Introduction
In tandem with the global obesity pandemic, type 2
diabetes mellitus (T2DM) has become a major public health issue
(1). In traditional glycemic
management based on a stepwise approach, insulin is considered as
only the final alternative. Nevertheless, emerging evidence
indicates that early initiation of insulin extended glycemic
remission, by preserving β-cell function, in newly onset T2DM
(2). Moreover, systemic insulin
sensitivity was also improved after early insulin administration
(3,4). The underlying mechanism, however,
remains obscure.
Lipotoxicity describes the deleterious effects of
ectopic fat deposition on glucose metabolism in insulin-responsive
tissues, playing a pivotal role in the pathogenesis of
obesity-associated insulin resistance (5). Insulin-mediated myocellular glucose
uptake is capital in maintaining systemic glucose homeostasis
(6). Along with the intramuscular
lipid accumulation, enriched lipid intermediates impede the insulin
signaling cascade and stimulate inflammation and endoplasmic
reticulum stress, causing muscular insulin resistance (7–9).
Triglyceride content relies on the equilibrium between de
novo lipogenesis and fatty acid oxidation. Sterol-regulated
element binding protein-1c (SREBP-1c) is a master regulator of
lipogenesis, controlling the transcription and expression of
lipogenic enzymes. It is well-established that the physiological
SREBP-1c expression is increased by insulin and nutrition
availability (10–12). On the other hand, β-oxidation is
modulated by carnitine palmitoyltransferase 1B (CPT1B), the
rate-limiting enzyme mainly expressed in skeletal muscle, to
transport fatty acids into mitochondria for oxidation. Individuals
with insulin resistance have reduced capacity of fatty acid
oxidation in skeletal muscle, resulting from suppressed CPT1B
activity (13–15). Both SREBP-1c and CPT1B are
regulated by AMP-activated protein kinase (AMPK), which responds to
various nutritional cues in order to balance the production of ATP
corresponding to the demand (16,17). Once activated, the phosphorylated
AMPK induces CPT1B to switch-on catabolism and inhibits SREBP-1c to
restrain anabolism. Different from the sustained AMPK activity in
the vastus lateralis of T2DM patients during a
euglycemic-hyperinsulinemic clamp (18), we previously uncovered that
continuous subcutaneous insulin intervention improved AMPK
phosphorylation in the gastrocnemius of diabetic mice (19).
The occurrence of obesity-associated insulin
resistance can be traced to a chronic state of energy surfeit
(20), which is detrimental to
AMPK activation. The adenylate energy charge, calculated as [ATP +
0.5 × ADP]/[ATP + ADP + AMP], is an integrated index of cellular
energy status. This value is increased with excessive calorie
intake or a sedentary lifestyle, while energy deficiency caused by
hunger and exercise abates it. Lipid biosynthesis is promoted to
store energy when increased delivery of calories overwhelms the
expenditure (5). Thus, uncoupling
protein 3 (UCP3) is a focus of intensive research for protecting
against lipid deposition in skeletal muscle, owing to its
inhibitory effect on ATP production (20,21). UCP3-mediated proton-leak across
the mitochondrial inner membrane eliminates the driving force of
ATP synthesis, and then creates a futile cycle of substrate
oxidation. Clinical observations have found that both mRNA and
protein expression of UCP3 were reduced in skeletal muscle of
pre-diabetic and T2DM populations (22–24). Additional experimental evidence
supports the protective role of UCP3 in conditions of energy
overload. UCP3 transgenic mice are lean and resistant to high-fat
diet (HFD)-induced obesity, which is attributable to decreased
metabolic efficiency (25–27).
Although accumulating evidence indicates that instant
insulin-stimulated muscular ATP synthesis is abolished in T2DM
patients (28,29), the impact of an early insulin
regimen on energy metabolism or UCP3 expression in skeletal muscle
with insulin resistance and its correlation to lipid amelioration
remain poorly understood.
Here, we investigated the effect of insulin on
muscular lipid metabolism and the underlying mechanisms in the
early phase of insulin resistance in vivo and in
vitro, with a focus on the regulation of energy balance. We
found that by recovering UCP3 activity, insulin blocked ATP surplus
and restored AMPK phosphorylation to alleviate lipid accumulation
in skeletal muscle cells exposed to palmitate (PA) and in
gastrocnemius in mice fed HFD. Our data provide insight into the
promising effects of insulin on lipid metabolism and insulin
sensitivity in the early course of diabetes.
Materials and methods
Reagents and antibodies
PA, insulin and carbobenzoxy-Leu-Leu-leucinal
(MG132) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Antibodies against voltage-dependent anion-selective channel 1
(rabbit polyclonal to VDAC1, ab34726, 1:5,000), NADH dehydrogenase
[ubiquinone] 1α subcomplex subunit 9 (rabbit monoclonal to NDUFA9,
ab181381, 1:1,000), ubiquinol cytochrome c reductase core
protein 2 (mouse monoclonal to UQCRC2, ab14745, 1:1,000), ATP
synthase subunit α (mouse monoclonal to ATP5A, ab110273, 1:4,000)
and UCP3 (rabbit polyclonal, ab3477, 1:2,000) were purchased from
Abcam (Cambridge, MA, USA). Anti-IgG (normal goat IgG, sc-2028,
1:1,000), antiubiquitin (rabbit polyclonal, sc-9133, 1:1,000),
anti-UCP3 for immunoprecipitation (goat polyclonal, sc-31387,
1:2,000) and anti-SREBP-1c antibodies (rabbit monoclonal, sc-8984,
1:5,000) were acquired from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Antibodies against succinate dehydrogenase complex
subunit A (rabbit monoclonal to SDHA, 11998, 1:2,000), cytochrome
c oxidase subunit IV (rabbit monoclonal to COX IV, 4850,
1:500), AMPKα (rabbit monoclonal, 2603, 1:2,000) and p-AMPKα
(Thr172) (rabbit monoclonal, 2535, 1:1,000) were obtained from Cell
Signaling Technology (Danvers, MA, USA). The anti-CPT1B antibody
was obtained from Absin Bioscience Inc. (Beijing, China) (rabbit
polyclonal, abs117445, 1:5,000).
Animal experiments
Male C57BL/6J mice were obtained from the
Comparative Medicine Centre of Yangzhou University (Yangzhou,
China) at 6 weeks of age. All mice were maintained under standard
light (12 h light/dark), temperature (22±2°C), and humidity
(40±10%) conditions with ad libitum access to food and
water. Mice were randomized into an HFD group (D12492, 60% calories
from fat, 21% calories from carbohydrates, and 19% calories from
protein; obtained from Guangdong Medical Laboratory Animal Center,
Guangzhou, China) at 8 weeks to establish a diet-induced obese
(DIO) model. The control mice were fed with chow diet (10% calories
from fat, 70% calories from carbohydrates and 20% calories from
protein). After an 8-week feeding, weight- and glucose-matched DIO
mice were further assigned randomly into a neutral protamine
Hagedorn (NPH) insulin (0.4–0.6 U/day, subcutaneous; Novo Nordisk
A/S, Maaloev, Denmark) or a saline treatment group for a total of 3
weeks, during which all animals continued on their original diets
(n=8/group). The NPH insulin was titrated to a glycemic target of
nonfasting blood glucose level <8 mmol/l in a week. Individual
body weight and fasting glucose concentration were monitored every
week. Three weeks later, mice were fasted for 14 h and challenged
with an intraperitoneal glucose tolerance test (IPGTT,
intraperitoneal D-glucose load, 2 g/kg body weight). Glucose levels
were assessed in duplicate by glucometer with tail vein blood
samples at 0, 30, 60 and 120 min after glucose administration.
After being deprived of food for 10 h without insulin intervention
overnight, mice were sacrificed for fasting blood and tissue
collection. Gastrocnemius was dissected from any visible adipose
tissue and stored at −80°C for further analysis. All of the animal
procedures were performed according to the National Institutes of
Health Guidelines and approved by the Animal Care Committee of Drum
Tower Hospital, which is affiliated with Nanjing University Medical
School, Nanjing, China.
