Introduction
Chronic myeloid leukemia (CML) is a malignant
hematopoietic stem cell disorder characterized by the reciprocal
translocation of chromosomes 9 and 22, resulting in the expression
of a constitutively active breakpoint cluster region (BCR)-Abelson
murine leukemia viral oncogene homolog 1 (ABL1) tyrosine kinase.
The introduction of tyrosine kinase inhibitors (TKIs), including
imatinib, nilotinib and dasatinib, has revolutionized the treatment
of patients with CML and markedly improved their quality of life
(1–3). A large proportion of patients with
CML subjected to long-term TKI treatment exhibit high-level and
stable molecular responses and, in certain cases, leads to
undetectable minimal residual disease (4). However, up to 40% of patients show
substantial resistance or intolerance to TKIs, typically during or
following treatment, which seriously limits the attainment of
treatment-free remission or a cure (5,6).
The underlying mechanisms of drug resistance remain to be
elucidated, although they are considered to be attributable mainly
to mutations in the kinase domain (KD) of BCR-ABL1 (7) and to signaling pathways activated by
BCR-ABL1, including RAS, phosphoinositide 3-kinase (PI3K), nuclear
factor-κB (NF-κB) and signal transducer and activator of
transcription 5 (STAT5) (8). In
particular, STAT5 has at least four key effects in the initiation
and progression of CML (9), and
may be considered a crucial modulator of imatinib responsiveness
(10).
Reactive oxygen species (ROS) are generated as a
by-product of the normal oxidative metabolism in eukaryotic cells
which, if generated to excess, can cause damage to cellular
molecules, including DNA, RNA and proteins, resulting in oxidative
stress. It is well established that ROS are important in a variety
of pathophysiological processes, including cell differentiation,
host defense, oxygen sensing, and cell proliferation and apoptosis.
The generation of ROS is regulated by the STAT5 transcription
factor, which is crucial in a number of hematological diseases and
commonly functions downstream of certain kinases, including
BCR-ABL1 in CML (11). It is
known that ROS can trigger genomic stress, secondary genome
instability and the potential accumulation of gene mutations within
the basal p53-mediated DNA damage response (DDR) pathway.
Therefore, there is scientific support for a link between STAT5,
ROS production and BCR-ABL1 mutations.
p53 is considered to be a universal sensor of
genotoxic stress, and is involved in different DNA repair
mechanisms and in cell cycle checkpoint regulation through various
signaling pathways. Generally, p53 and its downstream signaling
pathways are involved in ROS-induced apoptosis. Numerous studies
have reported crosstalk between p53 signaling and ROS metabolism
during normal physiological conditions, based on the feedback loop
between ROS and p53, whereby mitochondria-generated ROS promote p53
translocation and, in turn, trigger oxidative stress. A previous
study also demonstrated that the progression of CML was frequently
accompanied by increased inactivation of p53, and that p53
mutations were seldom observed in chronic phase CML, but were
detected in ~30% of patients presenting with myeloid blast crisis
(12). However, the mechanism of
the interaction between p53 and STAT5-mediated ROS production in
the resistance of CML to TKIs remains to be elucidated. Thus, in
this study, we aimed to examine the role of STAT5-mediated ROS
production and aberrant p53 apoptotic signaling in the resistance
of CML to imatinib.
Materials and methods
Cell culture
The human K562 CML cell line was purchased from the
Shanghai Institutes for Biological Sciences of the Chinese Academy
of Sciences (Shanghai, China) and imatinib-resistant K562 cells
(K562/G) were obtained from the Department of Pharmacology at the
Institute of Hematology, Chinese Academy of Medical Sciences
(Tianjin, China). The cells were cultured in Iscove's modified
Dulbecco's medium (IMDM) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1%
penicillin and 1% streptomycin, in a 5% CO2 incubator at
37°C. The cells were cultured in the presence or absence of 1
µM imatinib. All cells were passaged every 2–3 days.
Clinical specimens
All patients were enrolled from Anhui Provincial
Hospital of Anhui Medical University (Hefei, China) between July,
2015 and September, 2016. The present study was approved by the
Ethics Committee of the Affiliated Provincial Hospital of Anhui
Medical University. Peripheral blood mononuclear cells (PBMCs) were
isolated from patients with CML during routine examinations
following the provision of informed consent from all patients, in
accordance with the Declaration of Helsinki. Aliquots of the PBMCs
were used for subsequent analysis. The molecular responses to
imatinib were assessed according to the BCR-ABL/ABL ratio,
standardized to the International Scale (13). According to the 2013 European
Leukemia Net guidelines (https://www.leukemia-net.org), responses were assessed
using standardized reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis at 3 and 6 months (14). BCR-ABL1 transcript levels ≤10% at
3 months and <1% at 6 months were defined as optimal responses,
whereas levels >10% at 6 months were defined as failed responses
(14). BCR-ABL KD mutations and
p53 mutations were assessed as described previously (15). The characteristics of the
patients, and the results of the BCR-ABL KD mutations and p53
mutations are presented in Table
I.
 | Table IClinical characteristics of
patients. |
Table I
Clinical characteristics of
patients.
| Characteristic | Chronic myeloid
leukemia cases (n=63) | Non-IM-resistant
(n=31) | IM-resistant
(n=32) |
|---|
| Age at diagnosis
(years) | | | |
| Median | 37 | 40 | 36 |
| Range | 20.4–83.6 | 22.3–79.5 | 20.4–83.6 |
| Sex | | | |
| Male | 42 | 21 | 21 |
| Female | 21 | 10 | 11 |
| White blood cells
(107/ml) | 49.16±69.68 | 12.17±6.55 | 84.99±83.52 |
| International
standard | | | |
| <1% | 31 | 31 | 0 |
| >1% | 32 | 0 | 32 |
| BCR/ABL mutation
rate (%) | 19.01 | 9.60 | 28.13 |
| p53 mutation rate
(%) | 0.00 | 0.00 | 0.00 |
DNA damage signaling pathway PCR
array
The K562 and K562/G cells lines (~1×107
cells) were collected, respectively. RNA extraction and first
strand cDNA synthesis were performed via a routine protocol
(16). A Human DNA Damage
Signaling Pathway RT2 Profiler™
PCR array was purchased from SABiosciences (Frederick, MD, USA).
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad,
CA, USA) and RNA samples (1 µg) were reverse transcribed
into cDNA using the RT2 PCR Array
First Strand Synthesis kit (Qiagen). Subsequently, 91 µl of
ddH2O was added to each 20 µl cDNA synthesis
reaction and mixed well. The following components were mixed in a
5-ml tube or a multi-channel reservoir: 1,050 µl of 2X
SuperArray PCR master mix, 105 µl of the diluted
first-strand cDNA synthesis reaction and 945 µl of
ddH2O. The cocktails were then added to the PCR array.
