Introduction
Excess proliferation of vascular smooth muscle cells
(VSMCs) results in vascular remodelling and serves a key role in
several vascular disorders, including restenosis (1,2),
atherosclerosis (3) and pulmonary
artery hypertension (4). However,
the molecular mechanism underlying VSMC proliferation remains
unclear.
Inflammation is a key response to the damage in
vascular diseases (5–8). Clinical studies have demonstrated
that inflammation is a marker for predicting internal carotid
artery restenosis following eversion endarterectomy (9). The transcription factor nuclear
factor-κB (NF-κB) is activated in and is a master regulator of
inflammatory responses (10). It
has been reported that NF-κB serves a key role in restenosis; NF-κB
decoy may suppress restenosis following percutaneous coronary
intervention (11) and attenuates
in-stent restenosis in hypercholesterolemic rabbits (12). However, the mechanism by which
VSMC proliferation is regulated by NF-κB during inflammation
remains unclear.
MicroRNA (miRNA) are a type of non-coding RNA that
regulate cellular functions (13). Studies have indicated that miRNA
may have roles in vascular restenosis (14,15). miRNA-21 mediates
thrombospondin-1-induced VSMC migration and proliferation (16) and serves a role in the
proliferation of platelet-derived growth factor-induced human
aortic vascular smooth muscle cells (17). miR-21 has been applied in attempts
to treat restenosis (18,19). miR-17 belongs to the miR-17-92
cluster (20,21). Studies have revealed that the
miR-17-92 cluster promotes VSMC proliferation in a murine model
(22) and mediates inhibition of
VSMC proliferation via bone morphogenetic protein receptor type II
(BMPR2) (23). The
Smad3/miR-17-92/BMPR2 pathway (23) and downregulation of the miR-17-92
cluster by thrombospondin-1 (24)
serve important roles in the regulation of VSMC proliferation and
function. However, it remains unknown how miRNA mediate the
regulation of VSMC proliferation during inflammation through
activation of NF-κB.
The present study investigated the molecular
mechanism responsible for VSMC proliferation under inflammation. It
was demonstrated that overexpression of miR-17 stimulated VSMC
proliferation, enhanced cell cycle G1/S transition and increased
levels of E2F1 and proliferating cell nuclear antigen (PCNA).
miR-17 targeted retinoblastoma (RB) protein, which is a key
regulator of cell cycle progression and proliferation (25,26). p65 controlled the expression of
miR-17 and suppressed RB expression, which was abrogated by miR-17
inhibitor. The present data indicated that activation of the NF-κB
p65/miR-17/RB pathway resulted in VSMC proliferation. This finding
may identify the mechanism responsible for the excess proliferation
of VSMCs under inflammation, and miR-17 may be a novel target for
controlling several vascular disorders, including restenosis,
atherosclerosis, and pulmonary artery hypertension.
Materials and methods
Cell culture and treatment
The VSMC (T/G HA-VSMC) and 293T cell lines were
obtained from the American Type Culture Collection (Manassas, VA,
USA). All types of cells were cultured on poly-(L-lysine)-coated
plates/format discs (Sumitomo Bakelite Co., Ltd., Tokyo, Japan) in
Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT,
USA), 100 U/ml penicillin and 100 mg/ml streptomycin in a
humidified atmosphere with 5% CO2 at 37°C.
Bioinformatics analyses
To examine which cell cycle-regulating factor is a
potential target of miR-17, cell cycle-regulating factors were
surveyed for potential sites targeted by miR-17 using RNAhybrid
(bibiserv.cebitec.uni-bielefeld.de/rnahybrid/),
miRBase (mirbase.org/) and TargetScan (targetscan.org/vert_71/). Transcription factor
prediction was performed using RNAhybrid. The potential target was
selected based on conservation of the binding region and the
strength of the predicted interaction.
Transfection and reporter luciferase
activity assay
VSMCs and 293T cells were seeded on 6-well plates at
a density of 6×105 cells/cm2 and cultured
overnight in a humidified atmosphere with 5% CO2 at
37°C. Cells were then transfected with 20 nM miR-17 mimic or 20 nM
inhibitor or 20 nM control miRNA (Guangzhou RiboBio Co., Ltd.,
Guangzhou, China) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
After 24 h, cells were collected for western blotting and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analyses.
To construct the pGL3-RB-3′ untranslated region
(3′UTR), the full length 3′UTR of the human RB mRNA was cloned into
the pGL3-control vector (Promega Corp., Madison, WI, USA). To
construct the miR-17 promoter reporter genes, the DNA fragment
upstream of the transcription start site of the miR-17 gene was
inserted into the pGL3-basic vector (Promega Corp.). For luciferase
reporter assays, 293T cells were transiently transfected with
miR-17 mimics and Renilla-luciferase reporter plasmids
(Promega Corp.) containing the pGL3-RB-3′UTR (with the binding
region: AGGCGCUU) or pGL3-RB-3′UTR MUT (with the mutant binding
region: UUCGUGAA) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). After 48 h, cells were collected and
reporter gene activity was determined using the dual-luciferase
assay system (Invitrogen; Thermo Fisher Scientific, Inc.).
Renilla luciferase activity was used for normalization of
transfection efficiency. Reporter luciferase activity assay was
also used to assess the function of NF-κB p65 regulating miR-17
promoter activity. There were three NF-κB p65 binding sites in the
regulatory sequence of the miR-17 gene.
MTT assay
Cells were seeded on 96-well plates at a density of
3×103 cells/well and allowed to grow in the growth
medium for 24 h in a humidified atmosphere with 5% CO2
at 37°C. Following transfection with miR-17 mimic or inhibitor and
control for 48 h, cells were incubated with 5 mg/ml MTT for 4 h.
Subsequently, dimethyl sulfoxide (100 µl/well) was added.
