Introduction
Diabetes is a metabolic disorder characterized by
hyperglycemia caused by pancreatic β-cells failing to produce
sufficient amounts of insulin to meet the body's needs, or receptor
insensitivity to endogenous insulin (1). Persistent hyperglycemia can lead to
the accumulation of methylglyoxal (MG), a reactive dicarbonyl
species, which has been implicated in various diabetic
complications (2).
MG is a highly reactive dicarbonyl metabolite
produced during glucose metabolism and is a major precursor of
advanced glycated end products (AGEs), which are involved in the
pathogenesis of diabetes and inflammation (3). It has been suggested that AGEs and
MG can generate pro-inflammatory cytokines through the activation
of receptor for AGE (RAGE), and that this is related to the
modulation of inflammatory molecules through oxidative stress
(3). AGEs also decrease insulin
synthesis in pancreatic β-cells by repressing the protein
expression of pancreatic duodenal homeobox-1 (PDX-1), which plays a
significant role in both pancreatic development and the maintenance
of β-cell functions (4). Under
physiological conditions, MG is degraded into D-lactate by the
glyoxalase system (5).
Glucotoxicity-induced oxidative stress is one of
many classic risk factors (6),
and damages the function of pancreatic islets and reduces insulin
secretion via the overproduction of reactive oxygen species (ROS)
(7). It has been suggested that
oxidative stress plays a key role in the onset of type 2 diabetes
(8). In addition, pancreatic
β-cells express low physiological levels of antioxidant enzymes,
such as superoxide dismutase (SOD), catalase (CAT) and glutathione
peroxidase (GPX) (9,10). Therefore, the maintenance of
pancreatic β-cell function and preventing expanding cell mass
represent the most feasible therapeutic strategies to control
hyperglycemia in pathophysiological states of nutrient excess
(11,12).
Insulin is used in the treatment of type 1 diabetes,
and many oral hypoglycemic agents are used in the treatment of type
2 diabetes (13). Due to their
adverse side-effects and alleviation of symptoms while not
targeting the cause, there have been persistent efforts to identify
compounds that could potentially cure diabetes, for example by
stimulating β-cell regeneration and preventing apoptosis, leading
to a return of the endogenous control of glucose homeostasis.
Studies on supplementation with natural products have demonstrated
effects of reducing the hyperglycemic status by preserving the
functions of pancreatic β-cells (14,15). Naturally occurring plant compounds
are attractive candidates as they are abundantly found in nature,
inexpensive to produce, and may have fewer side-effects than
currently used pharmaceutical compounds. Flavonoids have been shown
to regulate carbohydrate digestion, insulin secretion, insulin
signaling and glucose uptake in insulin-sensitive tissues through
various intracellular signaling pathways (16). Flavonoids exert their effects by
influencing pancreatic β-cell mass and function, as well as energy
metabolism and insulin sensitivity in peripheral tissues. The
anti-diabetic effects of flavonoids may be due to antioxidant,
enzyme inhibitory, or receptor agonist or antagonist activity, or
due to novel mechanisms which have yet to be elucidated (17).
Black cohosh (Cimicifuga racemosa) has a long
history of medicinal use dating back to Native North American
indigenous groups (18). It is
used to reduce the frequency and intensity of hot flashes and it
has been reported that there is an improvement in psychological
complaints among users (18). To
date, >20 of these triterpene glycosides have been isolated from
this plant, of which 23-epi-26-deoxyactein (27-deoxyactein) is one
of the major constituents.
In our previous studies, we reported that
deoxyactein isolated from black cohosh promoted the function of
osteoblastic MC3T3-E1 cells and reduced antimycin A-induced cell
damage by preventing mitochondrial dysfunction and oxidative stress
(19,20). Model studies using RIN-m5F cells,
a lineage of pancreatic β-cells, challenged with MG (21) and hydrogen peroxide (22), have begun to shed some light on
the molecular mechanisms implicated in MG- and hydrogen
peroxide-induced cytotoxicity. Given the mounting evidence for a
major role of pancreatic β-cells in the pathogenesis of type 2
diabetes, in this study, we investigated the effects of deoxyactein
on the MG-induced oxidative cell damage of pancreatic β-cells and
the underlying mechanisms.
Materials and methods
Reagents
Deoxyactein, isolated from black cohosh
(Cimicifuga racemosa), was purchased from ChromaDex Inc.
(Irvine, CA, USA). This was dissolved in dimethyl sulfoxide (DMSO)
and then diluted with culture medium [final DMSO concentration
≤0.05% (v/v)]. α-modified minimal essential medium (α-MEM) and
fetal bovine serum (FBS) were purchased from Gibco-BRL (Grand
Island, NY, USA). Other reagents were purchased from Sigma Chemical
(St. Louis, MO, USA).
Cell culture
RIN-m5F cells derived from rat pancreatic β-cells
were purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). RIN-m5F cells were maintained in RPMI-1640
supplemented with 10% FBS and 1% penicillin/streptomycin solution
under conditions of saturated humidity in an atmosphere containing
5% CO2 at 37°C. The medium was renewed every 3 days.
Forty-eight hours after seeding, the cells were pre-incubated for 1
h in medium containing 0.1% FBS and deoxyactein (0.01–10 mM) prior
to exposure to MG (30–500 mM) for 48 h. Aminoguanidine (AG, 400
µM), a carbonyl scavenger, was used as a positive control in
our experiments.
Cell viability
The cells were seeded in 24-well culture plates at a
density of 2×104 cells/well. After 48 h of seeding, the
cells were incubated for 1 h with deoxyactein prior to treatment
with MG for 48 h. Cell viability was assessed by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT)method. A total of 20 µl of MTT in phosphate-buffered
salt solution, pH 7.4 (5 mg/ml), was added to each well, and the
plates were incubated for an additional 2 h. After the removal of
solution from the well, DMSO was added to dissolve the formazan
products, and the plates were shaken for 5 min. The absorbance was
measured with a Zenyth 3100 multimode detector (Anthos Labtec
Instruments, Wals/Salzburg, Austria) at 570 nm. The cells incubated
with culture medium alone were used to define 100% viability and
were included as a control in all experiments to allow for the
estimation of the percentage viability of the cell samples.
Measurements of insulin secretion
The cells were seeded in 24-well culture plates at a
density of 2×104 cells/well. The culture conditions were
the same as those described for the cell viability assay.
Supernatants were collected for the measurement of secreted insulin
using a high-range rat insulin enzyme-linked immunosorbent assay
(ELISA) kit (Mercodia Inc., Uppsala, Sweden) according to the
manufacturer's instructions. Protein concentrations were determined
using Bio-Rad protein assay reagent.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
The cells were seeded in 100-mm culture dishes at a
density of 5×105 cells/dish. The culture conditions were
the same as those described for the cell viability assay. Total RNA
was extracted from each well of RIN-m5F cells using an RNeasy mini
kit (Qiagen NV, Venlo, The Netherlands), and complementary DNA
(cDNA) was synthesized using a PrimeScript First Strand DNA
Synthesis kit (Takara Biotech, Otsu, Japan) according to the
manufacturer's instructions. Quantitative PCR (qPCR) was performed
using a SYBR Premix ExTaq kit (Takara Biotech, Dalian, China) and
the ABI Prism 7500 sequence detection system (Applied Biosystems,
Foster City, CA, USA) to determine the gene expression levels. Each
PCR reaction was performed in a 20 µl solution containing
0.8 µl (10 µM) of each pair of forward and reverse
primers, 10 µl of Premix Ex Taq DNA polymerase, 0.4
µl of ROX reference dye, 6 µl of dH2O, and
2 µl of reverse transcription reaction products. The qRT-PCR
primers used in the experiment are lissted in Table I. All experiments were performed
in quadruplicate. Relative expression was deter-mined using the
2−ΔΔCq method using the housekeeping gene, glucose
6-phosphate dehydrogenase (G6PD), as the internal control, and the
fold change was calculated in comparison with the corresponding
control group.
 | Table IPrimer sequences used in this
study. |
Table I
Primer sequences used in this
study.
| Gene | Accession no. | Forward primer | Reverse primer |
|---|
| Insulin-2 | NM_019130.2 | 5′-CGA AGT GGA GGA
CCC ACA-3′ | 5′-TGC TGG TGC AGC
ACT GAT-3′ |
| PDX-1 | NM_022852.3 | 5′-ACC CGT ACA GCC
TAC ACT CG-3′ | 5′-GCC GGG AGA TGT
ATT TGT TAA A-3′ |
| SOD1 | NM_017050.1 | 5′-TAA GAA ACA
TGGCGGTCC A-3′ | 5′-TGG ACA CAT TGG
CCA CAC-3′ |
| GPX1 | NM_030826.3 | 5′-AGA AGG CTC ACC
CGC TCT-3′ | 5′-GGA TCG TCA CTG
GGT GCT-3′ |
| G6PD | NM_017006.2 | 5′-TGC AGC AGC TGT
CCT CTA TG-3′ | 5′-ACT TCA GCT TTG
CGC TCA TT-3′ |
Measurement of interleukin (IL)-1β
levels
The cells were seeded in a 24-well plate at a
density of 2×104 cells/well in culture medium. After 48
h, the cells were exposed to deoxyactein or AG prior to exposure to
300 µM MG for 48 h. Cellular IL-1β contents were measured
using an enzyme immunoassay system (R&D Systems Inc.,
Minneapolis, MN, USA) according to the manufacturer's instructions.