Real-time PCR detection was performed under the following
thermocycling conditions: 95°C for 10 min; 40 cycles of 95°C for 15
sec and 60°C for 1 min. RT-qPCR and data analyses via the
2−ΔΔCq method were performed according to the
manufacturer's protocol. The expression data were normalized to
that of the housekeeping gene GAPDH. Differences in the levels of
gene expression are presented as the fold increase/decrease,
relative to the levels of the housekeeping gene (Table II).
| Table IImRNA fold-differences in DNA damage
response genes between K562 and K562G cells. |
Table II
mRNA fold-differences in DNA damage
response genes between K562 and K562G cells.
| Gene | Abbreviation | K562G/K562
(fold-difference) |
|---|
| Bloom syndrome,
RecQ helicase-like | BLM | −1.53 |
| Calcium and
integrin binding 1 (calmyrin) | CIB1 | −2.02 |
| Growth arrest and
DNA-damage-inducible, α | GADD45A | −1.51 |
| Nth endonuclease
III-like 1 | NTHL1 | −1.89 |
| Cell cycle
checkpoint protein RAD17 | RAD17 | −1.55 |
| Tumor protein
p53 | TP53 | −2.56 |
| Tumor protein p53
binding protein 1 | TP53BP1 | −1.93 |
| Hypoxanthine
phosphoribosyltransferase 1 | HPRT1 | −1.73 |
Cell counting assay
A cell counting kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Shanghai, China) was used to determine the
survival rate of the K562 and K562/G cells following incubation
with imatinib. The cells were seeded in a 96-well plate at a
density of 5×103 cells/well in IMDM containing 10% FBS.
Subsequently, various concentrations of imatinib (0.02–4 µM
for K562 cells and 0.2–400 µM for K562/G cells) were added.
Following incubation of the cells at 37°C in 5% CO2 for
24 h, 10 µl of CCK-8 solution was added to each well and the
cells were incubated for another 4 h. The absorbance was measured
at 450 nm with a microplate reader. A well containing medium and
CCK-8 solution only was used as a blank control. A control group of
cells were incubated with cell culture medium and CCK-8 solution.
The half maximal inhibitory concentration (IC50) values
of imatinib were determined as the mean of three independent
experiments.
Cell apoptosis assessment using Annexin
V-FITC and propidium iodide (PI) staining
Briefly, the cells were harvested and washed with
cooled PBS at 4°C. Cell suspensions (5×104 cells each)
were incubated with Annexin V-FITC (2 µg/ml) and PI (0.5
µg/ml) at room temperature for 15 min in the dark, and
subsequently analyzed using flow cytometry (17). The cells were considered to be
apoptotic if they exhibited Annexin V+/PI−
(early apoptotic) or Annexin V+/PI+ (late
apoptotic) staining. The data are presented as the mean of three
separate experiments, each performed in duplicate.
Detection of intracellular ROS
The accumulation of intracellular ROS was detected
using 2′,7′-dichlorofluorescindiacetate (DCFH-DA) probes (Molecular
Probes®; Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. In brief,
2×105 cells/ml of K562 and K562/G cells were stained
with 2.5 µM DCFH-DA at 37°C for 15 min. The samples were
then washed and resuspended in phosphate-buffered saline (PBS), and
the fluorescence intensity was analyzed using flow cytometry
(FACSCanto II) using BD FACSDiva software v6.1.3 (both from BD
Biosciences, San Jose, CA, USA). Additionally, K562/G cells were
pretreated with 5 mM N-acetylcysteine (NAC; Meilunbio, Dalian,
China) or 20 µM SH-4–54 (Selleck Chemicals, Houston, TX,
USA) for 24 h in a humidified atmosphere at 37°C and 5%
CO2, and then subjected to the same analysis. For each
sample, ~104 cells contained in the gated regions were
counted. ROS-positive cells were stained with DCFH-DA. The
experiments were repeated three times, and all data were analyzed
with BD FACSDiva software.
RT-qPCR analysis
Total RNA was extracted from the cultured cell lines
and PBMCs of the patients with CML using TRIzol reagent (Thermo
Fisher Scientific, Inc.) and quantified with a NanoDrop
spectrophotometer (Amoy Diagnostics, Xiamen, China). A total of 0.5
µg of the isolated RNA was used for cDNA synthesis and qPCR
analysis was subsequently performed on a 7500 Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with a
SYBR-Green reaction kit (Invitrogen; Thermo Fisher Scientific,
Inc.). GAPDH was used as a housekeeping gene to normalize the
levels of target gene expression. The changes in mRNA expression
levels were calculated using the comparative Cq method, as follows:
Fold change = 2−ΔΔCq = [(Cq gene of interest-Cq internal
control) sample A-(Cq gene of interest-Cq internal control) sample
B] (18). The primer sequences
used are listed in Table
III.
| Table IIIPrimer sequences used in gene
expression analysis. |
Table III
Primer sequences used in gene
expression analysis.
| Gene | Primer sequence
(5′-3′) | Product length
(bp) |
|---|
| GAPDH | F:
GGAGCGAGATCCCTCCAAAAT
R: GGCTGTTGTCATACTTCTCATGG | 197 |
| STAT5A | F:
GCAGAGTCCGTGACAGAGG
R: CCACAGGTAGGGACAGAGTCT | 106 |
| STAT5B | F:
CAGAACACGTATGACCGCTG
R: CTGGAGAGCTACCATTGTTGG | 106 |
| p53 | F:
CCAGCAGCTCCTACACCGGC
R: GAAACCGTAGCTGCCCTG | 99 |
| Bcl-2 | F:
CATGTGTGTGGAGAGCGTCAA
R: GCCGGTTCAGGTACTCAGTCA | 147 |
| Bax | F:
GCGTCCACCAAGAAGCTGAG
R: ACCACCCTGGTCTTGGATCC | 113 |
| MDM2 | F:
GAATCATCGGACTCAGGTACATC
R: TCTGTCTCACTAATTGCTCTCCT | 167 |
Immunofluorescence analysis of the
expression of phosphorylated H2AX (γ-H2AX)
γ-H2AX forms microscopically visible foci, and the
number of γ-H2AX foci has been found to correlate well with the
number of DNA double-strand breaks (DSBs) (19). The K562 and K562/G cells
(5×104) were plated on glass slides via a cell
concentrator and fixed with 4% paraformaldehyde for 30 min at room
temperature. Following three washes with PBS containing 0.2%
Tween-20, the cells were permeabilized with PBS containing 0.3%
Triton X-100 at room temperature for 30 min, and then blocked with
blocking buffer containing 5% goat serum (SL038; Solarbio, Beijing,
China) and 0.3% Triton X-100 in PBS for 1 h at room temperature.
