Introduction
Gliomas account for 29% of all primary brain tumors
and are classified by the World Health Organization (WHO) into 4
grades of malignancy (1). Grade
IV astrocytoma [glioblastoma multiforme (GBM)] is the most
aggressive form of brain tumor in adults and is characterized by
rapid cell proliferation, immunosuppressive capability and high
malignancy (2). GBM is typically
resistant to surgery, radiation, chemotherapy and drug treatment.
Thus, there is an urgent need to elucidate the underlying molecular
mechanisms involved in the genesis and progression of glioma, and
to develop effective therapies.
Transforming growth factor-β (TGF-β) has been
demonstrated to function as an oncogenic factor in high-grade
glioma, and an increased TGF-β expression can enhance glioma
outgrowth and prevent glioma-initiating cell (GIC) differentiation
(3,4). The phenomenon has also been shown to
be associated with a high expression of leukemia-inhibitory factor
(LIF) induced by the TGF-β/Smad signaling pathway (5). In general, TGF-β functions by
binding to type I and II TGF-β receptors on the cell membrane. Upon
binding TGF-β, type II receptors phosphorylate and activate type I
receptors, which then propagate the signal by phosphorylating the
receptor-regulated Smads (R-Smads), Smad2 and Smad3. These R-Smad
members can form a complex with Smad4, which subsequently
translocates to the nucleus and governs target gene expression
(6). Therefore, these signal
transducing proteins of the TGF-β pathway are potential therapeutic
targets for effectively treating patients with glioma.
MicroRNAs (miRNAs or miRs) comprise a class of small
non-coding RNAs, which regulate gene expression during both normal
and neoplastic development. miRNAs are generated by RNA polymerase
II as longer precursor RNAs in the nucleus and contain hairpins.
Subsequently, precursor RNAs are transported to the cytoplasm and
are processed into 21–23 nucleotide-long RNA molecules by the RNase
III enzymes Drosha and Dicer (7,8).
Mature miRNAs regulate gene expression by inhibiting mRNA
translation or inducing mRNA degradation after binding to the
complementary sequences in the 3′-untranslated regions (3′-UTRs) of
target mRNAs. Emerging data have indicated that miRNAs are involved
in tumor proliferation and apoptosis and can function either as
oncogenes or tumor suppressor genes (9,10).
A number of miRNAs have been identified, including some that are
involved in the malignant progression of glioma (11,12). In particular, miR-124 plays a
crucial role in neurogenesis and stimulates neuronal
differentiation. The expression level of miR-124 is high in the
central nervous system, but is low or even absent in high-grade
glioma (13,14). The loss of miR-124 expression has
been shown to enhance stem cell-like traits and to increase the
invasive ability of glioma cells (15). Furthermore, miR-124 overexpression
in GBM has been shown to result in decreased migration and invasion
(16,17). In addition, miR-124 has been shown
to inhibit the signal transducer and activator of transcription 3
(Stat3)-signaling pathway to promote T cell-mediated immune
clearance of glioma (18).
Therefore, miR-124 may serve as an effective therapeutic target if
its functional network is clarified.
Using target-prediction software programs
(TargetScan and Segal Lab), we noted that putative pairing regions
for miR-124-3p exist within the 3′-UTR of rat and human Smad4 mRNA.
In this study, our primary aim was to explore the associations
between miR-124 and Smad4, as well as their effects on glioma.
Establishing the mechanism whereby miR-124 regulates glioma
progression may provide fundamental supportive evidence for novel
clinical therapies for glioma.
Materials and methods
Animals
All animals were supplied by the Experimental Animal
Center of Jilin University (Changchun, China). All procedures
involving animals were approved by the Ethics Committee of Jilin
University and conformed to regulatory standards.
Cell culture
Rat C6 glioma (Cell Bank of the Chinese Academy of
Science in Shanghai, China) were cultured in DMEM/F12 supplemented
with 10% fetal bovine serum (FBS; both from Gibco, Grand Island,
NY, USA), 100 U/ml penicillin and 100 µg/ml streptomycin
(Genview, Calimesa, CA, USA), at 37°C in a humidified atmosphere
with 5% CO2. The cells were passaged at a ratio of 1:3
when they became confluent.
Astrocytes were isolated from the cortices of
neonatal rats (1–3 days old, n=6) as previously described by
Schwartz and Wilson with some modifications (19). Following the removal of all
meninges carefully from the brain surface, the rat cortex tissue
was dissected from brain and completely triturated using
fire-polished Pasteur pipettes. The destroyed tissues were then
treated with 0.25% Trypsin/EDTA for 30 min, and passed through 40
µm nylon mesh and centrifuged to collect the enzymatically
dissociated cells. The collected cells were suspended in the
above-mentioned complete culture medium, and plated in 25T flasks
and cultured for almost 2 weeks; the medium was replaced every 2
days.
When the cells were confluent, the flasks were
shaken at 250 rpm overnight at 37°C. After replacing with fresh
medium, the cells were treated with cytosine arabinoside (5
µM; Sigma, St. Louis, MO, USA) for 48 h. Th cells were then
subcultured to the next passage. The percentage of GFAP-expressing
cells at passage 1 in these cultures was found to be >95%.
Cell transfection
To perform cell transfection, the C6 cells were
plated at 2×105 cells/well. After 24 h, the cells were
transfected with the miR-124 mimic, negative control (NC), small
interfering RNA against Smad4 (siR-Smad4), or an siR-NC (50 nM;
GenePharma, Shanghai, China), using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA), after which they were incubated at 37°C for 5
h. Subsequently, the supernatant was replaced with fresh medium,
and the cells were cultured for a further 24 h.
RNA extraction and RT-qPCR
Total RNA was extracted using TRIzol reagent
(Invitrogen). To analyze miRNA-124 expression, total RNA was
polyadenylated and reverse transcribed using the All-in-One™ miRNA
First-Strand cDNA Synthesis kit (GeneCopoeia, Rockville, MD, USA).
The level of miRNA-124 (RmiRQP0074; GeneCopoeia) produced in the
cells and tissues was detected using the All-in-OneTM miRNA RT-qPCR
Detection kit (GeneCopoeia). Specific primers (GeneCopoeia) were
used to amplify miR-124-3p, and RnU6 was used as a normalization
control for miRNA expression.