The absorbance at 570 nm was then measured using an enzyme-linked
immunosorbent assay (ELISA) reader (Autobio Diagnostics Co., Ltd.,
Zhengzhou, China). Experiments were repeated at least three
times.
RT-qPCR analysis
Total RNA was isolated from experimental cells using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. A cDNA library was generated through
reverse transcription using 5X reaction buffer, RNase-free water,
dNTPs, enzyme inhibitor, MMLV reverse transcriptase (Takara Bio,
Inc., Otsu, Japan) and specific RT primers of miRNA (Guangzhou
RiboBio Co., Ltd.). The RT conditions were as follows: 42°C for 60
min followed by 95°C for 5 min, then immediate cooling to 4°C. The
expression levels of mature miRNA were determined using ExiLENT
SYBR®-Green master mix (Takara Bio, Inc.). U6 small
nuclear RNA was used as an internal control. Primers of miR-17 and
U6 were purchased from Guangzhou RiboBio Co., Ltd. The qPCR
conditions used were as follows: 95°C 10 min for polymerase
activation/denaturation, 95°C for 10 sec and 60°C for 1 min with
ramp rate 1.6°C/sec for 40 amplification cycles. To determine the
relative levels of RB transcripts, RT-qPCR was performed under the
following conditions, with glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) as an endogenous control: RT conditions of 25°C for 10 min,
50°C 15 min, 85°C for 5 min, followed by immediate cooling to 4°C;
qPCR conditions of 95°C for 10 min for polymerase
activation/denaturation, 95°C for 10 sec and 60°C for 1 min for 40
amplification cycles. An ABI PRISM 7500 Fast Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
amplify and detect the specific products. The following PCR primers
were used: RB forward, 5′-AAGGAGACAAGTTCGCATGT-3′ and reverse,
5′-GCCGGTAATTGTCGTAGTTT-3′; and GAPDH forward,
5′-CCATGTTCGTCATGGGTGTGAACCA-3′ and reverse,
5′-GCCAGTAGAGGCAGGGATGATGTTC-3′. The fold change for each miRNA and
RB transcript relative to the control was calculated using the
2−ΔΔCq method (27).
Western blotting
Nuclear extracts were isolated from human VSMCs
using a Nuclear and Cytoplasmic Protein Extraction kit (Nanjing
Keygen Biotech Co., Ltd., Nanjing, China). For western blot
analysis, VSMCs were homogenized in radioimmunoprecipitation assay
buffer and nM phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology, Haimen, China). The protein concentration was
determined according to a bicinchoninic assay (BCA) standard curve
using a BCA Protein αssay kit (Beyotime Institute of
Biotechnology). Proteins (30–50 µg) from each cell lysate
were mixed (4:1) with 5X sample buffer (Beyotime Institute of
Biotechnology). A total of 30 µl protein and buffer mixture
were loaded and resolved using 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins
were transferred onto polyvinylidene difluoride membranes by
electroblotting. Membranes were blocked with QuickBlock (Beyotime
Institute of Biotechnology) at room temperature for 15 min and then
incubated with an appropriate dilution of p65, RB, PCNA, E2F1, H3
and GAPDH antibodies (1:1,000–1:2,000) overnight at 4°C. The
membranes were then incubated with secondary antibodies conjugated
to horseradish peroxidase at room temperature for 1 h. Antibody
details are presented in Table I.
Detection was performed using an enhanced chemiluminescence kit
(cat. no. 32209; Thermo Fisher Scientific, Inc.). Proteins were
detected with Chemi Genius 2 via GeneSnap software 7.02 (Syngene,
Frederick, MD, USA), and relative intensities of the protein bands
were analysed using ImageJ 1.47 software (National Institutes of
Health, Bethesda, MD, USA).
 | Table IPrimary and secondary antibodies used
in western blotting. |
Table I
Primary and secondary antibodies used
in western blotting.
| Type of
antibody | Antibody | Supplier
details | Cat. no. |
|---|
| Primary | p65 | Cell Signaling
Technology, Inc., | 8242 |
| RB | Danvers, MA,
USA | Ab181616 |
| PCNA | Abcam, Cambridge,
MA, USA | Ab92552 |
| E2F1 | | Ab179445 |
| H3 | Cell Signaling
Technology, Inc., | 4499 |
| GAPDH | Danvers, MA,
USA | Ab8245 |
| Secondary | Goat anti-rabbit
IgG H&L (HRP) | Abcam, Cambridge,
MA, USA | Ab6721 |
| Rabbit anti-mouse
IgG H&L (HRP) | Abcam, Cambridge,
MA, USA | Ab6728 |
Flow cytometric analysis
All experimental cells were harvested by
trypsinization, washed in ice-cold phosphate-buffered saline (PBS)
three times, and fixed in 75% ice-cold ethanol in PBS overnight.
Before staining, the cells were centrifuged (200 × g for 5 min at
4°C), and then resuspended in ice-cold PBS containing RNase A (0.2
mg/ml). Cells were incubated at 37°C for 30 min and then stained
for 10 min at room temperature with 400 µl propidium iodide
(Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). Finally,
samples of 10,000 cells were analysed on a FACSCalibur cytometer
and data collected using CellQuest 3.1 software (BD Biosciences,
San Jose, CA, USA).