Incubation with primary γ-H2AX antibody (1:200; monoclonal rabbit
anti-H2AX; cat. no. 9718; Cell Signaling Technology, Inc., Danvers,
MA, USA) was performed in blocking solution overnight at 4°C. The
cells were then washed three times and incubated with anti-rabbit
IgG antibodies (1:600; cat. no. A-1101; AlexaFluor V®
488 goat anti-mouse; Molecular Probes®) at room
temperature for 1 h. The nuclei were stained with
4′,6-diamidino-2-phenylindole and images were captured with a laser
scanning confocal microscope (DMI6000B TCS SP5; Leica Microsystems
GmbH, Mannheim, Germany), using microscope imaging software (LAS
AF6500; Leica Microsystems GmbH, Wetzlar, Germany).
Western blot analysis
The K562 and K562/G cells were collected and lysed
with RIPA buffer containing a protease inhibitor cocktail. The
lysates were centrifuged at 12,000 × g for 10 min, 4°C and the
supernatant was collected. The total protein concentration in the
supernatant was determined using a BCA protein assay. Equal
quantities of protein (30 µg) were subjected to 10–12%
SDS-polyacrylamide gel electrophoresis at a constant voltage of 80
V for 30 min and 120 V for another 1.5 h. The resolved proteins
were electrophoretically transferred onto PVDF membranes (EMD
Millipore, Billerica, MA, USA), and the membranes were blocked in
5% skimmed milk for 1 h. Subsequently, the membranes were incubated
overnight at 4°C with primary monoclonal antibodies at 1:1,000
dilutions. The following day, the membranes were exposed to
horseradish peroxidase-conjugated anti-mouse or anti-rabbit
secondary antibodies (1:1,000 dilution; A21010, A21020-1; Abbkine,
Wuhan, China) for 2 h at room temperature. To visualize the protein
bands, the membranes were treated with an enhanced
chemiluminescence kit solution (GE Healthcare Life Sciences;
Chalfont, UK) and digitalized by scanning (Fusion Solo3 v 16.12;
Fusion FX Vilber Lourmat, France). The primary antibodies against
STAT5 (94205S), phosphorylated (p-)STAT5 (9359S), p53 (2524S),
B-cell lymphoma-2 (Bcl-2) (2870S), mouse double minute 2 homolog
(MDM2; 86934) and Bcl-2-associated X protein (Bax) (5023T) were
obtained from Cell Signaling Technology, Inc., and anti-β-actin was
purchased from Santa Cruz Biotechnology, Inc. (sc-47778; Dallas,
TX, USA)
Statistical analysis
All data in the present study are presented as the
mean ± standard deviation of three independent experiments unless
stated otherwise. The results were analyzed using one-way analysis
of variance and unpaired Student's t-tests to evaluate the
significance of differences between groups. P<0.05 was
considered to indicate a statistically significant difference. Data
analyses were performed using Prism software version 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA).
Results
Elevated levels of STAT5 correlate with
BCR-ABL1 mutation and imatinib sensitivity in vivo and in
vitro
Several previous studies have reported that the
activation of STAT5 contributes to imatinib resistance in BCR/ABL1
CML. In a previous study, Warsch et al revealed that
imatinib-resistant patients had upregulated levels of STAT5 in
leukemic cells (10). In the
present study, it was found that patients with CML and imatinib
resistance exhibited higher mRNA expression levels of STAT5A and
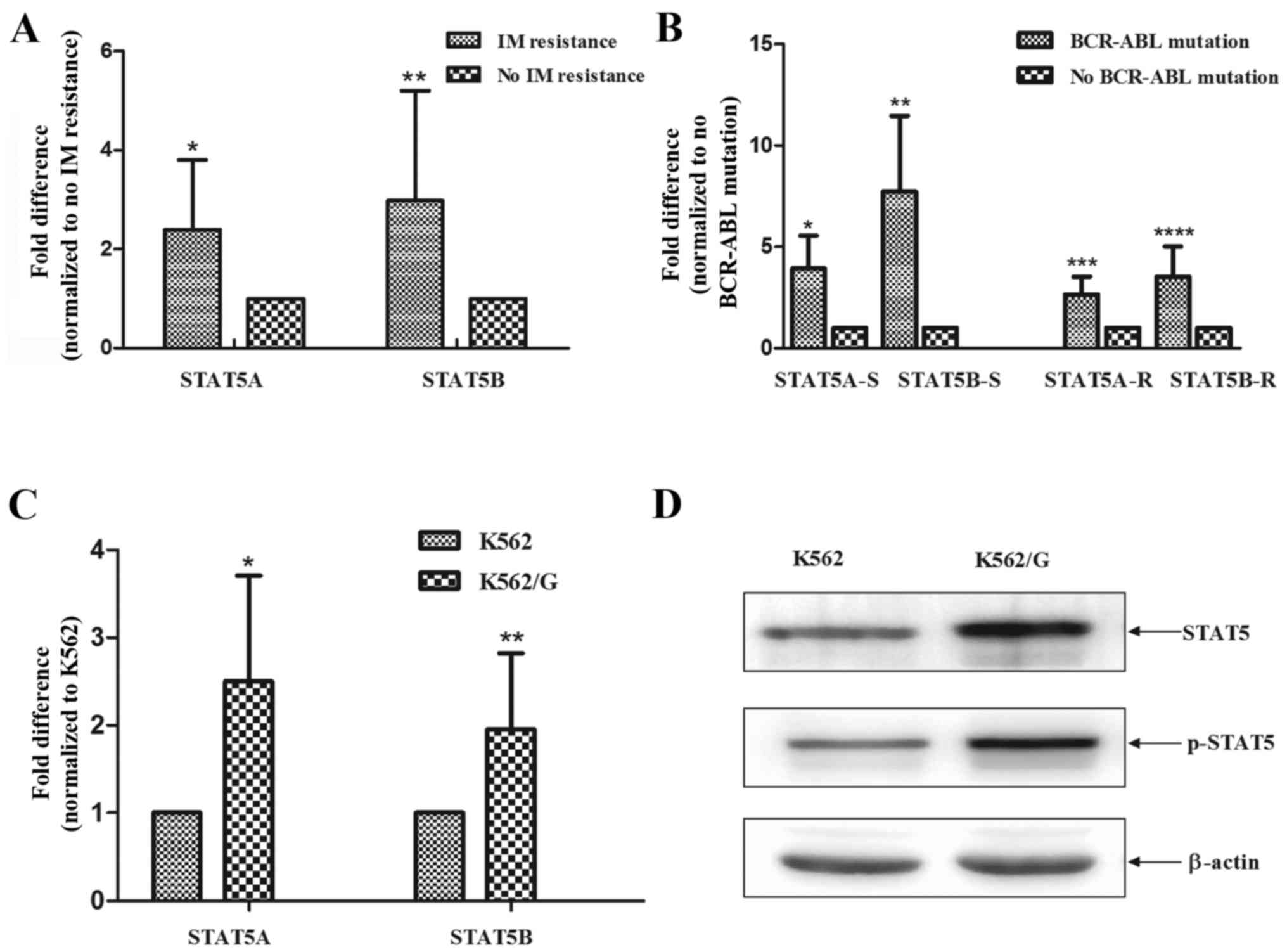
STAT5B (P=0.0093 and P=0.0091) (Fig.
