Introduction
The renin-angiotensin-aldosterone system (RAAS) has
an important role in the pathogenesis of cardiovascular diseases,
including hypertension (1) and
atherosclerosis (2). Angiotensin
II (Ang II) is the main active peptide hormone of the RAAS and has
a major role in endothelial dysfunction, vascular remodeling and
vascular inflammation, which are closely associated with
hypertension and atherosclerosis (3,4).
Previous study indicated that Ang II mediates an anti-growth effect
via stimulation of the Ang II receptor type 2 in endothelial cells
(5). Accumulating evidence
demonstrated that Ang II could induce apoptosis of human umbilical
vein endothelial cells (HUVECs) via a different pathway (6). However, there are limited studies on
the effect of Ang II on the proliferation of HUVECs.
Mitochondria are the 'power plants' in cells that
generate a large amount of adenosine triphosphate (ATP) to maintain
normal cell function. Investigating the role of mitochondria in
various cardiovascular diseases is currently an area of great
interest (7). Mitochondrial
damage and dysfunction has an important role in the atherosclerotic
process, which seems to be reactive oxygen species (ROS)/reactive
nitrogen species-dependent and may cause loss of bioenergetic
control (8,9). Mitochondria have a critical function
in regulating the redox state, energy metabolism, proliferation,
apoptosis and intracellular signaling (10,11). In most studies, polarographic
techniques have been used to examine mitochondria isolated from
cells, however this methodology can not measure bioenergetic
function in intact cells. Furthermore, this methodology has certain
disadvantages, such as it can result in anoikis and increased
oxidative stress (12). In this
study, the effects of Ang II on cellular bioenergetic function were
examined using a non-invasive technology, Seahorse Bioscience XF24
extracellular flux analyzer. This technology made it possible to
determine the impact of Ang II on mitochondrial respiration and
glycolysis in intact cells, and to characterize the changes in
bioenergetics.
Statins, including atorvastatin, simvastatin,
rosuvastatin and others, have exerted a variety of protective
effects including upregulation of endothelial nitric oxide (NO)
expression and antioxidant effects, which is independent of
lowering cholesterol concentrations (13). Studies from both basic and
clinical trial have demonstrated that atorvastatin has exhibited an
antihypertensive effect by improving endothelial function,
resisting oxidation and increasing vascular NO stores (14–16). However, further studies are
required to elucidate the underlying mechanism how atorvastatin is
involved in regulating stable endothelial function.
In the present study, it was aimed to explore
whether Ang II inhibits proliferation of HUVECs by altering
mitochondrial energy metabolism and whether atorvastatin has a
protective role via restoration of endothelial function.
Materials and methods
Cell culture and treatments
HUVECs (obtained from the Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) were cultured in Dulbecco's
modified Eagle's medium (DMEM; Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) containing 10% fetal bovine serum (FBS;
Hyclone; GE Healthcare Life Sciences) and penicillin-streptomycin
(100 U/ml-100 µg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), in humidified atmosphere with 5% CO2 at 37°C.
Medium was replaced every 2 days or as necessary. For each
experiment, the cells were treated with 1 µM Ang II alone or
in combination with 10 µM atorvastatin (both from
Sigma-Aldrich; Merck KGaA) for 24 h. Trypsin (0.25%)
(Sigma-Aldrich; Merck KGaA) was used for digestion.
Cell viability analysis
Cell viability was determined by measuring the
metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT), as described in our previous study (17). Briefly, HUVECs were plated in a
96-well plate at density of 2,000 cells/well. The medium was
changed after 1 day. The cells were incubated with fresh medium
containing 1 µM Ang II alone or with 10 µM
atorvastatin for 24 h. Subsequently, 10 µl sterile MTT
solution (final concentration, 0.5 mg/ml) was added to each well.
After incubation at 37°C for 4 h in the dark, the culture media
containing MTT was removed and replaced with 100 µl DMSO.
The plate was gently rotated on a linear and orbital shaker for 3–5
min to completely dissolve the precipitation. The absorbance was
measured with microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) at 570 nm. The percentage of cell viability was
calculated according to the following formula: Cell viability (%) =
optical density (OD) of the treatment group/OD of the control group
×100.
Cell proliferation assay
Cells were seeded into 6-well plates at a density of
10,000 cells/well in 2 ml medium supplemented with 10% FBS. The
medium was changed every 2 days or in necessary. Cell number at the
indicated time-points (1, 2, 3 and 4 days) was determined by
counting using a hemocytometer.
Growth curve assays using real-time cell
analyzer (RTCA)
To analyze the cell proliferation continuously over
time, growth curve assays were performed in RTCA in quadruplicate
with the xCELLigence system (ACEA Bioscience, Inc., San Diego, CA,
USA) according to the methods as previously described (18). Briefly, 5,000 cells/well were
seeded in RTCA E-plates (ACEA Bioscience, Inc.), the electrical
impedance in each well was measured continuously. The shift of the
electrical impedance was expressed as the cell index, which was a
parameter of cell viability.
5-ethynyl-2′-deoxyuridine (EdU)
incorporation analysis
As described by our previous studies (17,19) and the manufacturer's instructions,
the DNA synthesis rate in HUVECs was determined by an EdU
incorporation analysis using Click-iT™ EdU Alexa Fluor 555 Imaging
kit (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Briefly, the cells were incubated with EdU-labeling solution for 8
h at 37°C, and then fixed with 4% cold formaldehyde for 30 min at
room temperature. After permeabilization with 1% Triton X-100 for
20 min, the cells were reacted with Click-iT reaction cocktails
(Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at room
temperature. Subsequently, the DNA contents of the cells were
stained with Hoechst 33342 (final concentration, 5 µg/ml)
for 30 min. Finally, EdU-labeled cells were counted using ImageJ
1.47 software (National Institutes of Health, Bethesda, MD, USA)
and normalized to the total number of Hoechst-stained cells. At
least 500 cells each experiment were counted, EdU-positive cells
were expressed as a percentage of the total cells.