Statistical analysis
Data were analysed using SPSS 16.0 (SPSS, Inc.,
Chicago, IL, USA). Results were presented as mean ± standard
deviation. Two-tailed Student's t-tests were used for comparing
data between two groups. One-way analysis of variance was used to
compare differences among groups, followed by least significant
difference post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
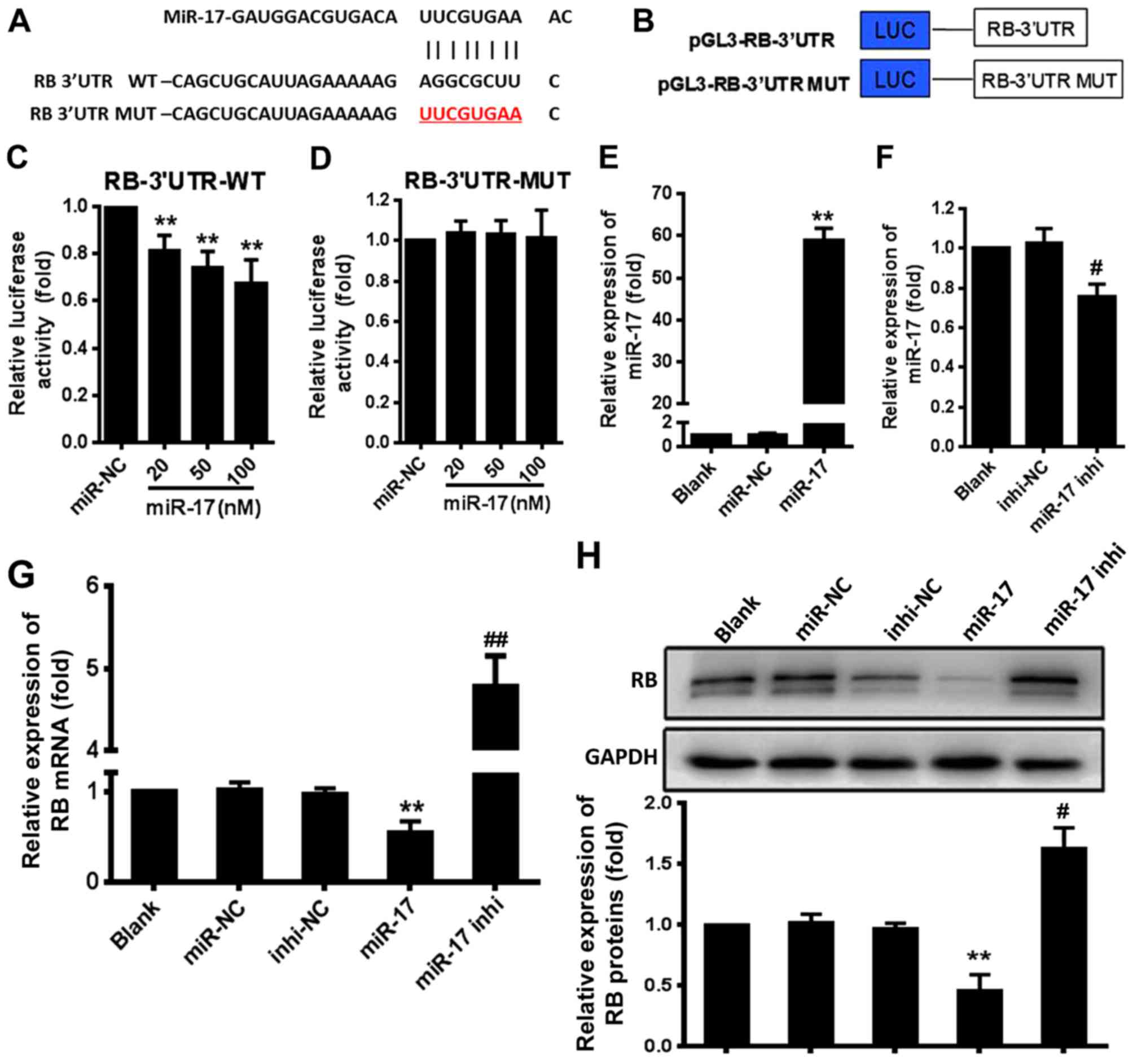
miR-17 directly targets the RB gene
The bioinformatics analyses indicated a potential
site for miR-17 in the RB mRNA-3′UTR (Fig. 1A). To confirm that this site was
an actual target of miR-17, 293T cells were co-transfected with
luciferase reporter plasmid constructs that contained the RB
mRNA-3′UTR or RB mRNA-3′UTR mutant (MUT) (Fig. 1B) and miR-17 mimic (miR-17) or
inhibitor (miR-17 inhi), and the luciferase activity was
determined. The results demonstrated that miR-17 significantly
suppressed the expression of the luciferase reporter gene directed
by the wild-type (WT) RB mRNA-3′UTR compared with the expression in
the miR-negative control (NC) group (P<0.01) (Fig. 1C). However, miR-17 did not
significantly suppress the expression of the luciferase reporter
gene directed by RB mRNA-3′UTR MUT compared with the miR-NC
(Fig. 1D). Overexpression of
miR-17 mimic resulted in a significant increase in the miR-17
expression level compared with the expression level induced by
miR-NC (P<0.01) (Fig. 1E), and
overexpression of miR-17 inhi significantly decreased the
endogenous miR-17 level in VSMCs compared with the level induced by
inhi-NC (P<0.05) (Fig. 1F).
The level of RB mRNA in VSMCs was significantly decreased by
treatment with miR-17 mimic compared with the levels in the miR-NC
group (P<0.01) and significantly increased by treatment with
miR-17 inhibitor compared with the levels in the inhi-NC group
(P<0.01) (Fig. 1G). This
finding was consistent with that of western blotting (Fig. 1H). These results indicated that
miR-17 suppressed RB expression by directly targeting the RB
mRNA-3′UTR in VSMCs.