1A). Of note, the expression levels of STAT5 were significantly
higher in patients with BCR-ABL1 mutations, compared with those
without BCR-ABL1 mutations (sensitive, P=0.02 and P=0.009;
resistant P=0.01 and P=0.009), and this effect was independent of
the state of imatinib resistance (Fig. 1B). Additionally, the K562/G cells
exhibited elevated mRNA levels of STAT5A and STAT5B, compared with
the imatinib-sensitive K562 cells (P<0.001 and P<0.001)
(Fig. 1C). Consistently, the
results of the western blot analysis demonstrated that the protein
expression levels of STAT5 and p-STAT5 were markedly increased in
the K562/G cells, compared with those in K562 cells (Fig. 1D).
ROS accumulation contributes to enhanced
levels of DNA DSBs in K562/G cells
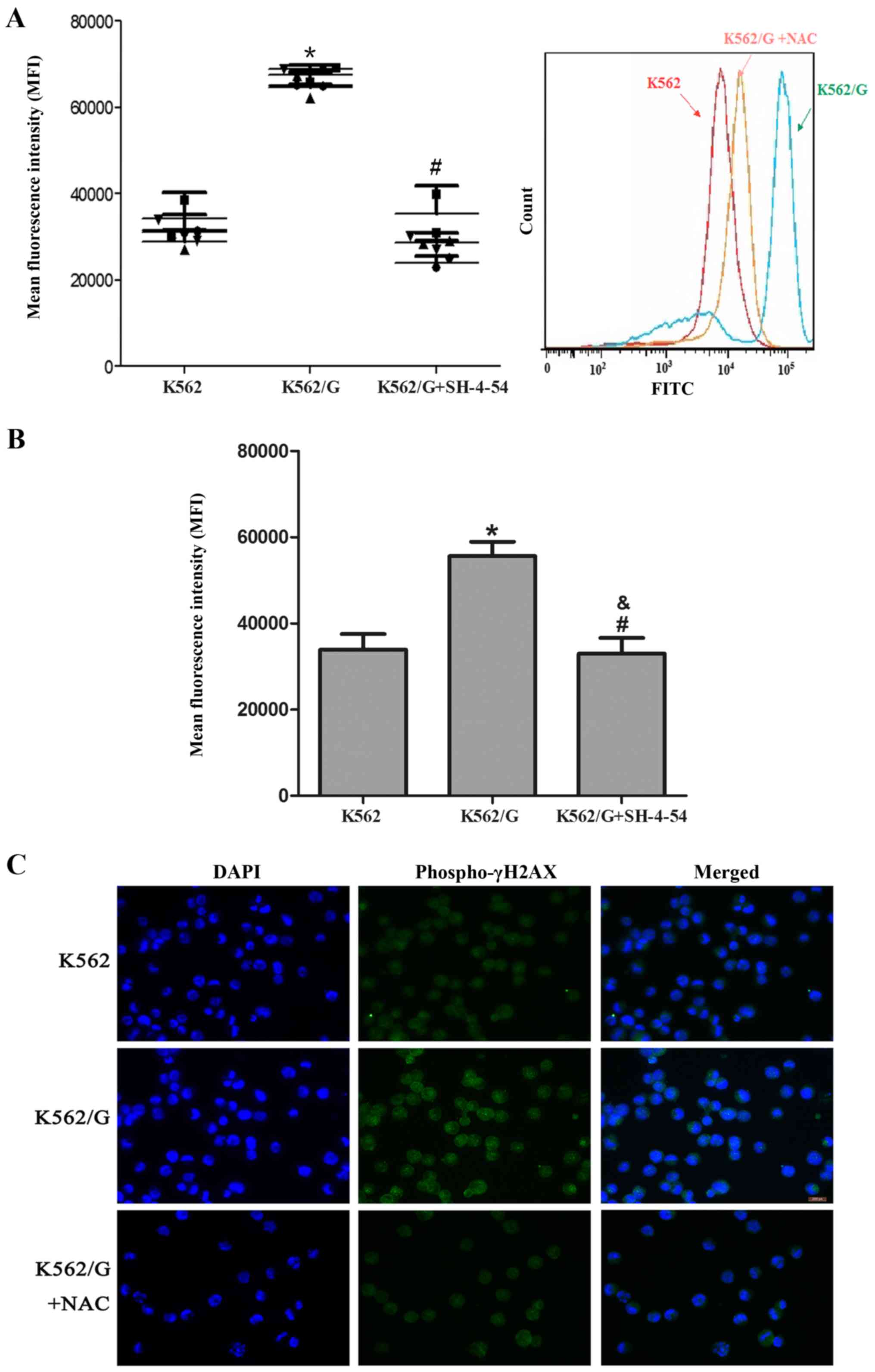
Significantly higher levels of ROS were detected in
the imatinib-resistant K562/G cells, compared with those in the
imatinib-sensitive K562 cells (P<0.001). Following treatment
with 5 mM NAC, a scavenger of ROS, the levels of ROS in the K562/G
cells were significantly decreased (P=0.323) (Fig. 2A). Additionally, the present study
investigated whether STAT5 contributed to the accumulation of ROS
by using the STAT5 inhibitor, SH-4-54. As shown in Fig. 2B, exposure to 20 µM SH-4-54
markedly reduced the levels of ROS in the K562/G cells (P=0.872 vs.
K562; P=0.01 vs. K562/G). It is well established that ROS can
induce DNA damage, including DNA DSBs, and is generally recognized
as an inducer of resistance mutation; γ-H2AX is frequently observed
in regions of histone lesions resulting from ROS damage and may be
used to quantify DSBs (20). The
present study showed that higher levels of γ-H2AX were detected in
the K562/G cells than in the K562 cells, which suggested that
higher levels of ROS and secondary DNA damage were present in the
K562/G cells. To confirm the link between STAT5-mediated ROS
production and DSB generation, the K562/G cells were pretreated
with NAC. As expected, NAC pretreatment decreased the level of
γ-H2AX staining (Fig. 2C).
