), which was asseessed by 3 independent experiments.
*p<0.05 vs. si-CTRL without PDGF-BB treatment;
#p<0.05 vs. si-CRTL with PDGF-BB treatment. |
Subsequently, miR-182 inhibitors and FGF9 siRNA were
both transfected into the SMCs. The downregulation of miR-182
increased SMC proliferation and migration, and decreased the
expression of dedifferentiated marker genes (Fig. 6A–C). However, FGF9 knockdown
interfered with the function of miR-182 inhibitors on SMC
proliferation and migration, but induced an increase in the
expression of SMA, SM22α and calponin. Overall, our data indicate
that FGF9 is a direct target gene of miR-182 in rat vascular SMCs,
and is critical in the process of miR-182-mediated rat vascular SMC
phenotypic modulation.
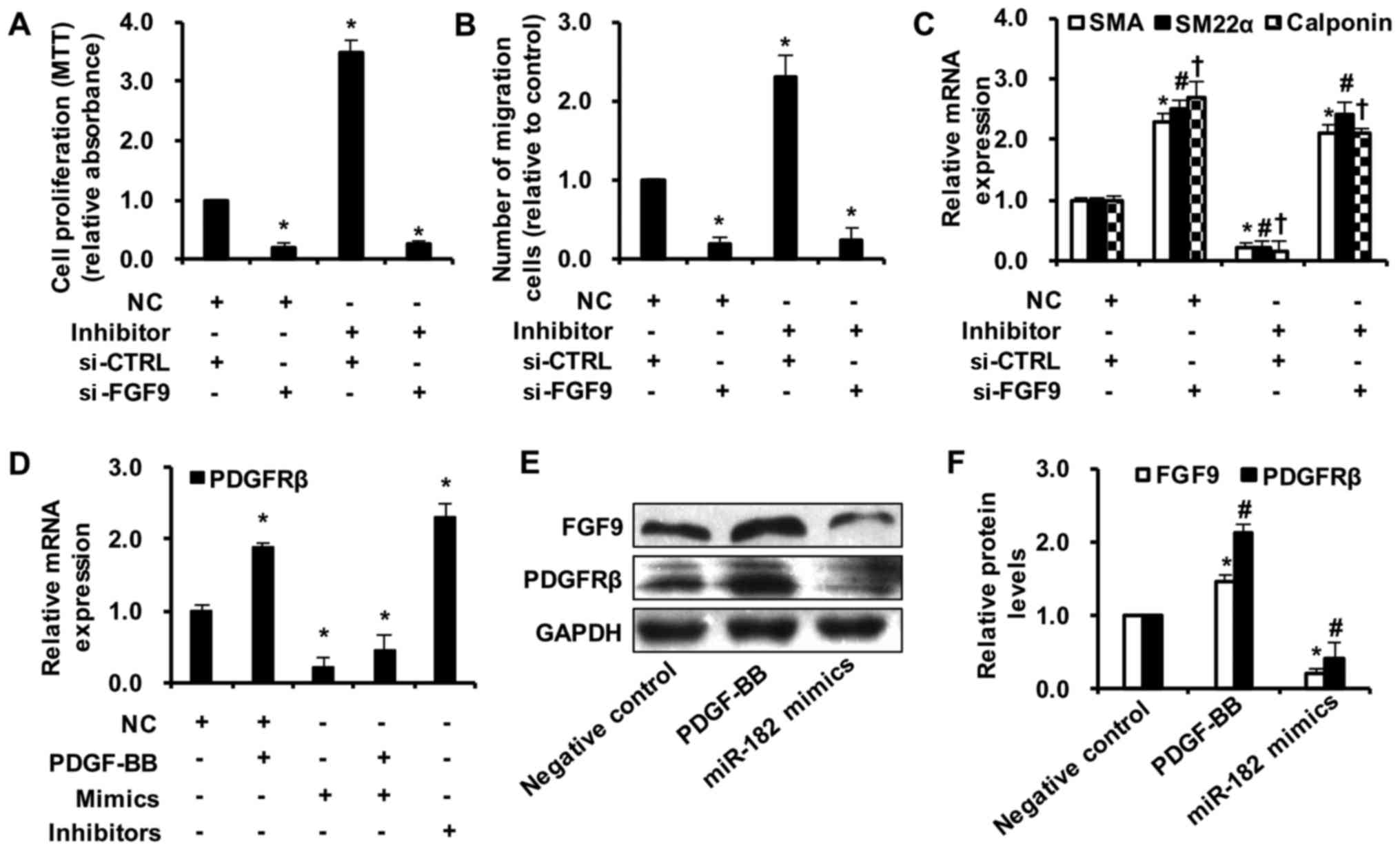 | Figure 6miR-182 mediates SMC phenotypic
modulation via fibroblast growth factor 9 (FGF9)/platelet-derived
growth factor receptor β (PDGFRβ) signaling. Smooth muscle cells
(SMCs) were transfected with either miR-182 inhibitors (80 nM), or
with si-FGF9 (100 nM), or both. (A) miR-182 inhibitor transfection
induced an increase in SMC proliferation; however, FGF9
downregulation inhibited miR-182 inhibitor-induced SMC
proliferation, as determined by MTT assay (n=5).