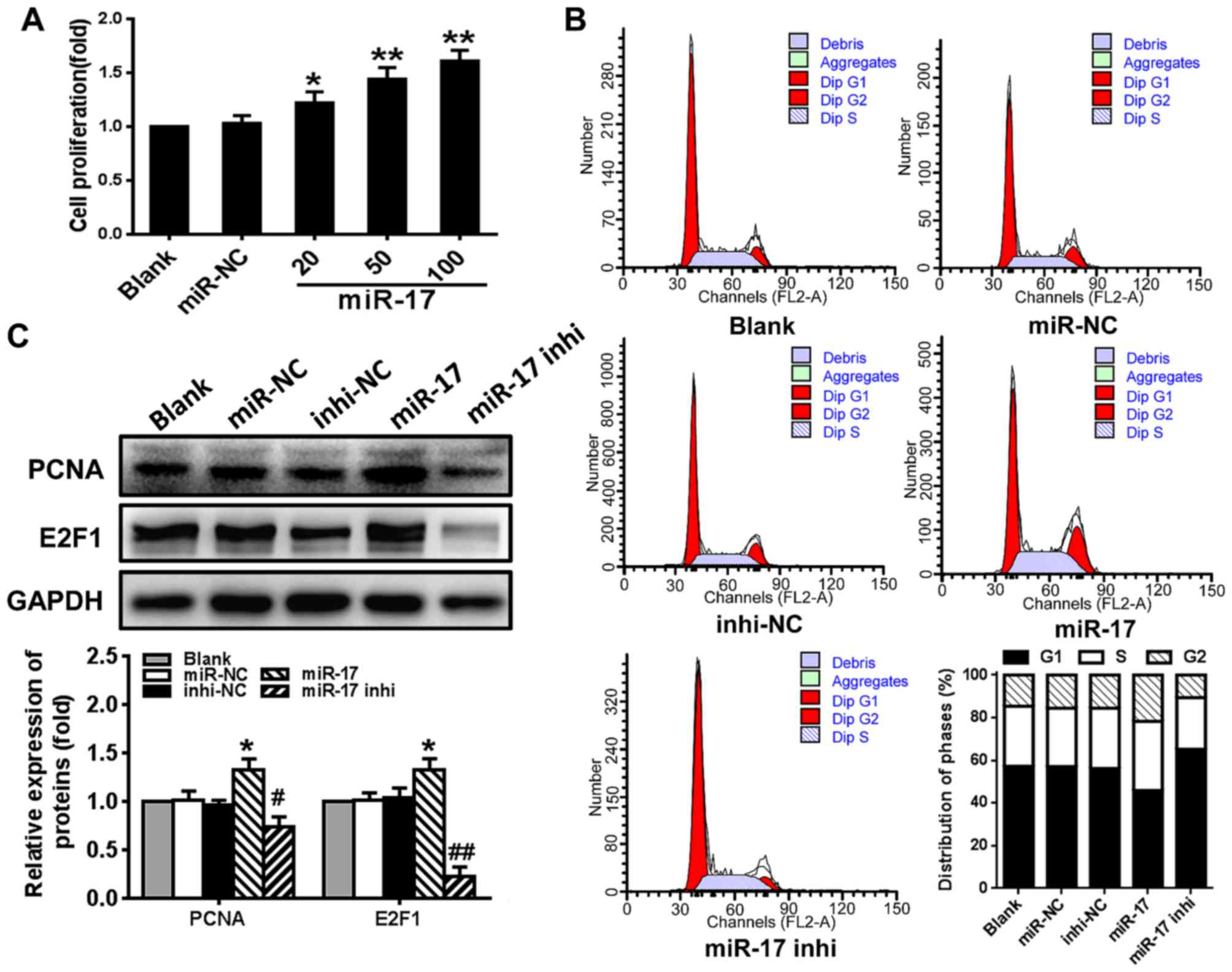
miR-17 stimulates VSMC proliferation and
enhances cell cycle G1/S transition
To determine whether miR-17 serves a role in
regulating VSMC proliferation, the effect of miR-17 on cell
viability, cell cycle progression and the expression of cell
cycle-regulating factors PCNA and E2F1 was examined. The results
demonstrated that VSMC proliferation was significantly stimulated
by miR-17 mimic in a dose-dependent manner compared with the
proliferation level in the miR-NC group (P<0.05) (Fig. 2A). Compared with the miR-NC group,
overexpression of miR-17 (miR-17 group) significantly increased the
number of cells that entered the S phase and decreased the number
of the cells in the G1 phase, whereas downregulating the expression
of miR-17 (miR-17 inhi group) markedly decreased the number of
cells that entered S phase and increased the number of cells in the
G1 phase (Fig. 2B). The levels of
PCNA and E2F1 protein were significantly increased in VSMCs
transfected with miR-17 mimic compared with the levels in those
transfected with miR-NC (P<0.05). The miR-17 inhibitor had the
opposite effect on the levels of these proteins, and significantly
reduced the expression levels compared with the levels in those
transfected with inhi-NC (P<0.05) (Fig. 2C). These results suggested that
overexpression of miR-17 stimulated VSMC proliferation and enhanced
cell cycle progression through promoting G1/S transition by
increasing the levels of PCNA and E2F1.
