Introduction
The heart is a highly aerobic organ (1,2).
Any cause of hypoxia may have a deep impact on cardiac function,
and the majority of heart diseases are associated with cardiac
hypoxia. Among the well-defined diseases associated with cardiac
hypoxia, congenital heart disease (CHD), particularly cyanotic
cardiac defects, including tetralogy of Fallot and Ebstein's
anomaly (3), is one of the most
common heart diseases associated with chronic and systemic
hypoxemia. In addition to myocardial infarction and cardiac
hypertrophy, local hypoxia promotes the progression of heart
failure. Therefore, understanding the molecular changes of
cardiomyocytes under chronic hypoxia may assist in the improvement
of the clinical treatment of heart disease.
In cardiac tissue, chronic hypoxia triggers
oxygen-sensitive transcriptional modulators to increase
carbohydrate metabolism, enhance mitochondrial respiratory capacity
and increase the efficiency of mitochondrial energy production.
Therefore, the mitochondria serve a pivotal role in cardiac
intrinsic regulation under hypoxia (4). Mitochondria take up at least 30% of
the volume of cardiomyocytes. It has been demonstrated that
peroxisome proliferator-activated receptor-γ coactivator-1α
(PGC-1α) is upregulated resulting in subsequent increase of
mitochondrial biogenesis under chronic hypoxia (5). However, in the process of
mitochondria increase, the level of reactive oxygen species (ROS)
is increased by mitochondria as byproducts, which can lead to
mitochondrial oxidative damage and mitochondrial dysfunction
(6). Defective mitochondria
enhance ROS emission, which may result in protein, nucleic acid and
liquid damage, inducing oxidative stress, cellular damage and
eventually cell death (7).
Therefore, mitochondrial quality control is considered to be
critical for the maintenance of the appropriate mitochondrial
quality and quantity for cellular health. However, the regulatory
mechanisms responsible for the mitochondrial quality control under
chronic hypoxia are complex and not completely understood.
Autophagy is a catabolic process that is responsible
for the degradation of cellular components via the lysosome. The
process serves pivotal cellular physiological functions, including
degradation of unwanted proteins, organelle turnover, response to
nutrient depletion and extension of lifespan (8). The selective targeting of
mitochondria by autophagy is known as mitophagy, and it eliminates
unwanted or damaged mitochondria to relieve oxidative stress,
thereby favoring adaptation in response to hypoxic stress (9). Accordingly, mitochondrial autophagy
or mitophagy may be one of the underlying mechanisms to promote
cardiomyocyte survival under hypoxic conditions.
Adenosine monophosphate-activated protein kinase
(AMPK), an energy sensor and regulator, serves an important role in
maintaining intracellular homoeostasis. AMPK activation can
increase the uptake of glucose, enhance fatty acid oxidation
(10) and inhibit protein
synthesis to reserve energy stores (11). Furthermore, it has been
demonstrated that hypoxia triggers AMPK activation through the
ROS-mediated opening of calcium release-activated calcium channels
(12). Importantly, AMPK can
regulate cellular survival through autophagic regulation. A recent
study reported that AMPK-mediated autophagy is essential for the
maintenance of skeletal muscle function, which contains long-lived
post-mitotic cells (13).
In the present study, we aim to confirm that
mitophagy is critical for the quality control of mitochondria and
cardiomyocyte survival, and AMPK is the key regulator of mitophagy
under chronic hypoxia. Furthermore, we want to demonstrate that
modulating the activity of AMPK is a therapeutic strategy for heart
disease under hypoxia.
Materials and methods
Patients
A total of 20 patients with CHD, 1–28 years old
(mean, 6.02 years old), were eligible for inclusion in the present
study. All patients were admitted to the Institute of
Cardiovascular Surgery, Xinqiao Hospital of the Third Military
Medical University (Chonqing, China). Of these, 10 patients
presented with a cyanotic defect and 10 with an acyanotic defect.
The local Ethics Review Board of the Third Military Medical
University Affiliated Hospital approved the study protocol and the
informed consent forms. This investigation conforms to the
principles outlined in the Declaration of Helsinki. Written
informed consent was obtained from the parents of all
participants.
Cardiac surgery and sampling of
myocardial biopsies
Human samples were sectioned and embedded as
previously described (14).
Briefly, a myocardial biopsy specimen was obtained from the right
ventricular outflow tracts during right ventricular outflow tract
reconstruction. The myocardial samples intended for western
blotting and quantitative polymerase chain reaction (qPCR) analysis
were immediately snap-frozen in liquid nitrogen and stored at −80°C
until analysis.
Cell culture and treatment
The embryonic rat heart-derived H9c2 cells purchased
from the American Type Culture Collection (CRL-1466) were grown in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc. Waltham, MA, USA) supplemented with 10% fetal
bovine serum (Biological Industries, Beti-Haemek, Israel) at 37°C
and exposed to 21% O2, 5% CO2 and 74%
N2. Following serum starvation overnight, cells in the
hypoxic group were placed in an in vivo 200 cultivator
(Ruskinn Technology, Ltd., Bridgend, UK) containing a gaseous
mixture of 94% N2, 5% CO2 and 1%
O2 at 37°C for 48 h. In addition, the AMPK activation
group was treated with AICAR (1 mM; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), an AMPK agonist, while the inhibition group
was exposed to compound C (1 μM; Merck KGaA), an AMPK
inhibitor. Cells in the normoxic groups were incubated under the
same conditions, except with 21% O2 concentrations.
qPCR analysis
Total DNA was isolated from H9c2 cells and
myocardial samples using DNAiso Plus (Takara Biotechnology Co.,
Ltd., Dalian, China) according to the manufacturer's instructions.