Biochemical assay
Fasting serum insulin and triglyceride contents, and
prepared gastrocnemius triglyceride samples (30) were analyzed with ELISA kits
(CEA448Mu and CEB687Ge; Cloud-Clone Corp., Houston, TX, USA)
accordingly.
Cell culture
L6 myoblasts were obtained from the Chinese Academy
of Sciences (Shanghai, China). They were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS) (vol/vol), and differentiated into myotubes with medium
containing 2% FBS (vol/vol), as previously described (31). L6 myotubes pre-incubated in
serum-free medium for 8 h were treated with 500 µmol/l PA
for 24 h, and then another 12 h with 100 nmol/l insulin. Where
indicated, differentiated L6 myotubes were pretreated with MG132 (5
µmol/l) for 48 h prior to PA (32). The appropriate vehicle served as
the control.
siRNA transfection
L6 myoblasts were transfected with siRNA
oligonucleotides (20 nmol/l, AM16708) using Lipofectamine RNAiMAX
(13778150) (both from Invitrogen, Grand Island, NY, USA). Cells
were cultured for 36 h prior to PA and insulin addition. The
sequence of the stealth RNAi against ucp3 is as follows:
5′-GGGACUUGGCCCAACAUCACAAGAA-3′. Stealth RNAi negative control with
low GC content (12935111; Invitrogen) was used as control.
Quantitative (real-time) PCR
The mtDNA copy number was evaluated from genomic DNA
prepared using the DNeasy Blood and Tissue kit (69504; Qiagen,
Hilden, Germany). Real-time PCR was used to quantify amplifications
of mtDNA with both sense and antisense primers (mtDNA forward,
5′-CCCCAGAGGATTAAACTCCAACGCA-3′ and reverse,
5′-GGGTGGGGTCAGGGGGT-3′; 18s RNA forward, 5′-TTGGGATGGGAAAGATGGG-3′
and reverse, 5′-CAGAGTAGGAGGGAACAAGTGT GAC-3′), at a final volume
of 20 µl using the QuantiNova SYBR-Green PCR kit (Qiagen)
with an ABI StepOne Real-Time PCR system (Applied Biosystems, Grand
Island, NY, USA). The thermal cycling conditions were as follows: 2
min at 95°C, and then 40 cycles of 5 sec at 95°C, 10 sec at 60°C.
Dissociation curves were plotted to ensure primer specificity. The
ΔΔCt method was applied for calculation (33).
Western blot analysis
L6 myotubes and gastrocnemius fragments were washed
twice with cold phosphate-buffered saline and lysed in RIPA lysis
buffer with Protease Inhibitor Cocktail (Roche, Basel,
Switzerland). Total protein concentrations were determined using
the bicinchoninic acid (BCA) method. Twenty micrograms of protein
per sample were separated on sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) gel and then transferred to
polyvinylidene difluoride membranes, which were blocked with 5%
nonfat milk (wt/vol) in Tris-buffered saline with 0.1% Tween-20
(TBST) and incubated with corresponding primary antibodies
overnight at 4°C. The membranes were washed with TBST and incubated
with an appropriate secondary antibody (goat anti-mouse IgG,
GAM0072, and rabbit anti-goat IgG, RAG0072; both from MultiSciences
Biotech, Suzhou, China; and goat anti-rabbit IgG, ZB-2301;
ZSGB-BIO, Beijing, China) at room temperature for ~2 h. The signals
were visualized by enhanced chemiluminescence (EMD Millipore,
Billerica, MA, USA) and quantified by densitometry (Quantity One;
Bio-Rad, Hercules, CA, USA).
ATP assay
AMP, ADP and ATP contents from cell samples were
measured with high performance liquid chromatography. Briefly, the
frozen cells were lysed in 500 µl ice-cold 0.6 mol/l
perchloric acid and centrifuged at 13,000 × g for 2 min at 4°C. The
supernatant was neutralized by 1 ml ice-cold freon/trioctylamine
(4:1 vol/vol). Then the upper aqueous layer was collected after
centrifugation (13,000 × g for 2 min at 4°C) for chromatographic
separation and analysis (34). As
to skeletal muscle tissues, ATP was measured with the ATP assay kit
(S0027; Beyotime Institute of Biotechnology, Shanghai, China)
immediately after tissue isolation. The supernatants were collected
following homogenate and centrifugation (12,000 × g for 5 min at
4°C), and then added to the reaction mixture. The luminescent
signals were detected by the luminometer (GloMax; Promega Corp.,
Madison, WI, USA). The ATP concentrations were normalized to the
protein concentration.
Isolation of mitochondria and detection
of mitochondrial membrane potential
L6 myotubes and gastrocnemius fragments were
suspended with ice-cold isolation agent containing PMSF (C3601;
Beyotime Institute of Biotechnology). The samples were then
homogenized gently and centrifuged twice at 4°C (1,000 × g for 10
min and then 3,500 × g for 10 min). Mitochondria were collected as
a pellet and resuspended for functional analysis.
Mitochondrial membrane potential was assessed by
JC-1 staining (GMS10013; Genmed Scientifics Inc., Wilmington, DE,
USA) immediately after mitochondrial isolation. Purified
mitochondria were mixed with the JC-1 reagent, and the relative
fluorescence unit (RFU) was detected at λ ex/em 490/590 nm on a
fluorescence microplate reader (SpectraMax M3; Molecular Devices,
Sunnyvale, CA, USA).
Respiratory complex activities
The activities of mitochondrial complexes in L6
myotubes were evaluated by a colorimetric method according to the
protocol of the manufacturer (Genmed Scientifics Inc.).
Co-immunoprecipitation
Skeletal muscle tissues were lysed with NP-40 lysis
buffer containing PMSF and cocktail. After homogenization and
centrifugation (12,000 × g for 15 min at 4°C), the supernatants
were pre-cleared with 10 µl protein A/G Plus agarose beads
(sc-2003; Santa Cruz Biotechnology, Inc.) for 2 h. Then the lysates
were incubated with anti-UCP3 antibody (2 µg/100–500
µg of total protein) overnight, followed by co-incubating
with 20 µl protein A/G Plus agarose beads for another 5 h.
The beads were washed three times with NP-40 lysis buffer with PMSF
and cocktail. All the procedures above were manipulated at 4°C. The
precipitated proteins were separated from beads by heating in
boiling water for 10 min, and analyzed by western blot
analysis.