To analyze mRNA expression, cDNA was synthesized
using the GoScript™ Reverse Transcription system (Promega, Madison,
WI, USA). Real-time PCR reactions were performed using SYBR Premix
Dimer-Eraser (Takara, Dalian, China). GAPDH was used as a
normalization control for mRNA expression. The qPCR primer
sequences were as follows: GAPDH forward,
5′-AGACAGCCGCATCTTCTTGT-3′ and reverse, 5′-CTTGCCGTGGGTAGAGTCAT-3′;
Smad4 forward, 5′-CCATCAGTCTGTCTGCTGCT-3′ and reverse,
5′-TGATGCTCTGTCTCGGGTAG-3′; LIF forward, 5′-GTGCCAATGCCCTCTTTATT-3′
and reverse, 5′-TGGTCTTCTCTGTCCCATTG-3′. Relative quantification
numbers were calculated using the 2−ΔΔCt method, which
is based on the ratio of gene expression between an experimental
group and a control group.
Immunofluorescence staining
The C6 cells were fixed in 4% paraformaldehyde.
Normal goat serum was used to block nonspecific binding. A rabbit
anti-Smad2/3 antibody (1:800; #8685; Cell Signaling Technology,
Inc., Danvers, MA, USA) and a mouse anti-Smad4 antibody (1:100;
sc-7966; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used
as a primary antibody. Following an overnight incubation with the
primary antibody at 4°C, the cells were further incubated with goat
anti-rabbit Alexa Fluor 594 (1:200; #111-585-144) and goat
anti-mouse Alexa Fluor 488 (1:200; #111-545-146)-conjugated
secondary antibodies (Jackson ImmunoResearch Inc., West Grove, PA,
USA) for 1 h at room temperature. Nuclei were stained with Hoechst
33342 (Invitrogen).
Western blot analysis
Total protein samples were collected from the C6
cells after lysing them in RIPA cell lysis buffer containing PMSF
and phosphorylase inhibitors (Beijing Dingguo Changsheng
Biotechnology Co., Ltd., Beijing, China). The lysates were
centrifuged at 12,000 rpm for 15 min at 4°C. Protein concentrations
were determined using the Bicinchoninic Acid Protein Assay kit
(Beyotime Institute of Biotechnology, Haimen, China). The proteins
were denatured for 5 min. The proteins were then concentrated in 5%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) stacking gels, separated on 10% SDS-PAGE gels, and
transferred onto PVDF membranes (Roche, Mannheim, Germany). The
membranes were then blocked in 5% non-fat milk and incubated with
antibodies against Smad4 (1:1,000; #38454), Stat3 (1:1,000;
#12640), phosphorylated-Stat3 (p-Stat3; 1:2,000; #9145), c-Myc
(1:1,000; #5605), cleaved caspase-3 (1:1,000; #9664) and GAPDH
(1:1,000; #5174) (all from Cell Signaling Technology, Inc.). The
PVDF membranes were then washed with Tris-buffered saline
containing 1% Tween-20 and probed with the secondary antibody
peroxidase-conjugated goat anti-rabbit IgG (H+L) (1:5,000; #111453;
Beijing Dingguo Changsheng Biotechnology Co., Ltd.) for 2 h at room
temperature. The signal was detected by chemiluminescence using the
ECL-Plus detection system (Amersham Pharmacia Biotech, Arlington
Heights, IL, USA). The densitometric intensities of protein bands
were semi-quantified using Bandscan 5.0 (Glyko Biomedical, Novato,
CA, USA) software and values were normalized to those of GAPDH for
each sample.
Cell proliferation assay
The viability of the C6 cells transfected with
miR-124 mimics, NC, siR-Smad4, or siR-NC was assessed at 3 time
points (days 1, 2, and 3) after seeding 2×103
transfected cells/well into 96-well culture plates. Cell
proliferation was measured using the MTT Assay kit (Beyotime
Institute of Biotechnology). According to the manufacturer's
instructions, the absorbance was measured at 450 nm using a
Infinite M200 PRO NanoQuant (Tecan, Männedorf, Switzerland). Three
independent experiments were performed for each group.
Enzyme-linked immunosorbent assays
(ELISAs)
The C6 cells were cultured in DMEM/F-12 supplemented
with 10% FBS until they reached 70% confluence. Subsequently, the
C6 cells were transfected with the miR-124 mimic, NC, siR-Smad4 or
siR-NC (50 nM final concentration), incubated at 37°C for 5 h, and
the supernatant was then replaced with fresh medium and the cells
were grown for a further 24 h before the medium was collected and
filter-sterilized. LIF secretion was quantified using an LIF ELISA
kit (Elabscience, Wuhan, China), according to the manufacturer's
instructions.
Target gene bioinformatic prediction and
luciferase reporter assay
The target site of rno-miR-124-3p within the 3′-UTR
region of SMAD4 mRNA was predicted using the target-prediction
software programs (http://www.targetscan.org/vert_71/) and Segal Lab
online microRNA prediction tool (https://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html).
For luciferase reporter assay, the 3′-UTR of Smad4
was cut from the PUC57-Smad4 vector (Promega) and inserted into the
psiCHECK-2 vector (Promega), using the XboI (New England
BioLabs, Ipswich, MA, USA) and NotI (New England BioLabs)
restriction enzyme sites, and the resulting vector was designated
as psiCHECK-Smad4. The predicted miR-124 binding site of the Smad4
3′-UTR was mutated and then inserted into the psiCHECK-2 vector,
which was designated as psiCHECK-Smad4-mut. Constructs were
verified by sequencing. 293 cells (Cell Bank of the Chinese Academy
of Sciences) were cultured in 48-well plates in DMEM/F-12
supplemented with 10% FBS without antibiotics until they reached
~80% confluency. The cells were then co-transfected with
rno-miR-124-3p or miR-NC (50 nM) and psiCHECK-Smad4 or
psiCHECK-Smad4-mut (100 pg), using Lipofectamine 2000. Following
transfection for 5 h, the medium was replaced with fresh mediaum
containing 10% FBS. Following a 48-h transfection, light emission
was measured. The luciferase assay was conducted using the Dual-Glo
Luciferase Assay system (Promega), following the manufacturer's
instructions. Light emission was measured using a
GloMax® 96 Microplate Luminometer (Promega). The ratios
of Renilla versus firefly luciferase signals served as a
measurement of reporter activity normalized for the transfection
efficiency.
Statistical analysis
Experimental data are presented as the means ±
standard deviation. All statistical analyses were performed and all
graphs were prepared using GraphPad Prism 6 statistical software
(GraphPad Software, Inc., La Jolla, CA, USA). Differences were
considered statistically significant with a value of P<0.05.