For the quantitative determination of mitochondrial DNA (mtDNA)
content relative to nuclear DNA (nDNA), primers specific for
amplification of mtDNA Cyt and nDNA-encoded GAPDH were selected.
The following primer sequences were used: Cyt sense,
5′-CCGGAGCAATCCAGGTCGGTT-3′ and antisense,
5′-TGGTTGGGAGCACTTATGGTAAGGA-3′; and GAPDH sense,
5′-TGGGGAAGGTGAAGGTCGGAGT-3′ and antisense,
5′-CCCGTTCTCAGCCTTGACGGTG-3′. SYBR® Premix Ex Taq™ II
(Takara Biotechnology Co., Ltd.) was used according to the
manufacturer's instructions, and qRT-PCR was performed using a
StepOnePlus™ Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The reaction conditions were as follows:
95°C for 30 sec, followed by 45 cycles of 95°C for 5 sec and 60°C
for 30 sec. Relative expression of target mtDNA was normalized to
GAPDH using the ΔΔCq method (15).
Immunofluorescence
The cells were treated with or without chloroquine
(Chl; 10 μM; Sigma-Aldrich; Merck KGaA), which inhibits
fusion of autophagosomes with lysosomes (16), for 4 h prior to incubation at 37°C
for 30 min in media containing MitoTracker (1:4,000; Life
Technologies; Thermo Fisher Scientific, Inc.), then fixed with 4%
formaldehyde for 15 min at room temperature and blocked with PBS
containing 10% goat serum, 0.3 M glycine, 1% BSA and 0.3% Triton
X-100 (Beyotime Institute of Biotechnology, Beijing, China) for 1 h
at room temperature. Staining of the treated cells with rabbit
anti-rat microtubule-associated proteins 1B light chain 3B (LC3B)
polyclonal antibody (1:200; L7543; Sigma-Aldrich) was performed
overnight at 4°C in PBS containing 1% BSA and 0.3% Triton X-100.
Alexa Fluor 488 anti-rabbit antibody (1:500; A0423; Beyotime
Institute of Biotechnology) was used as the secondary antibody
incubated for 1 h at room temperature. A confocal laser scanning
microscope (Leica TCS SP2; Leica Microsystems, Wetzlar, Germany)
was used to image stained cells, and the co-localization dots were
analyzed using ImageJ software (v1.6.0; National Institutes of
Health, Bethesda, MD, USA). Each group contained three dependent
samples, and every sample was analyzed in five random fields.
Mitochondrial potential analysis
The adherent cells were incubated with MitoTracker
for 30 min as aforementioned, followed by treatment with rhodamine
(5 μM; Beyotime Institute of Biotechnology) for 20 min. A
confocal laser scanning microscope was used to image the stained
cells. Five random fields were analyzed per sample.
JC-1 staining was also used to detect alterations of
mitochondrial transmembrane potential (ΛΨm). Cells were collected,
washed and suspended in JC-1 binding buffer (Beyotime Institute of
Biotechnology). JC-1 dye (1:100; Beyotime Institute of
Biotechnology) was added and the cells were incubated for 20 min at
37°C in the dark, followed by washing 2 times with binding buffer.
The cell suspensions were analyzed by flow cytometry.
Apoptosis analysis
Cell apoptosis was determined by Annexin
V-fluorescein 5-isothiocyanate (Annexin V-FITC) assay. H9c2 cells
(1×107 cells in 100 μl) were collected, washed
and suspended in Annexin V binding buffer (Wanleibio Co., Ltd.,
Shanghai, China). FITC-conjugated Annexin V and propidium iodide
(PI) (1:100; Wanleibio Co., Ltd.) were added to the binding buffer
that contained suspended cells, both incubated for 15 min following
the manufacturer's protocols. Following incubation, the cells were
analyzed by flow cytometry (MoFloTMXDP; Beckman Coulter, Co.,
Fullerton, CA, USA).
Cell proliferation assay
H9c2 cells were collected from the cultures and
replated into a 96-well plate (1×105 cells/ml). The
cells in the hypoxic group, the AMPK activation group and the AMPK
inhibition group were treated as described above. Cell
proliferation was measured by Cell Counting Kit-8 (CCK-8; Wuhan
Bioengineering Institute, Wuhan, China) assays according to the
manufacturer's protocols. Prior to reading the absorbance at 450
nm, 10 μl CCK-8 solution and 100 μl complete medium
were sequentially added to each well.
Western blotting
H9c2 cells and myocardial samples were lysed in
ice-cold lysis buffer (radioimmunoprecipitation assay), with a
protease inhibitor (0.5 mM PMSF) (both Beyotime Institute of
Biotechnology) and phosphatase inhibitor (1 tablet/10ml; Roche
Diagnostics GmbH, Mannheim, Germany). The lysates were further
centrifuged at 15,000 × g for 15 min at 4°C. Proteins from the H9c2
cells and myocardial samples lysates were measured using the
Bradford method. Cell proteins (20 μg) were extracted with
SDS lysis buffer and separated by 10% SDS-PAGE gel (both from
Beyotime Institute of Biotechnology). Subsequently, protein samples
were transferred to 0.22-μm polyvinylidene difluoride
membranes (Roche Diagnostics GmbH), which were blocked with 5% BSA
for 1 h at room temperature, followed by incubation with primary
antibodies at 4°C overnight. The primary antibodies included the
following: Rabbit anti-mouse AMPKα polyclonal antibody (1:1,000;
#5831), rabbit ant-mouse p-AMPKα polyclonal antibody (1:1,000;
#2535) (both Sigma-Aldrich; Merck KGaA) and rabbit anti-mouse
β-actin monoclonal antibody (1:1,000; bs-0061R; Bioss, Beijing,
China). Next, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:5,000; A0208) for 1 h
at 37°C and detected using an enhanced chemiluminescence kit (both
from Beyotime Institute of Biotechnology), and densitometry signals
were quantified using ImageQuant TL software (General Electric,
Co., Fairfield, CT, USA).