Statistical analysis
Data are presented as means ± SEM. Differences
between multiple groups were analyzed by one-way ANOVA with the
least significant difference (LSD) or Dunnett's T3 post hoc
comparison analysis as appropriate. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Early insulin therapy attenuates
triglyceride deposition in skeletal muscle in vivo
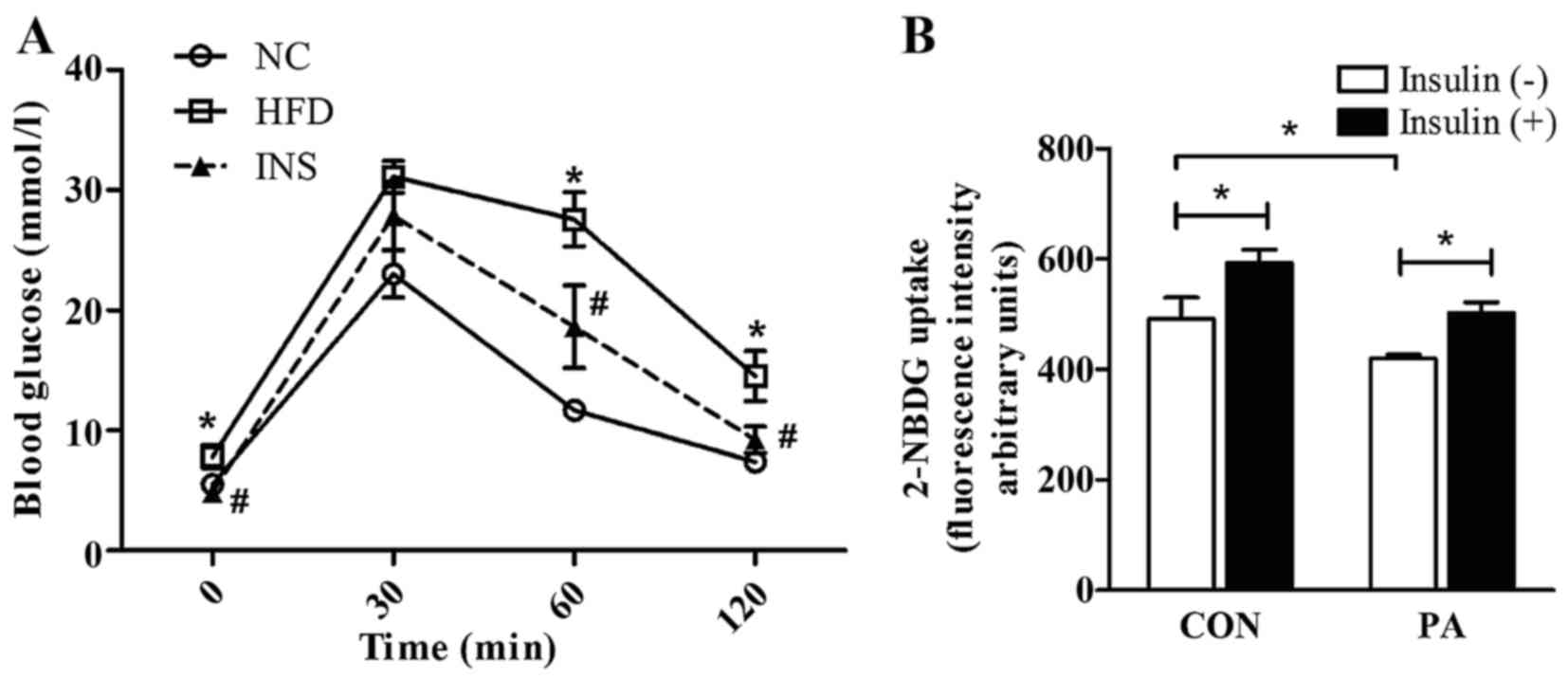
Compared with the HFD-fed mice, insulin treatment
for 3 weeks markedly decreased the fasting blood glucose (FBG),
fasting serum insulin level, and HOMA-IR despite the stabilized
body weight (Table I). In
addition, the IPGTT showed that systemic glucose clearance was
improved by insulin (Fig. 1A).
Concomitantly, the glucose areas under the curve (AUC) were
decreased after insulin therapy (Table I). In vitro, insulin also
ameliorated the impaired capacity of glucose uptake in the
PA-pretreated L6 myotubes (Fig.
1B).
 | Table ICharacteristics of the experimental
animals at the end of the intervention study. |
Table I
Characteristics of the experimental
animals at the end of the intervention study.
| NC | HFD | INS |
|---|
| Body weight
(g) | 26.17±0.67 | 33.02±0.84a | 32.29±0.86 |
| Fasting blood
glucose (mmol/l) | 5.53±0.52 | 7.82±0.34a | 4.82±0.31b |
| Fasting insulin
(pg/ml) | 1,341.53±52.57 | 1,177.94±119.4 |
511.08±85.22b |
| HOMA-IR | 1.000±0.13 | 1.34±0.16 | 0.48±0.09b |
| AUC for IPGTT
(mmol/l × min) | 1,520.5±80.73 |
2,726.4±42.28a |
2022.5±76.02b |
| Fasting
triglyceride (µg/ml) | 49.91±3.60 | 73.41±14.09 | 35.29±3.23c |
| Muscle triglyceride
(µg/ml × mg wet tissue) | 4.29±0.33 | 6.02±0.38a | 4.83±0.18c |
In contrast to the normal chow (NC) group, HFD mice
demonstrated a prominent triglyceride accumulation in the
gastrocnemius, which was notably alleviated by insulin. Moreover,
the insulin-treated mice had lower serum triglyceride level than
that noted in the HFD-fed mice (Table
I).
Insulin restores AMPK phosphorylation to
facilitate fatty acid oxidation and suppress lipogenesis
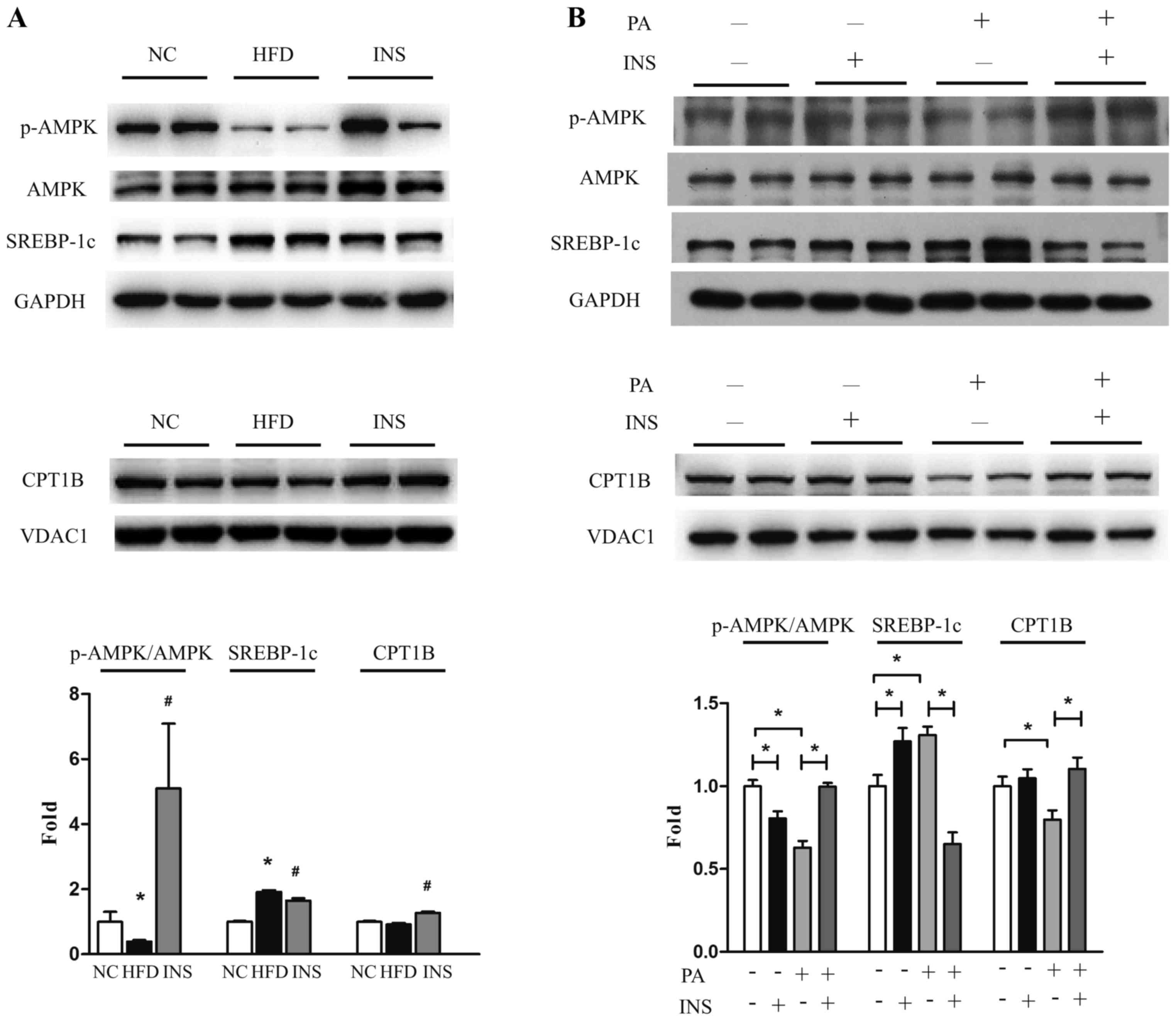
The disturbance between lipogenesis and fatty acid
oxidation contributes to lipid deposition. Thus, AMPK (the
metabolic modulator), SREBP-1c and CPT1B were examined in skeletal
muscle cells. In comparison to NC, HFD-fed mice had reduced
muscular AMPK phosphorylation, increased SREBP-1c and similar CPT1B
expression. Strikingly, after insulin administration, the
expression of CPT1B was upregulated and that of SREBP-1c was
suppressed, accompanied by recovered AMPK phosphorylation (Fig. 2A). Likewise, in vitro,
insulin restored p-AMPK and CPT1B expression, and attenuated
SREBP-1c expression in L6 myotubes after exposure to PA (Fig. 2B). These results indicated that
AMPK-associated SREBP-1c and CPT1B regulation participated in the
interplay of saturated fatty acid and insulin on triglyceride
deposition in skeletal muscle.