*p<0.05 vs. control. (B) FGF9 knockdown also
interfered with the function of miR-182 inhibitors on SMC
migration, as determined by scratch-wound healing assay (n=5).
*p<0.05 vs. control. (C) si-FGF9 transfection
increased differentiated marker gene expression. However, miR-182
inhibitor transfection induced a decrease in SMC-specific gene
expression. When si-FGF9 and miR-182 inhibitors were both
transfected into SMCs, the expression of SMA, smooth muscle 22α
(SM22α) and calponin was significantly increased, as determined by
RT-qPCR (n=5). *p<0.05 vs. the control group of SMA;
#p<0.05 vs. control of SM22α; †p<0.05
vs. control of calponin. (D) PDGF-BB treatment induced an increase
in PDGFRβ expression, while this increase was suppressed by miR-182
mimic transfection and was enhanced by miR-182 inhibitor
transfection (n=5). *p<0.05 vs. control. (E and F)
The protein level of PDGFRβ was increased by PDGF-BB stimulation,
but was decreased by miR-182 overexpression, as determined by (E)
western blot analysis and (F) densitometric analysis (n=5)
*p<0.05 vs. negative control of FGF9;
#p<0.05 vs. negative control of PDGFRβ. |
We hypothesized that the inhibition of SMC
phenotypic modulation via the upregulation of miR-182 or the
downregulation of FGF9 was related to PDGFRβ signaling. PDGF-BB
induced an increase in PDGFRβ gene expression, while transfection
with miR-182 mimics significantly inhibited PDGFRβ expression with
or without PDGF-BB stimulation (Fig.
6D). Conversely, the gene levels of PDGFRβ were upregulated
following transfection with miR-182 inhibitors. PDGF-BB- induced a
significant increase in the PDGFRβ protein level, while miR-182
overexpression markedly suppressed PDGFRβ expression (Fig. 6E and F). In addition, transfection
with FGF9 siRNA decreased PDGFRβ expression (Fig. 5A). Thus, these data indicate that
FGF9/PDGFRβ signaling is critical for the miR-182-mediated
prevention of the SMC phenotypic switch.
Discussion
In the present study, to the very best of our
knowledge, we demonstrate for the first time miR-182 is a novel
marker which has the capacity to modulate the rat vascular SMC
phenotype. The critical target gene of miR-182 is FGF9, and
miR-182-mediates the differentiation, proliferation and migration
of rat vascular SMCs via FGF9/PDGFRβ signaling.
Despite great advances in vascular SMC biology, the
molecular mechanisms responsible for the SMC phenotypic modulation
remain unclear. miR-182 has been reported to exhibit an altered
expression in various types of cancers, which may be a useful
prognostic biomarker for cancer (21). Previous studies have revealed that
miR-182 may function as an oncogenic miRNA to enhance cancer cell
proliferation, aggressiveness, tumorigenesis and drug resistance
(22–25). However, other studies have
reported conflicting results, showing that miR-182 suppressed cell
growth and aggression, and increased miR-182 levels were shown to
correlate with clinical treatment benefits (13,26–28). These results suggest the existence
of highly complex mechanisms linking miR-182, cell growth and
motility. In this study, we thus investigated the association
between miR-182 expression and the phenotype of vascular SMCs. In
this study, the expression of SMC-specific genes in rat vascular
SMCs declined eventually with prolonged incubation, which was
consistent with the findings of previous studies that dispersed
SMCs cultured in vitro exhibited rapidly downregulated
SMC-specific gene expression (29,30). During the period that the SMC
phenotype switches from a contractile to a synthetic type, the
expression of miR-182 was also demonstrated to gradually decrease.
Additionally, the miR-182 levels significantly decreased with
PDGF-BB stimulation in a dose-dependent manner. For the first time,
to the best of our knowledge, we established that miR-182 is a
novel phenotypic marker for SMCs.