Statistical analysis
Data from at least three independent experiments are
expressed as the mean ± SD. SPSS 16.0 software (SPSS, Inc.,
Chicago, IL, USA) was used for the statistical analysis. The
difference in means between 2 groups was evaluated using Student's
t-test, and one-way analysis of variance was used for comparisons
between ≥2 groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Hypoxia increases mitochondrial
biogenesis, ΛΨm loss and cell apoptosis in H9c2 cells
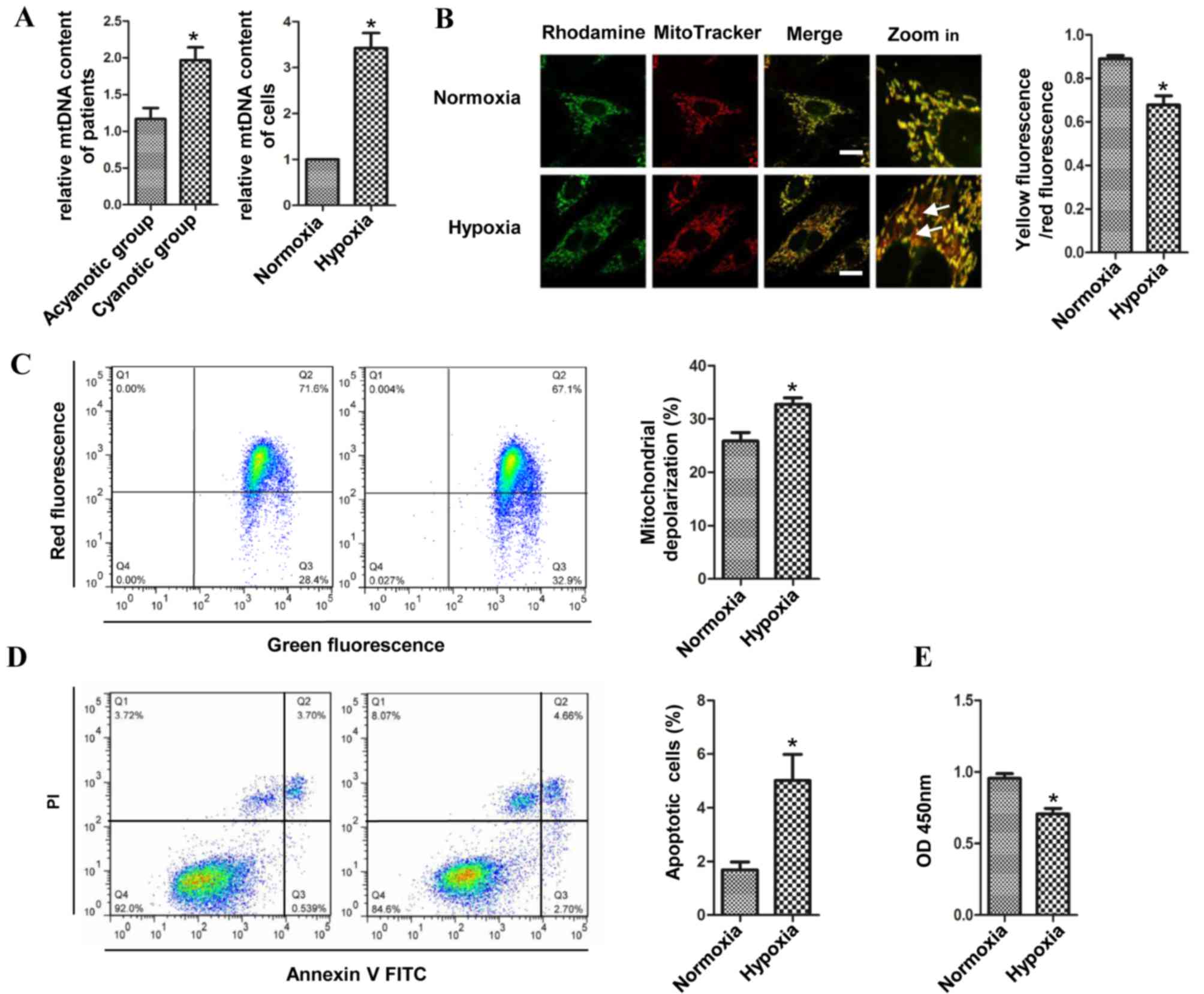
The epidemiological data of the patients is
summarized in Table I. Patients
with cyanotic heart disease had significantly lower preoperative
arterial oxygen saturation (SaO2) than patients with
acyanotic heart disease. Comparing the cyanotic patients with the
acyanotic patients, the relative mtDNA/nDNA ratio was found to be
higher in the cyanotic group. Similarly, the ratio of H9c2 cells in
the hypoxic group was higher than that in the H9c2 cells cultured
under normoxic conditions. The copies of mtDNA Cyt were
significantly increased in the cyanotic patients and the H9c2 cells
of hypoxic group when compared with the acyanotic patients and the
H9c2 cells of the normoxic group, respectively (Fig. 1A). Fluorescence microscopy was
performed to detect ΛΨm in the cells cultivated under hypoxic and
normoxic conditions. Cells were co-stained with rhodamine 123 and
MitoTracker, and it was found that the transmembrane potential was
decreased in hypoxic cardiomyocytes, as the co-localization of
rhodamine 123 and MitoTracker was decreased (Fig. 1B). Moreover, after the cells were
stained with JC-I, which more specifically reduces mitochondrial
ΛΨm, the loss of ΛΨm was increased and the number of H9c2 cells
with intact ΛΨm was reduced compared with the normoxic H9c2 cells
(Fig. 1C). Loss of the ΛΨm is a
key step in the intrinsic apoptotic pathway (17). Therefore, the apoptosis of H9c2
cells was evaluated using flow cytometry following staining of the
cells with Annexin V-FITC in combination with PI. A significantly
higher percentage of apoptotic cells were detected in the hypoxic
group (Fig. 1D). The CCK-8 assay
showed that the proliferation of the H9c2 cells was significantly
decreased in the hypoxic group compared with that in the normoxic
group (Fig. 1E). The results
revealed that compared with that in acyanotic patients,
mitochondrial biogenesis was upregulated in patients with cyanotic
heart disease and in the H9c2 cells cultivated in the chronic
hypoxic condition. In addition to the upregulated mitochondrial
biogenesis, the loss of ΛΨm and the apoptosis of the cells were
also significantly increased compared with the normoxic cells.