In brief, IL-6 present was bound by immobilized antibody pre-coated
onto a microplate. After washing away any unbound substances, an
enzyme linked polyclonal antibody specific for IL-1β was added to
the well. Following a wash to remove any unbound antibody-enzyme
reagent, a substrate solution was added to the wells. The enzyme
reaction yielded a blue product that turned yellow when stop
solution was added. The intensity of the color measured was in
proportion to the amount of IL-1β bound. Cellular IL-1β contents
were measured using an enzyme immunoassay system (R&D Systems
Inc.) according to the manufacturer's instructions.
Measurement of intracellular ROS
levels
The cells were seeded in 24-well culture plates at a
density of 2×104 cells/well. The culture conditions were
the same as those described for the cell viability assay. Formation
of intracellular ROS was measured using
2′,7′-dichlorodihydrofluorescin diacetate (H2DCFDA)
(23). Viable cells can
deacetylate H2DCFDA to the non-fluorescent derivative,
2′,7′-dichlorofluorescin (DCF), which reacts with oxygen species
and can be measured to provide an index of intracellular oxidant
production. In order to load the cells with the fluorescent dye,
the cells were incubated with H2DCFDA (Sigma Chemical)
in Hank's solution at a final concentration of 10 µM for 45
min at 37°C in the dark. Following washing with DPBS, the
fluorescence intensity was measured (excitation 485 nm, emission
515 nm) using a Zenyth 3100 multimode detector (Anthos Labtec
Instruments).
Measurement of cardiolipin
peroxidation
The cells were seeded in 24-well culture plates at a
density of 2×104 cells/well. The culture conditions were
the same as those described for the cell viability assay.
10-N-nonyl-acridine orange (NAO; Molecular Probes, Inc., Eugene,
OR, USA), which binds to mitochondrial cardiolipin, was used for
the measurement of cardiolipin. Decreases in the fluorescence of
NAO in cells reflect the peroxidation of intracellular cardiolipin
as the fluorochrome loses its affinity for peroxidised cardiolipin.
The cells were labeled with 5 µM NAO for 20 min. After
washing, fluorescence was measured at an excitation wavelength of
485 nm and an emission wavelength of 530 nm using a Zenyth 3100
multimode detector.
Measurement of phosphorylated sirtuin 1
(SIRT1) and peroxisome proliferator-activated receptor-γ
co-activator-1α (PGC-1α)
The cells were seeded in 24-well culture plates at a
density of 2×104 cells/well. The culture conditions were
the same as those described for the cell viability assay. The cells
were rinsed in ice-cold DPBS and homogenized in DPBS with a glass
Dounce homogenizer (Taylor Scientific, USA) on ice. The resulting
suspension was subjected to two freeze-thaw cycles to further break
the cell membranes. Cell homogenates were centrifuged at 13,000 × g
for 15 min at 4°C and the supernatant was used for ELISA and
protein content measurement. SIRT1 was measured using a Sirtuin 1
ELISA kit (Cloud-Clone, Houston, TX, USA). PGC-1α was measured
using a mouse peroxisome proliferator activated receptor-γ
coactivator-1α (PGC-1α) ELISA kit (MyBioSource, San Diego, CA,
USA). The kits were used according to the manufacturers'
instructions.
Quantification of MG-modified proteins
(adducts)
The quantification of MG-modified proteins
(MG-protein adducts) was determined using an ELISA kit purchased
from Cell BioLabs, Inc. (San Diego, CA, USA). The MG protein
adducts present in the sample or standard were probed with an
anti-MG specific monoclonal antibody, followed by a horseradish
peroxidase (HRP) conjugated secondary antibody (both contained in
the ELISA kit, cat. no. STA-811). The quantity of MG adducts in the
protein samples was determined by comparing their absorbances with
that of a known MG-BSA standard curve.
Glyoxalase I activity
The cells were rinsed in ice-cold DPBS and
homogenized them in DPBS with a glass Dounce homogenizer (Taylor
Scientific) on ice. The resulting suspension was subjected to two
freeze-thaw cycles to further break the cell membranes. Cell
homogenates were centrifuged at 13,000 × g for 15 min at 4°C and
the supernatant was used for glyoxalase I activity assay and
protein content measurement. Glyoxalase I activity was measured
using a modification of a previously published method (24). To measure glyoxalase I activity,
50 µl of sample was loaded onto a UV microplate (Microtiter;
Thermo Fisher Scientific, Waltham, MA, USA) and 200 µl of
reaction mix were added. The reaction mix consisted of 60 mm sodium
phosphate buffer, pH 6.6, containing 4 mm GSH and 4 mm MG, and was
pre-incubated for 10 min at 37°C. S-Lactoylglutathione synthesis
was followed by the measurement of the absorbance at 240 nm for 5
min at 25°C.
Statistical analysis
The results are expressed as the means ± SEM.
Statistical significance was determined by analysis of variance and
subsequently applying Dunnett's t-test. A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of deoxyactein on the viability
of RIN-m5F cells
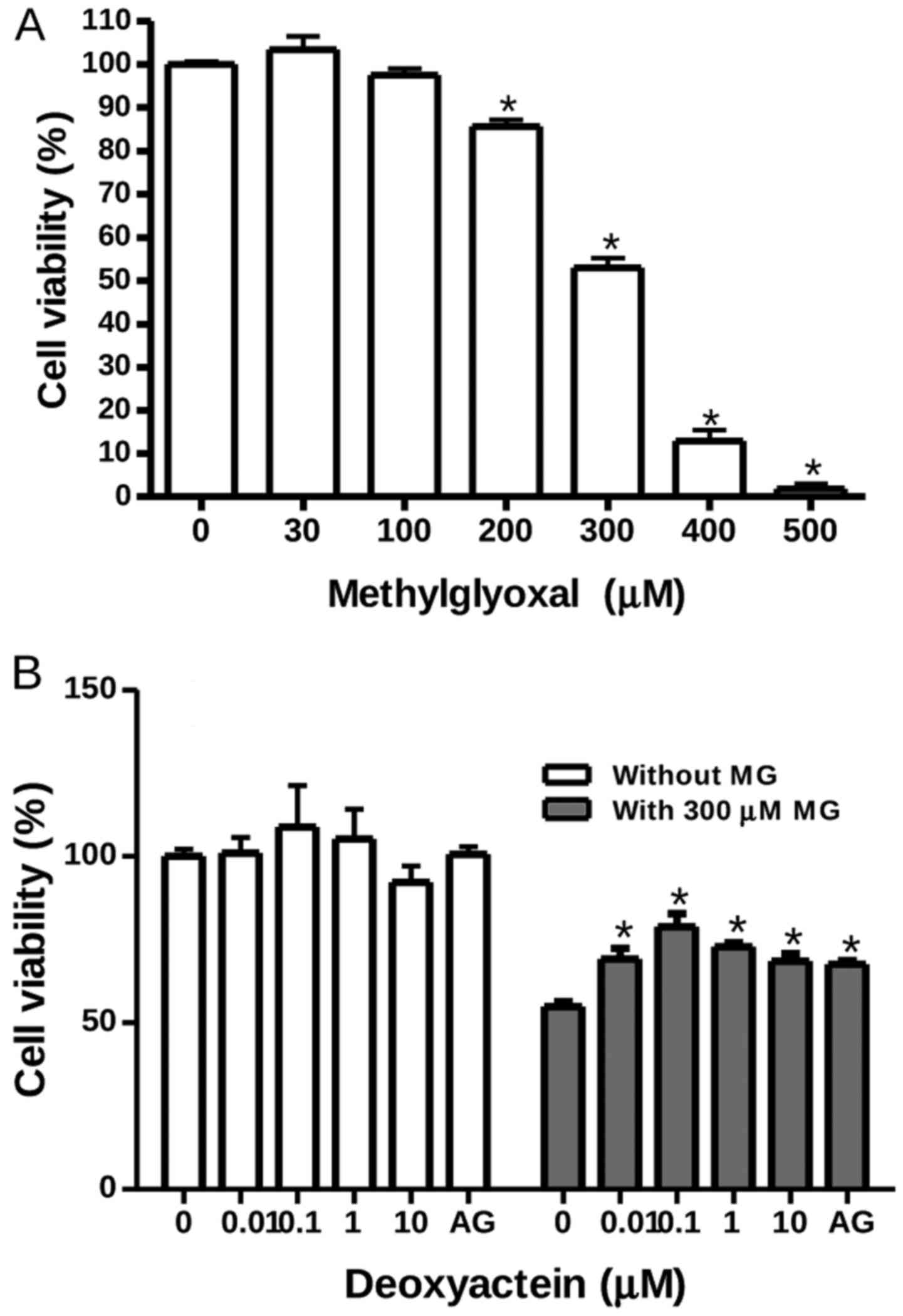
RIN-m5F cells were exposed to MG at concentrations
ranging from 30 to 500 µM for 48 h. Thereafter, cell
viability was measured by MTT assay. Our results revealed that MG
at concentrations of ≤100 µM had no effect on the viability
of RIN-m5F cells; however, it decreased their viability at
concentrations of ≥200 µM. At 300 µM, MG decreased
cell viability by ~50% (Fig. 1A).