Results
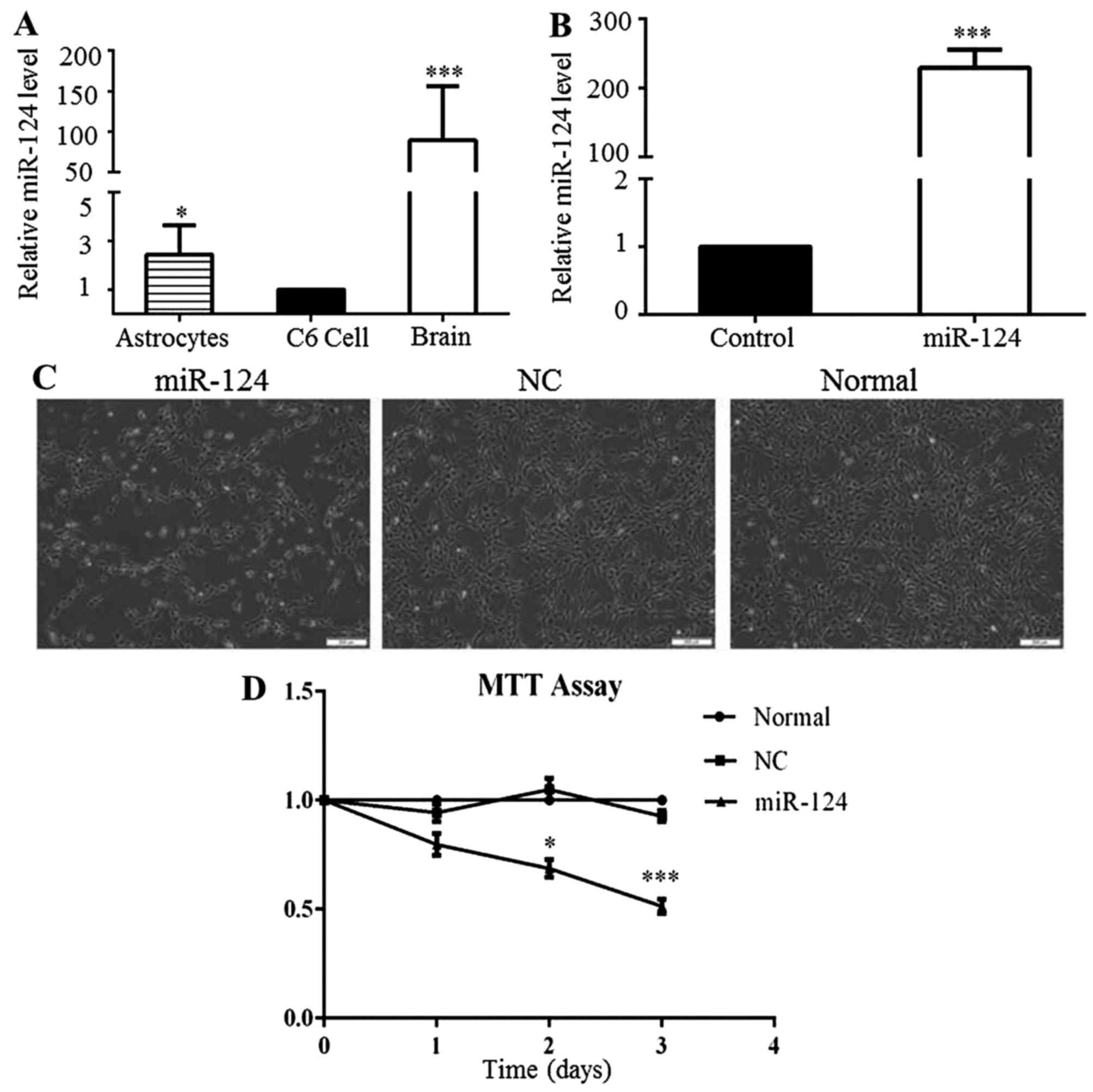
miR-124 expression is low in C6 cells and
the overexpression of miR-124 inhibits cell proliferation
To explore the functional roles of miR-124 in the
proliferation and apoptosis of glioma cells, miR-124 expression was
analyzed in C6 cells, non-tumor rat brain tissues and normal rat
astrocytes by RT-qPCR. The level of miR-124 expression in the C6
cells was significantly lower than that in the non-tumor rat brain
tissues and astrocytes (Fig. 1A).
To overexpress miR-124, the C6 cells were transfected with miR-124
mimics for 48 h, and the control cells were transfected separately
with negative control (NC) for 48 h. The level of miR-124
distinctly increased in the miR-124 mimic-transfected group,
compared to that observed in the NC group (Fig. 1B). In addition, we observed that
C6 cell growth decreased following miR-124 overexpression (Fig. 1C). MTT assays were conducted to
confirm the changes in the proliferation of the C6 cells following
the overexpression of miR-124. The results demonstrated that
miR-124 upregulation significantly inhibited the proliferation of
C6 cells for up to 3 days (Fig.
1D). These results implied that low miR-124 expression may play
a crucial role in GBM progression.