These data suggested that chronic hypoxia promoted mitochondrial
biogenesis and induced mitochondrial damage and cell apoptosis at
the same time.
 | Table IClinical characteristics of included
patients. |
Table I
Clinical characteristics of included
patients.
| Characteristic | Acyanotic | Cyanotic |
|---|
| n | 10 | 10 |
| Age at surgery,
yearsa | 6.17±5.98 | 5.86±8.64 |
| Gender
(male/female), na | 4/6 | 3/7 |
| SaO2,
%a | 97.30±1.89 | 71.30±11.83b |
| Hb, g/dla | 12.31±0.55 | 16.41±3.32b |
| Hct, %a | 37.65±1.64 | 51.24±9.48b |
| Pathology, n | | |
| TOF | | 8 |
| VSD and PA | | 2 |
| VSD and RVOS | 10 | |
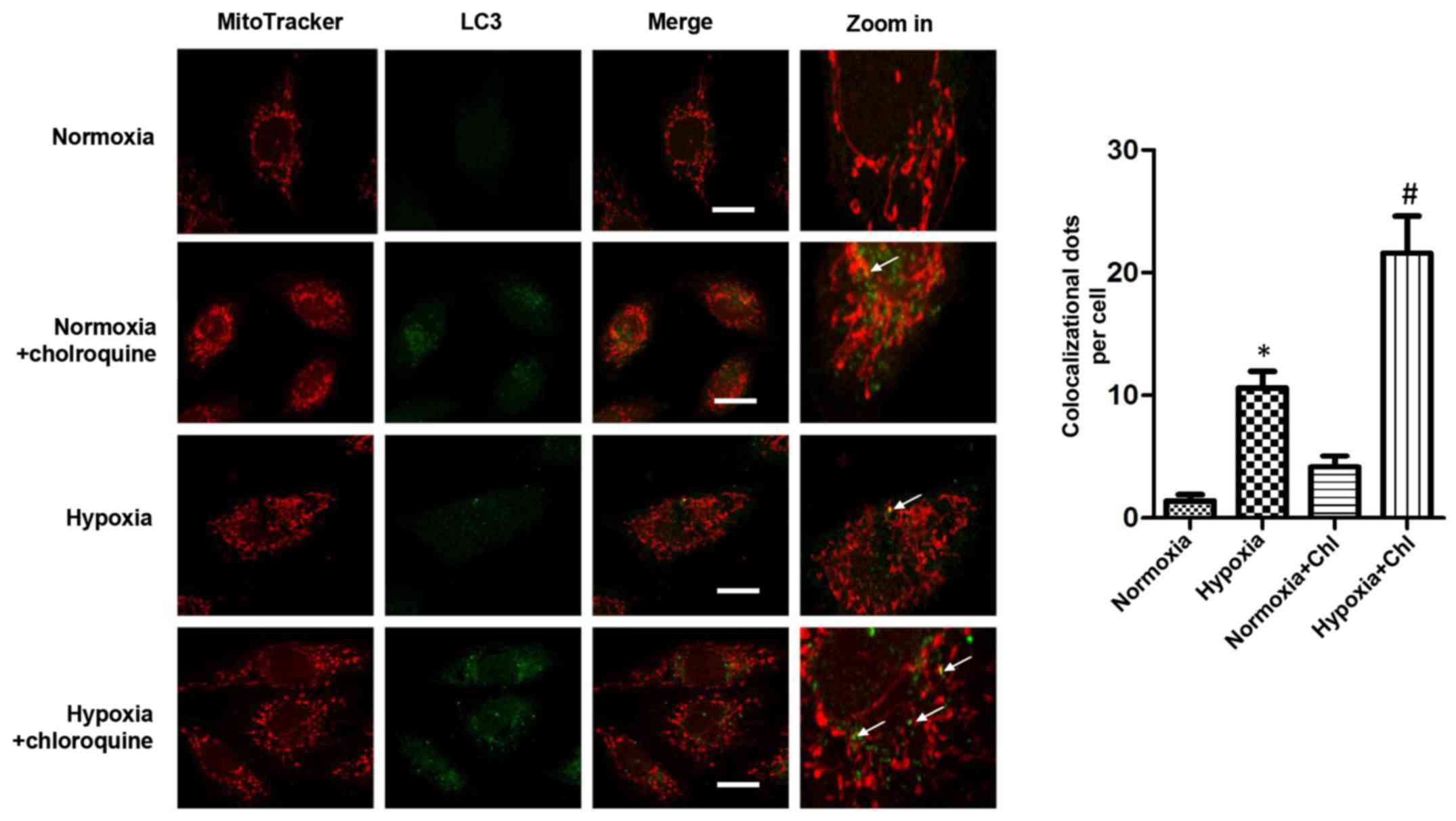
Hypoxia facilitates mitophagy
As the damaged and dysfunctional mitochondria
increased in the H9c2 cells exposed to long periods of hypoxia,
selective mitochondrial autophagy was detected in cells exposed to
hypoxia. After staining the cells with antibody to LC3B and
immunofluorescence assessment, it was found that LC3B rarely
localized in the mitochondria, confirming the presence of selective
mitochondrial autophagy, also known as mitophagy, in the
cardiomyocytes. Notably, the expression of LC3 was increased in the
mitochondria when the H9c2 cells were subjected to long periods of
hypoxia compared with that in cardiomyocytes cultured under