To determine whether deoxyactein exerts a protective effect against
MG-induced cytotoxicity, the cells were pre-incubated with
deoxyactein for 1 h and then cultured with 300 µM MG for 48
h. Deoxyactein at concentrations of ≤10 µM had no effect on
the viability of RIN-m5F cells in the absence of MG (Fig. 1B). However, pre-treatment with
deoxyactein (0.01–10 µM) inhibited MG-induced cytotoxicity
significantly. AG (400 µM), a carbonyl scavenger, also
inhibited the cytotoxicity induced by MG.
Deoxyactein relieves MG-abrogated insulin
secretion and increases the gene expression of INS2 and PDX-1
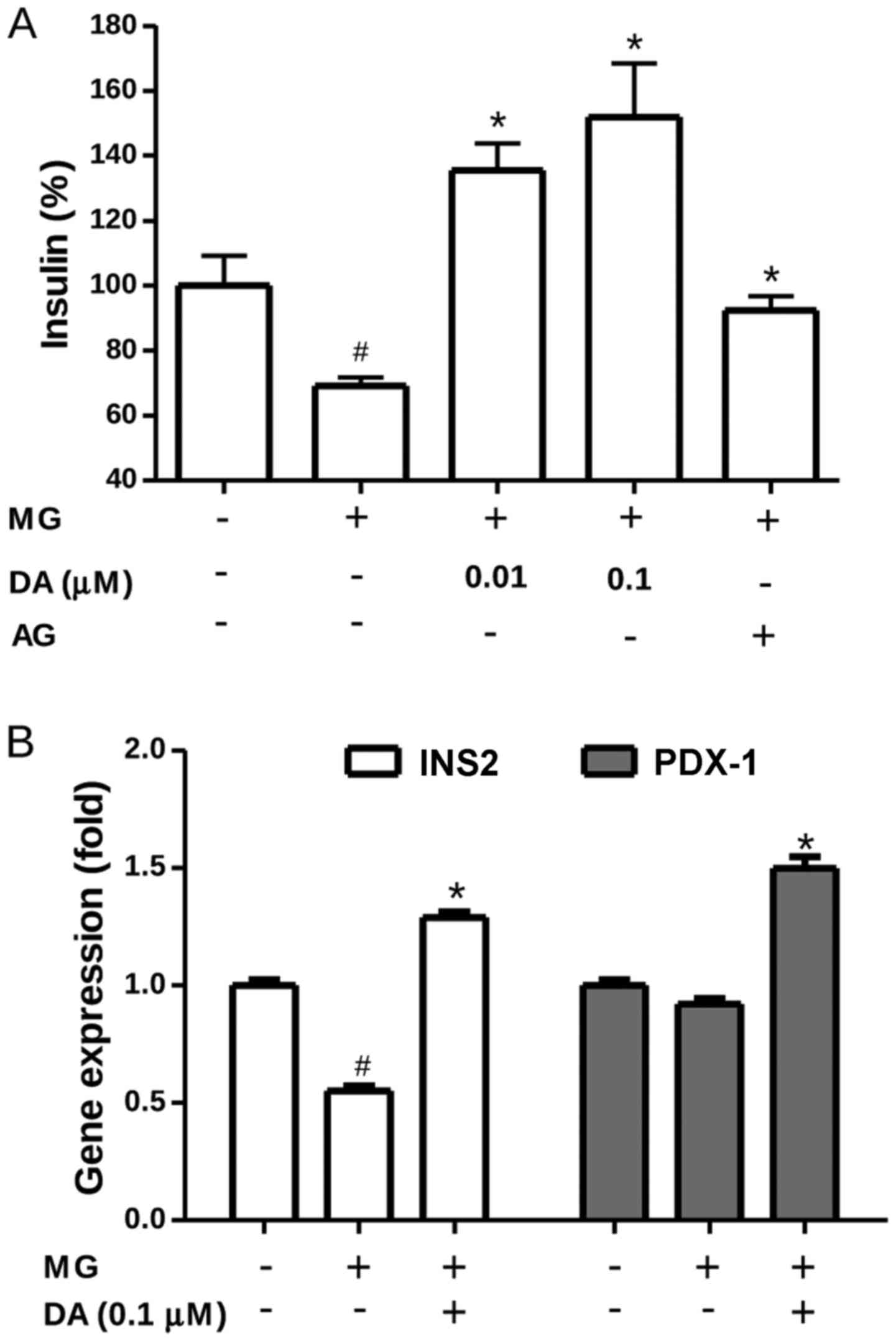
Insulin secretion was evaluated in order to examine
the protective effects of deoxyactein on β-cell functionality.
Exposure to 300 µM MG resulted in a marked decrease in
insulin secretion by the RIN-m5F cells (Fig. 2A), indicating a stress-induced
loss of insulin functionality. Most interestingly, insulin
secretion by the RIN-m5F cells exposed to MG was completely
restored when these cells were pre-treated with deoxyactein
(0.01–0.1 µM) or AG. These results indicated that
deoxyactein preserved not only RIN-m5F cell viability, but also the
most important β-cell function, insulin secretion. Alterations in
insulin signal transduction can lead to β-cell dysfunction,
contributing to the pathogenesis of type 2 diabetes (25). Thus, to investigate whether
deoxyactein modulates the expression profile of different genes
important for β-cell functioning, we compared the gene expression
levels of a series of regulators of insulin secretion processes in
pancreatic β-cells. As shown in Fig.
2B, the results of RT-qPCR revealed that MG significantly
down-regulated the transcription level of the INS2 gene compred to
that in the control cells (P<0.05). Pre-treatment with 0.1
µM deoxyactein significantly mitigated the decrease in the
gene expression of INS2 induced by exposure to MG. In addition
deoxyactein increased the expression of PDX1 in the cells.
Deoxyactein decreases the production of
IL-1β in MG-exposed RIN-m5F cells
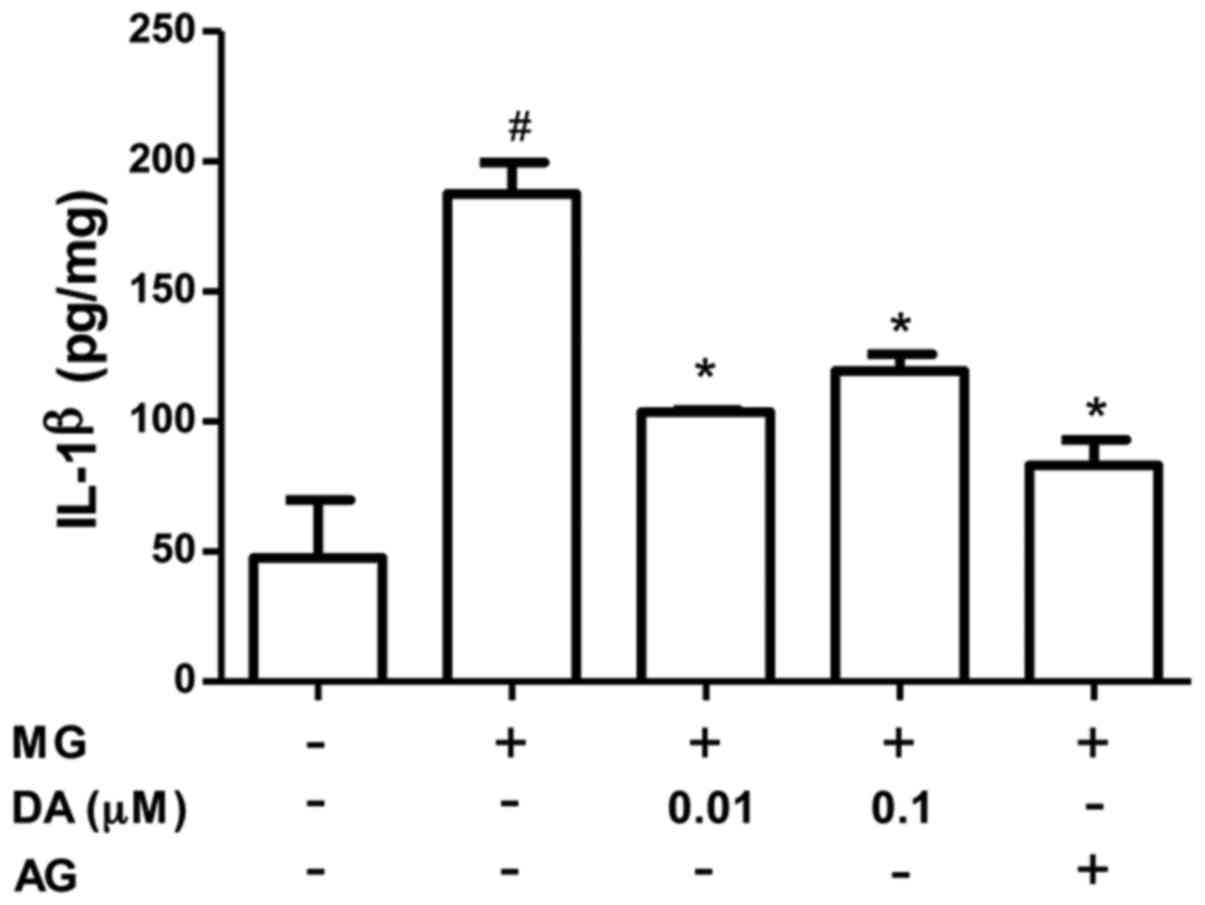
MG promotes the formation of pro-inflammatory
cytokines in various cell types (26–28). Thus, we also investigated whether
deoxyactein modulates the production of IL-1β in MG-exposed cells
(Fig. 3). When 300 µM MG
was added to the cell medium, the production of IL-1β increased
significantly. However, the MG-induced production of IL-1β was
significantly inhibited by pre-treatment with deoxyactein at
concentrations of 0.01–0.1 µM. AG also decreased the
MG-induced production of IL-1β.