Increasing evidence indicates the important roles of
miR-182 in cell biology. miR-182 has been shown to predict survival
in patients with various types of cancer (21). Kouri et al found that
miR-182 targeted Bcl-2-like 12, c-Met and hypoxia-inducible factor
2α to regulate the apoptosis, growth and differentiation programs
of glioma-initiating cells (31).
miR-182 has also been shown to inhibit Schwann cell proliferation
and invasion following nerve injury and repair (13). In the present study, the
overexpression of miR-182 inhibited the dedifferentiation of
cultured vascular SMCs, as well as SMC proliferation and migration.
Additionally, miR-182 overexpression prevented the SMCs from
switching from a contractile morphology to a synthetic phenotype.
Thus, miR-182 is a novel phenotypic modulator of SMC
differentiation, proliferation and migration.
miRNAs exert their biological functions by
inhibiting the transcription or translation of target genes
(4,6). By using the microRNA.org database, we identified that FGF9 is one
of the target genes of miR-182 across species. FGF9 combined with
its cognate receptors is able to induce cell dedifferentiation and
maturation (32). In addition, Yu
et al proved that miR-182 inhibited FGF9 expression by
binding to the 3′-UTR in rat Schwann cells using Luciferase
reporter assay (13). These data
suggest that FGF9 may be a direct target of miR-182 in rat vascular
SMCs. This study revealed a dose-dependent decrease in FGF9 protein
expression in miR-182 mimic-transfected SMCs. Accordingly, FGF9 is
the target gene of miR-182 in rat vascular SMCs.
The downregulation of FGF9 or upregulation of
miR-182 induced a similarly protective effect on the SMC phenotype
and an inhibitory effect on proliferation and migration. miR-182
downregulation induced SMC differentiation, proliferation and
migration; however, these processes were prevented by FGF9
knockdown. Moreover, the inhibition of FGF9 itself was able to
suppress the dedifferentiation, proliferation and migration of rat
SMCs. Although FGF9 was only one of the target genes, the results
suggest that FGF9 is critical in the process of miR-182-mediated
rat vascular SMC phenotypic modulation.
The activation of PDGFRβ is essential for the
proliferation and migration of a variety of cells, and FGF9 has
been implicated in upregulating of PDGFRβ expression (16,18). Frontini et al reported that
the FGF9-induced migration of cultured SMCs and neovascularization
required both PDGFRβ and SHH, but not ERK1/2 (16). PDGF-AB or -BB binding with the
PDGFRβ or the receptor overexpression results in the activation of
PI3K/Akt, PLCγ1 and MAPK pathways (33–36). PDGFRβ is a critical downstream
target of FGF9 and an important signaling for FGF9-induced mural
cell recruitment and vessel maturation (16). Thus, we hypothesized that the
inhibition of SMC phenotypic modulation via the upregulation of
miR-182 or the downregulation of FGF9 was related to PDGFRβ
signaling. In this study, the expression of PDGFRβ was demonstrated
to be markedly altered by regulating the expression of miR-182 and
FGF9. Indeed, the downregulation of FGF9 decreased PDGFRβ
expression, whereas the upregulation of FGF9 increased the PDGFRβ
levels in rat vascular SMCs. In addition, PDGF-BB induced an
increase in the expression of PDGFRβ, which was significantly
inhibited by miR-182 overexpression, but was upregulated following
miR-182 knockdown. Thus, FGF9/PDGFRβ signaling is critical in the
miR-182-mediated SMC phenotypic switch.
In conclusion, the present study demonstrated that
the miR-182 level was altered markedly during the phenotypic
transformation of rat vascular SMCs in vitro. The
upregulation of miR-182 inhibited SMC dedifferentiation,
proliferation and migration under both basal conditions and PDGF-BB
stimulation. FGF9 was the critical target gene of miR-182 in the
process of miR-182-mediated rat vascular SMC phenotypic modulation.
The downregulation of FGF9 increased SMC-specific contractile
marker expression, and inhibited the proliferation and migration of
SMCs via PDGFRβ signaling. This study suggests that miR-182 is a
novel phenotypic marker, which modulates the differentiation,
proliferation and migration of rat vascular SMCs via FGF9/PDGFRβ
signaling. miR-182 may thus play a potential role in the diagnosis
and treatment of vascular proliferative diseases.
Acknowledgments
The author Nana Dong has received grants from the
Open Foundation in China's Ministry of Education (KF201401) and
from the Graduate Student Innovation Foundation of Heilongjiang
Province (YJSCX2012-217HLJ). This study was also supported by the
National Key Project Foundation of China (no. 81330033). The
authors would like to thank MedSci Editing for the revision of the
manuscript.