normoxic conditions. Moreover, when the cardiomyocytes were treated
with Chl, an inhibitor of autophagosome-lysosome fusion, the
expression of LC3 in the mitochondria was significantly increased
(Fig. 2). These results suggested
that mitophagosomes were increased in the H9c2 cells under hypoxic
conditions and that the mitophagy was induced by chronic
hypoxia.
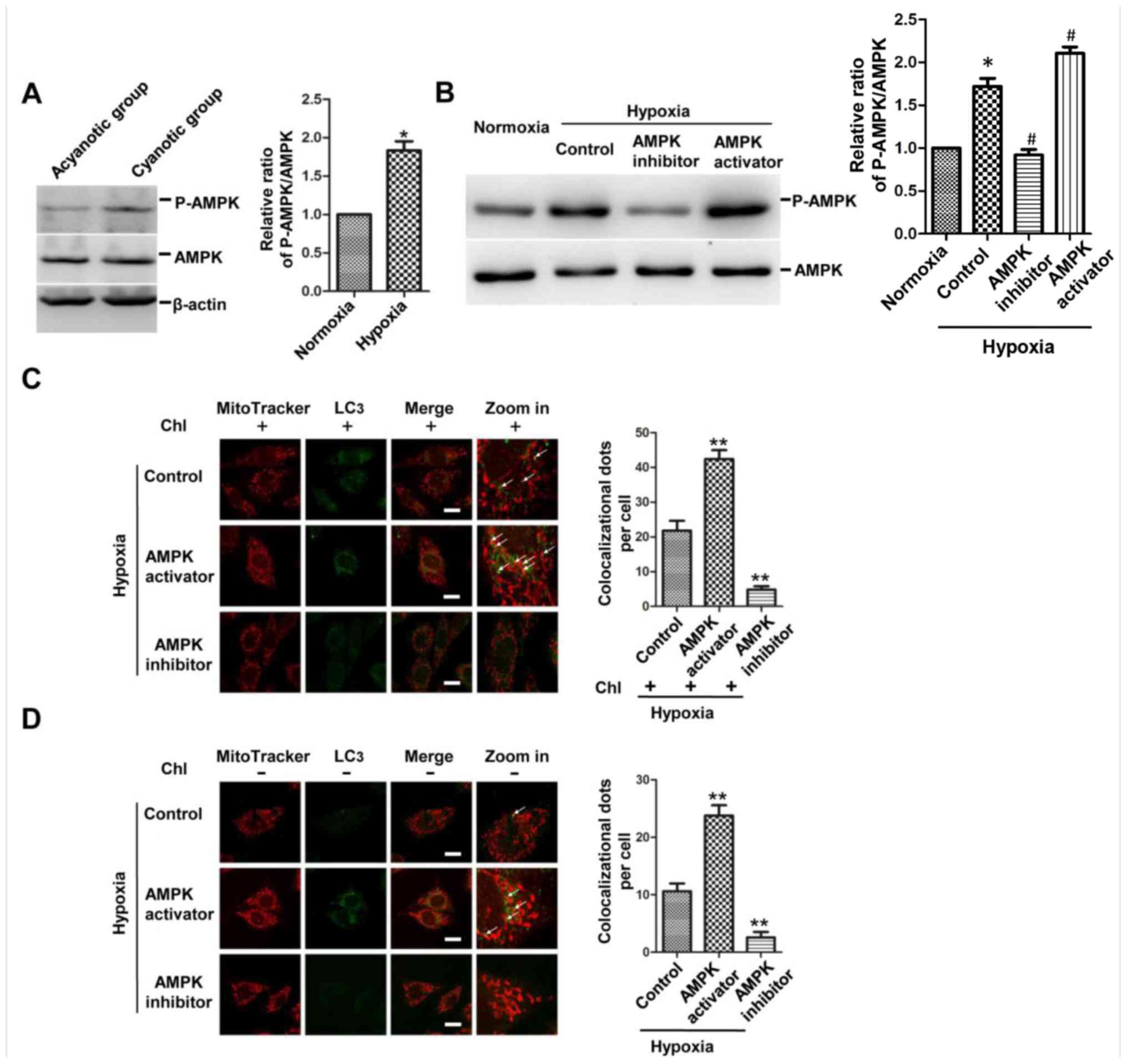
Hypoxia induces the activation of AMPK,
and inhibited AMPK activation decreases mitophagy in
cardiomyocytes
Since AMPK plays a significant role in maintaining
intracellular homoeostasis during hypoxia, the present study first
investigated the impact of chronic hypoxia on AMPK activation. As
expected, western blotting results confirmed that the level of
phosphorylation of AMPK was increased in the ventricular myocardium
of cyanotic patients and in the H9c2 cells cultured under hypoxic
conditions (Fig. 3A and B). To
test further whether AMPK was necessary in regulating mitophagy,
the activation of AMPK was determined following different
treatments, including AMPK activator AICAR and AMPK inhibitor
compound C, and it was found that the level of phosphorylation of
AMPK was increased in the AMPK activation group and decreased in
the AMPK inhibition group compared with that in the hypoxic control
group (Fig. 3B).