Involvement of the p53 apoptotic pathway
in the reduced apoptotic rate and resistance of K562/G cells to
imatinib
Imatinib, as a first generation TKI, is used as a
principle treatment for patients with CML (4). A significant difference in the rate
of ABL mutation was observed between the imatinib-resistant group
and the non-resistant group (28.13 vs. 9.60%) (Table I). The imatinib-resistant K562/G
CML cell line was used in the present study, and the resistance of
the K562/G cells was first characterized and compared with that of
normal K562 cells. The K562 and K562/G cells were treated with
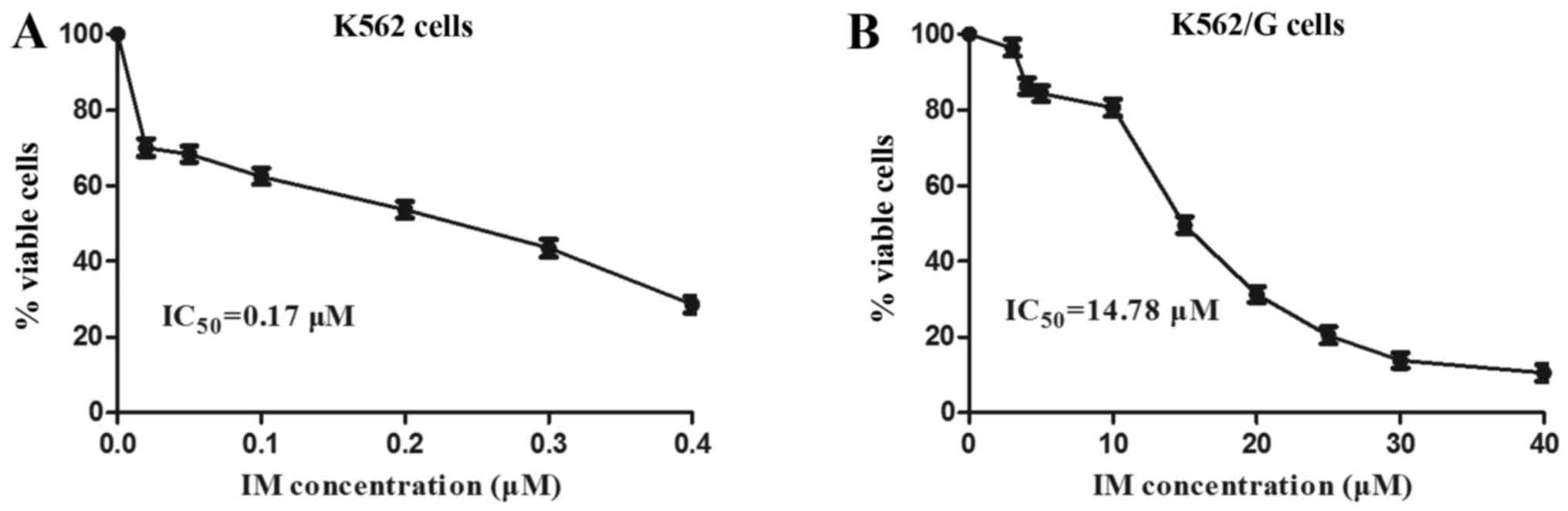
imatinib, and a CCK-8 assay was used to assess the cytotoxic
effects of various concentrations of imatinib on the cell lines,
for which the IC50 values were determined. The results
showed that the ratio of IC50 values between the K562/G
and K562 cells was 87:1 (14.78±0.43 vs. 0.17±0.07) (Fig. 3). Therefore, the K562/G cell line
was resistant to imatinib and maintained a high level of drug
resistance, which confirmed the efficacy of the imatinib-resistant
cell model. The apoptotic rates of the K562 and K562/G cells in
response to different concentrations of imatinib (0.1, 1, 5, 10 and
15 µM) were measured using flow cytometric analysis. The
results showed that the rate of apoptosis in the K562/G cells was
significantly lower, compared with that in the K562 cells at all
concentrations of imatinib (P<0.05). This effect was evident in
early and late apoptosis (Fig.
4A–C). Previous evidence indicates that ROS-induced apoptosis
is characterized by the upregulation of tumor suppressor protein
p53 (21), delocalization of
cytochrome c, caspase activation, DNA fragmentation, and suppressed
expression of histone deacetylasein CML cells (22). In the present study, using a human
DNA damage signaling pathway array, the downregulated transcription
levels of DDR genes were found in K562/G cells. Among these
downregulated genes, tumor protein p53 and tumor protein
p53-binding protein 1 were involved in the apoptotic pathway
(Table II). Although the K562
and K562/G cells exhibited low protein expression levels of p53,
the mRNA level of p53 was significantly lower in the K562/G cells,
compared with that in the K562 cells (P<0.01) (Fig. 4D and E). Similarly, the expression
levels of Bax were significantly decreased in the K562/G cells
(P<0.01), whereas those of Bcl-2 and MDM2 were significantly
increased (P<0.01) (Fig. 4D).
The p53 mutation was not found in the CML patients (Table I).