Deoxyactein decreases ROS production and
increases the gene expression of SOD and GPX in MG-exposed
cells
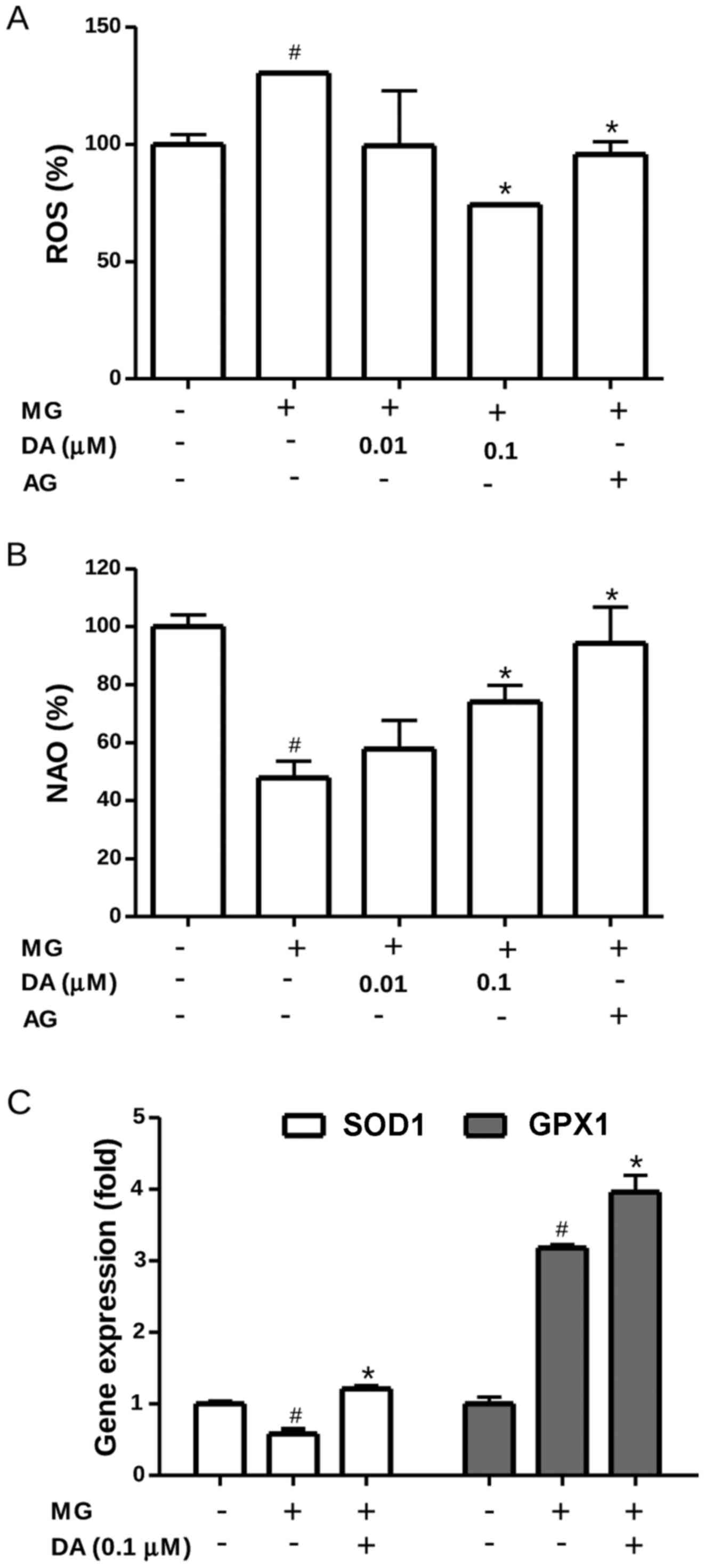
Intracellular ROS levels were measured using the
oxidation-sensitive probe, DCFH-DA. As shown in Fig. 4A, exposure to 300 µM MG
significantly increased the ROS levels compared with those in the
control cells (P<0.05). Under conditions of MG toxicity,
deoxyactein (0.1 µM) or AG suppressed the increase of ROS
significantly (P<0.05), suggesting that the cytoprotective
effects of deoxyactein are partly attributable to its regulation of
ROS production. To obtain further evidence for oxidative stress
within the mitochondria, we assessed the oxidation of cardiolipin,
as this phospholipid exists in association with cytochrome c
on the outer surface of the inner mitochondrial membrane (29). As the fluorescent dye, NAO, binds
to the non-oxidized form, but not to the oxidized form of
cardiolipin, measurements of NAO fluorescence allow us to monitor
the oxidation of cardiolipin in the mitochondria (30). The results revealed that exposure
to 300 µM MG decreased NAO fluorescence, indicating the
induction of cardiolipin peroxidation (Fig. 4B). However, deoxyactein (0.1
µM) or AG reduced cardiolipin peroxidation induced by MG.
These data indicated that deoxyactein reduced MG-induced ROS
generation and oxidative stress within the mitochondria. The
effects of deoxyactein on the expression of the SOD and GPX genes
in the RIN-M5F β-cells were also examined. As shown in Fig. 4C, the mRNA expression level of SOD
decreased in the RIN-M5F β-cells exposed to MG cells compared to
that in the control cells. However, pre-treatment with deoxyactein
(0.1 µM) significantly mitigated the decreased gene
expression of SOD observed in the MG-exposed cells. We found also
that the expression of GPX increased significantly in the
MG-exposed cells compared to the control cells (Fig. 4C). Pre-treatment of the RIN-m5F
β-cells with deoxyactein (0.1 µM) further increased the gene
expression of GPX compared to the MG-exposed cells. These findings
suggest that deoxyactein is capable of enhancing the antioxidant
status by upregulating the mRNA expression of SOD and GPX in
MG-exposed RIN-M5F β-cells.
Effects of deoxyactein on mitochondrial
metabolic factors in MG-exposed RIN-M5F cells
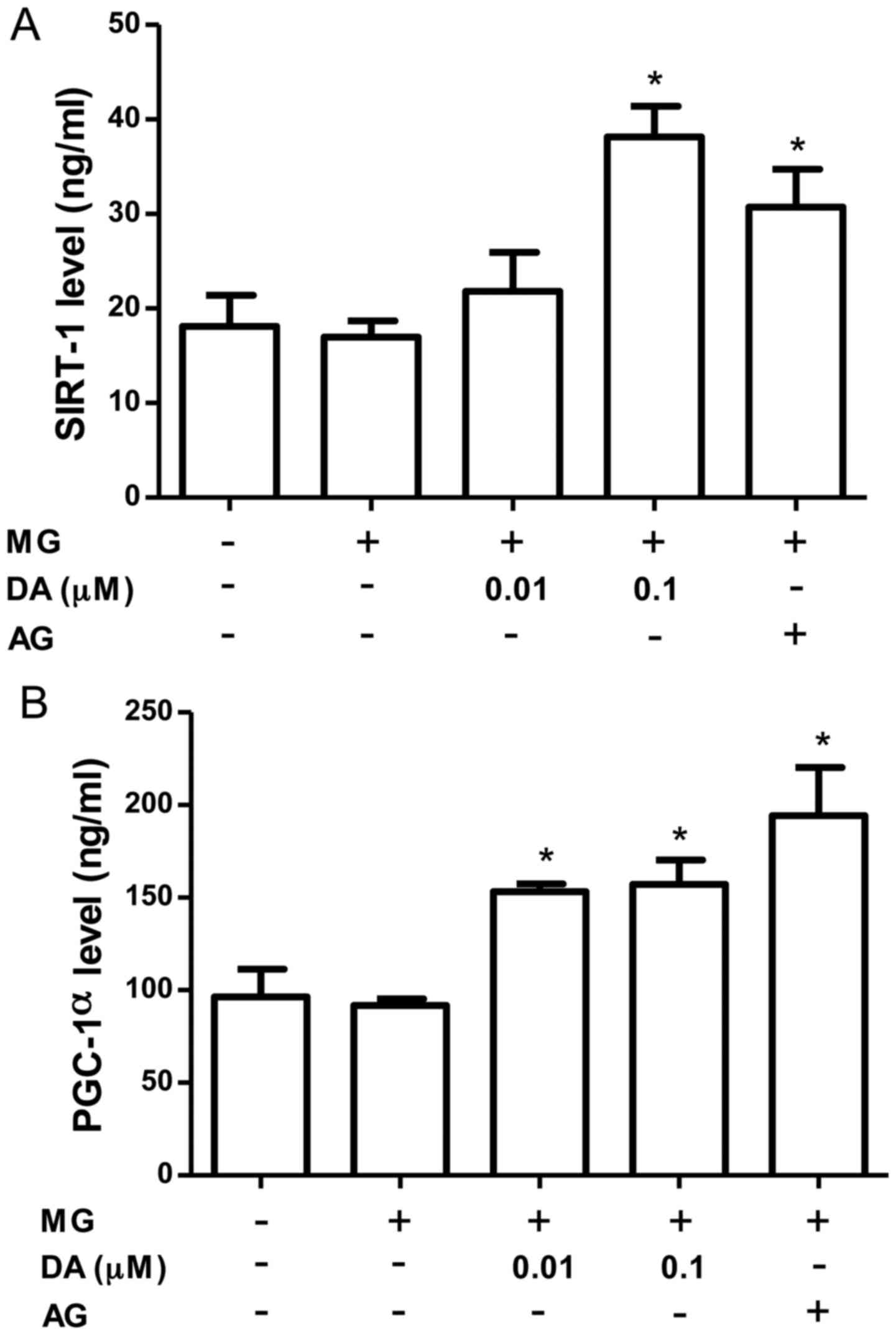
SIRT1 likely regulates multiple pathways involved in
mitochondrial biogenesis. As shown in Fig. 5A, the SIRT-1 levels were increased
by treatment with deoxyactein (0.1 µM) or AG. PGC-1α is also
considered to be a key regulator of mitochondrial biogenesis in
multiple tissues (31). Since
SIRT1 may also directly deacetylate PGC-1α and increase its
activity (32), we measured the
PGC-1α levels in the pancreatic β-cells. As shown in Fig. 5B, the level of PGC-1α was
significantly increased by deoxyactein (0.01–0.1 µM) or AG
treatment. These data indicate that deoxyactein enhances pancreatic
β-cell function due to the activation of the SIRT1-PGC-1α pathway.