Insulin alleviates energy surfeit through
dissipating mitochondrial membrane potential in skeletal muscle
cells exposed to excessive fatty acid
AMPK activation is sensitive to the ratio of AMP:ATP
in targeting metabolic processes, thus serving as an energy sensor.
To address the effect of saturated fatty acid and insulin on
muscular adenylate energy metabolism, AMP, ADP and ATP contents
were detected and the adenylate energy charge was calculated. In L6
myotubes, the PA exposure promoted a significant augmentation of
ATP content and energy charge, leading to a decline in the AMP:ATP
ratio. Insulin was observed to abrogate the increase in ATP and
energy charge, thereby raising the AMP:ATP ratio (Fig. 3A). Similarly, insulin alleviated
the ATP overload in the gastrocnemius of HFD-fed mice (Fig. 3B). Moreover, a positive
correlation between ATP and muscle triglyceride was observed
(r=0.745; P=0.013).
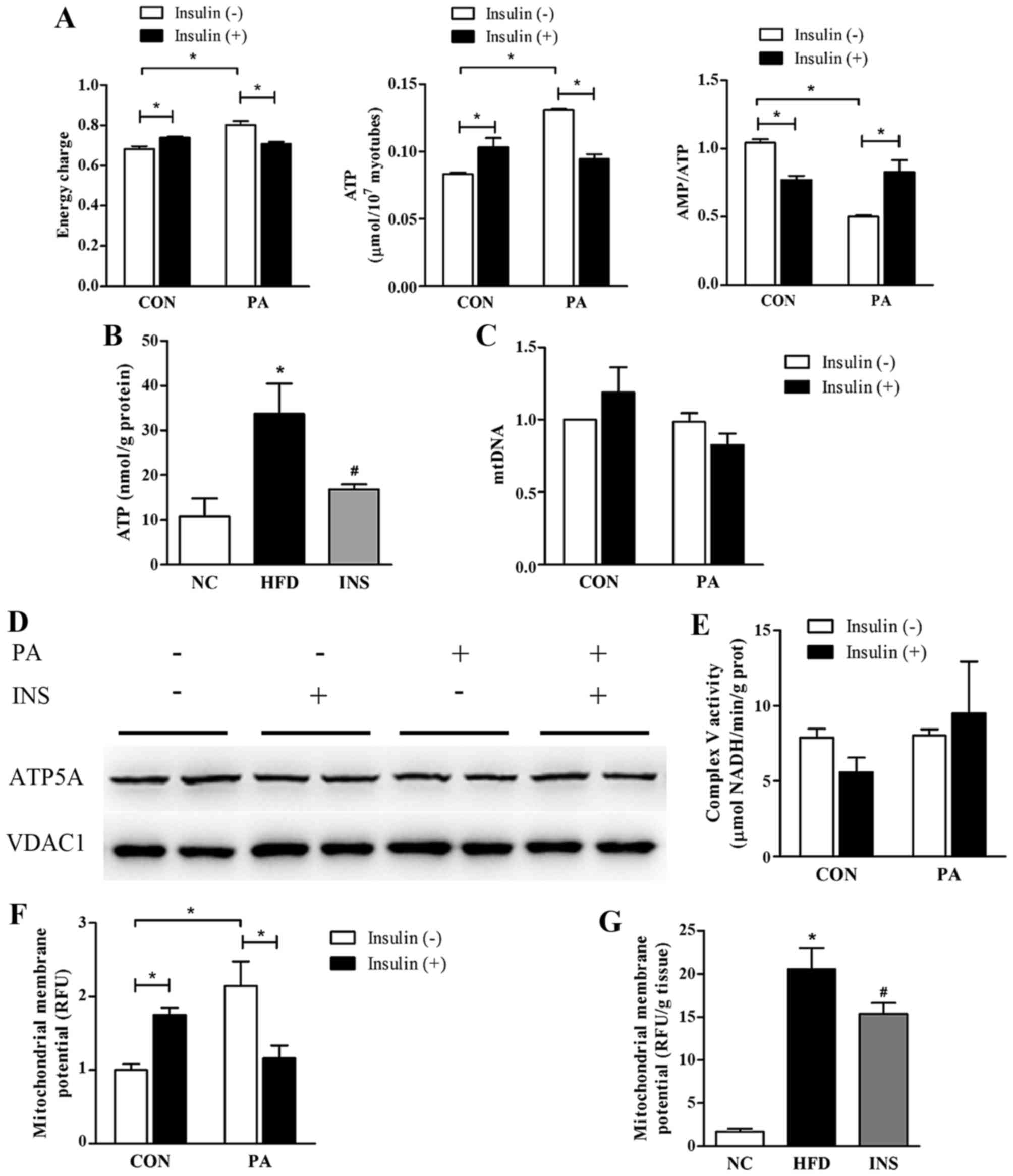 | Figure 3Insulin alleviates the energy surplus
and mitochondrial membrane potential stimulation in skeletal muscle
cells with a surfeit of fatty acid. (A) The cellular adenylate
energy charge level, ATP content, and AMP:ATP ratio were assessed
in L6 myotubes with PA and insulin intervention (n=3/group). (B)
The ATP content of gastrocnemius was determined in the three groups
of mice (n=5–6/group). (C-E) PA and insulin had little effects on
mitochondrial quantity (evaluated by mtDNA) or ATP synthase in L6
myotubes (n=4/group). (F and G) Mitochondrial membrane potential
levels were evaluated in L6 myotubes (n=3/group) and in
gastrocnemius of mice (n=3–6/group). (H and I) The effects of PA
and insulin on protein expression and activities of respiratory
complexes in L6 myotubes (n=3/group). For (A, C, E and F): CON,
BSA-induced L6 myotubes; PA, PA-induced L6 myotubes.
*P<0.05 as indicated. For (B and G): NC, normal chow
diet; HFD, high-fat diet-fed mice with saline; INS, HFD-fed mice
treated with insulin. *P<0.05 vs. NC;
#P<0.05 vs. HFD. For (I): PA, palmitate; INS,
insulin. *P<0.05 as indicated. Values are expressed
as means ± SEM. |
Given that mitochondrial content, mitochondrial
membrane potential and ATP synthase are the major determinants of
ATP synthesis in the organelle, these were estimated to identify
the precise mechanism of ATP regulation. Results showed that
neither PA nor insulin exerted any significant effect on mtDNA
content in the L6 myotubes (Fig.
3C). Both the protein expression and the activity of ATP
synthase were not significantly altered after PA or insulin
administration (Fig. 3D and E).
However, notably, the muscular level of mitochondrial membrane
potential was stimulated in the presence of fatty acid but
predominantly declined after insulin intervention in vivo
and in vitro (Fig. 3F and
G).