Western blotting
Western blotting was performed as previously
described (17,20). The cells were lysed in lysis
buffer (20 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA and 1% Triton X-100)
containing a protease inhibitor cocktail (Sigma-Aldrich; Merck
KGaA). The total protein concentrations were measured by a BCA
Protein Assay kit (Sigma-Aldrich; Merck KGaA). After
heat-denaturing, equal quantities of proteins (20 µg) were
separated by NuPAGE Novex 12% Bis-Tris Gel and electrophoresed in
the XCell SureLock™ Mini-Cell (both from Invitrogen; Thermo Fisher
Scientific, Inc.), and then transferred to polyvinylidene
difluoride membranes and blocked with 5% non-fat milk in
Tris-buffered saline (TBS) for 1.5 h at the room temperature. The
membranes were incubated with primary antibodies against OXPHOS
(ab110413; 1:1,000; Abcam, Cambridge, UK) or GAPDH (AB2302;
1:1,000; EMD Millipore, Billerica, MA, USA) overnight at 4°C. After
washing three times with 1X TBS, the membranes were incubated with
secondary antibodies [anti-mouse (AP181R) and anti-rabbit
antibodies (AP187R); 1:10,333; EMD Millipore] for 1.5 h at room
temperature. After washing steps, immunoreactive binding was
detected with ECL detection reagent (Amersham Biosciences,
Piscataway, NJ, USA) with MicroChemi 4.2. The band intensity was
quantified using ImageJ 1.47 software.
Measurement of mitochondrial membrane
potential
We used tetramethylrhodamine (TMRE; Molecular
Probes; Thermo Fisher Scientific, Inc.) to detect changes in
mitochondrial membrane potential, as described previously (21,22). Briefly, unfixed live cells were
incubated with 100 nM TMRE in the dark for 30 min at 37°C and 5%
CO2. After washing, cells were images using a
fluorescence microscope.
Bioenergetic measurements using Seahorse
mitochondrial flux analyses
To measure the rate of oxidative phosphorylation
(OXPHOS) and glycolysis in HUVECs, a Seahorse metabolic flux
analyzer was used, according to methods previously described
(12,23). For measuring oxygen consumption
rate (OCR), XF Cell Mito Stress Test kit (Agilent Technologies,
Inc., Santa Clara, CA, USA) including different pharmacological
inhibitors were used to probe the function of individual components
in the respiratory chain. The cells were incubated in XF24 culture
microplates with culture medium for 24 h as well as the sensor
cartridge hydrated in XF Calibrant at 37°C in a non-CO2
incubator overnight. Prior to all bioenergetic assays, the culture
medium was replaced 1 h before with unbuffered DMEM (pH 7.4)
supplemented with 4 mM L-glutamine. To estimate the basal OCR
coupled to ATP synthesis, 1 µM oligomycin was injected to
inhibit the ATP synthase (complex V). Typically, the decreased OCR
in response to oligomycin indicated the cells were using
mitochondria to generate ATP. The remaining OCR could be ascribed
to either proton leakage or the demand on the mitochondrial proton
gradient for the movement of ions or metabolites. To determine the
maximal OCR that the cells could sustain, 0.5 µM carbonyl
cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) was injected,
which made the mitochondrial inner membrane permeable to protons.
Finally, 0.5 µM antimycin A (Anti-A) and rotenone was
injected to inhibit electron flux. The remaining OCR could be
ascribed to oxygen consumption due to the formation of
mitochondrial ROS and non-mitochondrial sources.
For measuring extracellular acidification rate
(ECAR), XF Glycolysis Stress Test kit (Agilent Technologies, Inc.)
including different pharmacological inhibitors were used to probe
the function of glycolysis. Initially, cells were incubated in
sugar or pyruvate-free glycolytic assay medium for 1 h before the
assay and then the first injection of a saturating concentration of
glucose was conducted. The cells may catabolize glucose into
pyruvate via glycolysis pathway, producing ATP, NADH, water and
protons. The extrusion of protons into surrounding medium leaded to
a sudden increase in ECAR, which was defined as basal glycolytic
capacity. The second injection was oligomycin that could shift
energy production to glycolysis by means of restraining
mitochondrial ATP production. Consequently, the sharp increase of
ECAR indicated the level of glycolytic capacity. The final
injection was 2-deoxy-D-glucose, a glucose analog, which inhibited
glycolysis through competitive binding to glucose hexokinase, the
first enzyme in the glycolytic pathway. The decrease in ECAR
confirmed that the ECAR produced in the experiment was caused by
glycolysis. The gap between glycolytic capacity and glycolysis was
defined as glycolytic reserve. ECAR, prior to glucose injection was
referred as non-glycolytic acidification and that may due to other
processes in cells.
First, the optimum number of cells needed for these
experiments was determined. HUVECs were seeded to a density of
10,000, 20,000, 40,000 or 80,000 cells/well. Oxygen consumption in
these cells was proportional to cell number within this range, and
a seeding density of 10,000 cells/well was selected for the
subsequent experiments. Subsequently, an assay was developed to
measure indices of mitochondrial function in HUVECs incubated with
Ang II and/or atorvastatin. HUVECs were seeded at a density of
10,000 cells/well in the 24-well Seahorse assay plates. Cells were
then serum-starved in 0.1% FBS DMEM for 24 h and subsequently 1
µM Ang II alone or with 10 µM atorvastatin were added
for 24 h. Medium was replaced with DMEM and then incubated for 1 h
at 37°C without CO2. XF Cell Mito Stress Test kit was
used to test the mitochondrial respiration and XF Glycolysis Stress
Test kit (Agilent Technologies, Inc.) was used to test glycolysis
function. To allow comparison between experiments, data are
expressed as the rate of OCR in pmol/min or the rate of ECAR in
mpH/min.
Statistical analysis
The statistical significance of the difference was
analyzed by analysis of variance and post hoc Dunnett's test. All
experiments were repeated for at least three times, and data were
expressed as the mean ±standard deviation. All analyses were
performed using SPSS 17.0 statistical software (SPSS, Inc.,
Chicago, IL, USA) and P<0.05 was considered to indicate a
statistically significant difference.