Immunofluorescence showed that the AMPK activator increased the
co-localization of LC3 dots (green) and mitochondria (red) in the
Chl-treated and untreated H9c2 cells, while the AMPK inhibitor
presented the opposite results (Fig.
3C and D). These results demonstrated that the activity of AMPK
was increased in the myocardium of infants with cyanotic cardiac
defects and in the H9c2 cells under hypoxia. In addition, mitophagy
was promoted in the cells of the AMPK activation group and
suppressed in the AMPK inhibition group, thereby suggesting that
activation of AMPK is a critical factor for mitophagy under chronic
hypoxia.
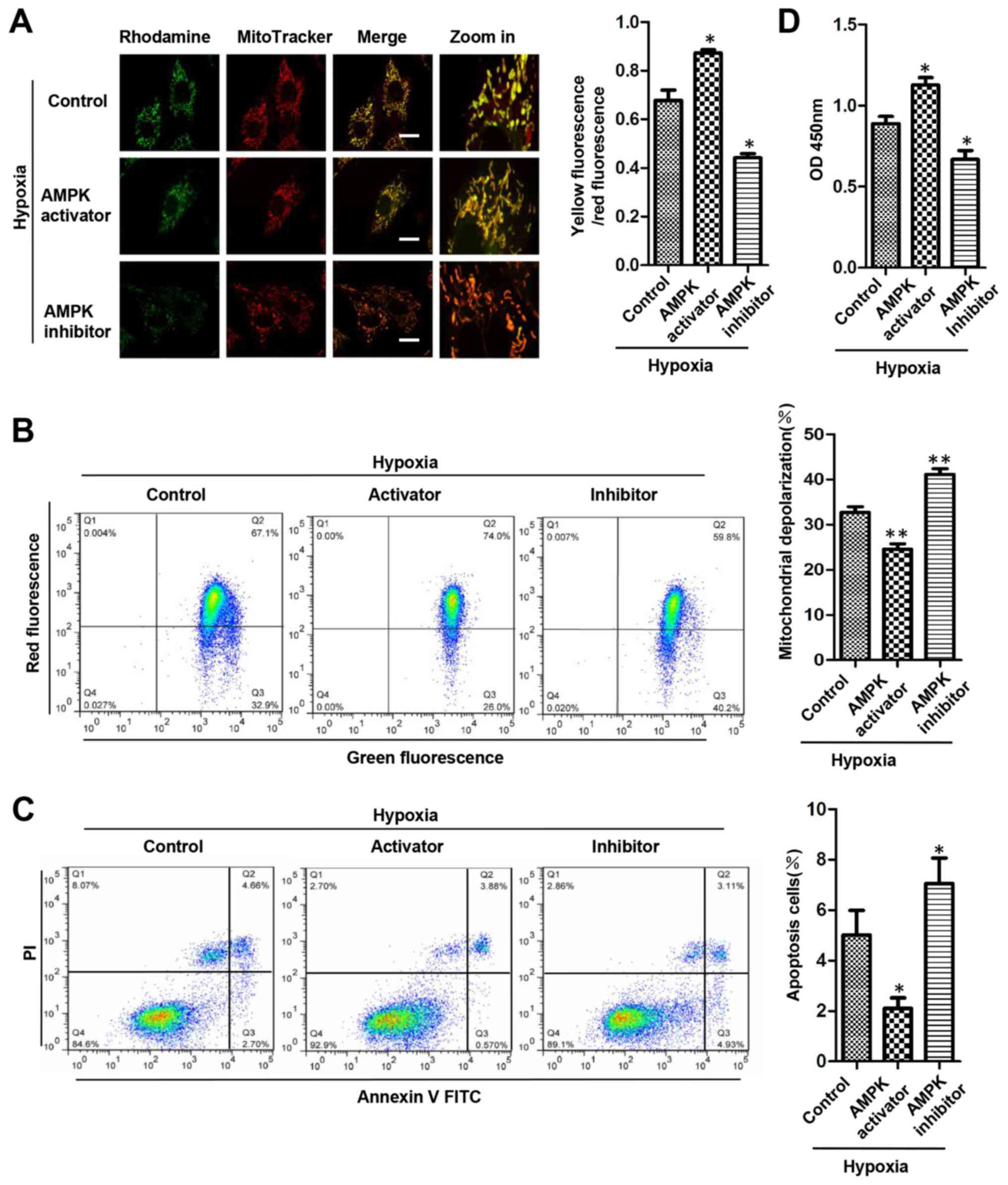
AMPK activation contributes to
mitochondrial quality control and promotes H9c2 cell survival
To determine whether AMPK contributes to
mitochondrial quality control and protects cardiomyocytes during
chronic hypoxia, ΛΨm was detected using fluorescence microscopy
following rhodamine 123 and MitoTracker staining. Compared with
that of the hypoxic control group, the percentage of dysfunctional
mitochondria was decreased in the AMPK activation group, but
increased in the AMPK inhibition group (Fig. 4A). Following the JC-1 staining,
flow cytometry revealed that the number of H9c2 cells with reduced
ΛΨm was decreased in the AMPK activation group and increased in the
AMPK inhibition group compared with that of the hypoxic group
(Fig. 4B). The apoptosis of H9c2
cells was detected using flow cytometry following staining with
Annexin V-FITC in combination with PI. It was observed that,
compared with that in the control group, the ratio of early
apoptotic cells was decreased following treatment of AICAR, but
increased following treatment with compound C (Fig. 4C). The proliferation of H9c2 cells
that was detected by CCK-8 assay was increased by AMPK activator,
but decreased by AMPK inhibitor (Fig.