Mitochondrial membrane potential is a proton
electrochemical gradient, which is established by respiratory
complexes. Therefore, we evaluated the protein expression and
activities of all the respiratory complexes in an attempt to
ascertain the molecules responsible for mitochondrial membrane
potential alteration. PA exerted a neutral impact on the protein
levels of mitochondrial complexes, consisting of NDUFA9 (complex I
subunit), SDHA (complex II subunit), UQCRC2 (complex III subunit),
and COX4 (complex IV subunit). In addition, only the activities of
complex III and IV were promoted by PA. Nonetheless, in the
presence of PA, neither the expression nor the activities of
respiratory complexes were affected by insulin (Fig. 3H and I).
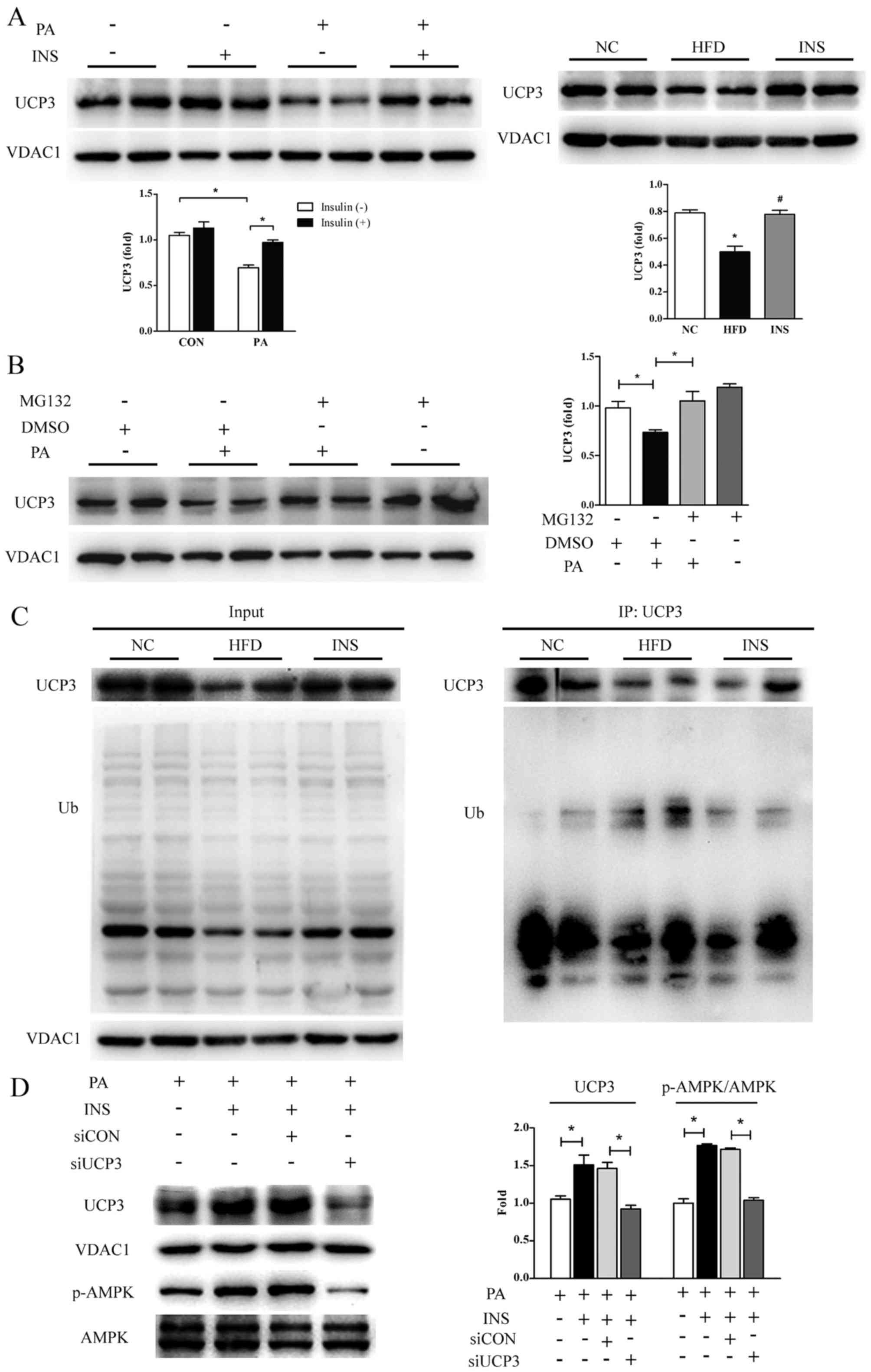
Insulin, by inhibiting degradation,
restores the UCP3 protein level which contributes to the
preservation of AMPK phosphorylation
Apart from respiratory complexes, mitochondrial
membrane potential could also be affected by UCP3 in skeletal
muscle. In vitro, the UCP3 protein level was attenuated ~40%
in the PA-treated L6 myotubes; however, this inhibition was
relieved markedly by insulin, thereby dissipating the proton motive
force. The restoration of UCP3 protein by insulin was also observed
in the gastrocnemius of mice provided with HFD (Fig. 4A).
Considering that the rapid turnover of UCP3 is
attributable to the ubiquitin-proteasome system (35,36), MG132, a pharmacological inhibitor
of 26S proteasome, was used to pretreat L6 myotubes prior to PA.
The results showed that the PA-induced decrease in UCP3 protein
expression was annulled in the presence of MG132 (Fig. 4B). Next, to investigate whether
UCP3 is indeed ubiquitinated in vivo, ubiquitination assays
based on co-immunoprecipitated UCP3 were carried out with the
skeletal muscles from the three groups of mice. As presented in
Fig. 4C, HFD efficiently
ubiquitinated UCP3, supporting that high-fat-intake did deteriorate
UCP3 protein stability by accelerating degradation; especially,
insulin was observed to greatly attenuate HFD-induced UCP3
ubiquitination, implying the potential involvement of the
ubiquitin-proteasome pathway in the recovery of UCP3 protein by
insulin.
Furthermore, to determine the role of UCP3 in the
restoration of AMPK phosphorylation by insulin, its expression was
knocked down with siRNA in L6 myotubes. Insulin-induced stimulation
of AMPK phosphorylation was profoundly impeded in the absence of
UCP3 (Fig. 4D). These data
suggested that the increased UCP3 protein expression was
responsible for the repression of mitochondrial membrane potential
and it mediated the recovered phosphorylation of AMPK elicited by
insulin.