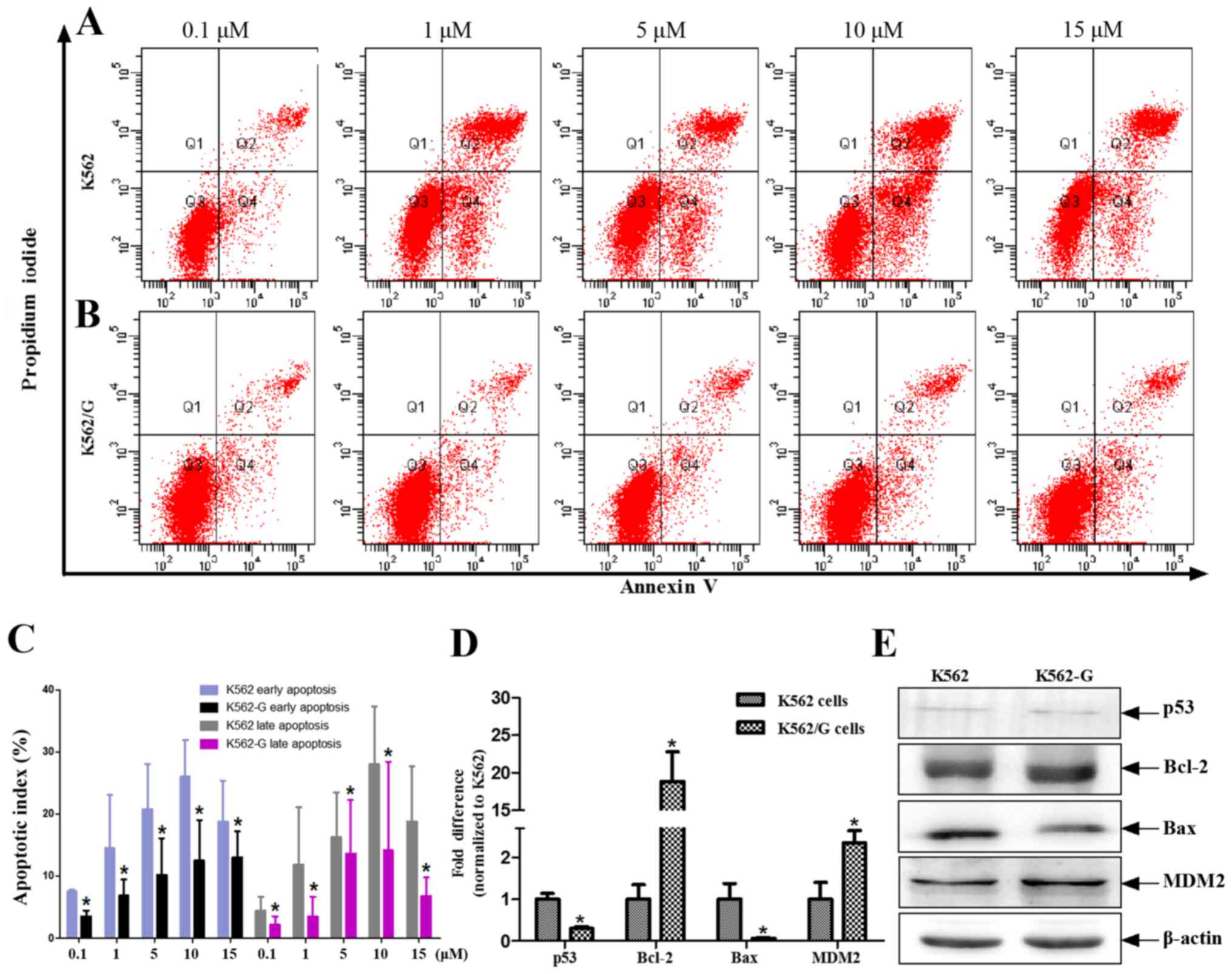 | Figure 4IM induces different rates of
apoptosis in K562 and K562/G cells. (A) K562 and (B) K562/G cells
were treated with different doses of IM for 24 h, following which
PI and Annexin V-FITC were added to the culture medium. Apoptotic
cells were detected by Annexin V-FITC/PI staining followed by flow
cytometric analysis. (C) Data are expressed as the mean ± standard
deviation of four independent experiments and were analyzed using
Student's t-test. *P<0.05 K562/G vs. K562. Analysis
of p53 and other apoptosis-related genes. (D) Reverse
transcription-quantitative polymerase chain reaction analysis was
performed to detect the mRNA expression levels of p53, Bcl-2, Bax
and MDM2. (E) Changes in protein expression levels of p53, Bax,
Bcl-2 and MDM2 in whole cell lysates (K562 and K562/G cells) were
detected using western blot analysis. Similar results were obtained
from six replicates (three independent experiments performed in
duplicate). *P<0.01 K562/G vs. K562. IM, imatinib;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; MDM2,
mouse double minute 2 homolog; PI, propidium iodide. |
Discussion
CML is commonly considered to be a typical model for
understanding the molecular pathogenesis of malignancy. Although
the treatment and prognosis of CML have markedly improved since the
development of TKIs, including imatinib, which is now used as a
standard first-line therapeutic agent, not all patients with CML
respond well. Even following long-term imatinib treatment, stem
cells in the majority of patients with CML continue to express
BCR-ABL mRNA. Additionally, following long-term therapy, it has
been reported that 60% of patients experience relapse at the
molecular level, whereas 15% of patients developed drug resistance
and subsequently discontinued imatinib following a sustained
complete molecular response (23). The most frequently reported cause
for TKI resistance is a gene mutation in the KD of BCR-ABL1, which
typically occurs at a frequency of 40–90% (24). Other mechanisms may include the
upregulation of BCR-ABL1, increased expression of drug transporter
ABCB1, elevated levels of granulocyte-macrophage colony-stimulating
factor, and inactivation of TP53 (25,26). In the present study, 63 blood
samples from imatinib-resistant (n=32) and non-resistant (n=31)
patients were analyzed by DNA sequencing, and 28% of the
imatinib-resistant cases were found to harbor mutations within the
KD of BCR-ABL1 (Table I).
It is generally accepted that STAT5 exerts key
effects in various hematological malignancies, including
anti-apoptotic effects and growth-stimulatory functions (27). To date, STAT5 has been confirmed
to support BCR-ABL1-triggered CML via four key mechanisms: Cell
cycle control, cell viability maintenance, ROS generation and TKI
resistance (28). The importance
of STAT5 in the pathogenesis of CML is further highlighted by
findings that STAT5 is upregulated during disease progression and
that high STAT5 levels can significantly decrease imatinib
sensitivity. In the present study, significantly increased
expression levels of STAT5 were consistently found in the blood
samples of imatinib-resistant patients and in imatinib-resistant
K562/G cells. This was in agreement with a previous study that
identified significantly increased mRNA levels of STAT5A in
patients presenting with secondary imatinib resistance without
BCR-ABL1 mutations, compared with newly diagnosed imatinib
responders (29). The present
study investigated the potential connection between the
transcription factor STAT5 and the occurrence of BCR-ABL1
mutations. The data showed that significantly higher levels of
STAT5 were expressed in the BCR-ABL1 mutation group, compared with
those in the non-BCR-ABL1 mutation group, regardless of TKI
resistance state.
ROS are considered to be potent signaling mediators,
and may interfere with gene regulatory pathways, including the
mitogen-activated protein kinase and hypoxia-responsive
element/hypoxia-inducible factor pathways, which are also known to
be regulated by STAT5 transcription factors. In the present study,
as expected, the STAT5 inhibitor SH-4-54 caused a marked reduction
in the levels of ROS in K562/G cells. However, under environmental
stress, ROS levels increase markedly, which can cause significant
damage to cell structures, and ultimately mutation. In
BCR-ABL1+ cells, high levels of STAT5 have been
associated with elevated levels of endogenous ROS (30). In the present study, the levels of
ROS in K562/G cells were elevated, compared with those in K562
cells. Previous studies have shown that certain chemotherapeutic
drugs can induce cell death by increasing the generation of
intracellular ROS (31,32). The production of ROS can lead to
DNA damage (33). DNA DSBs are
considered to be the most destructive form of DNA damage, and have
a high probability of resulting in cumulative mutations. In the
present study, it was observed that ROS increased the levels of
γ-H2AX, which was alleviated by NAC.