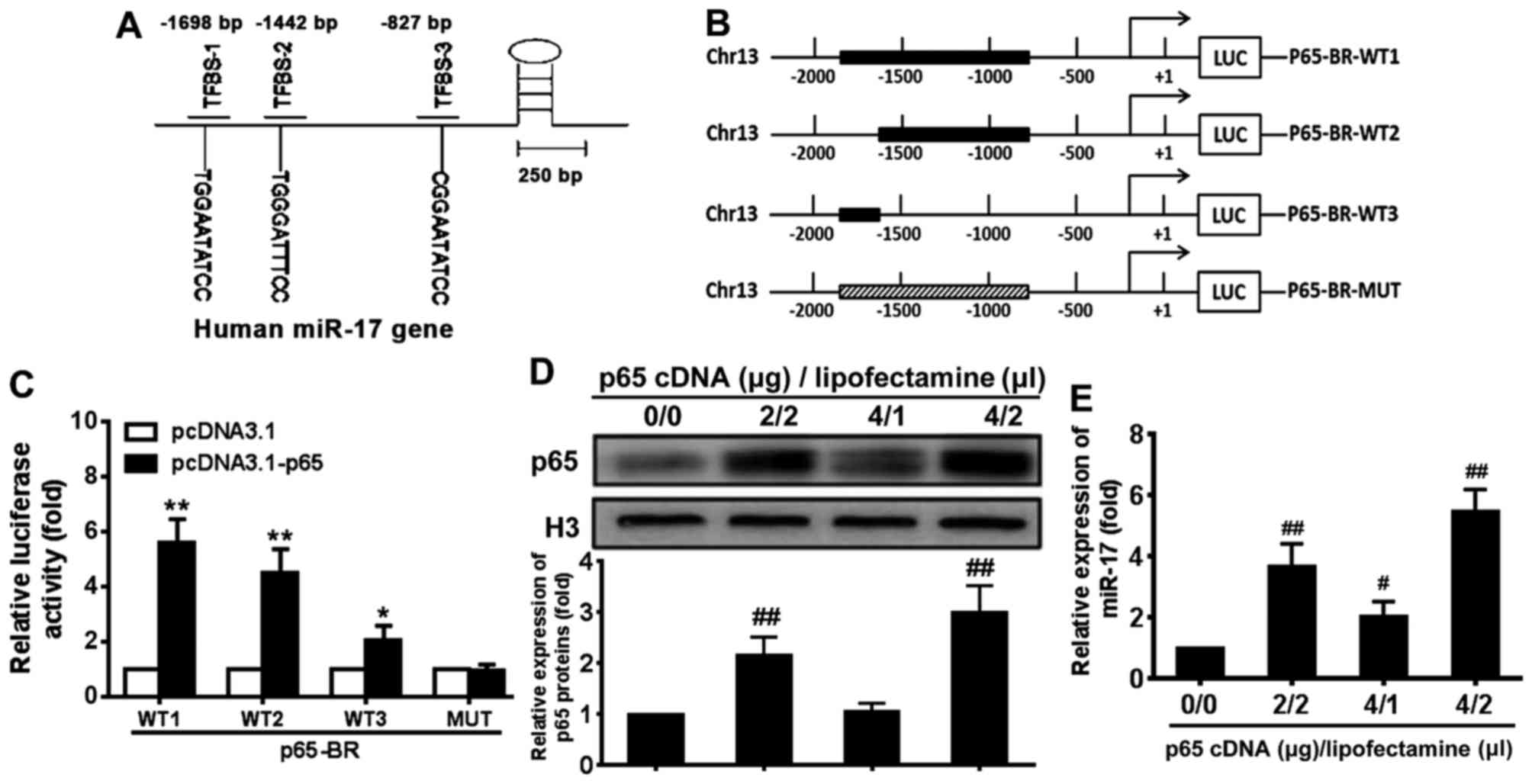
NF-κB p65 subunit induces miR-17
transcription by binding its promoter elements
Previous studies have demonstrated that exogenous
miR-17 mimic and inhibitor regulated RB protein level and VSMC
proliferation (28,29). To further investigate the
endogenous mechanism of regulation of miR-17, the potential binding
sites for p65 in the upstream regulatory region of miR-17 were
investigated and three potential p65 binding sites were identified
around −1698, −1442 and −827 bp (relative to the transcription
start site) in the regulatory sequence of the miR-17 gene (Fig. 3A). To determine whether these
sites mediated the regulation of miR-17 expression by p65,
luciferase reporter genes driven by the upstream regulatory DNA of
miR-17 that contained all or some of the potential p65 binding
sites were constructed (Fig. 3B).
These constructs and p65cDNA WT expression plasmid or p65cDNA
loss-of-function MUT expression plasmid were co-transfected into
293T cells, and the reporter luciferase activity was determined.
The results demonstrated that the expression of the reporter genes
containing p65 binding sites was significantly upregulated by
pcDNA3.1-p65 compared with the levels induced by pcDNA3.1 in 293T
cells (P<0.05) (Fig. 3C). The
magnitude of the upregulation was related to the number of
potential p65 binding sites contained in the reporter
constructs.
Following transfection with different amounts of p65
cDNA and Lipofectamine, the level of p65 protein was upregulated
compared with the level in the control group (Fig. 3D). To determine whether NF-κB p65
signalling regulates miR-17 expression, the miR-17 levels in VSMCs
transfected with p65 cDNA were examined (Fig. 3E). The miR-17 expression level was
also significantly increased in VSMCs transfected with p65 cDNA in
a dose-dependent manner compared with 0 µg p65 cDNA/0
µl Lipofectamine group (P<0.05) (Fig. 3E). These results indicated that
activation of NF-κB p65 signal-ling induced an increase in miR-17
levels in VSMCs.