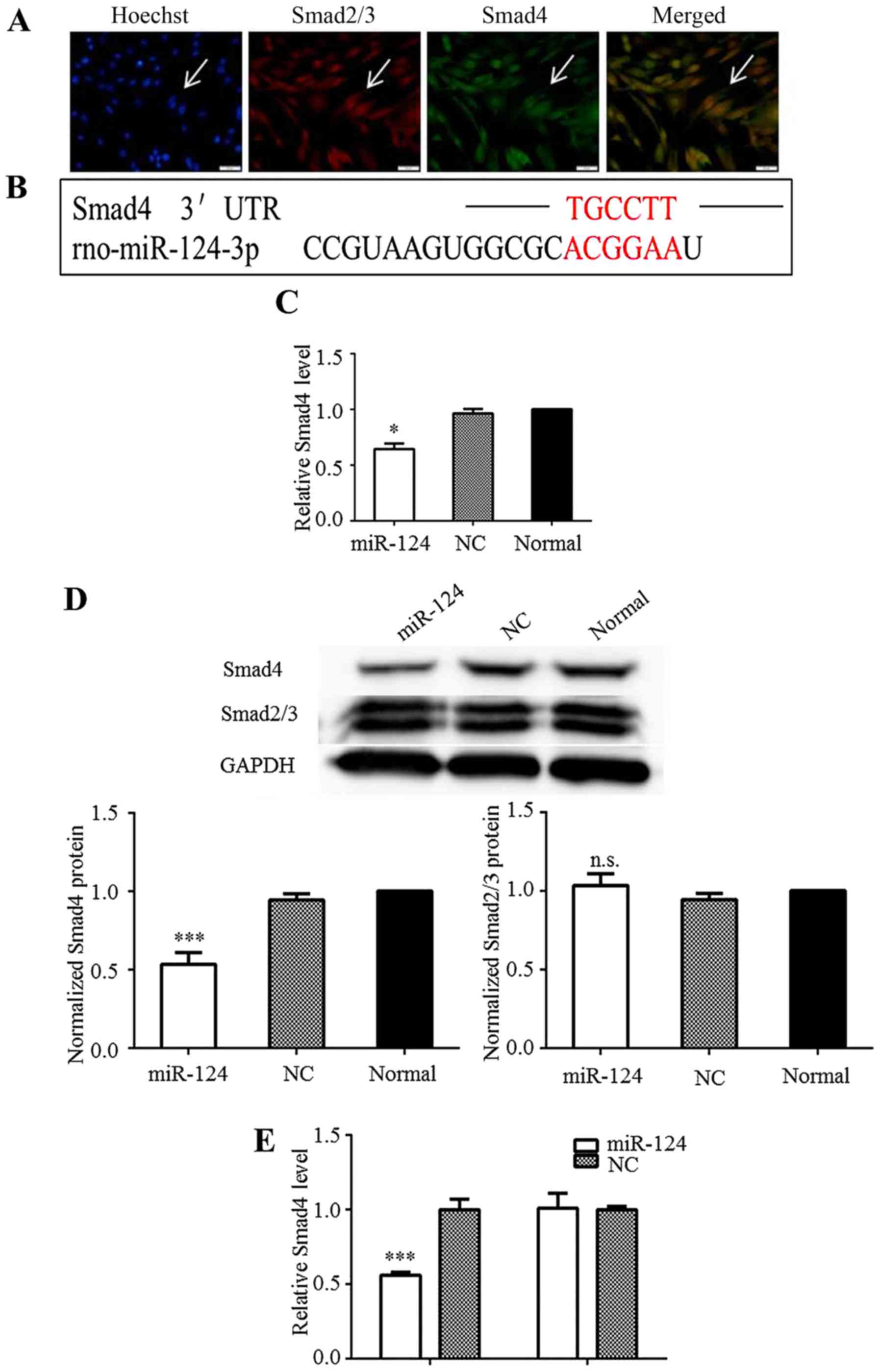
Smad4 is a direct target of miR-124
The TGF-β/Smad pathway is considered a potential
target for the clinical treatment of glioma. As shown in this
study, the Smad2, Smad3, and Smad4 proteins were strongly expressed
in the C6 cells under normal culture conditions, indicating that
the TGF-β/Smad pathway was activated (Fig. 2A). Using target-prediction
software programs (TargetScan and Segal Lab), the 1921bp–1926bp of
nucleotides sequence within 3′-UTR of the rat Smad4 mRNA
(NM_019275.3) was identified as a theoretical seed site of miR-124
(Fig. 2B). Hence, miR-124 may
affect downstream Smad4 effector functions in the cytoplasm of C6
cells by regulating Smad4 expression. RT-qPCR analysis revealed
that the Smad4 mRNA level was significantly decreased in the C6
cells upon the overexpression of miR-124 (Fig. 2C). The transient overexpression of
miR-124 in the C6 cells also downregulated Smad4 at the protein
level (Fig. 2D). The protein
levels of Smad2/3 were measured to determine whether miR-124
regulates Smad2/3 expression. However, no marked differences in
their expression were observed (Fig.
2D). These results implied that miR-124 regulated Smad4
expression without influencing Smad2/3 expression.
To confirm that Smad4 is indeed directly targeted by
miR-124, we examined whether miR-124 recognized the 3′-UTR of Smad4
mRNA by performing Dual-Luciferase reporter assays. The 3′-UTR of
Smad4 was inserted into the the psiCHECK-2 vector, after which
several constructs were sequenced. Smad4-mut was created as a
control vector. The data indicated that miR-124 overexpression
significantly inhibited the luciferase activity of psiCHECK-smad4,
but not that of psiCHECK-smad4-mut (Fig. 2E). These results confirmed that
miR-124 targets the 3′-UTR of Smad4 to directly suppress its
expression.
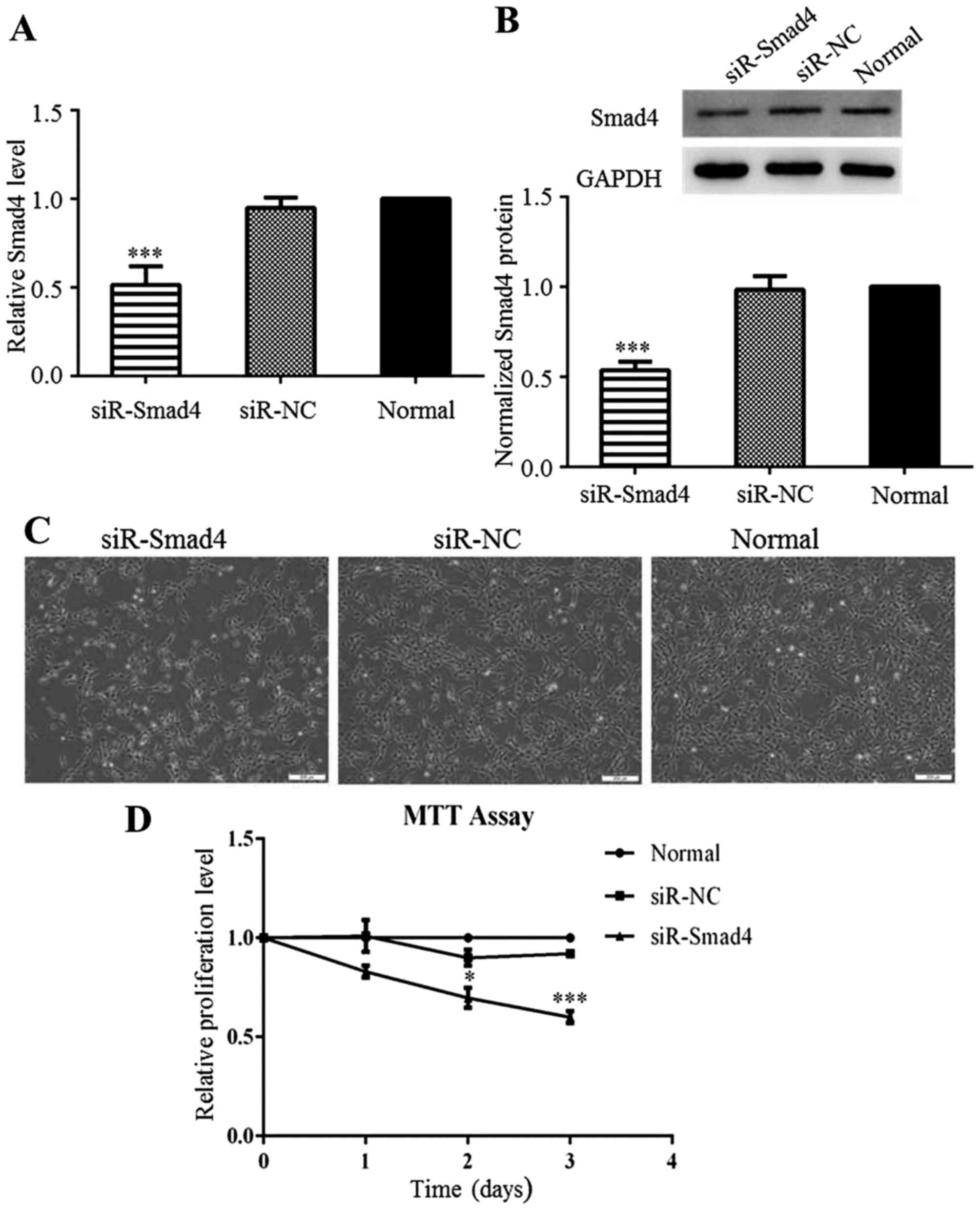
Downregulation of Smad4 inhibits C6 cell
proliferation
The complete pathological significance of Smad4 in
glioma remains unclear. Thus, in this study, to explore the
potential role of Smad4 in C6 cells, Smad4 was knocked down by
transfection with siR-Smad4 or siR-NC. AT 24 h after transfection,
Smad4 expression was significantly decreased in the siR-Smad4
group, compared to that in the siR-NC group (Fig. 3A and B). As with miR-124
overexpression, Smad4 downregulation also restricted the
proliferation of the C6 cells (Fig.