Results
Atorvastatin attenuates the Ang
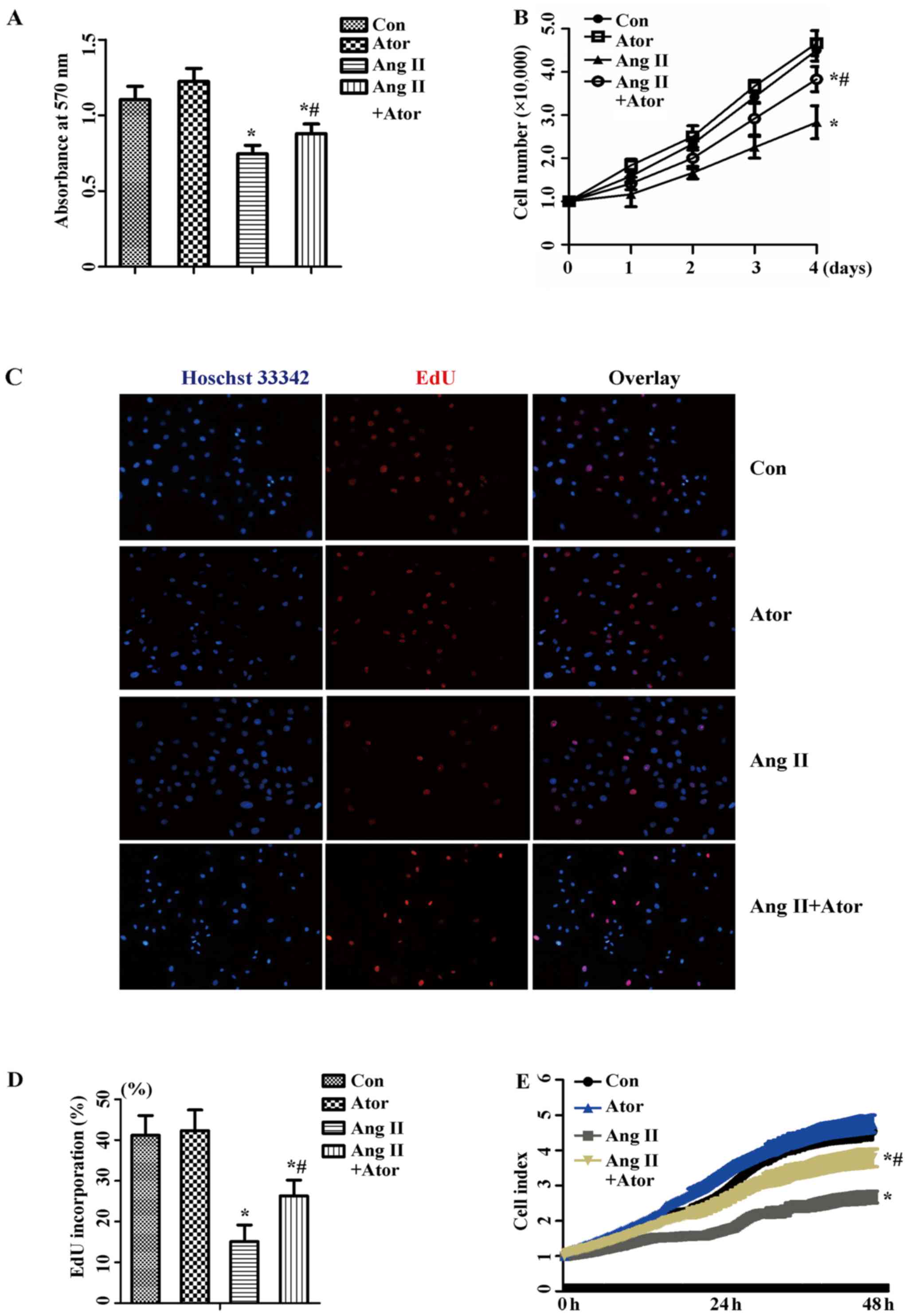
II-induced inhibition of the proliferation of HUVECs
To investigate how atorvastatin affected Ang
II-inhibited proliferation in HUVECs, cells were cultured with 1
µM Ang II alone or with 10 µM atorvastatin for 24 h.
An MTT assay to measure the cellular activity, and the
proliferation level was determined by cell counting, EdU staining
and RTCA. As presented in Fig. 1,
1 µM Ang II exhibited an
inhibitory effect on cellular activity (Fig. 1A) and the proliferation of HUVECs
(Fig. 1B–D). Co-treatment with 10
µM atorvastatin recovered cellular activity, and similar
results were observed with the proliferation level. The result was
further confirmed by the results of RTCA (Fig. 1E). Therefore, these results
suggested that atorvastatin attenuated Ang II-induced inhibition of
proliferation in HUVECs.
Measurement of mitochondrial function in
endothelial cells
To assess cellular bioenergetics in intact
endothelial cells, the Seahorse metabolic flux analyzer was used to
determine rates of oxygen consumption and glycolysis (12,23). The flow charts of the experimental
design are presented in Fig. 2A and
B, and the experimental principles and methods are detailed
described in the Materials and methods. In the first series of
experiments, the optimal number of HUVECs required to obtain
measurable OCR and ECAR was established, as shown in Fig. 2C and D. Both OCR and ECAR
responses were proportional to cell number. For subsequent
experiments, a seeding density of 10,000 cells/well was selected to
detect the changes in OCR and ECAR due to exposure to Ang II and/or
atorvastatin.
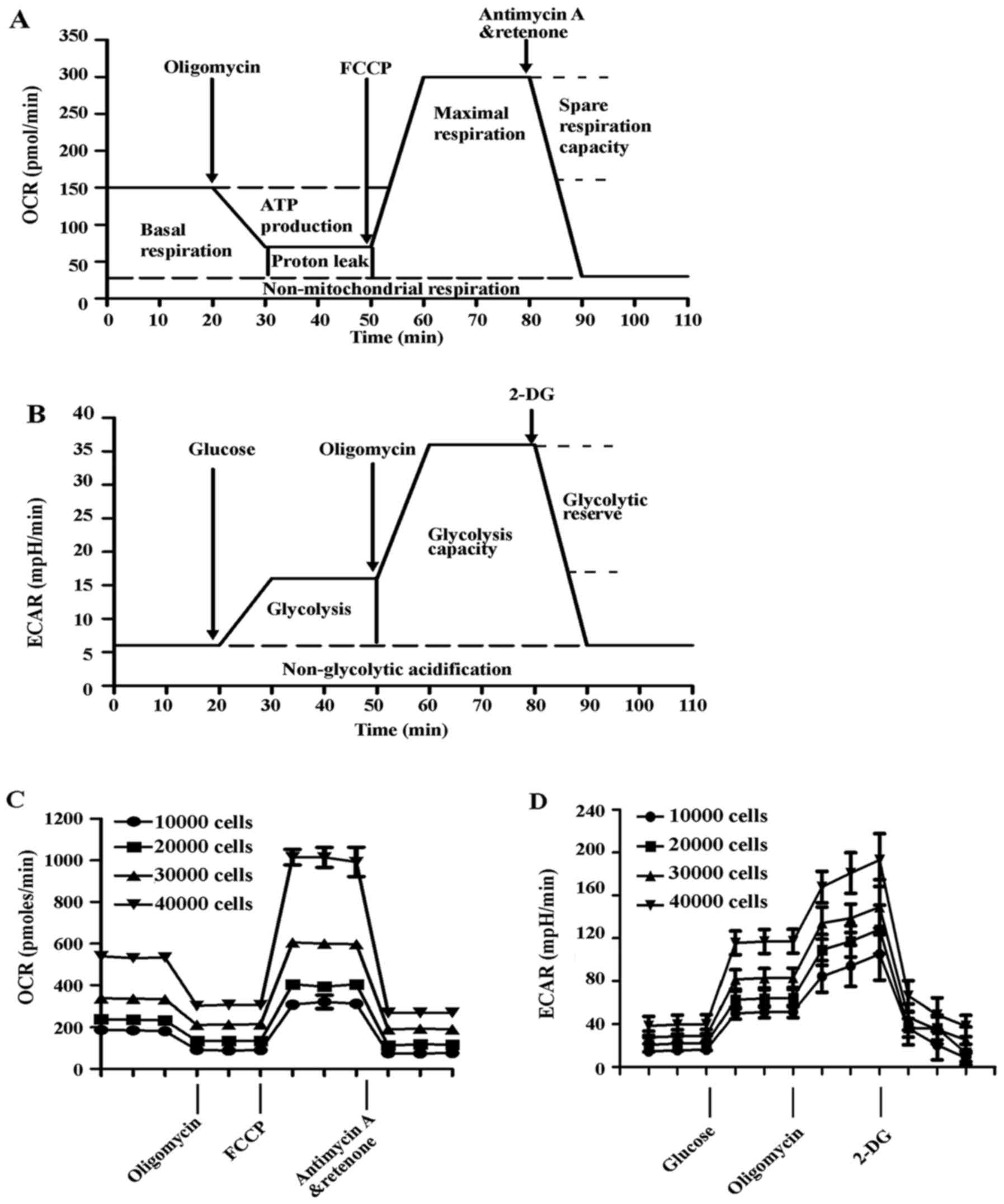 | Figure 2Measurement of mitochondrial function
in HUVECs. The experiments were performed using the XF24
extracellular flux analyzer, and the flow chart showed the
measurement of (A) OCR and (B) ECAR. The optimal number (10,000,
20,000, 40,000 and 80,000 cells/well) of HUVECs were seeded in the
Seahorse Bioscience microplates. (C) OCR and (D) ECAR were measured
and plotted as a function of cell seeding number. Results are
presented as the mean ± standard deviation (n=4). HUVECs, human
umbilical vein endothelial cells; OCR, oxygen consumption rate;
FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; ECAR,