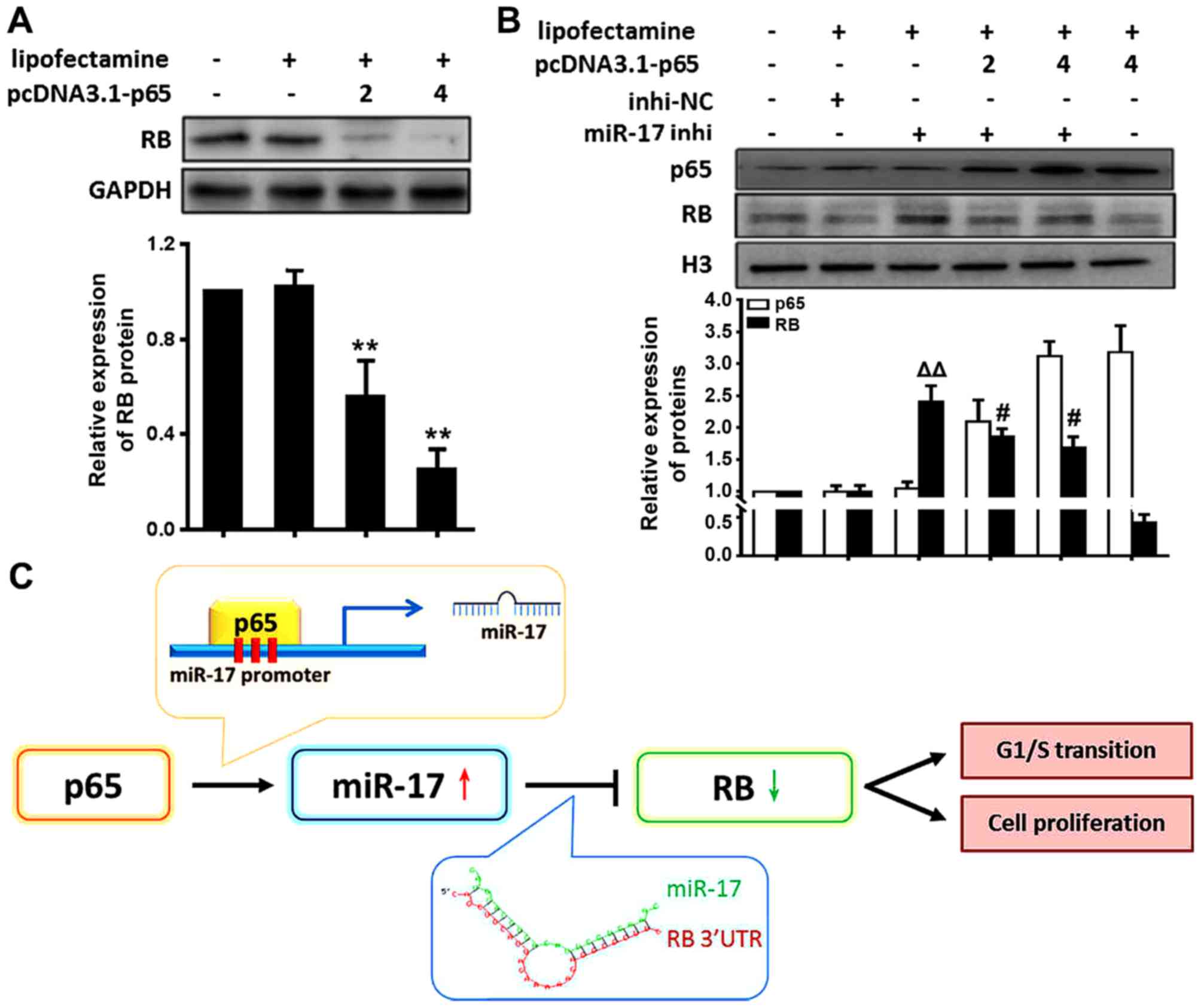
NF-κB/p65 activation decreases RB level
through miR-17 in VSMCs
The present study demonstrated that the expression
of miR-17 is directly regulated by NF-κB p65 activation and that
miR-17 suppressed the RB levels by direct targeting. These data
suggested that NF-κB p65 signalling decreased the RB levels in
VSMCs through induction of miR-17 expression. To determine whether
RB expression was actually regulated by NF-κB p65, RB expression in
VSMCs transfected with p65 cDNA was examined. The results
demonstrated that RB levels were significantly decreased in VSMCs
treated with p65 cDNA compared with the levels in those treated
with Lipofectamine only (P<0.01) (Fig. 4A). RB levels were decreased in
VSMCs by activating NF-κB p65. These results suggested that
activation of NF-κB p65 reduced RB levels.
In order to further verify whether p65 activation
inhibited RB expression through miR-17, the p65 and RB protein
levels in VSMCs following treatment with miR-17 inhi and p65 cDNA
were examined. The results revealed that RB protein levels were
significantly increased in VSMCs following treatment with miR-17
inhi (miR-17 inhi vs. inhi-NC, P<0.01); however, this trend was
weakened following overexpression of p65 (miR-17 inhi vs. miR-17
inhi + pcDNA3.1-p65, P<0.05) (Fig.
4B). These results suggested that decreases in miR-17 levels
elevated RB levels suppressed by NF-κB p65 activation, supporting a
model that activation of NF-κB p65 reduced RB levels through miR-17
in VSMCs under inflammation, as represented by the schematic in
Fig. 4C.
Discussion
Excess proliferation of VSMCs and inflammation have
a key role in several vascular disorders; however, the underlying
mechanism remains unclear. The present study demonstrated that
miR-17 was the most highly induced miRNA in proliferating VSMCs and
overexpression of miR-17 stimulated VSMC proliferation, enhanced
cell cycle G1/S transition and increased levels of E2F1 and PCNA.
NF-κB p65 signalling activated the expression of miR-17, and miR-17
targeted RB, which is a key regulator of cell cycle progression and
proliferation (25,26). p65 activation resulted in
suppression of RB expression, which was abrogated by miR-17
inhibitor. These data suggest that NF-κB p65/miR-17/RB pathway
activation leads to the proliferation of VSMCs. These findings may
indicate a novel mechanism for restenosis and suggest a novel
target for restenosis treatment or prevention.
Transcription factor E2F1 activates the expression
of genes that promote DNA synthesis during cell cycle progression
(30,31). PCNA is important for DNA synthesis
and DNA repair (32). It is well
known that RB is a negative regulator of E2F function in the
regulation of cell cycle progression and proliferation (33). Activation of E2F1, E2F2 and E2F3a
promotes cell cycle progression and proliferation by stimulating
the expression of S phase genes (25,26,34,35). In the present study, miR-17
suppressed RB expression by directly targeting the RB mRNA-3′UTR.
Overexpression of miR-17 decreased RB levels, releasing E2F1, which
in turn promoted the transition from G1 to S phase of the cell
cycle. Consistently, a previous study indicated that E2F3 promotes
VSMC proliferation (36). How
miR-17 increases the levels of E2F1 and PCNA requires further
investigation.