3C). The results from MTT assay also confirmed that C6 cell
proliferation was inhibited by the knockdown of Smad4 (Fig. 3D). We can thus conclude that Smad4
downregulation inhibited C6 cell proliferation, and thus the
inhibitory effects of miR-124 on C6 cell growth interfered with
Smad4 expression.
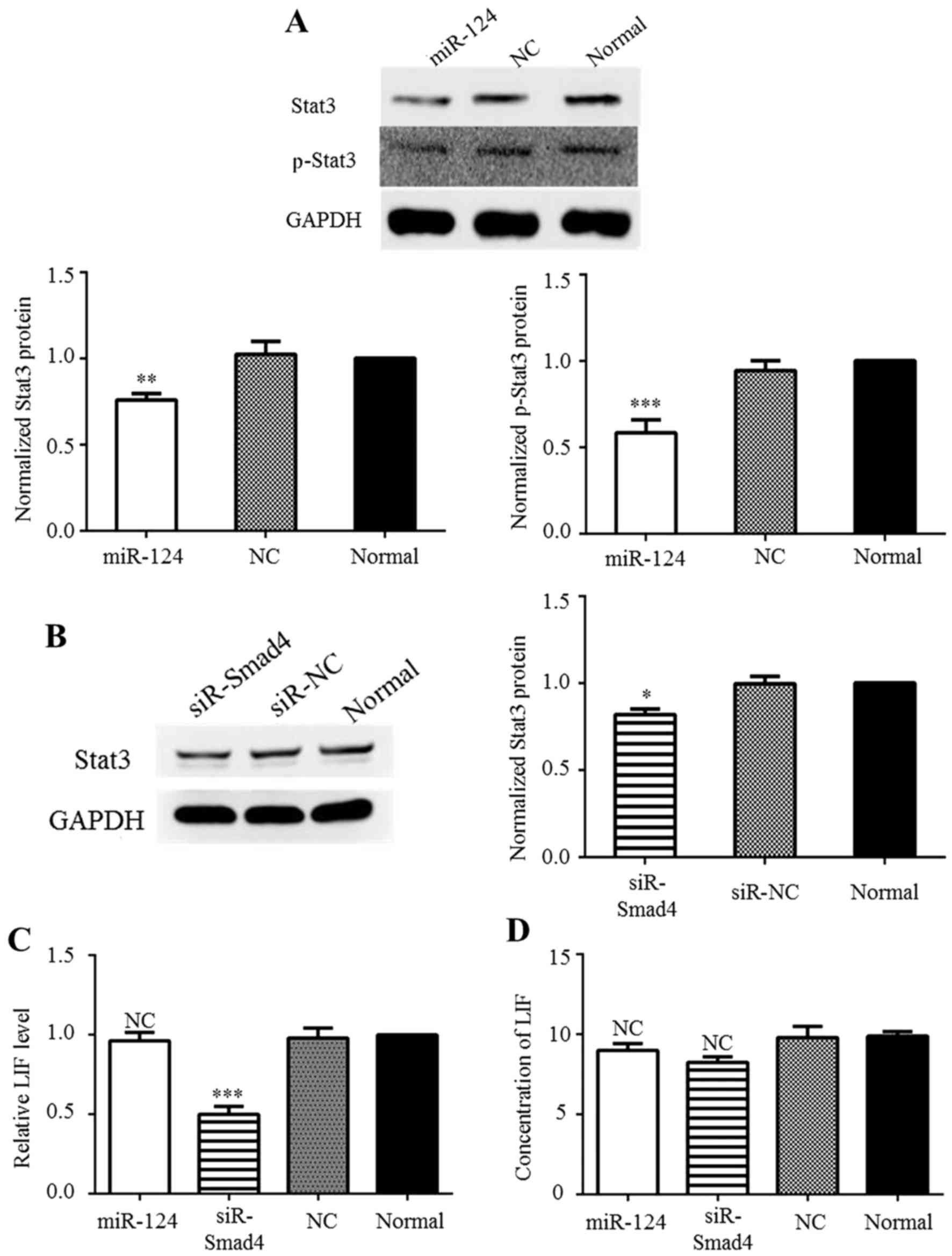
miR-124 reduces Stat3 expression by
directly targeting Stat3 and through the modulation of Smad4
expression
Based on the TargetScan results (www.targetscan.com) and previous research (18), miR-124 binds to the 3′-UTR of
Stat3. Theoretically, miR-124 can inhibit Stat3 expression and thus
overcome Stat3-mediated downstream suppression in GBM. Western blot
analysis was performed to confirm the effects of miR-124 on the
Jak/Stat pathway. In agreement with a previous study (18), Stat3 and p-Stat3 production was
inhibited at the protein level by miR-124 overexpression (Fig. 4A). Previously, Peñuelas et
al demonstrated that the TGF-β/Smad complex can activate the
Jak/Stat Pathway via the induction of LIF in patient-derived GICs
(5). Thus, in this study, to
determine whether differences in Smad4 expression affect Stat3
expression, the C6 cells were transfected with siR-Smad4 or siR-NC,
after which western blot analysis was conducted. The results
revealed that reducing Smad4 expression significantly decreased
Stat3 expression (Fig. 4B). In
addition, to determine whether Stat3 expression was also regulated
by miR-124 through LIF induction, the level of LIF was detected by
RT-qPCR and ELISA. The RT-qPCR results demonstrated that siR-Smad4
reduced the mRNA level of LIF (Fig.