References
|
1
|
Duband JL, Gimona M, Scatena M, Sartore S
and Small JV: Calponin and SM 22 as differentiation markers of
smooth muscle: Spatiotemporal distribution during avian embryonic
development. Differentiation. 55:1–11. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davis-Dusenbery BN, Wu C, Hata A and Sessa
WC: Micromanaging vascular smooth muscle cell differentiation and
phenotypic modulation. Arterioscler Thromb Vasc Biol. 31:2370–2377.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim VN: MicroRNA biogenesis: Coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Niwa R and Slack FJ: The evolution of
animal microRNA function. Curr Opin Genet Dev. 17:145–150. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hammond SM: RNAi, microRNAs, and human
disease. Cancer Chemother Pharmacol. 58(Suppl 1): s63–s68. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davis BN, Hilyard AC, Nguyen PH, Lagna G
and Hata A: Induction of microRNA-221 by platelet-derived growth
factor signaling is critical for modulation of vascular smooth
muscle phenotype. J Biol Chem. 284:3728–3738. 2009. View Article : Google Scholar :
|
|
9
|
Wang YS, Wang HY, Liao YC, Tsai PC, Chen
KC, Cheng HY, Lin RT and Juo SH: MicroRNA-195 regulates vascular
smooth muscle cell phenotype and prevents neointimal formation.
Cardiovasc Res. 95:517–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li P, Zhu N, Yi B, Wang N, Chen M, You X,
Zhao X, Solomides CC, Qin Y and Sun J: MicroRNA-663 regulates human
vascular smooth muscle cell phenotypic switch and vascular
neointimal formation. Circ Res. 113:1117–1127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stenvold H, Donnem T, Andersen S, Al-Saad
S, Busund LT and Bremnes RM: Stage and tissue-specific prognostic
impact of miR-182 in NSCLC. BMC Cancer. 14:1382014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mihelich BL, Khramtsova EA, Arva N,
Vaishnav A, Johnson DN, Giangreco AA, Martens-Uzunova E, Bagasra O,
Kajdacsy-Balla A and Nonn L: miR-183-96-182 cluster is
overexpressed in prostate tissue and regulates zinc homeostasis in
prostate cells. J Biol Chem. 286:44503–44511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu B, Qian T, Wang Y, Zhou S, Ding G, Ding
F and Gu X: miR-182 inhibits Schwann cell proliferation and
migration by targeting FGF9 and NTM, respectively at an early stage
following sciatic nerve injury. Nucleic Acids Res. 40:10356–10365.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Naruo K, Seko C, Kuroshima K, Matsutani E,
Sasada R, Kondo T and Kurokawa T: Novel secretory heparin-binding
factors from human glioma cells (glia-activating factors) involved
in glial cell growth. Purification and biological properties. J
Biol Chem. 268:2857–2864. 1993.PubMed/NCBI
|
|
15
|
Miyakawa K, Hatsuzawa K, Kurokawa T, Asada
M, Kuroiwa T and Imamura T: A hydrophobic region locating at the
center of fibroblast growth factor-9 is crucial for its secretion.
J Biol Chem. 274:29352–29357. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frontini MJ, Nong Z, Gros R, Drangova M,
O'Neil C, Rahman MN, Akawi O, Yin H, Ellis CG and Pickering JG:
Fibroblast growth factor 9 delivery during angiogenesis produces
durable, vasoresponsive microvessels wrapped by smooth muscle
cells. Nat Biotechnol. 29:421–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hellström M, Kalén M, Lindahl P, Abramsson
A and Betsholtz C: Role of PDGF-B and PDGFR-beta in recruitment of
vascular smooth muscle cells and pericytes during embryonic blood
vessel formation in the mouse. Development. 126:3047–3055.
1999.PubMed/NCBI
|
|
19
|
Metz RP, Patterson JL and Wilson E:
Vascular smooth muscle cells: Isolation, culture, and
characterization. Methods Mol Biol. 843:169–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Wang Y, Du W, Liu W, Liu F, Zhang
L, Zhang M, Hou M, Liu K, Zhang S and Yu B: Wnt1 inhibits hydrogen
peroxide-induced apoptosis in mouse cardiac stem cells. PLoS One.