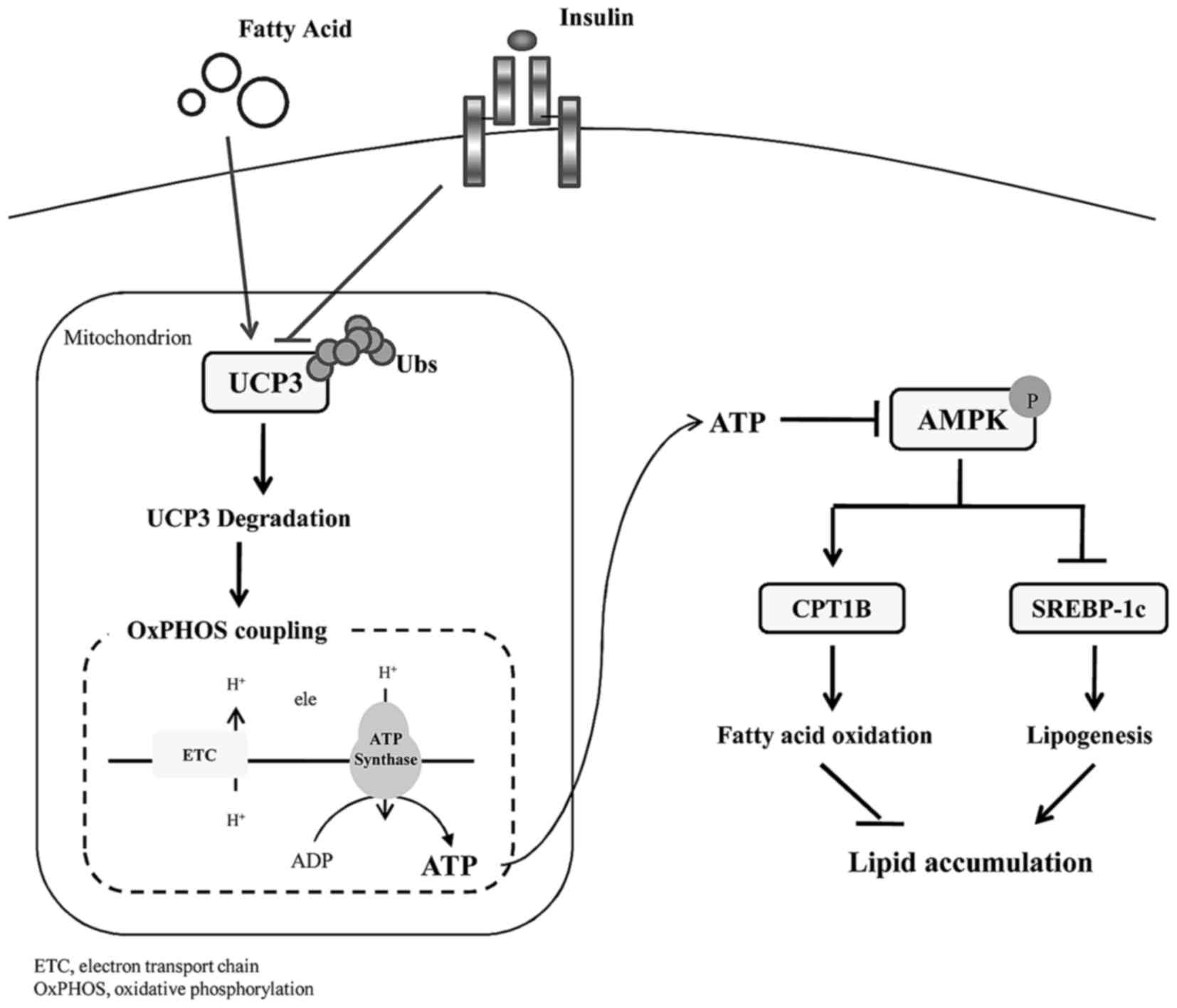
Altogether, insulin protected UCP3 from
ubiquitination degradation, thereby uncoupling mitochondrial
oxidative phosphorylation with a decline in cellular ATP.
Sequentially, increased AMPK phosphorylation underlies the
promising effect of early insulin therapy on lipid homeostasis in
skeletal muscle (Fig. 5).
Discussion
In contrast to the conventional perspective that
insulin promotes lipid deposition physiologically, a plethora of
clinical data and animal models imply that early insulin
administration improves the lipid profile and reduces lipid content
in metabolically active tissues of diabetic subjects (4,30,37). However, the molecular mechanism of
insulin action is not yet fully understood. The present study
provided mechanistic insight into the protective action of insulin
in sustaining muscular lipid homeostasis, which was in correlation
with abating energy surplus evoked by fatty acid overload, followed
by AMPK-mediated augmentation of fatty acid oxidation and
attenuation of lipogenesis. The recovery of UCP3 expression was
found to be responsible for the decline in the cellular energy
charge of insulin. This suggests an active role of insulin in
improving lipotoxicity of skeletal muscle, especially in the early
development of insulin resistance.
Mitochondria have received increased attention in
order to understand the etiology of obesity-associated insulin
resistance owing to its central role in lipid utilization and
energy production. Several pieces of evidence have linked a
compromised mitochondrial function to the development of insulin
resistance (38,39), which is recognized as a dynamic
alteration of mitochondrial function adapted to oscillations in
energy demand and substrate availability, termed as mitochondrial
plasticity (39). The initial
stage of HFD-induced obesity is continually associated with
mitochondrial over-activation and concomitant energy surplus
(40). Consistently, we observed
a high energy charge and ATP content, which was generated from
stimulating activities of complex III and IV and coupling process
in skeletal muscle cells treated with excessive fatty acid in
vivo and in vitro; increased ATP inhibits the
phosphorylation of AMPK. The anabolic pathways are driven by
utilizing ATP to store glycogen and lipid as energy sources and
balance the energy supply with demand. Nevertheless, this is
detrimental to insulin sensitivity with prolonged duration. Thus,
blocking excessive ATP synthesis provides a latent approach to
alleviate insulin resistance.
Multiple anti-diabetic agents, involving metformin
and resveratrol, have been demonstrated to improve insulin
sensitivity via declining cellular energy state. The underlying
mechanisms by which metformin and berberine diminish
glyconeogenesis in the liver and triglyceride accumulation in
skeletal muscle was found to include interruption of the activity
of mitochondrial complex I, resulting in decreased ATP production
(41–45). Resveratrol was documented to block
ATP synthase activity, thereby inhibiting mitochondrial ATP
synthesis (46). Nevertheless,
the conclusions concerning the effect of insulin on mitochondrial
oxidative phosphorylation are yet controversial. During
hyperinsulinemic-euglycemic clamp, insulin was found to promote ATP
production, accompanied by upregulated complex IV activity in
skeletal muscles of healthy subjects (29) while patients with overt T2DM and
their first-degree relatives failed to show an increased
mitochondrial activity (28,47). Conversely, intensive insulin
therapy for 10 days increased muscular mRNA levels of complex I,
III and V in diabetics (48).
Taken together, the impact of insulin on mitochondria, at least
partially, depends on the concentration, method, and duration of
administration. Moreover, the duration of T2DM at the time of
intervention may also influence the impact of insulin on energy
metabolism owing to long-term glycemic remission in patients with
newly diagnosed rather than prolonged T2DM, induced by insulin
therapy (49). In our rodent
model of early-stage diabetes, insulin mitigated the muscular
energy surplus, subsequently, improving AMPK signaling. However,
whether this action of insulin persists in the later event of
diabetes needs to be clarified in the future. Additionally, the
convergence of insulin and metformin, both targeting a decreased
energy state, may potentially provide a synergistic therapeutic
effect on insulin resistance, which still necessitates further
evaluation.
Different from metformin or resveratrol, our study
indicated that the protective role of early insulin intervention in
muscular bioenergy was disassociated from the expression or
activities of respiratory complexes. On the other hand, insulin, by
restoring UCP3 protein expression, enhanced the cellular uncoupling
process in rodent myocytes provided with abundant fatty acid.
Mitochondrial uncoupling serves to reduce the efficiency of ATP
synthesis by decreasing the proton motive force. Likewise, in this
study, the elevation of mitochondrial membrane potential in
insulin-resistant skeletal muscle cells was annulled after insulin
administration, despite the lack of comprehensive evaluation for
systematic energy expenditure and respiratory control. Currently,
uncoupling has become an appealing therapeutic target to facilitate
energy turnover, improving insulin sensitivity as a consequence.
Hence, exploring chemical mitochondrial uncouplers and evaluating
their potential clinical application are critical for T2DM
prevention and treatment. Dinitrophenol, a non-selective
mitochondrial uncoupler, effectively boosts the energy expenditure
without generating ATP and was approved for treating obesity in the
1930s (50). However, at high
dosages, dinitrophenol causes hyperthermia that precludes its
therapeutic usage. Until recently, one study reported that a
liver-targeted derivative of dinitrophenol alleviated fatty liver
and whole-body insulin resistance in HFD rats with a wide
therapeutic index, thereby providing a basis for exploiting
tissue-targeted mitochondrial uncoupling agents (51). Alternatively, oral niclosamide
ethanolamine, derived from niclosamide which uncouples mitochondria
of parasitic worms and serves as an anthelmintic drug, was observed
to improve the hepatic steatosis and glycemic control with little
impact on body temperature in insulinresistant mice (52). Moreover, curcumin has been shown
to uncouple the oxidative phosphorylation at low concentrations in
isolated rat liver mitochondria (53,54). The resulting increase in the
AMP:ATP ratio could explain its activation in AMPK signaling.