extracellular acidification rate; 2-DG, 2-deoxy-D-glucose. |
Effect of Ang II and/or atorvastatin on
mitochondrial aerobic metabolism
The inhibitory effects of statins on Ang II-induced
endothelial dysfunction has described previously (24). Although statins played a
protective effect for endothelial dysfunction induced by Ang II,
the underlying mechanism was still not clear. We speculate that it
may be associated with cellular energy metabolism.
To further investigate the changes of mitochondrial
aerobic metabolism that occurred in HUVECs in response to Ang II
and/or atorvastatin, a Cell Mito Stress assay kit was used to
detect the OCR. HUVECs were treated with 1 µM Ang II and/or
10 µM atorvastatin for 24 h before exposure to 1 µM
oligomycin, 0.5 µM FCCP and 0.5 µM rotenone and
Anti-A. As demonstrated in Fig.
3A, Ang II reduced the OCR in HUVECs, suggesting that Ang II
inhibits mitochondrial aerobic respiration. These data are used to
calculate an apparent respiratory control ratio (RCR) for various
metabolic conditions for mitochondria in cells (12).
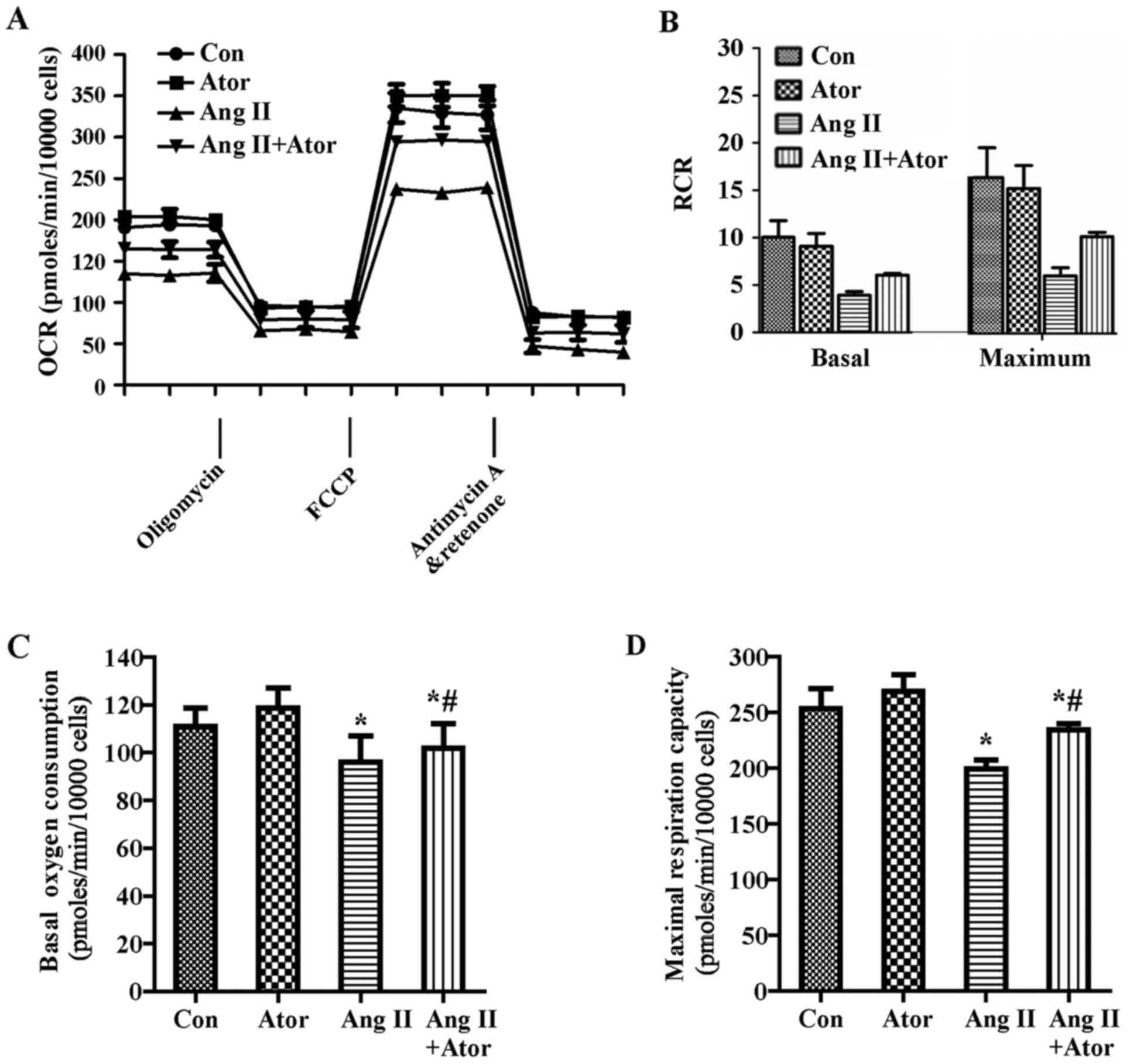 | Figure 3Effect of Ang II and/or atorvastatin
on mitochondrial aerobic metabolism in HUVECs. HUVECs were seeded
in the Seahorse Bioscience microplates (10,000 cells/well). (A) The
OCR was conducted using Mito Stress Test kit. After adherence for 4
h, 1 µM Ang II and/or 10 µM atorvastatin was added
into the microplates for co-incubation with cells for 24 h with
subsequent injection of 1 µM oligomycin, 0.5 µM FCCP
and 0.5 µM rotenone and antimycin A. (B) The contribution of
associated parameters including RCR, (C) basal oxygen consumption,
(D) maximal oxygen consumption, (E) spare respiration capacity, (F)
ATP-linked oxygen consumption, (G) proton leak and (H)
non-mitochondrial oxygen consumption to the total cellular oxygen
consumption was plotted, respectively. Each data point represented
an OCR measurement. Results shown represent mean ± standard
deviation (n=4). *P<0.05 vs. control;
#P<0.05 vs. Ang II. HUVECs, human umbilical vein
endothelial cells; Con, control; Ang II, angiotensin II; Ator,
atorvastatin; OCR, oxygen consumption rate; RCR, respiratory
control ratio. |
RCRbasal=(basal−Anti-A)/(Oligo−Anti-A)
RCRmax=(FCCP−Anti-A)/(Oligo−Anti-A)
RCR under the basal condition was 10.0±1.8 and under