4C), but the ELISA results revealed that no significant change
occurred at the protein level (Fig.
4D) These results may be due to the basal level of LIF
secretion which is low in C6 cells. Taken together, these results
indicate that Stat3 expression can be directly suppressed by
miR-124 and by Smad4-dependent suppression in C6 cells, but not
through LIF induction.
miR-124 inhibits the proliferation of C6
cells and decreases c-Myc expression
Zhu et al reported that TGF-β activates both
Smad-dependent and Smad-independent pathways, recruiting Smad4 to
the Smad-binding element (SBE) of the c-Myc promoter, thereby
promoting cell proliferation (20). In this study, c-Myc expression
significantly decreased in the presence of miR-124 mimics and
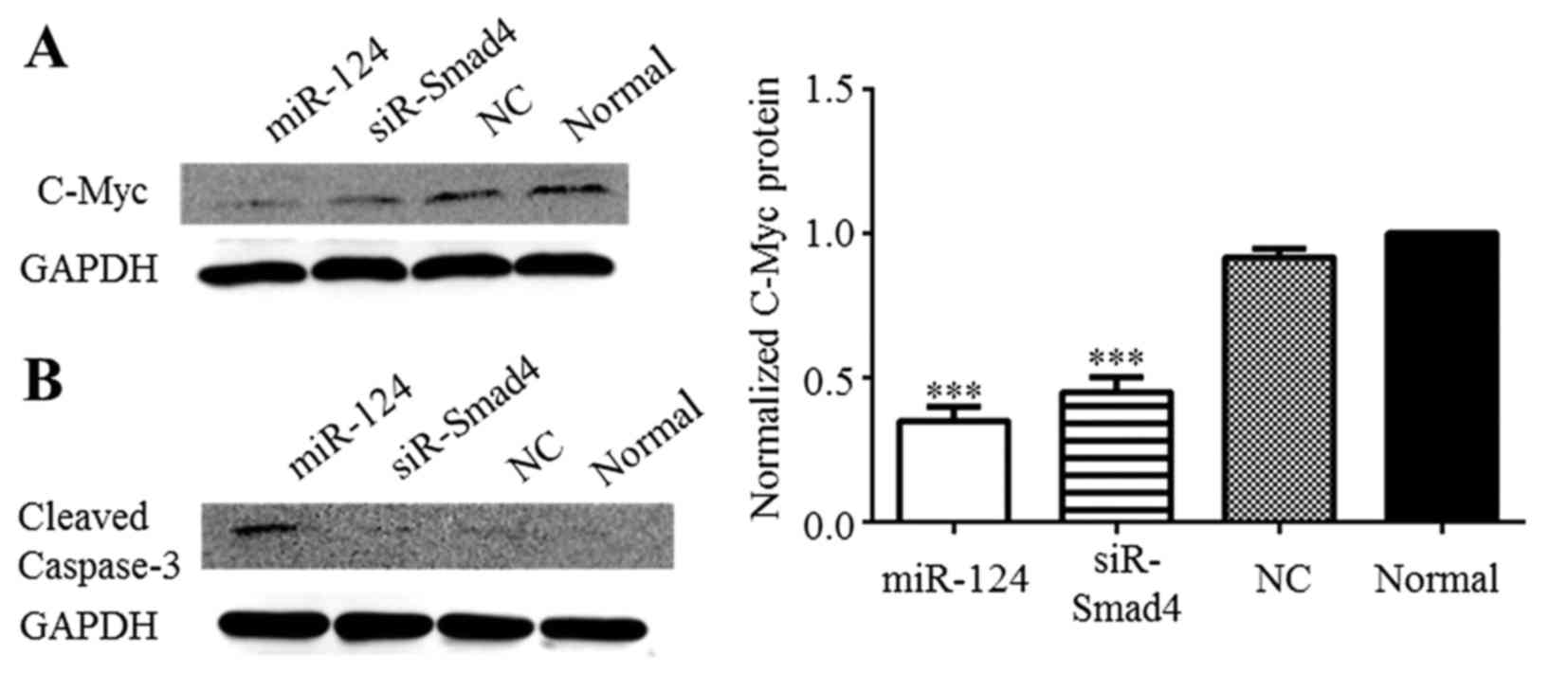
siR-Smad4 in C6 cells (Fig. 5A).
Cleaved caspase-3 was detected by western blot anaysis to determine
whether apoptosis occurs in the C6 cells after miR-124
overexpression. Cleaved caspase-3 was only distinctly expressed in
the C6 cells after miR-124 overexpression (Fig. 5B). Based on these results, we
suggest that miR-124 may influence the apoptosis of C6 glioma
cells, but not through Smad4 modulation. Furthermore, the
overexpression of miR-124 suppressed Smad4 production, leading to
c-Myc downregulation, which inhibited the proliferation of C6
cells.
Discussion
In the present study, we confirmed the following: i)
miR-124 expression was significantly downregulated in C6 glioma
cells compared to in normal rat brain tissue and astrocytes, which
was consistent with a previous report (15); ii) restoring miR-124 expression
inhibited C6 cell proliferation, which correlated with Smad4
downregulation; and iii) Smad4 is a novel direct target gene of
miR-124 that controls c-Myc expression in C6 cells.
Glioma is the most aggressive tumor type of the
nervous system, and recurrence is high following therapy (21,22). To date, many aspects of the
molecular mechanisms of glioma tumorigenesis and progression have
been investigated. miR-124 has been classified as a tumor
suppressor in several types of human cancers (23–25). A number of studies have generated
data indicating that miR-124 expression is significantly suppressed
in glioma cells compared with normal brain tissue, and miR-124
expression levels negatively correlate with the pathological grade
of glioma (15,16,18,26–28). Several genes that control the
biological behavior of glioma cells, such as self-renewal
(proliferation), migration and invasion are direct targets of
miR-124, including Stat3, IQGAP1, ROCK1, SOS1, SLC16A1, and CDK4
(18,26–30). These results indicate that in
glioma cells, miR-124 functions through the post-transcriptional
downregulation of multiple target genes. As with other miRNAs,
miR-124 has numerous potential target genes and the underlying
molecular mechanisms whereby miR-124 controls glioma cell
proliferation are not limited to those target genes mentioned
above.