The function of p53 as a tumor suppressor has been
attributed to its ability to promote cell death or permanently
inhibit cell proliferation. p53 is subject to a wide range of
post-translational modifications, including phosphorylation,
acetylation, methylation and ubiquitination. Regulation of the gene
expression of p53 usually occurs mainly at the protein level
(34). Somatic TP53 mutations
have been identified in several types of cancer, at various
frequencies depending on the cancer type. Overall, TP53 mutations
are found in 5–10% of de novo cases of myelodysplastic
syndrome MDS and acute myeloid leukemia cases, and have been
associated with complex karyotypes and reduced survival rates
(35,36). p53 mutations are considered to be
high-risk factors for the development of leukemia, and indicators
of a poor response to chemotherapy and poor prognosis (37). In the patients with CML enrolled
in the present study, no p53 mutations were detected. However,
whether the contributions of p53-associated TKI resistance occur
mainly via mutations or epigenetic modifications remains to be
elucidated. In a previous study, the deletion of p53 was associated
with the progression of CML, and the wild-type p53 protein was
present in the KBM5 chronic phase cell line, whereas the low
expression or absence of p53 was observed in K562 cells (an
advanced stage CML cell line) (38). In the present study, the results
of the Human DNA Damage Signaling Pathway Array revealed
downregulated transcription levels of DDR genes, including p53 and
p53-binding protein 1, in the K562/G cells. The expression levels
of p53, Bcl-2 and Bax, proteins of the p53 apoptotic pathway, were
assessed using western blot analysis, and the downregulation of Bax
was observed in K562/G cells. These experimental results are
consistent with the findings of a previous study involving the
deletion of p53 in K562 and K562/G cells (38).
In conclusion, the present study identified elevated
levels of STAT5 in patients with imatinib-resistant CML and K562/G
cells. In addition, the high levels of STAT5 and subsequent
accumulation of ROS were associated with increased chronic
oxidative damage to DNA, as evidenced by increased DSBs within the
DNA of K562/G cells, which may be a cause of gene mutations
associated with resistance. It was also found that aberrant
regulation and expression of the p53 pathway was involved in
imatinib resistance. The expression of p53 can be modulated by
changes in transcriptional and translational events, and the
activity and protein levels of p53 are negatively regulated by the
E3 ubiquitin ligase MDM2 (39).
The present study detected lower mRNA and protein expression levels
of MDM2 in K562 cells, compared with those in the K562/G cells.
Therefore, a time profile of the induced expression of p53 and its
epigenetic modification (methylation status, post transcriptional
regulation by microRNAs) during the establishment of resistance to
imatinib require further examination in future investigations.
Acknowledgments
This study was supported by the Anhui Provincial
Natural Science Foundation (grant no. 1708085MH223)
References
|
1
|
Hochhaus A, Saglio G, Hughes TP, Larson
RA, Kim DW, Issaragrisil S, le Coutre PD, Etienne G,
Dorlhiac-Llacer PE, Clark RE, et al: Long-term benefits and risks
of frontline nilotinib vs imatinib for chronic myeloid leukemia in
chronic phase: 5-year update of the randomized ENESTnd trial.
Leukemia. 30:1044–1054. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hochhaus A, Rosti G, Cross NC, Steegmann
JL, le Coutre P, Ossenkoppele G, Petrov L, Masszi T, Hellmann A,
Griskevicius L, et al: Frontline nilotinib in patients with chronic
myeloid leukemia in chronic phase: Results from the European
ENEST1st study. Leukemia. 30:57–64. 2016. View Article : Google Scholar :
|
|
3
|
Breccia M, Stagno F, Luciano L, Abruzzese
E, Annunziata M, D'Adda M, Maggi A, Sgherza N, Russo-Rossi A,
Pregno P, et al: Dasatinib first-line: Multicentric Italian
experience outside clinical trials. Leuk Res. 40:24–29. 2016.
View Article : Google Scholar
|
|
4
|
Hughes TP and Ross DM: Moving
treatment-free remission into mainstream clinical practice in CML.
Blood. 128:17–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vaidya S, Vundinti BR, Shanmukhaiah C,
Chakrabarti P and Ghosh K: Evolution of BCR/ABL gene mutation in
CML is time dependent and dependent on the pressure exerted by
tyrosine kinase inhibitor. PLoS One. 10:e01148282015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parker WT, Yeung DT, Yeoman AL, Altamura
HK, Jamison BA, Field CR, Hodgson JG, Lustgarten S, Rivera VM,
Hughes TP, et al: The impact of multiple low-level BCR-ABL1
mutations on response to ponatinib. Blood. 127:1870–1880. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soverini S, Hochhaus A, Nicolini FE,
Gruber F, Lange T, Saglio G, Pane F, Müller MC, Ernst T, Rosti G,
et al: BCR-ABL kinase domain mutation analysis in chronic myeloid
leukemia patients treated with tyrosine kinase inhibitors:
Recommendations from an expert panel on behalf of European
LeukemiaNet. Blood. 118:1208–1215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Turhan AG: STAT5 as a CML target: STATinib
therapies? Blood. 117:3252–3253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warsch W, Grundschober E and Sexl V:
Adding a new facet to STAT5 in CML: Multitasking for leukemic
cells. Cell Cycle. 12:1813–1814. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Warsch W, Kollmann K, Eckelhart E, Fajmann
S, Cerny-Reiterer S, Hölbl A, Gleixner KV, Dworzak M, Mayerhofer M,
Hoermann G, et al: High STAT5 levels mediate imatinib resistance
and indicate disease progression in chronic myeloid leukemia.
Blood. 117:3409–3420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Walz C, Ahmed W, Lazarides K, Betancur M,
Patel N, Hennighausen L, Zaleskas VM and Van Etten RA: Essential
role for Stat5a/b in myeloproliferative neoplasms induced by
BCR-ABL1 and JAK2(V617F) in mice. Blood. 119:3550–3560. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carter BZ, Mak PY, Mak DH, Ruvolo VR,
Schober W, McQueen T, Cortes J, Kantarjian HM, Champlin RE,
Konopleva M, et al: Synergistic effects of p53 activation via MDM2
inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in
CD34+ proliferating and quiescent chronic myeloid
leukemia blast crisis cells. Oncotarget. 6:30487–30499. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hughes T, Deininger M, Hochhaus A,
Branford S, Radich J, Kaeda J, Baccarani M, Cortes J, Cross NC,
Druker BJ, et al: Monitoring CML patients responding to treatment
with tyrosine kinase inhibitors: Review and recommendations for
harmonizing current methodology for detecting BCR-ABL transcripts
and kinase domain mutations and for expressing results. Blood.