Notwithstanding these encouraging results in animal models, the
hypothesis that chemical uncouplers may contribute to improved
insulin sensitivity and their safety in clinical practice
necessitate further substantiation by experimental data in
humans.
In conclusion, UCP3-induced energy collapse,
accompanied by a relief in AMPK inhibition, was found to be
associated with the positive effect of insulin on muscular
lipotoxicity. This study suggests that obstruction of an energy
surplus is involved in the benefits of early insulin therapy on
lipid regulation, which supports the idea that energy homeostasis
could be a therapeutic target for obesity and T2DM.
Abbreviations:
|
AMPK
|
AMP-activated protein kinase
|
|
CPT1B
|
carnitine palmitoyltransferase 1B
|
|
FBG
|
fasting blood glucose
|
|
HFD
|
high-fat diet
|
|
HOMA-IR
|
homeostasis model assessment of the
insulin resistance index
|
|
MG132
|
carbobenzoxy-Leu-Leu-leucinal
|
|
NPH
|
neutral protamine Hagedorn
|
|
PA
|
palmitate
|
|
SREBP-1c
|
sterol-regulated element binding
protein-1c
|
|
T2DM
|
type 2 diabetes mellitus
|
|
UCP3
|
uncoupling protein 3
|
|
VDAC1
|
voltage-dependent anion-selective
channel 1, ATP, adenosine triphosphate
|
|
ADP
|
adenosine diphosphate
|
|
AMP
|
adenosine monophosphate
|
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81570736, 81570737,
81370947, 81270906, 81600632, 81600637, 81500612 and 81400832), the
National Key Research and Development Program of China (no.
2016YFC1304804), the Project of National Key Clinical Division,
Jiangsu Province's Key Discipline of Medicine (no. XK201105), China
Diabetes Young Scientific Talent Research Project, the Key Research
and Development Program of Jiangsu Province of China (nos.
BE2015604 and BE2016606), the Key Provincial Talents Program of
Jiangsu Province of China (no. RC2011011), the Jiangsu Province's
Key Laboratory for Molecular Medicine (no. BM2007208), the Nanjing
Science and Technology Development Project (no. 201605019) and the
Key Project of Nanjing Clinical Medical Science.
References
|
1
|
Guariguata L, Whiting DR, Hambleton I,
Beagley J, Linnenkamp U and Shaw JE: Global estimates of diabetes
prevalence for 2013 and projections for 2035. Diabetes Res Clin
Pract. 103:137–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kramer CK, Zinman B and Retnakaran R:
Short-term intensive insulin therapy in type 2 diabetes mellitus: A
systematic review and meta-analysis. Lancet Diabetes Endocrinol.
1:28–34. 2013. View Article : Google Scholar
|
|
3
|
Hu Y, Li L, Xu Y, Yu T, Tong G, Huang H,
Bi Y, Weng J and Zhu D: Short-term intensive therapy in newly
diagnosed type 2 diabetes partially restores both insulin
sensitivity and β-cell function in subjects with long-term
remission. Diabetes Care. 34:1848–1853. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D,
Hu Y, Zhou Z, Yan X, Tian H, et al: Effect of intensive insulin
therapy on beta-cell function and glycaemic control in patients
with newly diagnosed type 2 diabetes: A multicentre randomised
parallel-group trial. Lancet. 371:1753–1760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samuel VT, Petersen KF and Shulman GI:
Lipid-induced insulin resistance: Unravelling the mechanism.
Lancet. 375:2267–2277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shulman GI, Rothman DL, Jue T, Stein P,
DeFronzo RA and Shulman RG: Quantitation of muscle glycogen
synthesis in normal subjects and subjects with
non-insulin-dependent diabetes by 13C nuclear magnetic resonance
spectroscopy. N Engl J Med. 322:223–228. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eckardt K, Taube A and Eckel J:
Obesity-associated insulin resistance in skeletal muscle: Role of
lipid accumulation and physical inactivity. Rev Endocr Metab
Disord. 12:163–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Itani SI, Ruderman NB, Schmieder F and
Boden G: Lipid-induced insulin resistance in human muscle is
associated with changes in diacylglycerol, protein kinase C, and
IkappaB-alpha. Diabetes. 51:2005–2011. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Samuel VT and Shulman GI: Mechanisms for
insulin resistance: Common threads and missing links. Cell.
148:852–871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guillet-Deniau I, Mieulet V, Le Lay S,
Achouri Y, Carré D, Girard J, Foufelle F and Ferré P: Sterol
regulatory element binding protein-1c expression and action in rat
muscles: Insulin-like effects on the control of glycolytic and
lipogenic enzymes and UCP3 gene expression. Diabetes. 51:1722–1728.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guillet-Deniau I, Pichard AL, Koné A,
Esnous C, Nieruchalski M, Girard J and Prip-Buus C: Glucose induces
de novo lipogenesis in rat muscle satellite cells through a
sterol-regulatory-element-binding-protein-1c-dependent pathway. J
Cell Sci. 117:1937–1944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsintzas K, Jewell K, Kamran M, Laithwaite
D, Boonsong T, Littlewood J, Macdonald I and Bennett A:
Differential regulation of metabolic genes in skeletal muscle
during starvation and refeeding in humans. J Physiol. 575:291–303.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bruce CR, Anderson MJ, Carey AL, Newman
DG, Bonen A, Kriketos AD, Cooney GJ and Hawley JA: Muscle oxidative
capacity is a better predictor of insulin sensitivity than lipid
status. J Clin Endocrinol Metab. 88:5444–5451. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim JY, Hickner RC, Cortright RL, Dohm GL
and Houmard JA: Lipid oxidation is reduced in obese human skeletal
muscle. Am J Physiol Endocrinol Metab. 279:E1039–E1044.
2000.PubMed/NCBI
|
|
15
|
Ukropcova B, Sereda O, de Jonge L, Bogacka
I, Nguyen T, Xie H, Bray GA and Smith SR: Family history of
diabetes links impaired substrate switching and reduced
mitochondrial content in skeletal muscle. Diabetes. 56:720–727.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X,
Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ronnett GV, Kleman AM, Kim EK, Landree LE
and Tu Y: Fatty acid metabolism, the central nervous system, and
feeding. Obesity (Silver Spring). 14(Suppl 5): 201S–207S. 2006.
View Article : Google Scholar
|
|
18
|
Kjøbsted R, Pedersen AJ, Hingst JR,
Sabaratnam R, Birk JB, Kristensen JM, Højlund K and Wojtaszewski
JF: Intact regulation of the AMPK signaling network in response to
exercise and insulin in skeletal muscle of male patients with type
2 diabetes: Illumination of AMPK activation in recovery from
exercise. Diabetes. 65:1219–1230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang S, Wu W, Tang W, Ge Z, Wang H, Hong
T, Zhu D and Bi Y: Suppression of Rho-kinase 1 is responsible for
insulin regulation of the AMPK/SREBP-1c pathway in skeletal muscle
cells exposed to palmitate. Acta Diabetol. Mar 7–2017.Epub ahead of
print. View Article : Google Scholar
|
|
20
|
Saltiel AR: New therapeutic approaches for
the treatment of obesity. Sci Transl Med. 8:323rv22016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Busiello RA, Savarese S and Lombardi A:
Mitochondrial uncoupling proteins and energy metabolism. Front
Physiol. 6:362015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Krook A, Digby J, O'Rahilly S, Zierath JR
and Wallberg-Henriksson H: Uncoupling protein 3 is reduced in
skeletal muscle of NIDDM patients. Diabetes. 47:1528–1531. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mensink M, Hesselink MK, Borghouts LB,
Keizer H, Moonen-Kornips E, Schaart G, Blaak EE and Schrauwen P:
Skeletal muscle uncoupling protein-3 restores upon intervention in
the prediabetic and diabetic state: Implications for diabetes
pathogenesis? Diabetes Obes Metab. 9:594–596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schrauwen P, Hesselink MK, Blaak EE,
Borghouts LB, Schaart G, Saris WH and Keizer HA: Uncoupling protein
3 content is decreased in skeletal muscle of patients with type 2
diabetes. Diabetes. 50:2870–2873. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clapham JC, Arch JR, Chapman H, Haynes A,
Lister C, Moore GB, Piercy V, Carter SA, Lehner I, Smith SA, et al:
Mice overexpressing human uncoupling protein-3 in skeletal muscle
are hyperphagic and lean. Nature. 406:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Costford SR, Chaudhry SN, Salkhordeh M and
Harper ME: Effects of the presence, absence, and overexpression of
uncoupling protein-3 on adiposity and fuel metabolism in congenic
mice. Am J Physiol Endocrinol Metab. 290:E1304–E1312. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Son C, Hosoda K, Ishihara K, Bevilacqua L,
Masuzaki H, Fushiki T, Harper ME and Nakao K: Reduction of
diet-induced obesity in transgenic mice overexpressing uncoupling
protein 3 in skeletal muscle. Diabetologia. 47:47–54. 2004.