TGF-β is a well-known growth factor and is generally
considered to function as a tumor suppressor. However, emerging
evidence has indicated that in some tumor types, specifically in
high-grade glioma, TGF-β acts an oncogenic factor by increasing
tumor cell motility and invasion (5,31–34). Therefore, TGF-β and its functional
pathways are considered as potential therapeutic targets for
glioma. In the Smad-dependent TGF-β signaling pathway, there are 3
types of signal-transducing proteins, including Smad2, Smad3 and
Smad4. Smad4 is also an indispensable element of the bone
morphogenetic protein (BMP)-signaling pathway. The BMP signaling
pathway has been shown to promote tumorigenesis in a murine model
of high-grade glioma (35). In
addition, Smad4 protein is highly expressed in glioma; therefore,
Smad4 is a crucial target gene in the treatment of glioma. Based on
the inverse correlation between miR-124 with Smad4 expression
within glioma cells, we hypothesized that the lower expression of
miR-124 promoted Smad4 expression. Recently, Zu et al
identified Smad4 as a novel target of miR-124 in human non-small
cell lung cancer (36). In the
present study, we confirmed that Smad4 was a novel direct target
gene of miR-124 in C6 glioma cells. These results imply that
miR-124 simultaneously influences the TGF-β/Smad and BMP signaling
pathways by attenuating the Smad4 protein level.
Through a Smad-dependent manner, TGF-β can activate
the Jak-Stat3 pathway and promote the self-renewal of human GICs
(5). Unexpectedly, we found that
in C6 cells, the enhanced miR-124 or decreased Smad4 expression did
not affect the secreted LIF protein levels, while Stat3 and p-Stat3
levels were decreased at the protein level. Furthermore, upon
transfection of the C6 cells with siRNAs against Smad4, Stat3
protein was downregulated. We therefore suggest that at least in C6
cells, TGF-β/Smad4 pathway signaling may activate the Jak/Stat3
pathway, possibly by stimulating other activators, such as TNF-α,
without depending on enhanced LIF expression. However, exactly
which activator was involved should be further addressed. Stat3 is
a direct target gene of miR-124 (18). Thus, after restoring miR-124
expression, Stat3 protein levels in C6 cells were downregulated in
2 ways. One way was through direct miR-124 binding to the Stat3
mRNA 3′-UTR, which inhibited Stat3 mRNA transcription. Another way
was through miR-124-dependent downregulation of Smad4 protein
expression. Therefore, in this study, although we did not evaluate
whether the miR-124 controls other target genes, e.g., CDK4 and
CDK6, which regulate cell cycle progression, it is plausible that
miR-124 partially inhibited C6 cell proliferation by downregulating
Stat3 protein expression.
The c-Myc transcription factor is a critical
regulator of cell growth, particularly of cell cycle progression
from the G1 to S phase. Hence, c-Myc expression levels are tightly
related to cell proliferation (37). In early-stage cancers, including
glioma, TGF-β suppresses cell proliferation, whereas in later
stages, TGF-β promotes cancer cell proliferation by enhancing c-Myc
gene expression (38). The
molecular mechanisms behing these dual roles in carcinogenesis
remain unclear. It has been shown that the c-Myc promoter has
several potential SBEs, namely TBE1, TBE2 and TIE. During palatal
growth, TGF-β3 promotes palatal mesenchymal cell proliferation by
enhancing Smad4 binding to TBE1 and activating c-Myc gene
expression (20). As shown in
this study, c-Myc protein expression was similarly downregulated
after enhancing miR-124 expression or silencing Smad4 in C6 cells.
These results indicated that the downregulation of Smad4 may cause
decreased c-Myc protein expression following the ectopic
overexpression of miR-124 in C6 cells. Controversially, other
researchers have presented data showing that an adenovirus encoding
Smad4 suppresses glioma cell proliferation (39). Therefore, if Smad4 directly
controls c-Myc expression in glioma cells, the associated
mechanisms should be investigated in greater detail. As is
well-known, Stat3 is abnormally overexpressed in various types of
cancer cells, including glioma cells, which induces c-Myc
expression and stimulates cell proliferation (40,41). Accordingly, we suggest that after
enhancing the miR-124 level, the protein-expression levels of Smad4
and Stat3 are suppressed and eventually the c-Myc protein level
becomes downregulated in C6 cells, representing another novel
mechanism for explaining how miR-124 inhibits C6 cell
proliferation.
In conclusion, in this study, we found that Smad4 is
a novel, direct target of miR-124 in C6 cells and demonstrated that
upon miR-124 upregulation Smad4 was downregulated, which may be a
major cause of the inhibition of C6 cell proliferation, The results
presented in this study confirm that miR-124 may be a potential
target in the treatment of glioma, as it effectively inhibits
glioma cell proliferation by targeting multiple genes.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81571199) and the Jilin
University Youth Science and Technology Innovation Fund (no.
450060507061).
References
|
1
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: Primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14(Suppl 5): v1–v49. 2012. View Article : Google Scholar :
|
|
2
|
Kong LY, Wu AS, Doucette T, Wei J, Priebe
W, Fuller GN, Qiao W, Sawaya R, Rao G and Heimberger AB:
Intratumoral mediated immunosuppression is prognostic in
genetically engineered murine models of glioma and correlates to
immunotherapeutic responses. Clin Cancer Res. 16:5722–5733. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Roberts AB and Wakefield LM: The two faces
of transforming growth factor beta in carcinogenesis. Proc Natl
Acad Sci USA. 100:8621–8623. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peñuelas S, Anido J, Prieto-Sánchez RM,
Folch G, Barba I, Cuartas I, García-Dorado D, Poca MA, Sahuquillo
J, Baselga J and Seoane J: TGF-beta increases glioma-initiating
cell self-renewal through the induction of LIF in human
glioblastoma. Cancer Cell. 15:315–327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
ten Dijke P and Hill CS: New insights into
TGF-beta-Smad signalling. Trends Biochem Sci. 29:265–273. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010.PubMed/NCBI
|
|
9
|
Chen CZ: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lawler S and Chiocca EA: Emerging
functions of microRNAs in glioblastoma. J Neurooncol. 92:297–306.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Novakova J, Slaby O, Vyzula R and Michalek
J: MicroRNA involvement in glioblastoma pathogenesis. Biochem
Biophys Res Commun. 386:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Silber J, James CD and Hodgson JG:
microRNAs in gliomas: Small regulators of a big problem.