4D). These data suggested that under chronic hypoxic
conditions, activation of AMPK contributed to the mitochondrial
quality and has a protective role for cell survival.
Discussion
In the present study, mitophagy was found to be
upregulated under hypoxic conditions, with AMPK activity being
increased accordingly. AMPK played a critical role in the
regulation of mitophagy under hypoxic conditions. Mitochondrial
function is key for maintaining cardiomyocyte function, and
extensive mitochondrial damage can induce apoptosis via different
pathways (18,19). Mitochondria are dynamic organelles
that have unique features, including their own genome and a
maternal mode of germ line transmission (20). To ensure the integrity of
mitochondria and their DNA, mitochondria undergo fusion-fission
dynamics, organelle transport, genetic selection of functional
genomes and mitochondrial autophagy or mitophagy (21). Mitochondrial fusion and fission
allows exchange of mitochondrial components and segregation of
terminally damaged mitochondria, to ensure more effective
degradation of unwanted and damaged mitochondria by specific
mitophagy (22). Mitophagy not
only recycles the components of the mitochondria, but also
selectively eliminates the damaged mitochondria to regulate their
quantity and quality (23). This
significantly contributes to cellular mitochondrial quality control
and maintaining mitochondrial function, and even cell survival.
In recent years, intensive studies on the regulation
and function of AMPK have indicated that AMPK acts as a 'fuel
gauge' in numerous types of cells. AMPK is important in maintaining
intracellular homoeostasis during many stressful challenges,
including hypoxia, and regulates the ROS/redox balance, cell
proliferation, cell apoptosis, autophagy, mitochondrial function
and genotoxic response (24). It
has been well established that AMPK can regulate ROS through the
regulation of autophagy (25). In
the heart, AMPK restores the adenosine triphosphate supply for
cardiomyocytes and plays an important role in physiological states
and stress conditions. A number of studies have indicated that AMPK
activation can protect cardiomyocytes and limit the damage from
myocardial ischemic injury (26,27).
It has been demonstrated that hypoxia leads to
mitochondrial damage and dysfunction, including decreased oxidative
phosphorylation, released apoptogenic factors, such as cytochrome
c oxidase, and increased ROS regeneration (7,28).
Hypoxia also triggers AMPK activation to deal with stress
challenge. The present study observations indicate that increased
AMPK activation could play a protective role in allowing
cardiomyocytes to survive under chronic hypoxic stress by
stimulating mitophagy, which ensures mitochondrial quality
control.
Under hypoxic conditions, hypoxia-inducible factor-1
is induced and mediates the induction of B-cell lymphoma 2
(Bcl-2)/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), which
is usually described as a pro-apoptotic member of the Bcl-2 family
that triggers selective mitochondrial autophagy (29). Moreover, it has been demonstrated
that several mitophagy receptors, including BNIP3, NIX (also known
as BNIP3L) and mitochondrial outer membrane protein FUN14 domain
containing 1 cooperate with the PTEN-induced putative kinase
1-Parkin pathway and play a significant role to fine-tune the
mitophagy process (9). A recent
study indicated that endogenous dynamin-related protein 1 serves an
important role in mediating mitochondrial autophagy in
cardiomyocytes (30). It is well
known that a molecular mechanism for the regulation of mammalian
autophagy involves UNK-51-like kinase 1 (ULK1), the mammalian
homolog of yeast protein kinase Atg1. ULK1 induces autophagy by
phosphorylating beclin 1 and activating Vps34 lipid kinase
(31). AMPK stimulates autophagy
or mitophagy by directly activating ULK1 through phosphorylation of
Ser317 and Ser777, and suppressing the mammalian target of
rapamycin complex 1, which inhibits autophagy by preventing ULK1
activation by phosphorylation Ulk1 Ser757 (32,33). In addition, another recent
investigation indicated that the AMPK-dependent phosphorylation of
ULK1 regulates the translocation of ULK1 to the mitochondria to
serve a critical role in the mitophagy response to hypoxic stress
(34).
In conclusion, chronic hypoxia induces the
activation of AMPK, which promotes selective mitophagy to maintain
mitochondrial quality and quantity for the cell. The present study
demonstrated that mitophagy induced by AMPK activation may play a
significant role in cardiac protection while cardiomyocytes undergo
chronic hypoxia. This study provides a possible mechanism for
metabolic adaptation and cardioprotection in response to chronic
hypoxia; therefore, it is worthy of clinical exploration as a
therapeutic strategy for heart disease caused by chronic hypoxia,
including cyanotic heart disease, high-altitude heart disease and
chronic cor pulmonale. The mechanism underlying AMPK-mediated
mitophagy requires future investigation.