maximal condition was 22.1±4.3 at a seeding density of 10,000
cells/well, which indicated that the mitochondria were tightly
coupled under normal physiological conditions in HUVECs.
Additionally, Ang II decreased the RCR (Fig. 3B). Furthermore, Ang II decreased
basal oxygen consumption, maximal respiration capacity, spare
respiration capacity, ATP-linked respiration and non-mitochondrial
respiration compared with the control group (Fig. 3C–F and H). By contrast, Ang II
increased the proton leak (Fig.
3G). Furthermore, atorvastatin significantly reversed the
dysfunction of aerobic metabolism in mitochondria induced by Ang II
(Fig. 3).
Effect of Ang II and/or atorvastatin on
glycolytic function
In addition to OCR, the Seahorse metabolic flux
analyzer can be used to measure the protons produced by the cells,
and the ECAR indicated lactate production and was therefore an
index of glycolysis (25). As
shown in Fig. 4A, ECAR increased
with the inhibition of Ang II on mitochondrial respiration, which
was attributable to the increased requirement for glycolysis to
generate ATP. The parameters of glycolytic function included
glycolysis, glycolytic capacity, glycolytic reserve and
non-glycolytic acidification. Following quantification, all the
parameters of glycolytic function besides glycolytic reserve were
increased in the Ang II-treatment group compared with the control
treatment group (Fig. 4B–E).
Furthermore, atorvastatin significantly reversed the glycolysis
dysfunction induced by Ang II.
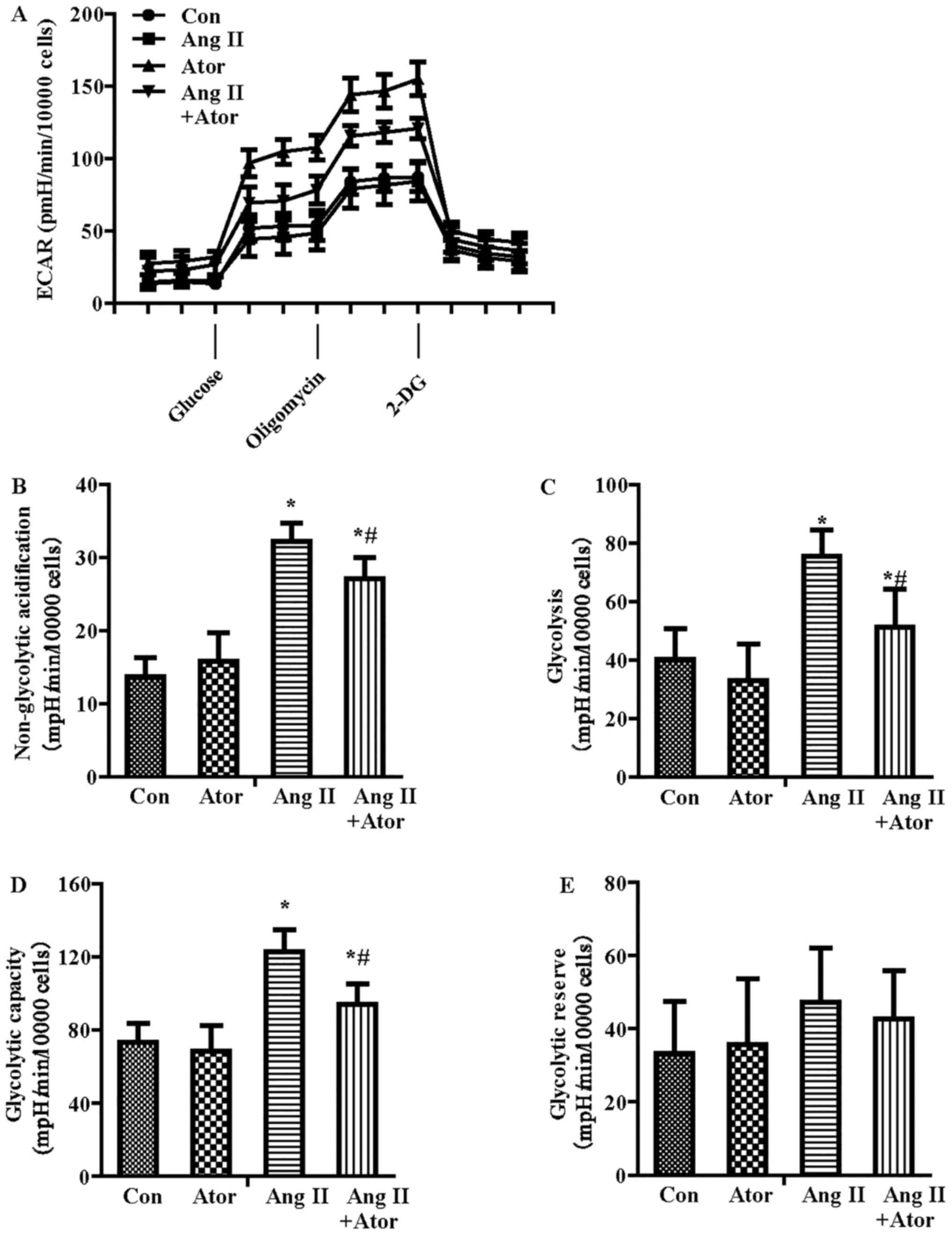 | Figure 4Effect of Ang II and/or atorvastatin
on glycolytic function in HUVECs. HUVECs were seeded in the
Seahorse Bioscience microplates (10,000 cells/well). (A) The ECAR
was conducted using XF Glycolysis Stress Test kit. After adherence
for 4 h, 1 µM Ang II and/or 10 µM atorvastatin was
added into the microplates for co-incubation with cells for 24 h
with subsequent injection of 15 mM glucose, 2 µM oligomycin
and 50 mM 2-DG. (B) The contribution of associated parameters
including non-glycolytic acidification, (C) glycolysis, (D)
glycolytic capacity and (E) glycolytic reserve to the total ECAR
was plotted, respectively. Each data point represented an ECAR
measurement. Results shown represent mean ± standard deviation
(n=4). *P<0.05 vs. control; #P<0.05 vs.