Neuromolecular Med. 11:208–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoo AS, Sun AX, Li L, Shcheglovitov A,
Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW and Crabtree
GR: MicroRNA-mediated conversion of human fibroblasts to neurons.
Nature. 476:228–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xia H, Cheung WK, Ng SS, Jiang X, Jiang S,
Sze J, Leung GK, Lu G, Chan DT, Bian XW, et al: Loss of
brain-enriched miR-124 microRNA enhances stem-like traits and
invasiveness of glioma cells. J Biol Chem. 287:9962–9971. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao X, Pfaff SL and Gage FH: A functional
study of miR-124 in the developing neural tube. Genes Dev.
21:531–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silber J, Lim DA, Petritsch C, Persson AI,
Maunakea AK, Yu M, Vandenberg SR, Ginzinger DG, James CD, Costello
JF, et al: miR-124 and miR-137 inhibit proliferation of
glioblastoma multiforme cells and induce differentiation of brain
tumor stem cells. BMC Med. 6:142008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei J, Wang F, Kong LY, Xu S, Doucette T,
Ferguson SD, Yang Y, McEnery K, Jethwa K, Gjyshi O, et al: miR-124
inhibits STAT3 signaling to enhance T cell-mediated immune
clearance of glioma. Cancer Res. 73:3913–3926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schwartz JP and Wilson DJ: Preparation and
characterization of type 1 astrocytes cultured from adult rat
cortex, cerebellum, and striatum. Glia. 5:75–80. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu X, Ozturk F, Liu C, Oakley GG and
Nawshad A: Transforming growth factor-β activates c-Myc to promote
palatal growth. J Cell Biochem. 113:3069–3085. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vehlow A and Cordes N: Invasion as target
for therapy of glioblastoma multiforme. Biochim Biophys Acta.
1836:236–244. 2013.PubMed/NCBI
|
|
23
|
Lv XB, Jiao Y, Qing Y, Hu H, Cui X, Lin T,
Song E and Yu F: miR-124 suppresses multiple steps of breast cancer
metastasis by targeting a cohort of pro-metastatic genes in vitro.
Chin J Cancer. 30:821–830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lang Q and Ling C: MiR-124 suppresses cell
proliferation in hepatocellular carcinoma by targeting PIK3CA.
Biochem Biophys Res Commun. 426:247–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia J, Wu Z, Yu C, He W, Zheng H, He Y,
Jian W, Chen L, Zhang L and Li W: miR-124 inhibits cell
proliferation in gastric cancer through down-regulation of SPHK1. J
Pathol. 227:470–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
An L, Liu Y, Wu A and Guan Y: microRNA-124
inhibits migration and invasion by down-regulating ROCK1 in glioma.
PLoS One. 8:e694782013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deng X, Ma L, Wu M, Zhang G, Jin C, Guo Y
and Liu R: miR-124 radiosensitizes human glioma cells by targeting
CDK4. J Neurooncol. 114:263–274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li KK, Pang JC, Ching AK, Wong CK, Kong X,
Wang Y, Zhou L, Chen Z and Ng HK: miR-124 is frequently
down-regulated in medulloblastoma and is a negative regulator of
SLC16A1. Hum Pathol. 40:1234–1243. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu SH, Jiang XJ, Xiao GL, Liu DY and Yuan
XR: miR-124a restoration inhibits glioma cell proliferation and
invasion by suppressing IQGAP1 and β-catenin. Oncol Rep.
32:2104–2110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lv Z and Yang L: miR-124 inhibits the
growth of glioblastoma through the downregulation of SOS1. Mol Med
Rep. 8:345–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Piek E, Westermark U, Kastemar M, Heldin
CH, van Zoelen EJ, Nistér M and Ten Dijke P: Expression of
transforming-growth-factor (TGF)-beta receptors and Smad proteins
in glioblastoma cell lines with distinct responses to TGF-beta1.
Int J Cancer. 80:756–763. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kjellman C, Olofsson SP, Hansson O, Von
Schantz T, Lindvall M, Nilsson I, Salford LG, Sjögren HO and
Widegren B: Expression of TGF-beta isoforms, TGF-beta receptors,
and SMAD molecules at different stages of human glioma. Int J
Cancer. 89:251–258. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rich JN: The role of transforming growth
factor-beta in primary brain tumors. Front Biosci. 8:e245–e260.
2003. View Article : Google Scholar
|
|
34
|
Golestaneh N and Mishra B: TGF-beta,
neuronal stem cells and glioblastoma. Oncogene. 24:5722–5730. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hover LD, Owens P, Munden AL, Wang J,
Chambless LB, Hopkins CR, Hong CC, Moses HL and Abel TW: Bone
morphogenetic protein signaling promotes tumorigenesis in a murine
model of high-grade glioma. Neuro Oncol. 18:928–938. 2016.
View Article : Google Scholar :
|
|
36
|
Zu L, Xue Y and Wang J, Fu Y, Wang X, Xiao
G, Hao M, Sun X, Wang Y, Fu G and Wang J: The feedback loop between
miR-124 and TGF-β pathway plays a significant role in non-small
cell lung cancer metastasis. Carcinogenesis. 37:333–343. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar
|
|
38
|
Singh G, Singh SK, König A, Reutlinger K,
Nye MD, Adhikary T, Eilers M, Gress TM, Fernandez-Zapico ME and
Ellenrieder V: Sequential activation of NFAT and c-Myc
transcription factors mediates the TGF-beta switch from a
suppressor to a promoter of cancer cell proliferation. J Biol Chem.
285:27241–27250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang Z, Zhong L, Zhong S, Xian R and Yuan
B: Adenovirus encoding Smad4 suppresses glioma cell proliferation
and increases apoptosis through cell cycle arrest at G1 phase. Int
Immunopharmacol. 25:169–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kiuchi N, Nakajima K, Ichiba M, Fukada T,
Narimatsu M, Mizuno K, Hibi M and Hirano T: STAT3 is required for
the gp130-mediated full activation of the c-myc gene. J Exp Med.
189:63–73. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin YM, Wang CM, Jeng JC, Leprince D and
Shih HM: HIC1 interacts with and modulates the activity of STAT3.
Cell Cycle. 12:2266–2276. 2013. View Article : Google Scholar : PubMed/NCBI
|