MG alone did not affect SIRT1 and PGC-1α protein expression. This
may be due to compensatory mechanisms maintaining the levels of
these proteins.
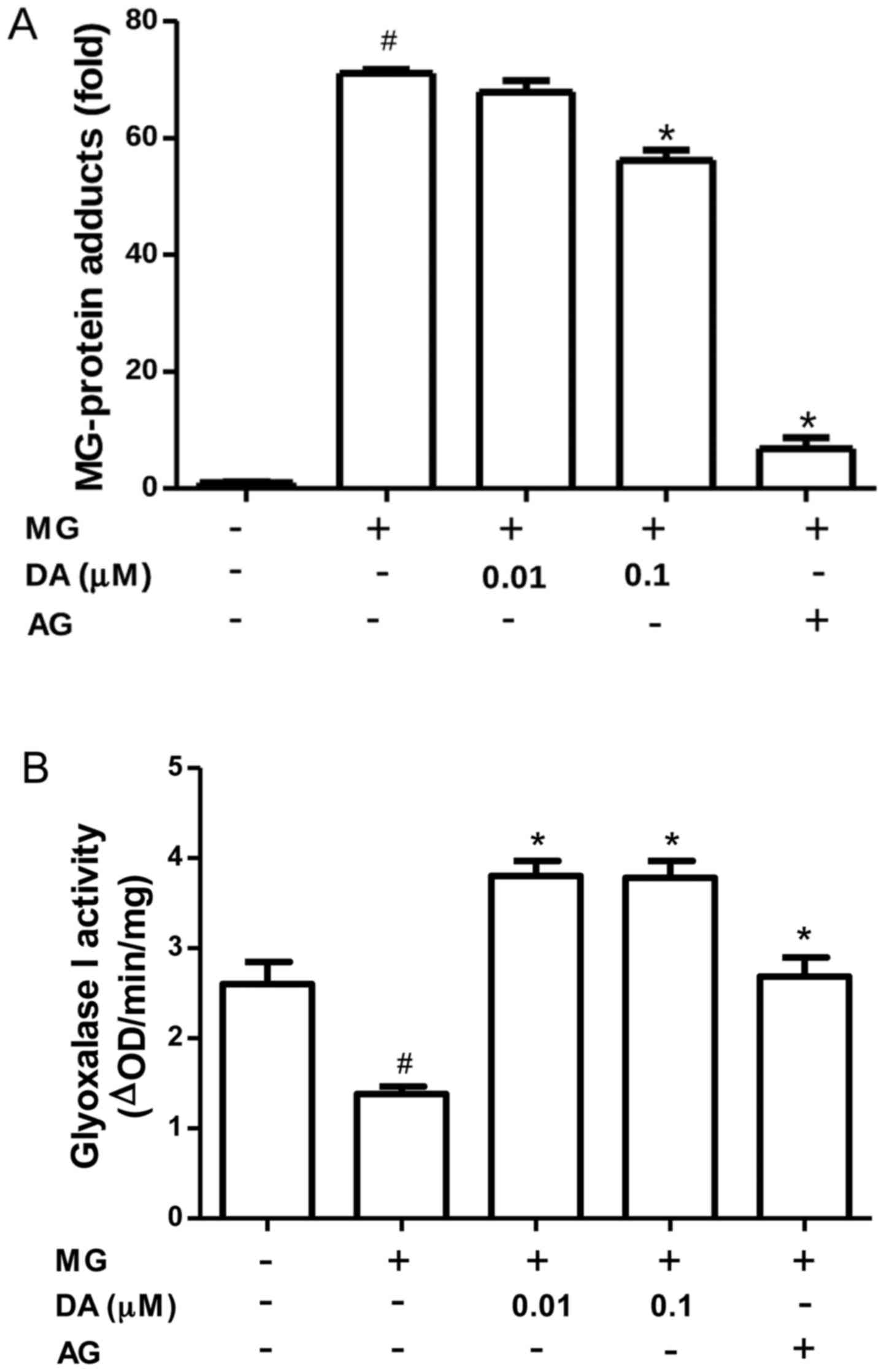
Inhibitory effect of deoxyactein on
MG-induced glycation in RIN-m5F cells
MG reacts with free amino groups and thiols to form
AGE protein adducts, thereby altering protein function (33). Thus, we investigated whether the
incubation of RIN-m5F β-cells with deoxyactein could reduce the
formation of protein adducts. As shown in Fig. 6A, protein adducts accumulated in
the cells exposed to 300 µM MG. However, pre-treatment with
deoxyactein (0.1 µM) or AG decreased protein adduct
formation induced by MG. These data indicated that deoxyactein
blocked the MG-derived protein glycation in RIN-m5F β-cells, which
may be part of the mechanisms responsible for its inhibitory effecs
on RIN-M5F β-cell death. MG is detoxified by the glyoxalase system.
Thus, we examined the effect of deoxyactein on the activity of
glyoxalase I in RIN-m5F β-cells. As shown in Fig. 6B, we found a significant decrease
in the glyoxalase I activity of RIN-m5F β-cells exposed to MG (300
µM). However, deoxyactein (0.01–0.1 µM) or AG
recovered the glyoxalase I activity inhibited by MG. These data
demonstrate that deoxyactein decreases MG-induced glycation in part
through an increase in glyoxalase I activity.
Discussion
Pancreatic β-cell dysfunction is the pivotal
physiological disorder in the development of diabetes. Decreased
viability of and damage to β-cells accelerates diabetic
pathogenesis, which is associated with an increased risk of
mortality. In the present study, we investigated the effects of
deoxyactein on pancreatic β-cells under MG-induced toxic conditions
and the underlying mechanisms. The results revealed that a decrease
in cell viability was detected when the MG concentration exceeded
300 µM. When the cells were pre-treated with deoxyactein,
pancreatic β-cell toxicity was attenuated. This study indicated
that incubation with 300 µM MG for 48 h significantly
impaired insulin secretion and the expression of the insulin gene
in RIN-m5F cells. In addition to insulin secretion, insulin gene
transcription is a pancreatic β-cell-specific function, and
inappropriate insulin biosynthesis may contribute to the
pathogenesis of diabetes mellitus (34). Previous studies have shown that MG
alters insulin secretion (21,35). The present study on pancreatic
β-cells further documents the inhibitory effect of MG on insulin
secretion. Moreover, pre-treatment with deoxyactein caused a
significant increase in insulin secretion and gene expression under
conditions of MG-induced toxicity. Among several transcription
factors binding to the promoter of the insulin gene, PDX-1 has been
shown to be important for the maintenance of insulin biosynthesis,
as well as β-cell mass (36).
Mice homozygous for a targeted disruption of the PDX-1 gene fail to
develop a pancreas, and the inactivation of PDX-1 specifically in
β-cells of mice decreases β-cell mass and insulin expression
(36). It has been shown that
insulin stimulates PDX-1 binding to the insulin promoter via PI3K
and SAPK2/p38 (37). PDX-1
deficiency contributes to impaired proliferation and enhanced
apoptosis via transcriptional mechanisms in models of type 2
diabetes (38). In this study,
PDX-1 gene expression was significantly increased by treatment with
deoxyactein. Therefore, it may be presumed that deoxyactein may
attenuate the inhibitory effect of MG on insulin secretion by
regulating insulin and PDX-1 gene expression.