8:e588832013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang F, Zhong S, Zhang H, Zhang W, Zhang
H, Wu X and Chen B: Prognostic value of MicroRNA-182 in cancers: A
meta-analysis. Dis Markers. 2015:4821462015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Qian P, Zhang X, Zhang M, Wang H,
Wu M, Kong X, Tan S, Ding K, Perry JK, et al: Autocrine/paracrine
human growth hormone-stimulated MicroRNA 96-182-183 cluster
promotes epithelial-mesenchymal transition and invasion in breast
cancer. J Biol Chem. 290:13812–13829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Zhang D, Wang X, Yao X, Ye C, Zhang
S, Wang H, Chang C, Xia H, Wang YC, et al: Hypoxia-inducible
miR-182 enhances HIF1α signaling via targeting HD2 and FIH1 in
prostate cancer. Sci Rep. 5:124952015. View Article : Google Scholar
|
|
24
|
Du C, Weng X, Hu W, Lv Z, Xiao H, Ding C,
Gyabaah OA, Xie H, Zhou L, Wu J and Zheng S: Hypoxia-inducible
miR-182 promotes angiogenesis by targeting RASA1 in hepatocellular
carcinoma. J Exp Clin Cancer Res. 34:672015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen L, Chu F, Cao Y, Shao J and Wang F:
Serum miR-182 and miR-3313p as diagnostic and prognostic markers in
patients with hepatocellular carcinoma. Tumour Biol. 36:7439–7447.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang L, Chen F, Pang EJ, Zhang ZQ, Jin BW
and Dong WF: MicroRNA-182 inhibits proliferation through targeting
oncogenic ANUBL1 in gastric cancer. Oncol Rep. 33:1707–1716.
2015.PubMed/NCBI
|
|
27
|
Sun Y, Fang R, Li C, Li L, Li F, Ye X and
Chen H: Hsa-miR-182 suppresses lung tumorigenesis through
downregulation of RGS17 expression in vitro. Biochem Biophys Res
Commun. 396:501–507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moskwa P, Buffa FM, Pan Y, Panchakshari R,
Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K,
Weinstock DM, et al: miR-182-mediated downregulation of BRCA1
impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell.
41:210–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chamley-Campbell J, Campbell GR and Ross
R: The smooth muscle cell in culture. Physiol Rev. 59:1–61.
1979.PubMed/NCBI
|
|
30
|
Shanahan CM and Weissberg PL: Smooth
muscle cell heterogeneity: Patterns of gene expression in vascular
smooth muscle cells in vitro and in vivo. Arterioscler Thromb Vasc
Biol. 18:333–338. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kouri FM, Hurley LA, Daniel WL, Day ES,
Hua Y, Hao L, Peng CY, Merkel TJ, Queisser MA, Ritner C, et al:
miR-182 integrates apoptosis, growth, and differentiation programs
in glioblastoma. Genes Dev. 29:732–745. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cinaroglu A, Ozmen Y, Ozdemir A, Ozcan F,
Ergorul C, Cayirlioglu P, Hicks D and Bugra K: Expression and
possible function of fibroblast growth factor 9 (FGF9) and its
cognate receptors FGFR2 and FGFR3 in postnatal and adult retina. J
Neurosci Res. 79:329–339. 2005. View Article : Google Scholar
|
|
33
|
Heldin CH, Ostman A and Rönnstrand L:
Signal transduction via platelet-derived growth factor receptors.
Biochim Biophys Acta. 1378:F79–F113. 1998.PubMed/NCBI
|
|
34
|
Cospedal R, Abedi H and Zachary I:
Platelet-derived growth factor-BB (PDGF-BB) regulation of migration
and focal adhesion kinase phosphorylation in rabbit aortic vascular
smooth muscle cells: Roles of phosphatidylinositol 3-kinase and
mitogen-activated protein kinases. Cardiovasc Res. 41:708–721.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song MC, Kim EC, Kim WJ and Kim TJ:
Meso-dihydroguaiaretic acid inhibits rat aortic vascular smooth
muscle cell proliferation by suppressing phosphorylation of
platelet-derived growth factor receptor beta. Eur J Pharmacol.
744:36–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Caglayan E, Vantler M, Leppänen O,
Gerhardt F, Mustafov L, Ten Freyhaus H, Kappert K, Odenthal M,
Zimmermann WH, Tallquist MD and Rosenkranz S: Disruption of
platelet-derived growth factor-dependent phosphatidylinositol
3-kinase and phospholipase Cγ1 activity abolishes vascular smooth
muscle cell proliferation and migration and attenuates neointima
formation in vivo. J Am Coll Cardiol. 57:2527–2538. 2011.
View Article : Google Scholar : PubMed/NCBI
|