Ang II. HUVECs, human umbilical vein endothelial cells; ECAR,
extracellular acidification rate; Con, control; Ang II, angiotensin
II; Ator, atorvastatin; 2-DG, 2-deoxy-D-glucose. |
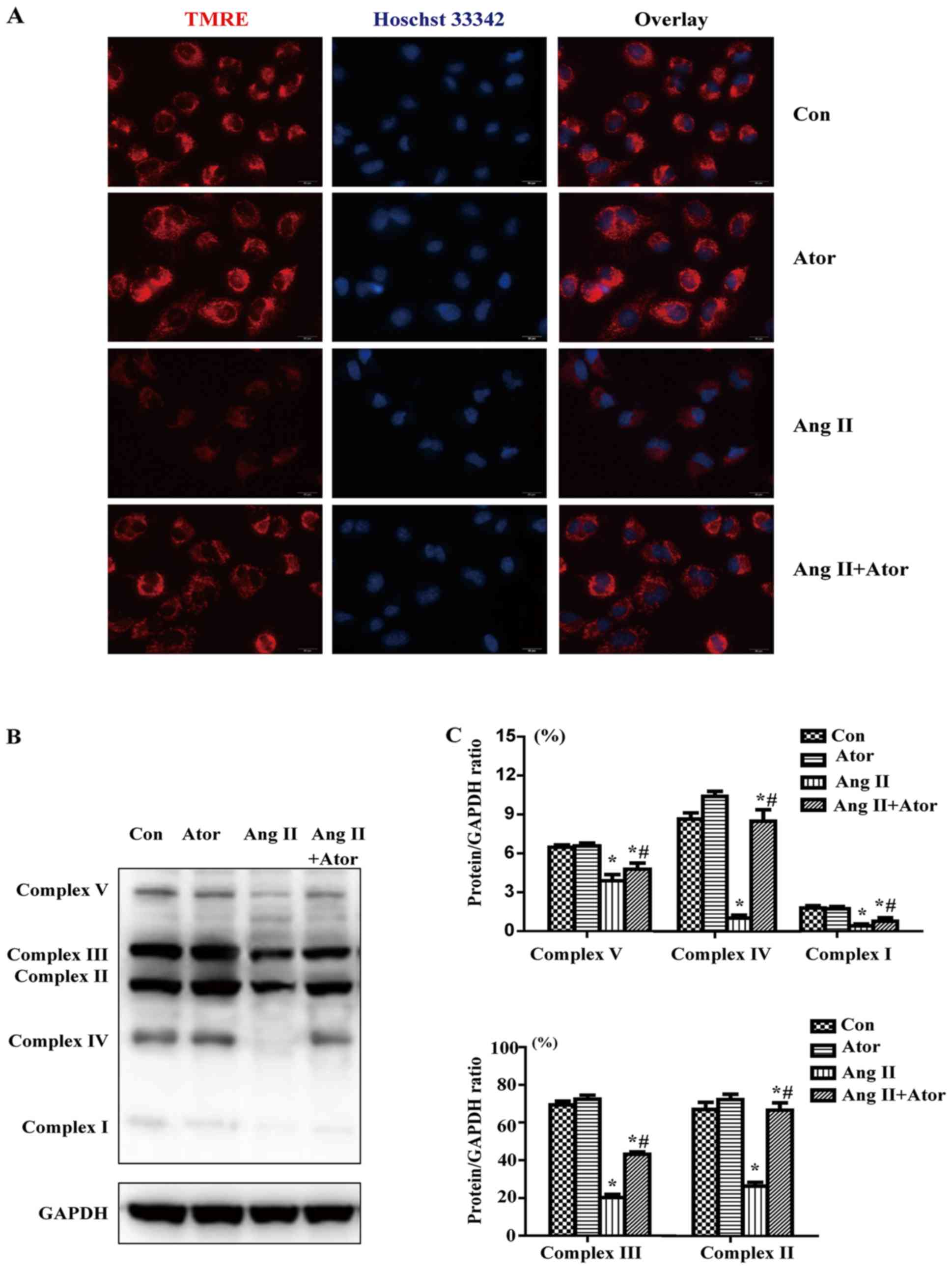
Effect of Ang II and/or atorvastatin on
mitochondrial membrane potential and mitochondrial respiration
chain complexes
To determine whether Ang II and/or atorvastatin had
effects on mitochondrial membrane potential, HUVECs were labeled
with TMRE (red) and the nuclei were stained with Hoechst 33342
(blue). TMRE (red) staining indicated that mitochondrial membrane
potential was reduced by Ang II; however co-treatment with
atorvastatin maintained the mitochondrial normal membrane potential
(Fig. 5A).
In order to investigate the effect of Ang II on
mitochondrial respiration chain complexes, western blotting was
performed to detect the protein level of OXPHOS components,
including complexes I–V. The expression of complexes I–V was
markedly decreased in the Ang II group compared with the control
treatment (Fig. 5B and C).
Furthermore, co-treatment with Ang II and atorvastatin rescued the
inhibition in OXPHOS protein expression induced by Ang II.