In response to acute elevation of glucose and
survival factors, such as insulin, PDX-1 is phosphorylated and
translocates to the nucleus (39). By contrast, stimuli associated
with diabetes, such as oxidative stress (40) and free fatty acids (41), cause nuclear exclusion of PDX-1
(41). Ardestani et al
(42) showed that the nuclear
localization of PDX-1 is essential for functional β-cells, which is
a novel mechanism of the protective effects of IL-1 receptor
antagonist in β-cell survival and function.
Treatment with an IL-1β antibody also improves
glycemic control in diet-induced obesity in mice (43). IL-1β is thought to be a critical
mediator of the impaired function of pancreatic β-cells during the
development of autoimmune type 1 diabetes (44). Furthermore, previous findings have
established the role of IL-1β in β-cell failure in type 2 diabetes
(45). The IL-1β-induced
dysfunction of the pancreatic β-cells is mainly due to the
activation of extracellular signal-regulated kinase (ERK) and
nuclear factor-κB (45). The
exposure of pancreatic islets to IL-1β decreases the mRNA levels of
pro-insulin-converting enzymes, indicating a decreased proinsulin
conversion (46). In this study,
we demonstrated a profound inhibitory effect of deoxyactein on
MG-induced IL-1β production by β-cells. The protective action
toward pancreatic β-cell function and viability by deoxyactein is
likely due to its inhibition of IL-1β. Therefore, the present study
shows that deoxyactein may protect pancreatic islets against the
noxious effects induced by IL-1β.
MG accumulates in oxidative conditions and may
contribute to oxidant-induced cellular damage (47). Increased MG modification of
proteins is a likely outcome of oxidative stress, and increased MG
modification of mitochondrial proteins may also induce oxidative
stress (48). Furthermore,
oxidative stress has been implicated in the deterioration of
insulin signaling in diabetes (49), in decreased insulin secretion and
in β-cell death (50). In this
study, to evaluate the role of free radicals in the protective
activity of deoxyactein, its effect on MG-induced ROS generation
was analyzed using H2DCFDA assays. Following
pre-treatment with deoxyactein, MG-induced ROS generation was
observed to decline, which may account for the observed
cytoprotective effects of deoxyactein. The increased generation of
ROS accelerates the oxidation of lipids, proteins, nucleic acids
and other molecules. Mitochondrial cardiolipin molecules are an
early target of ROS attack, either due to their high content of
unsaturated fatty acids or because of their location in the inner
mitochondrial membrane near the site of ROS production (51). It seems likely that enhanced ROS
production could lead to cardiolipin oxidative damage, which would
negatively impact the biochemical function of the mitochondrial
membranes, altering membrane fluidity and ion permeability. In
deoxyactein-pre-treated cells, MG-induced cardiolipin peroxidation
was significantly decreased. The inhibitory effect of deoxyactein
on cardiolipin peroxidation in the mitochondria can be explained by
its ability to inhibit the peroxidation of linoleic fatty acid
constituents of mitochondrial cardiolipin molecules. These data
strongly indicate that deoxyactein modulates ROS generation via the
reduction of MG-induced mitochondrial membrane lipid
peroxidation.
Overabundant ROS are scavenged by endogenous
anti-oxidant enzymes. In cells, SOD catalyzes the conversion of
superoxide to hydrogen peroxide, which is further reduced to
H2O by the activity of CAT or GPX. In the present study,
MG significantly decreased the expression of SOD and increased that
of GPX in pancreatic β-cells. Antioxidant defenses are affected by
oxidative challenge, evoking a marked increase in GPX activity to
cope with elevated ROS. The induction of GPX is an essential
mechanism of the cell defense against oxidative insults and
consequently plays a major role in overcoming ROS production
(52). In the study,
pre-treatment with deoxyactein increased the gene expression of
these antioxidant enzymes in MG-exposed pancreatic β-cells,
indicating that deoxyactein is able to reduce MG-induced oxidative
stress. This finding should be relevant to therapies directed
toward pancreatic β-cells due to the low antioxidant enzyme gene
expression in pancreatic tissue compared to other tissues (53). Our results mentioned above
indicate that the treatment of pancreatic β-cells with deoxyactein
creates conditions favorable for combating the increased generation
of ROS induced by MG, and consequently the maintenance of cell
function and viability. Muscogiuri et al (54) reported that the genetic ablation
of SOD caused glucose intolerance, which was associated with
reduced in vivo insulin secretion by pancreatic β-cells and
decreased β-cell volume, which suggests that oxidative stress
caused by SOD ablation leads to glucose intolerance secondary to
β-cell dysfunction. Studies have reported that the overexpression
of SOD provides a protective effect to insulin-secreting cells
(55) and against
streptozotocin-induced diabetes (56). In addition, the over expression of
GPX has been shown to confer a protective effect against
ROS-induced oxidative stress by increasing the activity of SOD
(57). Therefore, the
deoxyactein-induced increase in the activities of GPX and SOD,
which participate in the defense against hydrogen peroxides and
superoxides, is essential to prevent ROS cytotoxicity induced by
MG.
The main source of ROS in diabetes is most probably
altered mitochondrial metabolism, which results in the
overproduction of superoxide by the electron transport chain
(58). The transcriptional
co-activator, PGC-1α, and the NAD+-dependent
deacetylase, SIRT1, are considered important inducers of
mitochondrial biogenesis as they regulate the transcription of
nucleus-encoded mitochondrial genes (59). SIRT1 acts as an important
regulator of metabolism by controlling the activity of key
transcription factors, such as PGC-1α, forkhead box protein O1
(FOXO1) and p53 (60,61). PGC-1α functions as an upstream
inducer of genes of mitochondrial metabolism by positively
affecting the activity of some hormone nuclear receptors and
nuclear transcription factors (e.g., NRF-1 and -2) (62). Additionally, NRF-1 regulates the
activation of the Tfam, Tfb1m and Tfb2m promoters and indirectly
affects the expression of Cox genes, Glut4 and PGC-1α itself
(63). Our data revealed that
deoxyactein increased the levels of SIRT1 and PGC-1α. Therefore,
the beneficial effect of deoxyactein is mediated by enhanced
mitochondrial biogenesis via the activation of the SIRT1-PGC-1α
pathway.
The MG-induced formation of protein adducts with
cell surface or intracellular targets has been shown to initiate
tyro-sine kinase signaling (64),
mitochondrial dysfunction (65)
and the activation of the caspase cascade (66). MG-protein adducts are generated by
irreversible nonenzymatic modification of free amino groups of
proteins, and carbonyl stress results from an imbalance between
reactive carbonyl species levels, the efficiency of scavenger and
detoxification pathways, and accumulation of MG-protein adducts
(67). The modification of
proteins and DNA by MG has emerged as an important endogenous
threat to the functional integrity of the proteome and genome.
Moreover, the crosslinking reaction that occurs during MG amino
acid glycation has been shown to yield the superoxide radical anion
(68). Protein modification by MG
is directed to functional sites where it is associated with
metabolic, structural and functional abnormalities: for example,
mitochondrial dysfunction with increased formation of ROS (66), cell detachment from the
extracellular matrix by decreased integrin binding to MG-modified
extracellular matrix proteins and anoikis (69), and the induction of accelerated
cell senescence (70). In the
present study, the exposure of pancreatic β-cells to MG increased
the formation of protein adducts to levels above those observed in
the controls. However, pre-treatment of the MG-exposed cells with
0.1 µM deoxyactein decreased the formation of protein
adducts significantly. The present data indicate that deoxyactein
may block MG-derived protein adduct formation in pancreatic
β-cells, which may be involved in the mechanism protecting them
against cell death. Therefore, deoxyactein may help prevent the
development of diabetic complications by blocking the MG-mediated
intracellular glycation system.
Under physiological conditions the glyoxalase
system, in which the enzyme glyoxalase I is the rate-limiting step,
efficiently detoxifies highly reactive carbonyls and the AGE
precursor MG to D-lactate and thereby inhibits the formation of
AGEs (5). We demonstrated that
the activity of glyoxalase I was markedly increased by deoxyactein
under conditions of MG-induced toxicity. Glyoxalase I is
ubiquitously distributed in cells and plays an important role in
the regulation of signals related to oxidative stress and AGE
formation (71). The
overexpression of glyoxalase I inhibits intracellular AGE formation
in bovine endothelial cells and prevents hyperglycemia-induced
increases in macromolecular endocytosis (72). By contrast, glyoxalase I
deficiency is associated with increased levels of AGEs (73). The overexpression of glyoxalase I
exerts protective effects in renal ischemia-reperfusion injury via
the reduction of MG accumulation in tubular cells (74). As excessive ROS production has
been implicated in the pathogenesis of diabetes (75), pharmacologic agents that increase
glyoxalase activity may have unique clinical efficacy in the
prevention and treatment of these conditions. In this study, we
demonstrated that deoxyactein significantly prevented the damage to
pancreatic β-cell function induced by MG, by preventing oxidative
stress or enhancing the MG-detoxifying system. Therefore,
deoxyactein may be employed to protect against diseases, such as
diabetes, in which excess the production of ROS has been implicated
as a causal or contributory factor.
In conclusion, the present study indicates that MG
negatively affects β-cell function, and that deoxyactein may
ameliorate MG-induced pancreatic β-cell damage. The mechanisms of
of action of deoxyactein likely involve the potentiation of
SIRT-1/PGC-1α signaling, the increased activity of glyoxalase I,
the elevated gene expression of PDX-1, INS2, SOD and GPX and
protection against detrimental oxidative and inflammatory damage.