108:28–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong HJ, Zhou LT, Zhu DX, Wang DM, Fang C,
Zhu HY, Zhuang Y, Miao KR, Xu W and Li JY: The prognostic
significance of TP53 mutations in Chinese patients with chronic
lymphocytic leukemia is independent of del(17p13). Ann Hematol.
90:709–717. 2011. View Article : Google Scholar
|
|
16
|
Lan X, Zhao C, Chen X, Zhang P, Zang D, Wu
J, Chen J, Long H, Yang L, Huang H, et al: Nickel pyrithione
induces apoptosis in chronic myeloid leukemia cells resistant to
imatinib via both Bcr/Abl-dependent and Bcr/Abl-independent
mechanisms. J Hematol Oncol. 9:1292016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S,
Huang H, Liu N, Liao S, Song W, et al: Gambogic acid induces
apoptosis in imatinib-resistant chronic myeloid leukemia cells via
inducing proteasome inhibition and caspase-dependent Bcr-Abl
downregulation. Clin Cancer Res. 20:151–163. 2014. View Article : Google Scholar :
|
|
18
|
Wu L, Chen X, Huang L, Tian J, Ke F, Xu J,
Chen Y and Zheng M: A Novobiocin derivative, XN4, inhibits the
proliferation of chronic myeloid leukemia cells by inducing
oxidative DNA damage. PLoS One. 10:e01233142015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ismail IH, Wadhra TI and Hammarsten O: An
optimized method for detecting gamma-H2AX in blood cells reveals a
significant interindividual variation in the gamma-H2AX response
among humans. Nucleic Acids Res. 35:e362007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren G, Luo W, Sun W, Niu Y, Ma DL, Leung
CH, Wang Y, Lu JJ and Chen X: Psoralidin induced reactive oxygen
species (ROS)-dependent DNA damage and protective autophagy
mediated by NOX4 in breast cancer cells. Phytomedicine. 23:939–947.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maillet A and Pervaiz S: Redox regulation
of p53, redox effectors regulated by p53: a subtle balance.
Antioxid Redox Signal. 16:1285–1294. 2012. View Article : Google Scholar
|
|
22
|
Banerjee K, Ganguly A, Chakraborty P,
Sarkar A, Singh S, Chatterjee M, Bhattacharya S and Choudhuri SK:
ROS and RNS induced apoptosis through p53 and iNOS mediated pathway
by a dibasic hydroxamic acid molecule in leukemia cells. Eur J
Pharm Sci. 52:146–164. 2014. View Article : Google Scholar
|
|
23
|
Chomel JC, Bonnet ML, Sorel N, Bertrand A,
Meunier MC, Fichelson S, Melkus M, Bennaceur-Griscelli A, Guilhot F
and Turhan AG: Leukemic stem cell persistence in chronic myeloid
leukemia patients with sustained undetectable molecular residual
disease. Blood. 118:3657–3660. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Branford S, Rudzki Z, Walsh S, Parkinson
I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, et
al: Detection of BCR-ABL mutations in patients with CML treated
with imatinib is virtually always accompanied by clinical
resistance, and mutations in the ATP phosphate-binding loop
(P-loop) are associated with a poor prognosis. Blood. 102:276–283.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raja T: Optimizing second-line therapy for
chronic myeloid leukemia. Indian J Cancer. 49:46–56. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Cai D, Brendel C, Barett C, Erben
P, Manley PW, Hochhaus A, Neubauer A and Burchert A: Adaptive
secretion of granulocyte-macrophage colony-stimulating factor
(GM-CSF) mediates imatinib and nilotinib resistance in
BCR/ABL+ progenitors via JAK-2/STAT-5 pathway
activation. Blood. 109:2147–2155. 2007. View Article : Google Scholar
|
|
27
|
Hoelbl A, Schuster C, Kovacic B, Zhu B,
Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, et
al: Stat5 is indispensable for the maintenance of bcr/abl-positive
leukaemia. EMBO Mol Med. 2:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hoelbl A, Kovacic B, Kerenyi MA, Simma O,
Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R and Sexl V:
Clarifying the role of Stat5 in lymphoid development and
Abelson-induced transformation. Blood. 107:4898–4906. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang WW, Cortes JE, Yao H, Zhang L, Reddy
NG, Jabbour E, Kantarjian HM and Jones D: Predictors of primary
imatinib resistance in chronic myelogenous leukemia are distinct
from those in secondary imatinib resistance. J Clin Oncol.
27:3642–3649. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mallette FA, Moiseeva O, Calabrese V, Mao
B, Gaumont-Leclerc MF and Ferbeyre G: Transcriptome analysis and
tumor suppressor requirements of STAT5-induced senescence. Ann NY
Acad Sci. 1197:142–151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reczek CR and Chandel NS: ROS-dependent
signal transduction. Curr Opin Cell Biol. 33:8–13. 2015. View Article : Google Scholar :
|
|
33
|
Brem R, Li F, Montaner B, Reelfs O and
Karran P: DNA breakage and cell cycle checkpoint abrogation induced
by a therapeutic thiopurine and UVA radiation. Oncogene.
29:3953–3963. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gu B and Zhu WG: Surf the
post-translational modification network of p53 regulation. Int J
Biol Sci. 8:672–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bejar R, Stevenson K, Abdel-Wahab O,
Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine
RL, Neuberg D, et al: Clinical effect of point mutations in
myelodysplastic syndromes. N Engl J Med. 364:2496–2506. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kulasekararaj AG, Smith AE, Mian SA,
Mohamedali AM, Krishnamurthy P, Lea NC, Gäken J, Pennaneach C,
Ireland R, Czepulkowski B, et al: TP53 mutations in myelodysplastic
syndrome are strongly correlated with aberrations of chromosome 5,
and correlate with adverse prognosis. Br J Haematol. 160:660–672.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perrotti D, Jamieson C, Goldman J and
Skorski T: Chronic myeloid leukemia: Mechanisms of blastic
transformation. J Clin Invest. 120:2254–2264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Woo SM, Choi YK, Kim AJ, Cho SG and Ko SG:
p53 causes butein mediated apoptosis of chronic myeloid leukemia
cells. Mol Med Rep. 13:1091–1096. 2016. View Article : Google Scholar
|
|
39
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|