Discussion
In the present study, a newly emerging assay was
applied to determine the effect of Ang II and atorvastatin on
mitochondrial function in HUVECs. The principal findings of this
study demonstrated that atorvastatin attenuated the inhibitory
effect of Ang II on the proliferation of HUVECs via the
mitochondria.
Under physiological conditions, endothelial cells
maintain low proliferation capacity to reduced the damage of shear
stress caused by blood flow. Ang II is the main active peptide
hormone of the RAAS and has a major role in hypertension and
atherosclerosis (3,4). A previous study demonstrated that
Ang II could induce apoptosis of HUVECs via different pathway
(6). However, there are limited
studies on the effect of Ang II on the proliferation of HUVECs. To
the best of our knowledge, the current study is the first to
demonstrate that Ang II inhibits the proliferation of HUVECs by
damaging mitochondria to decrease aerobic metabolism, and making
energy conversion to glycolysis. To regulate redox state, energy
metabolism, apoptosis and intracellular signaling, cells need large
amounts of ATP, which is predominantly generated by mitochondria
(10,11). Glucose metabolism and glycolysis
are two important sources of ATP in cells. Notably, the calculated
RCR values in HUVECs indicated that the mitochondria were tightly
coupled under normal physiological conditions. However, Ang II
treatment of HUVECs decreased RCR, which affected the coupling
efficiency of OXPHOS. In addition, Ang II decreased the basal and
ATP-linked oxygen consumption, which may decrease the generation of
ATP necessary to the proliferation of HUVECs. Furthermore, loss of
mitochondrial maximal and reverse respiration capacity lead to a
decrease ability to respond to secondary energetic stressors, and
failure to maintain organ function and cellular repair (12,26). Additionally, Ang II increased
proton leak and lead to the collapse of mitochondrial membrane
potential. The increased level of proton leak across the inner
mitochondrial membrane is partially dependent on uncoupling
proteins or leakage through damaged respiratory complexes (7). In addition, Ang II could reduced the
expression of OXPHOS proteins (i.e., respiratory complexes
including complexes I–V). Dysfunction in the mitochondrial
respiration chain caused reduced basal respiration capacity,
reduced ATP-linked oxygen consumption and s reduced pare
respiration capacity, resulting in a decreased in ATP production
(26). Previous studies
demonstrated that through Ang II receptor, Ang II was implicated in
the generation of ROS, the accumulation of which may cause
mitochondrial dysfunction in HUVECs (27). On the other hand, inhibition of
the electron transport chain in mitochondria can increase ROS
generation (26). Therefore,
mitochondrial dysfunction and ROS may promote each other in a
feedback look and ultimately lead to cell death.
A previous study demonstrated that high glycolytic
flux could yield more ATP in a short time than OXPHOS when energy
supply is deficient in cells (28). Other studies also proved that the
stress injury of stimuli significantly augmented glycolysis
response to the decreased OCR in myocardial cells or bovine aortic
endothelial cells (7,12). Consistently, the results of the
present study indicated that Ang II stimulated ECAR in HUVECs in
response to energy demand and expenditure, which caused energy
metabolism to switch from OXPHOS to glycolysis.
Studies from both basic research and clinical trials
have demonstrated that atorvastatin has an antihypertensive effect
by improving endothelial function, resisting oxidation and
increasing vascular NO stores (14–16). Notably, there are also studies
that indicate that stains had side-effects in skeletal muscle,
ranging from mild weakness to severe rhabdomyolysis (29,30). It is well-known that statins
competitively inhibit 3-hydroxy-3-methylglutaryl-coenzyme (HMG-CoA)
reductase, which catalyzes the rate limiting step in cholesterol
biosynthesis (30). With the
inhibition of HMG-CoA reductase, ubiquinol biosynthesis is also
suppressed, which is an essential component of normal mitochondrial
electron transport (31).
Ubiquinol reduction may contribute to rhabdomyolysis. Furthermore,
a previous study indicated that treatment with simvastatin reduces
mitochondrial content, cell metabolism and cell viability in muscle
cells (32). Furthermore,
treatment with simvastatin significantly suppresses basal oxidative
reliance in muscle cells (32).
Notably, simvastatin had no effect on oxygen consumption in HepG2
cells (33). The difference in
susceptibility to toxicity of simvastatin between liver HepG2 cells
and skeletal muscle C2C12 myotubes was associated with the activity
of the insulin-like growth factor-1/Akt signaling pathway (33). A previous study indicated that
compared with hydrophilic rosuvastatin, lipophilic atorvastatin and
simvastatin had a more pronounced effect on ubiquinol production in
muscle tissue (34). However, the
results of the current study indicated that atorvastatin had no
toxicity on the mitochondrial aerobic metabolism, mitochondrial
membrane potential and mitochondrial respiration chain complexes.
In addition, atorvastatin attenuated Ang II-induced mitochondrial
dysfunction. This difference in tolerance to toxicity of stains may
reflect a difference in the activity of certain signaling pathways
in different cell lines.
The current study focused on the protective effect
of atorvastatin on Ang II-inhibited proliferation of HUVECs.
Investigation into cellular bioenergetics demonstrated that
atorvastatin prevented cellular energy metabolism switching from
OXPHOS to glycolysis induced by Ang II. Atorvastatin may attenuate
the Ang-II-induced damage of the mitochondrial respiratory chain
complexes in HUVECs. The results of the current study may enhance
understanding of the effects of Ang II/atorvastatin on the
proliferation of HUVECs and further provide potential therapeutic
or preventive targets for hypertension and atherosclerosis.
Acknowledgments
This study was supported by National Natural Science
Foundation of China (grant no. 81470417) and Natural Science
Foundation of Liaoning Province (grant no. 2013021090).
References
|
1
|
Whaley-Connell A, Johnson MS and Sowers
JR: Aldosterone: role in the cardiometabolic syndrome and resistant
hypertension. Prog Cardiovasc Dis. 52:401–409. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aroor AR, Demarco VG, Jia G, Sun Z,
Nistala R, Meininger GA and Sowers JR: The role of tissue
renin-angiotensin-aldosterone system in the development of
endothelial dysfunction and arterial stiffness. Front Endocrinol
Lausanne: 4. pp. 1612013, View Article : Google Scholar
|
|
3
|
Schiffrin EL and Touyz RM: From bedside to
bench to bedside: role of renin-angiotensin-aldosterone system in
remodeling of resistance arteries in hypertension. Am J Physiol
Heart Circ Physiol. 287:H435–H446. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Touyz RM: The role of angiotensin II in
regulating vascular structural and functional changes in
hypertension. Curr Hypertens Rep. 5:155–164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stoll M, Steckelings UM, Paul M, Bottari
SP, Metzger R and Unger T: The angiotensin AT2-receptor mediates
inhibition of cell proliferation in coronary endothelial cells. J
Clin Invest. 95:651–657. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sata M and Fukuda D: Crucial role of
renin-angiotensin system in the pathogenesis of atherosclerosis. J
Med Invest. 57:12–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hill BG, Dranka BP, Zou L, Chatham JC and
Darley-Usmar VM: Importance of the bioenergetic reserve capacity in
response to cardiomyocyte stress induced by 4-hydroxynonenal.