Deoxyactein may allow the preservation and/or improvement of β-cell
function in diabetics associated with elevated circulating levels
of toxic aldehydes due to chronic hyperglycemia.
Acknowledgments
This study was supported by a grant from the Korea
Health Technology R&D Project through the Korea Helath Industry
Development Institute (KHIDI), funded by the Ministry of Health and
Welfare, Republic of Korea (grant no. HI14C-2700-020014) and by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education (no.
NRF-2013R1A1A2A10004361).
References
|
1
|
Brownlee M: The pathobiology of diabetic
complications: A unifying mechanism. Diabetes. 54:1615–1625. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kalapos MP: The tandem of free radicals
and methylglyoxal. Chem Biol Interact. 171:251–271. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu CH, Huang SM, Lin JA and Yen GC:
Inhibition of advanced glycation endproduct formation by
foodstuffs. Food Funct. 2:224–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shu T, Zhu Y, Wang H, Lin Y, Ma Z and Han
X: AGEs decrease insulin synthesis in pancreatic β-cell by
repressing Pdx-1 protein expression at the post-translational
level. PLoS One. 6:e187822011. View Article : Google Scholar
|
|
5
|
Thornalley PJ: Glyoxalase I - structure,
function and a critical role in the enzymatic defence against
glycation. Biochem Soc Trans. 31:1343–1348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Solomon TPJ, Knudsen SH, Karstoft K,
Winding K, Holst JJ and Pedersen BK: Examining the effects of
hyperglycemia on pancreatic endocrine function in humans: Evidence
for in vivo glucotoxicity. J Clin Endocrinol Metab. 97:4682–4691.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brownlee M: A radical explanation for
glucose-induced beta cell dysfunction. J Clin Invest.
112:1788–1790. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakuraba H, Mizukami H, Yagihashi N, Wada
R, Hanyu C and Yagihashi S: Reduced beta-cell mass and expression
of oxidative stress-related DNA damage in the islet of Japanese
Type II diabetic patients. Diabetologia. 45:85–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanaka Y, Tran PO, Harmon J and Robertson
RP: A role for glutathione peroxidase in protecting pancreatic beta
cells against oxidative stress in a model of glucose toxicity. Proc
Natl Acad Sci USA. 99:12363–12368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Welsh N, Margulis B, Borg LA, Wiklund HJ,
Saldeen J, Flodström M, Mello MA, Andersson A, Pipeleers DG,
Hellerström C, et al: Differences in the expression of heat-shock
proteins and antioxidant enzymes between human and rodent
pancreatic islets: Implications for the pathogenesis of
insulin-dependent diabetes mellitus. Mol Med. 1:806–820.
1995.PubMed/NCBI
|
|
11
|
Bonora E: Protection of pancreatic
beta-cells: Is it feasible? Nutr Metab Cardiovasc Dis. 18:74–83.
2008. View Article : Google Scholar
|
|
12
|
Chang-Chen KJ, Mullur R and
Bernal-Mizrachi E: Beta-cell failure as a complication of diabetes.
Rev Endocr Metab Disord. 9:329–343. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Philippe J and Raccah D: Treating type 2
diabetes: How safe are current therapeutic agents? Int J Clin
Pract. 63:321–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SH, Park MH, Park SJ, Kim J, Kim YT,
Oh MC, Jeong Y, Kim M, Han JS and Jeon YJ: Bioactive compounds
extracted from Ecklonia cava by using enzymatic hydrolysis protects
high glucose-induced damage in INS-1 pancreatic β-cells. Appl
Biochem Biotechnol. 167:1973–1985. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chakraborty D, Samadder A, Dutta S and
Khuda-Bukhsh AR: Antihyperglycemic potentials of a threatened
plant, Helonias dioica: Antioxidative stress responses and the
signaling cascade. Exp Biol Med (Maywood). 237:64–76. 2012.
View Article : Google Scholar
|
|
16
|
Hanhineva K, Törrönen R, Bondia-Pons I,
Pekkinen J, Kolehmainen M, Mykkänen H and Poutanen K: Impact of
dietary polyphenols on carbohydrate metabolism. Int J Mol Sci.
11:1365–1402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Babu PV, Liu D and Gilbert ER: Recent
advances in understanding the anti-diabetic actions of dietary
flavonoids. J Nutr Biochem. 24:1777–1789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKenna DJ, Jones K, Humphrey S and Hughes
K: Black cohosh: Efficacy, safety, and use in clinical and
preclinical applications. Altern Ther Health Med. 7:93–100.
2001.PubMed/NCBI
|
|
19
|
Choi EM: Deoxyactein isolated from
Cimicifuga racemosa protects osteoblastic MC3T3-E1 cells against
antimycin A-induced cytotoxicity. J Appl Toxicol. 33:488–494. 2013.
View Article : Google Scholar
|
|
20
|
Choi EM: Deoxyactein stimulates osteoblast
function and inhibits bone-resorbing mediators in MC3T3-E1 cells. J
Appl Toxicol. 33:190–195. 2013. View Article : Google Scholar
|
|
21
|
Sheader EA, Benson RS and Best L:
Cytotoxic action of methylglyoxal on insulin-secreting cells.
Biochem Pharmacol. 61:1381–1386. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tiedge M, Lortz S, Munday R and Lenzen S:
Complementary action of antioxidant enzymes in the protection of
bioengineered insulin-producing RINm5F cells against the toxicity
of reactive oxygen species. Diabetes. 47:1578–1585. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jakubowski W and Bartosz G:
2,7-dichlorofluorescin oxidation and reactive oxygen species: What
does it measure? Cell Biol Int. 24:757–760. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thornalley PJ and Tisdale MJ: Inhibition
of proliferation of human promyelocytic leukaemia HL60 cells by
S-D-lactoylglutathione in vitro. Leuk Res. 12:897–904. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rains JL and Jain SK: Oxidative stress,
insulin signaling, and diabetes. Free Radic Biol Med. 50:567–575.
2011. View Article : Google Scholar
|
|
26
|
Suh KS, Rhee SY, Kim YS and Choi EM:
Inhibitory effect of apocynin on methylglyoxal-mediated glycation
in osteoblastic MC3T3-E1 cells. J Appl Toxicol. 35:350–357. 2015.
View Article : Google Scholar
|
|
27
|
Vulesevic B, McNeill B, Giacco F, Maeda K,
Blackburn NJ, Brownlee M, Milne RW and Suuronen EJ:
Methylglyoxal-induced endothelial cell loss and inflammation
contribute to the development of diabetic cardiomyopathy. Diabetes.
65:1699–1713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun YP, Gu JF, Tan XB, Wang CF, Jia XB,
Feng L and Liu JP: Curcumin inhibits advanced glycation end
product-induced oxidative stress and inflammatory responses in
endothelial cell damage via trapping methylglyoxal. Mol Med Rep.
13:1475–1486. 2016.PubMed/NCBI
|
|
29
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Multiple pathways of cytochrome c release from mitochondria in
apoptosis. Biochim Biophys Acta. 1757:639–647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Petit JM, Maftah A, Ratinaud MH and Julien
R: 10N-nonyl acridine orange interacts with cardiolipin and allows
the quantification of this phospholipid in isolated mitochondria.
Eur J Biochem. 209:267–273. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jornayvaz FR and Shulman GI: Regulation of
mitochondrial biogenesis. Essays Biochem. 47:69–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nemoto S, Fergusson MM and Finkel T: SIRT1
functionally interacts with the metabolic regulator and
transcriptional coactivator PGC-1{alpha}. J Biol Chem.
280:16456–16460. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Salahuddin P, Rabbani G and Khan RH: The
role of advanced glycation end products in various types of
neurodegenerative disease: a therapeutic approach. Cell Mol Biol
Lett. 19:407–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sherry NA, Tsai EB and Herold KC: Natural
history of β-cell function in type 1 diabetes. Diabetes. 54(Suppl
2): S32–S39. 2005. View Article : Google Scholar
|
|
35
|
Dhar A, Dhar I, Jiang B, Desai KM and Wu
L: Chronic methylglyoxal infusion by minipump causes pancreatic
beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley
rats. Diabetes. 60:899–908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ahlgren U, Jonsson J, Jonsson L, Simu K
and Edlund H: beta-cell-specific inactivation of the mouse
Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and
maturity onset diabetes. Genes Dev. 12:1763–1768. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu H, MacFarlane WM, Tadayyon M, Arch JR,
James RF and Docherty K: Insulin stimulates pancreatic-duodenal
homoeobox factor-1 (PDX1) DNA-binding activity and insulin promoter
activity in pancreatic beta cells. Biochem J. 344:813–818.
1999.PubMed/NCBI
|
|
38
|
Leibowitz G, Ferber S, Apelqvist A, Edlund
H, Gross DJ, Cerasi E, Melloul D and Kaiser N: IPF1/PDX1 deficiency
and beta-cell dysfunction in Psammomys obesus, an animal With type
2 diabetes. Diabetes. 50:1799–1806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Macfarlane WM, Shepherd RM, Cosgrove KE,
James RF, Dunne MJ and Docherty K: Glucose modulation of insulin
mRNA levels is dependent on transcription factor PDX-1 and occurs
independently of changes in intracellular Ca2+.