View Article : Google Scholar
|
|
28
|
Petersen KF, Dufour S and Shulman GI:
Decreased insulin-stimulated ATP synthesis and phosphate transport
in muscle of insulin-resistant offspring of type 2 diabetic
parents. PLoS Med. 2:e2332005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stump CS, Short KR, Bigelow ML, Schimke JM
and Nair KS: Effect of insulin on human skeletal muscle
mitochondrial ATP production, protein synthesis, and mRNA
transcripts. Proc Natl Acad Sci USA. 100:7996–8001. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bi Y, Cai M, Liang H, Sun W, Li X, Wang C,
Zhu Y, Chen X, Li M and Weng J: Increased carnitine palmitoyl
transferase 1 expression and decreased sterol regulatory
element-binding protein 1c expression are associated with reduced
intramuscular triglyceride accumulation after insulin therapy in
high-fat-diet and streptozotocin-induced diabetic rats. Metabolism.
58:779–786. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bi Y, Wu W, Shi J, Liang H, Yin W, Chen Y,
Tang S, Cao S, Cai M, Shen S, et al: Role for sterol regulatory
element binding protein-1c activation in mediating skeletal muscle
insulin resistance via repression of rat insulin receptor
substrate-1 transcription. Diabetologia. 57:592–602. 2014.
View Article : Google Scholar
|
|
32
|
Xie B, Chen Q, Chen L, Sheng Y, Wang HY
and Chen S: The Inactivation of RabGAP function of AS160 promotes
lysosomal degradation of GLUT4 and causes postprandial
hyperglycemia and hyperinsulinemia. Diabetes. 65:3327–3340. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Bhatt DP, Chen X, Geiger JD and
Rosenberger TA: A sensitive HPLC-based method to quantify adenine
nucleotides in primary astrocyte cell cultures. J Chromatogr B
Analyt Technol Biomed Life Sci. 889–890:110–115. 2012. View Article : Google Scholar
|
|
35
|
Azzu V, Mookerjee SA and Brand MD: Rapid
turnover of mitochondrial uncoupling protein 3. Biochem J.
426:13–17. 2010. View Article : Google Scholar
|
|
36
|
Mookerjee SA and Brand MD: Characteristics
of the turnover of uncoupling protein 3 by the ubiquitin proteasome
system in isolated mitochondria. Biochim Biophys Acta.
1807:1474–1481. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bi Y, Sun WP, Chen X, Li M, Liang H, Cai
MY, Zhu YH, He XY, Xu F and Weng JP: Effect of early insulin
therapy on nuclear factor kappaB and cytokine gene expressions in
the liver and skeletal muscle of high-fat diet,
streptozotocin-treated diabetic rats. Acta Diabetol. 45:167–178.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pagel-Langenickel I, Bao J, Pang L and
Sack MN: The role of mitochondria in the pathophysiology of
skeletal muscle insulin resistance. Endocr Rev. 31:25–51. 2010.
View Article : Google Scholar :
|
|
39
|
Szendroedi J, Phielix E and Roden M: The
role of mitochondria in insulin resistance and type 2 diabetes
mellitus. Nat Rev Endocrinol. 8:92–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y and Ye J: Mitochondrial inhibitor
as a new class of insulin sensitizer. Acta Pharm Sin B. 2:341–349.
2012. View Article : Google Scholar
|
|
41
|
Bridges HR, Jones AJ, Pollak MN and Hirst
J: Effects of metformin and other biguanides on oxidative
phosphorylation in mitochondria. Biochem J. 462:475–487. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Foretz M, Hébrard S, Leclerc J,
Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F and
Viollet B: Metformin inhibits hepatic gluconeogenesis in mice
independently of the LKB1/AMPK pathway via a decrease in hepatic
energy state. J Clin Invest. 120:2355–2369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Owen MR, Doran E and Halestrap AP:
Evidence that metformin exerts its anti-diabetic effects through
inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J. 348:607–614. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Turner N, Li JY, Gosby A, To SW, Cheng Z,
Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, et al:
Berberine and its more biologically available derivative,
dihydroberberine, inhibit mitochondrial respiratory complex I: A
mechanism for the action of berberine to activate AMP-activated
protein kinase and improve insulin action. Diabetes. 57:1414–1418.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xia X, Yan J, Shen Y, Tang K, Yin J, Zhang
Y, Yang D, Liang H, Ye J and Weng J: Berberine improves glucose
metabolism in diabetic rats by inhibition of hepatic
gluconeogenesis. PLoS One. 6:e165562011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gledhill JR, Montgomery MG, Leslie AG and
Walker JE: Mechanism of inhibition of bovine F1-ATPase by
resveratrol and related polyphenols. Proc Natl Acad Sci USA.
104:13632–13637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Szendroedi J, Schmid AI, Chmelik M, Toth
C, Brehm A, Krssak M, Nowotny P, Wolzt M, Waldhausl W and Roden M:
Muscle mitochondrial ATP synthesis and glucose
transport/phosphorylation in type 2 diabetes. PLoS Med. 4:e1542007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sreekumar R, Halvatsiotis P, Schimke JC
and Nair KS: Gene expression profile in skeletal muscle of type 2
diabetes and the effect of insulin treatment. Diabetes.
51:1913–1920. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Retnakaran R and Zinman B: Short-term
intensified insulin treatment in type 2 diabetes: Long-term effects
on β-cell function. Diabetes Obes Metab. 14(Suppl 3): 161–166.
2012. View Article : Google Scholar
|
|
50
|
Parascandola J: Dinitrophenol and
bioenergetics: An historical perspective. Mol Cell Biochem.
5:69–77. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Perry RJ, Kim T, Zhang XM, Lee HY, Pesta
D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, et al:
Reversal of hypertriglyceridemia, fatty liver disease, and insulin
resistance by a liver-targeted mitochondrial uncoupler. Cell Metab.
18:740–748. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tao H, Zhang Y, Zeng X, Shulman GI and Jin
S: Niclosamide ethanolamine-induced mild mitochondrial uncoupling
improves diabetic symptoms in mice. Nat Med. 20:1263–1269. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ligeret H, Barthélémy S, Bouchard Doulakas
G, Carrupt PA, Tillement JP, Labidalle S and Morin D: Fluoride
curcumin derivatives: New mitochondrial uncoupling agents. FEBS
Lett. 569:37–42. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lim HW, Lim HY and Wong KP: Uncoupling of
oxidative phosphorylation by curcumin: Implication of its cellular
mechanism of action. Biochem Biophys Res Commun. 389:187–192. 2009.
View Article : Google Scholar : PubMed/NCBI
|