Biochem J. 424:99–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ballinger SW: Mitochondrial dysfunction in
cardiovascular disease. Free Radic Biol Med. 38:1278–1295. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Madamanchi NR and Runge MS: Mitochondrial
dysfunction in atherosclerosis. Circ Res. 100:460–473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryan MT and Hoogenraad NJ:
Mitochondrial-nuclear communications. Annu Rev Biochem. 76:701–722.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Picard M, Taivassalo T, Gouspillou G and
Hepple RT: Mitochondria: isolation, structure and function. J
Physiol. 589:4413–4421. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dranka BP, Hill BG and Darley-Usmar VM:
Mitochondrial reserve capacity in endothelial cells: the impact of
nitric oxide and reactive oxygen species. Free Radic Biol Med.
48:905–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davignon J: Beneficial cardiovascular
pleiotropic effects of statins. Circulation. 109(Suppl 1):
III39–III43. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guimarães DA, Rizzi E, Ceron CS, Pinheiro
LC, Gerlach RF and Tanus-Santos JE: Atorvastatin and sildenafil
lower blood pressure and improve endothelial dysfunction, but only
atorvastatin increases vascular stores of nitric oxide in
hypertension. Redox Biol. 1:578–585. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou MS, Tian R, Jaimes EA and Raij L:
Combination therapy of amlodipine and atorvastatin has more
beneficial vascular effects than monotherapy in salt-sensitive
hypertension. Am J Hypertens. 27:873–880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma Y, Chen Z, Zou Y and Ge J: Atorvastatin
represses the angiotensin 2-induced oxidative stress and
inflammatory response in dendritic cells via the I3K/Akt/Nrf2
pathway. Oxid Med Cell Longev. 148798:2014. View Article : Google Scholar
|
|
17
|
Chen S, Liu B, Kong D, Li S, Li C, Wang H
and Sun Y: Atorvastatin calcium inhibits phenotypic modulation of
PDGF-BB-induced VSMCs via down-regulation the Akt signaling
pathway. PLoS One. 10:e01225772015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ke N, Wang X, Xu X and Abassi YA: The
xCELLigence system for real-time and label-free monitoring of cell
viability. Methods Mol Biol. 740:33–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma M, Guo X, Chang Y, Li C, Meng X, Li S,
Du ZX, Wang HQ and Sun Y: Advanced glycation end products promote
proliferation and suppress autophagy via reduction of cathepsin D
in rat vascular smooth muscle cells. Mol Cell Biochem. 403:73–83.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Berk M, McIntyre TM, Gores GJ and
Feldstein AE: The lysosomal-mitochondrial axis in free fatty
acid-induced hepatic lipotoxicity. Hepatology. 47:1495–1503. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lei T, Guo N, Tan MH and Li YF: Effect of
mouse oocyte vitrification on mitochondrial membrane potential and
distribution. J Huazhong Univ Sci Technolog Med Sci. 34:99–102.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hill BG, Higdon AN, Dranka BP and
Darley-Usmar VM: Regulation of vascular smooth muscle cell
bioenergetic function by protein glutathiolation. Biochim Biophys
Acta. 1797:285–295. 2010. View Article : Google Scholar :
|
|
24
|
Alvarez E, Rodiño-Janeiro BK,
Ucieda-Somoza R and González-Juanatey JR: Pravastatin counteracts
angiotensin II-induced upregulation and activation of NADPH oxidase
at plasma membrane of human endothelial cells. J Cardiovasc
Pharmacol. 55:203–212. 2010. View Article : Google Scholar
|
|
25
|
Ferrick DA, Neilson A and Beeson C:
Advances in measuring cellular bioenergetics using extracellular
flux. Drug Discov Today. 13:268–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sansbury BE, Jones SP, Riggs DW,
Darley-Usmar VM and Hill BG: Bioenergetic function in
cardiovascular cells: the importance of the reserve capacity and
its biological regulation. Chem Biol Interact. 191:288–295. 2011.
View Article : Google Scholar :
|
|
27
|
Li P, Guo X, Lei P, Shi S, Luo S and Cheng
X: I3K/Akt/uncoupling protein 2 signaling pathway may be involved
in cell senescence and apoptosis induced by angiotensin II in human
vascular endothelial cells. Mol Biol Rep. 41:6931–6937. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eelen G, de Zeeuw P, Simons M and
Carmeliet P: Endothelial cell metabolism in normal and diseased
vasculature. Circ Res. 116:1231–1244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Graham DJ, Staffa JA, Shatin D, Andrade
SE, Schech SD, La Grenade L, Gurwitz JH, Chan KA, Goodman MJ and
Platt R: Incidence of hospitalized rhabdomyolysis in patients
treated with lipid-lowering drugs. JAMA. 292:2585–2590. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sathasivam S: Statin induced myotoxicity.
Eur J Intern Med. 23:317–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mabuchi H, Higashikata T, Kawashiri M,
Katsuda S, Mizuno M, Nohara A, Inazu A, Koizumi J and Kobayashi J:
Reduction of serum ubiquinol-10 and ubiquinone-10 levels by
atorvastatin in hypercholesterolemic patients. J Atheroscler
Thromb. 12:111–119. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vaughan RA, Garcia-Smith R, Bisoffi M,
Conn CA and Trujillo KA: Ubiquinol rescues simvastatin-suppression
of mitochondrial content, function and metabolism: implications for
statin-induced rhabdomyolysis. Eur J Pharmacol. 711:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mullen PJ, Zahno A, Lindinger P, Maseneni
S, Felser A, Krähenbühl S and Brecht K: Susceptibility to
simvastatin-induced toxicity is partly determined by mitochondrial
respiration and phosphorylation state of Akt. Biochim Biophys Acta.
1813:2079–2087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carnicka S, Adameova A, Nemcekova M,
Matejikova J, Pancza D and Ravingerova T: Distinct effects of acute
pretreatment with lipophilic and hydrophilic statins on myocardial
stunning, arrhythmias and lethal injury in the rat heart subjected
to ischemia/reperfusion. Physiol Res. 60:825–830. 2011.PubMed/NCBI
|