Acknowledgments
This study was supported by the National Natural and
Science Foundation of China (grant no. 81270228).
References
|
1
|
Schaper J, Meiser E and Stammler G:
Ultrastructural morphometric analysis of myocardium from dogs,
rats, hamsters, mice, and from human hearts. Circ Res. 56:377–391.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vásquez-Trincado C, García-Carvajal I,
Pennanen C, Parra V, Hill JA, Rothermel BA and Lavandero S:
Mitochondrial dynamics, mitophagy and cardiovascular disease. J
Physiol. 594:509–525. 2016. View
Article : Google Scholar
|
|
3
|
Driscoll DJ: Evaluation of the cyanotic
newborn. Pediatr Clin North Am. 37:1–23. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kornosky JL and Salihu HM: Getting to the
heart of the matter: epidemiology of cyanotic heart defects.
Pediatr Cardiol. 29:484–497. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv
Z, Gu Z, Zhang J, Wang J, Zen K, et al: Hypoxia induces PGC-1α
expression and mitochondrial biogenesis in the myocardium of TOF
patients. Cell Res. 20:676–687. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fischer F, Hamann A and Osiewacz HD:
Mitochondrial quality control: an integrated network of pathways.
Trends Biochem Sci. 37:284–292. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Held NM and Houtkooper RH: Mitochondrial
quality control pathways as determinants of metabolic health.
BioEssays. 37:867–876. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu H and Chen Q: Hypoxia activation of
mitophagy and its role in disease pathogenesis. Antioxid Redox
Signal. 22:1032–1046. 2015. View Article : Google Scholar
|
|
10
|
Hardie DG and Sakamoto K: AMPK: a key
sensor of fuel and energy status in skeletal muscle. Physiology
(Bethesda). 21:48–60. 2006.
|
|
11
|
Liu L, Cash TP, Jones RG, Keith B,
Thompson CB and Simon MC: Hypoxia-induced energy stress regulates
mRNA translation and cell growth. Mol Cell. 21:521–531. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mungai PT, Waypa GB, Jairaman A, Prakriya
M, Dokic D, Ball MK and Schumacker PT: Hypoxia triggers AMPK
activation through reactive oxygen species-mediated activation of
calcium release-activated calcium channels. Mol Cell Biol.
31:3531–3545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bujak AL, Crane JD, Lally JS, Ford RJ,
Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD, et
al: AMPK activation of muscle autophagy prevents fasting-induced
hypoglycemia and myopathy during aging. Cell Metab. 21:883–890.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jian Z, Li JB, Ma RY, Chen L, Zhong QJ,
Wang XF, Wang W, Hong Y and Xiao YB: Increase of macrophage
migration inhibitory factor (MIF) expression in cardiomyocytes
during chronic hypoxia. Clin Chim Acta. 405:132–138. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Webster KA: Mitochondrial membrane
permeabilization and cell death during myocardial infarction: roles
of calcium and reactive oxygen species. Future Cardiol. 8:863–884.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gustafsson AB and Gottlieb RA: Heart
mitochondria: gates of life and death. Cardiovasc Res. 77:334–343.
2008. View Article : Google Scholar
|
|
19
|
Chiong M, Wang ZV, Pedrozo Z, Cao DJ,
Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA and
Lavandero S: Cardiomyocyte death: mechanisms and translational
implications. Cell Death Dis. 2:e2442011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H and Chan DC: Mitochondrial dynamics
- fusion, fission, movement, and mitophagy - in neurodegenerative
diseases. Hum Mol Genet. 18:R169–R176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mishra P and Chan DC: Mitochondrial
dynamics and inheritance during cell division, development and
disease. Nat Rev Mol Cell Biol. 15:634–646. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011. View Article : Google Scholar
|
|
24
|
Wang S, Song P and Zou MH: AMP-activated
protein kinase, stress responses and cardiovascular diseases. Clin
Sci (Lond). 122:555–573. 2012. View Article : Google Scholar
|
|
25
|
Roach PJ: AMPK -> ULK1 -> autophagy.
Mol Cell Biol. 31:3082–3084. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Young LH: AMP-activated protein kinase
conducts the ischemic stress response orchestra. Circulation.
117:832–840. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Russell RR 3rd, Li J, Coven DL, Pypaert M,
Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ and Young LH:
AMP-activated protein kinase mediates ischemic glucose uptake and
prevents postischemic cardiac dysfunction, apoptosis, and injury. J
Clin Invest. 114:495–503. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Semenza GL: Mitochondrial autophagy: life
and breath of the cell. Autophagy. 4:534–536. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ikeda Y, Shirakabe A, Maejima Y, Zhai P,
Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et
al: Endogenous Drp1 mediates mitochondrial autophagy and protects
the heart against energy stress. Circ Res. 116:264–278. 2015.
View Article : Google Scholar
|
|
31
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tian W, Li W, Chen Y, Yan Z, Huang X,
Zhuang H, Zhong W, Chen Y, Wu W, Lin C, et al: Phosphorylation of
ULK1 by AMPK regulates translocation of ULK1 to mitochondria and
mitophagy. FEBS Lett. 589:1847–1854. 2015. View Article : Google Scholar : PubMed/NCBI
|