It has been demonstrated that p65 disrupts the
physical interaction between the activator E2Fs and the histone
acetyltransferase cofactor transactivation/transformation-domain
associated protein, resulting in a reduction in E2F-responsive gene
expression in normal human fibroblasts. This leads to inhibition of
cell cycle progression in an RB-independent manner (37). In the present study, it was
indicated that NF-κB p65 signalling directly activated the
expression of miR-17, which directly targeted RB expression in
VSMCs. Therefore, NF-κB p65 signalling may regulate cell cycle
progression and proliferation through RB-dependent and -independent
pathways.
Interplay between NF-κB and miRNA has important
roles in cell growth and proliferation (13,38). As a transcription factor, NF-κB
regulates the expression of miR-143, which enhances hepatocellular
carcinoma metastasis by repressing fibronectin expression (39). In the present study, it was
demonstrated that miR-17 levels were increased by activation of
NF-κB p65 signalling and decreased by inactivation of NF-κB p65
signalling in VSMCs. NF-κB p65 signalling directly regulates the
miR-17 promoter activity, likely through potential p65 binding
sites in the upstream regulatory region of the miR-17 gene. These
data indicated that miR-17 expression is directly regulated by
NF-κB p65 signalling. The present study also indicated that
activation of p65 decreases the levels of RB, which is a direct
target of miR-17. Furthermore, suppression of RB expression by
activation of p65 was abrogated by miR-17 inhibitor in the present
study. These findings support that p65 may regulate gene
expression, and cell growth and proliferation by modulating the
expression levels of miRNA, such as miR-17.
Taken together, the results of the present study
suggest that miR-17 expression is highly induced in proliferating
VSMCs. Overexpression of miR-17 stimulates the proliferation of
VSMCs, likely via enhancing cell cycle G1/S transition by
increasing the levels of E2F1 and PCNA. p65 activates the
expression of miR-17 at the transcription level, and miR-17
directly targets RB, a key regulator of cell cycle progression and
proliferation, through interaction with E2Fs. NF-κB p65 signalling
actually decreases the RB level in VSMCs, which is abrogated by
miR-17 inhibitor. These data support that proliferation of VSMCs is
regulated by activation of the NF-κB p65/miR-17/RB pathway. The
transcription factor NF-κB is activated in and is a master
regulator of the inflammatory response, which is a characteristic
of restenosis. The present findings may provide insight into the
mechanism responsible for the excessive proliferation of VSMCs
under inflammation during restenosis, adding to the regulation of
proliferation of VSMCs by the Smad3/miR-17-92/BMPR2 pathway
(23) and
thrombospondin-1-mediated downregulation of the miR-17-92 cluster
(24) in pathophysiological
conditions, including restenosis, atherosclerosis and pulmonary
artery hypertension. miR-17 may be a potential novel target for the
treatment of restenosis.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81170295 and
81000136), Liaoning Provincial Science and Technology Department
(grant no. 2013225086) and Liaoning Provincial Special Program of
Medical Science (grant no. LNCCC-C03-2015).
References
|
1
|
Bauters C and Isner JM: The biology of
restenosis. Prog Cardiovasc Dis. 40:107–116. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yahagi K, Otsuka F, Sakakura K, Sanchez
OD, Kutys R, Ladich E, Kolodgie FD, Virmani R and Joner M:
Pathophysiology of superficial femoral artery in-stent restenosis.
J Cardiovasc Surg Torino: 55. pp. 307–323. 2014
|
|
3
|
McNamara CA, Sarembock IJ, Bachhuber BG,
Stouffer GA, Ragosta M, Barry W, Gimple LW, Powers ER and Owens GK:
Thrombin and vascular smooth muscle cell proliferation:
Implications for atherosclerosis and restenosis. Semin Thromb
Hemost. 22:139–144. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sheikh AQ, Lighthouse JK and Greif DM:
Recapitulation of developing artery muscularization in pulmonary
hypertension. Cell Reports. 6:809–817. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Satoh K, Satoh T, Kikuchi N, Omura J,
Kurosawa R, Suzuki K, Sugimura K, Aoki T, Nochioka K, Tatebe S, et
al: Basigin mediates pulmonary hypertension by promoting
inflammation and vascular smooth muscle cell proliferation. Circ
Res. 115:738–750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Donners MM, Daemen MJ, Cleutjens KB and
Heeneman S: Inflammation and restenosis: Implications for therapy.
Ann Med. 35:523–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Drachman DE and Simon DI: Inflammation as
a mechanism and therapeutic target for in-stent restenosis. Curr
Atheroscler Rep. 7:44–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schillinger M and Minar E: Restenosis
after percutaneous angioplasty: The role of vascular inflammation.
Vasc Health Risk Manag. 1:73–78. 2005. View Article : Google Scholar
|
|
9
|
Tanaskovic S, Isenovic ER and Radak D:
Inflammation as a marker for the prediction of internal carotid
artery restenosis following eversion endarterectomy - evidence from
clinical studies. Angiology. 62:535–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parker M, Mohankumar KM, Punchihewa C,
Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN,
Thiruvenkatam R, et al: C11orf95-RELA fusions drive oncogenic NF-κB
signalling in ependymoma. Nature. 506:451–455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki J, Tezuka D, Morishita R and Isobe
M: An initial case of suppressed restenosis with nuclear
factor-kappa B decoy transfection after percutaneous coronary
intervention. J Gene Med. 11:89–91. 2009. View Article : Google Scholar
|
|
12
|
Ohtani K, Egashira K, Nakano K, Zhao G,
Funakoshi K, Ihara Y, Kimura S, Tominaga R, Morishita R and
Sunagawa K: Stent-based local delivery of nuclear factor-kappaB
decoy attenuates in-stent restenosis in hypercholesterolemic
rabbits. Circulation. 114:2773–2779. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Beitzinger M and Meister G: Preview.