Diabetes. 49:418–423. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kawamori D, Kaneto H, Nakatani Y, Matsuoka
TA, Matsuhisa M, Hori M and Yamasaki Y: The forkhead transcription
factor Foxo1 bridges the JNK pathway and the transcription factor
PDX-1 through its intracellular translocation. J Biol Chem.
281:1091–1098. 2006. View Article : Google Scholar
|
|
41
|
Hagman DK, Hays LB, Parazzoli SD and
Poitout V: Palmitate inhibits insulin gene expression by altering
PDX-1 nuclear localization and reducing MafA expression in isolated
rat islets of Langerhans. J Biol Chem. 280:32413–32418. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ardestani A, Sauter NS, Paroni F,
Dharmadhikari G, Cho JH, Lupi R, Marchetti P, Oberholzer J, Conte
JK and Maedler K: Neutralizing interleukin-1beta (IL-1beta) induces
beta-cell survival by maintaining PDX1 protein nuclear
localization. J Biol Chem. 286:17144–17155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Osborn O, Brownell SE, Sanchez-Alavez M,
Salomon D, Gram H and Bartfai T: Treatment with an Interleukin 1
beta antibody improves glycemic control in diet-induced obesity.
Cytokine. 44:141–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mandrup-Poulsen T: The role of
interleukin-1 in the pathogenesis of IDDM. Diabetologia.
39:1005–1029. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maedler K, Sergeev P, Ris F, Oberholzer J,
Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA and Donath MY:
Glucose-induced beta cell production of IL-1beta contributes to
glucotoxicity in human pancreatic islets. J Clin Invest.
110:851–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Börjesson A and Carlsson C: Altered
proinsulin conversion in rat pancreatic islets exposed long-term to
various glucose concentrations or interleukin-1beta. J Endocrinol.
192:381–387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Abordo EA, Minhas HS and Thornalley PJ:
Accumulation of alpha-oxoaldehydes during oxidative stress: A role
in cytotoxicity. Biochem Pharmacol. 58:641–648. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Miyazawa N, Abe M, Souma T, Tanemoto M,
Abe T, Nakayama M and Ito S: Methylglyoxal augments intracellular
oxidative stress in human aortic endothelial cells. Free Radic Res.
44:101–107. 2010. View Article : Google Scholar
|
|
49
|
Newsholme P, Haber EP, Hirabara SM,
Rebelato EL, Procopio J, Morgan D, Oliveira-Emilio HC, Carpinelli
AR and Curi R: Diabetes associated cell stress and dysfunction:
Role of mitochondrial and non-mitochondrial ROS production and
activity. J Physiol. 583:9–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Robertson RP: Chronic oxidative stress as
a central mechanism for glucose toxicity in pancreatic islet beta
cells in diabetes. J Biol Chem. 279:42351–42354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Petrosillo G, Di Venosa N, Pistolese M,
Casanova G, Tiravanti E, Colantuono G, Federici A, Paradies G and
Ruggiero FM: Protective effect of melatonin against mitochondrial
dysfunction associated with cardiac ischemia-reperfusion: Role of
cardiolipin. FASEB J. 20:269–276. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Martín MA, Serrano AB, Ramos S, Pulido MI,
Bravo L and Goya L: Cocoa flavonoids up-regulate antioxidant enzyme
activity via the ERK1/2 pathway to protect against oxidative
stress-induced apoptosis in HepG2 cells. J Nutr Biochem.
21:196–205. 2010. View Article : Google Scholar
|
|
53
|
Robertson RP and Harmon JS: Pancreatic
islet beta-cell and oxidative stress: The importance of glutathione
peroxidase. FEBS Lett. 581:3743–3748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Muscogiuri G, Salmon AB, Aguayo-Mazzucato
C, Li M, Balas B, Guardado-Mendoza R, Giaccari A, Reddick RL, Reyna
SM, Weir G, et al: Genetic disruption of SOD1 gene causes glucose
intolerance and impairs β-cell function. Diabetes. 62:4201–4207.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Moriscot C, Pattou F, Kerr-Conte J,
Richard MJ, Lemarchand P and Benhamou PY: Contribution of
adenoviral-mediated superoxide dismutase gene transfer to the
reduction in nitric oxide-induced cytotoxicity on human islets and
INS-1 insulin-secreting cells. Diabetologia. 43:625–631. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kubisch HM, Wang J, Bray TM and Phillips
JP: Targeted overexpression of Cu/Zn superoxide dismutase protects
pancreatic beta-cells against oxidative stress. Diabetes.
46:1563–1566. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mysore TB, Shinkel TA, Collins J, Salvaris
EJ, Fisicaro N, Murray-Segal LJ, Johnson LE, Lepore DA, Walters SN,
Stokes R, et al: Overexpression of glutathione peroxidase with two
isoforms of superoxide dismutase protects mouse islets from
oxidative injury and improves islet graft function. Diabetes.
54:2109–2116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nishikawa T, Edelstein D, Du XL, Yamagishi
S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP,
et al: Normalizing mitochondrial superoxide production blocks three
pathways of hyperglycaemic damage. Nature. 404:787–790. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rodgers JT, Lerin C, Gerhart-Hines Z and
Puigserver P: Metabolic adaptations through the PGC-1 alpha and
SIRT1 pathways. FEBS Lett. 582:46–53. 2008. View Article : Google Scholar
|
|
60
|
Cheng HL, Mostoslavsky R, Saito S, Manis
JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW and Chua KF:
Developmental defects and p53 hyperacetylation in Sir2 homolog
(SIRT1)-deficient mice. Proc Natl Acad Sci USA. 100:10794–10799.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et
al: Stress-dependent regulation of FOXO transcription factors by
the SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Finkel T: Cell biology: A clean energy
programme. Nature. 444:151–152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hock MB and Kralli A: Transcriptional
control of mitochondrial biogenesis and function. Annu Rev Physiol.
71:177–203. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Akhand AA, Kato M, Suzuki H, Liu W, Du J,
Hamaguchi M, Miyata T, Kurokawa K and Nakashima I: Carbonyl
compounds cross-link cellular proteins and activate
protein-tyrosine kinase p60c-Src. J Cell Biochem. 72:1–7. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rosca MG, Monnier VM, Szweda LI and Weiss
MF: Alterations in renal mitochondrial respiration in response to
the reactive oxoaldehyde methylglyoxal. Am J Physiol Renal Physiol.
283:F52–F59. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Akhand AA, Hossain K, Mitsui H, Kato M,
Miyata T, Inagi R, Du J, Takeda K, Kawamoto Y, Suzuki H, et al:
Glyoxal and methylglyoxal trigger distinct signals for map family
kinases and caspase activation in human endothelial cells. Free
Radic Biol Med. 31:20–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Stitt AW, Jenkins AJ and Cooper ME:
Advanced glycation end products and diabetic complications. Expert
Opin Investig Drugs. 11:1205–1223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yim HS, Kang SO, Hah YC, Chock PB and Yim
MB: Free radicals generated during the glycation reaction of amino
acids by methylglyoxal. A model study of protein-cross-linked free
radicals. J Biol Chem. 270:28228–28233. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pedchenko VK, Chetyrkin SV, Chuang P, Ham
AJ, Saleem MA, Mathieson PW, Hudson BG and Voziyan PA: Mechanism of
perturbation of integrin-mediated cell-matrix interactions by
reactive carbonyl compounds and its implication for pathogenesis of
diabetic nephropathy. Diabetes. 54:2952–2960. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sejersen H and Rattan SI:
Dicarbonyl-induced accelerated aging in vitro in human skin
fibroblasts. Biogerontology. 10:203–211. 2009. View Article : Google Scholar
|
|
71
|
Barati MT, Merchant ML, Kain AB, Jevans
AW, McLeish KR and Klein JB: Proteomic analysis defines altered
cellular redox pathways and advanced glycation end-product
metabolism in glomeruli of db/db diabetic mice. Am J Physiol Renal
Physiol. 293:F1157–F1165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shinohara M, Thornalley PJ, Giardino I,
Beisswenger P, Thorpe SR, Onorato J and Brownlee M: Overexpression
of glyoxalase-I in bovine endothelial cells inhibits intracellular
advanced glycation endproduct formation and prevents
hyperglycemia-induced increases in macromolecular endocytosis. J
Clin Invest. 101:1142–1147. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Miyata T, van Ypersele de Strihou C,
Imasawa T, Yoshino A, Ueda Y, Ogura H, Kominami K, Onogi H, Inagi
R, Nangaku M, et al: Glyoxalase I deficiency is associated with an
unusual level of advanced glycation end products in a hemodialysis
patient. Kidney Int. 60:2351–2359. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kumagai T, Nangaku M, Kojima I, Nagai R,
Ingelfinger JR, Miyata T, Fujita T and Inagi R: Glyoxalase I
overexpression ameliorates renal ischemia-reperfusion injury in
rats. Am J Physiol Renal Physiol. 296:F912–F921. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
McLellan AC, Thornalley PJ, Benn J and
Sonksen PH: Glyoxalase system in clinical diabetes mellitus and
correlation with diabetic complications. Clin Sci (Lond). 87:21–29.
1994. View Article : Google Scholar
|