MicroRNAs: From decay to decoy. Cell. 140:612–614. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gareri C, De Rosa S and Indolfi C:
MicroRNAs for restenosis and thrombosis after vascular injury. Circ
Res. 118:1170–1184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lv J, Wang L, Zhang J, Lin R, Wang L, Sun
W, Wu H and Xin S: Long noncoding RNA H19-derived miR-675
aggravates restenosis by targeting PTEN. Biochem Biophys Res
Commun: Jan. 4:2017Epub ahead of print. View Article : Google Scholar
|
|
16
|
Stein JJ, Iwuchukwu C, Maier KG and Gahtan
V: Thrombospondin-1-induced vascular smooth muscle cell migration
and proliferation are functionally dependent on microRNA-21.
Surgery. 155:228–233. 2014. View Article : Google Scholar
|
|
17
|
Li Y, Yan L, Zhang W, Hu N, Chen W, Wang
H, Kang M and Ou H: MicroRNA-21 inhibits platelet-derived growth
factor-induced human aortic vascular smooth muscle cell
proliferation and migration through targeting activator protein-1.
Am J Transl Res. 6:507–516. 2014.PubMed/NCBI
|
|
18
|
Muthiah M, Islam MA, Cho CS, Hwang JE,
Chung IJ and Park IK: Substrate-mediated delivery of microRNA-145
through a polysorbitol-based osmotically active transporter
suppresses smooth muscle cell proliferation: Implications for
restenosis treatment. J Biomed Nanotechnol. 10:571–579. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santulli G, Wronska A, Uryu K, Diacovo TG,
Gao M, Marx SO, Kitajewski J, Chilton JM, Akat KM, Tuschl T, et al:
A selective microRNA-based strategy inhibits restenosis while
preserving endothelial function. J Clin Invest. 124:4102–4114.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mogilyansky E and Rigoutsos I: The
miR-17/92 cluster: A comprehensive update on its genomics,
genetics, functions and increasingly important and numerous roles
in health and disease. Cell Death Differ. 20:1603–1614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Z, Wu J, Yang C, Fan P, Balazs L,
Jiao Y, Lu M, Gu W, Li C, Pfeffer LM, et al: DiGeorge syndrome
critical region 8 (DGCR8) protein-mediated microRNA biogenesis is
essential for vascular smooth muscle cell development in mice. J
Biol Chem. 287:19018–19028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo T, Cui S, Bian C and Yu X: Crosstalk
between TGF-β/Smad3 and BMP/BMPR2 signaling pathways via miR-17-92
cluster in carotid artery restenosis. Mol Cell Biochem.
389:169–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maier KG, Ruhle B, Stein JJ, Gentile KL,
Middleton FA and Gahtan V: Thrombospondin-1 differentially
regulates microRNAs in vascular smooth muscle cells. Mol Cell
Biochem. 412:111–117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burke JR, Hura GL and Rubin SM: Structures
of inactive retinoblastoma protein reveal multiple mechanisms for
cell cycle control. Genes Dev. 26:1156–1166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
28
|
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T,
Wang H, Xuan Q, Chen P, Xu J, et al: miR-17-3p contributes to
exercise-induced cardiac growth and protects against myocardial
ischemia-reperfusion injury. Theranostics. 7:664–676. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen T, Zhou Q, Tang H, Bozkanat M, Yuan
JX, Raj JU and Zhou G: miR-17/20 controls prolyl hydroxylase 2
(HD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary
artery smooth muscle cell proliferation. J Am Heart Assoc.
5:e0045102016. View Article : Google Scholar
|
|
30
|
Blais A and Dynlacht BD: Hitting their
targets: An emerging picture of E2F and cell cycle control. Curr
Opin Genet Dev. 14:527–532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wong JV, Dong P, Nevins JR, Mathey-Prevot
B and You L: Network calisthenics: Control of E2F dynamics in cell
cycle entry. Cell Cycle. 10:3086–3094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Essers J, Theil AF, Baldeyron C, van
Cappellen WA, Houtsmuller AB, Kanaar R and Vermeulen W: Nuclear
dynamics of PCNA in DNA replication and repair. Mol Cell Biol.
25:9350–9359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fischer M and Müller GA: Cell cycle
transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev
Biochem Mol Biol. Aug 11–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Korenjak M and Brehm A: E2F-Rb complexes
regulating transcription of genes important for differentiation and
development. Curr Opin Genet Dev. 15:520–527. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schaal C, Pillai S and Chellappan SP: The
Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and
metastasis. Adv Cancer Res. 121:147–182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Giangrande PH, Zhang J, Tanner A, Eckhart
AD, Rempel RE, Andrechek ER, Layzer JM, Keys JR, Hagen PO, Nevins
JR, et al: Distinct roles of E2F proteins in vascular smooth muscle
cell proliferation and intimal hyperplasia. Proc Natl Acad Sci USA.
104:12988–12993. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Araki K, Kawauchi K and Tanaka N:
IKK/NF-kappaB signaling pathway inhibits cell-cycle progression by
a novel Rb-independent suppression system for E2F transcription
factors. Oncogene. 27:5696–5705. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kravtsova-Ivantsiv Y, Shomer I,
Cohen-Kaplan V, Snijder B, Superti-Furga G, Gonen H, Sommer T, Ziv
T, Admon A, Naroditsky I, et al: KPC1-mediated ubiquitination and
proteasomal processing of NF-κB1 105 to 50 restricts tumor growth.
Cell. 161:333–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang X, Liu S, Hu T, Liu S, He Y and Sun
S: Up-regulated microRNA-143 transcribed by nuclear factor kappa B
enhances hepatocarcinoma metastasis by repressing fibronectin
expression. Hepatology. 50:490–499. 2009. View Article : Google Scholar : PubMed/NCBI
|