Parkinson's disease (PD) is a common
neurodegenerative disorder, characterized by the progressive
degeneration and death of dopaminergic neurons, and by the
expression of Lewy bodies in the surviving neurons of the
substantia nigra (SN) (1). The
exact mechanism of dopaminergic neuron damage remains poorly
understood, but several lines of evidence implicate mitochondrial
dysfunction as a possible primary cause for the cell death in PD
(2–5). Mitochondria are pleiotropic
organelles affecting a wide range of molecular and cellular
processes. Besides generation of cellular energy in the form of
adenosine triphosphate (ATP) by oxidative phosphorylation, they
play a crucial role in calcium homeostasis, reactive oxygen species
(ROS) production and intrinsic cell death pathway regulation, thus
determining cell survival and death (2,5,6).
Mitochondria are therefore vital for normal cellular function,
including intracellular metabolic activities and signal
transduction of various cellular pathways. Mitochondrial
dysfunction has been considered as a central event and primary
initiator responsible for the progressive loss of dopaminergic
neurons in PD (6–8). A better understanding of the
molecular mechanisms underlying the pathogenesis will provide
potential targets acting as blocking or reversing factors to limit
PD progression.
Mitochondria are important cytoplasmic organelles
with double lipid membranes: The outer mitochondrial membrane (OMM)
and the inner mitochondrial membrane (IMM). The elaborate
structures of the mitochondria are required for the maintenance of
their normal functions (9). The
OMM contains numerous channels formed by the protein, which allow
the passage of ions and low molecular-weight substances from the
relatively permeable membrane (10). The voltage-dependent anion channel
(VDAC) is essential for the exchange of metabolites between the
cytosol and intermembrane space bordered by the OMM and the IMM
(11). A large hydrophilic pore
of the VDAC provides a structural device for the translocation of
ions and a variety of metabolites, including ATP and adenosine
diphosphate (12). The OMM is
involved in mitochondria-mediated cell death via various different
pathways through the interaction with a number of regulator
proteins. Bcl-2-associated X protein (Bax), for example,
translocates from the cytosol into the OMM in response to the
apoptotic signals, subsequently oligomerizing at the OMM and
promoting the release of apoptotic factors cytochrome c and
apoptosis-inducing factor, and other pro-apoptotic mediators
(13).
The IMM is a convoluted structure formed by a large
number of infoldings known as mitochondrial cristae, and is almost
impermeable, thus providing an efficient barrier to form an
electron gradient and a relatively closed inner matrix for the
electron transport chain (ETC), which is required for oxidative
phosphorylation in ATP formation (14). The specialized cation transporters
and exchangers mediate the cation transmembrane fluxes that are
essential for the maintenance of mitochondrial bioactivities.
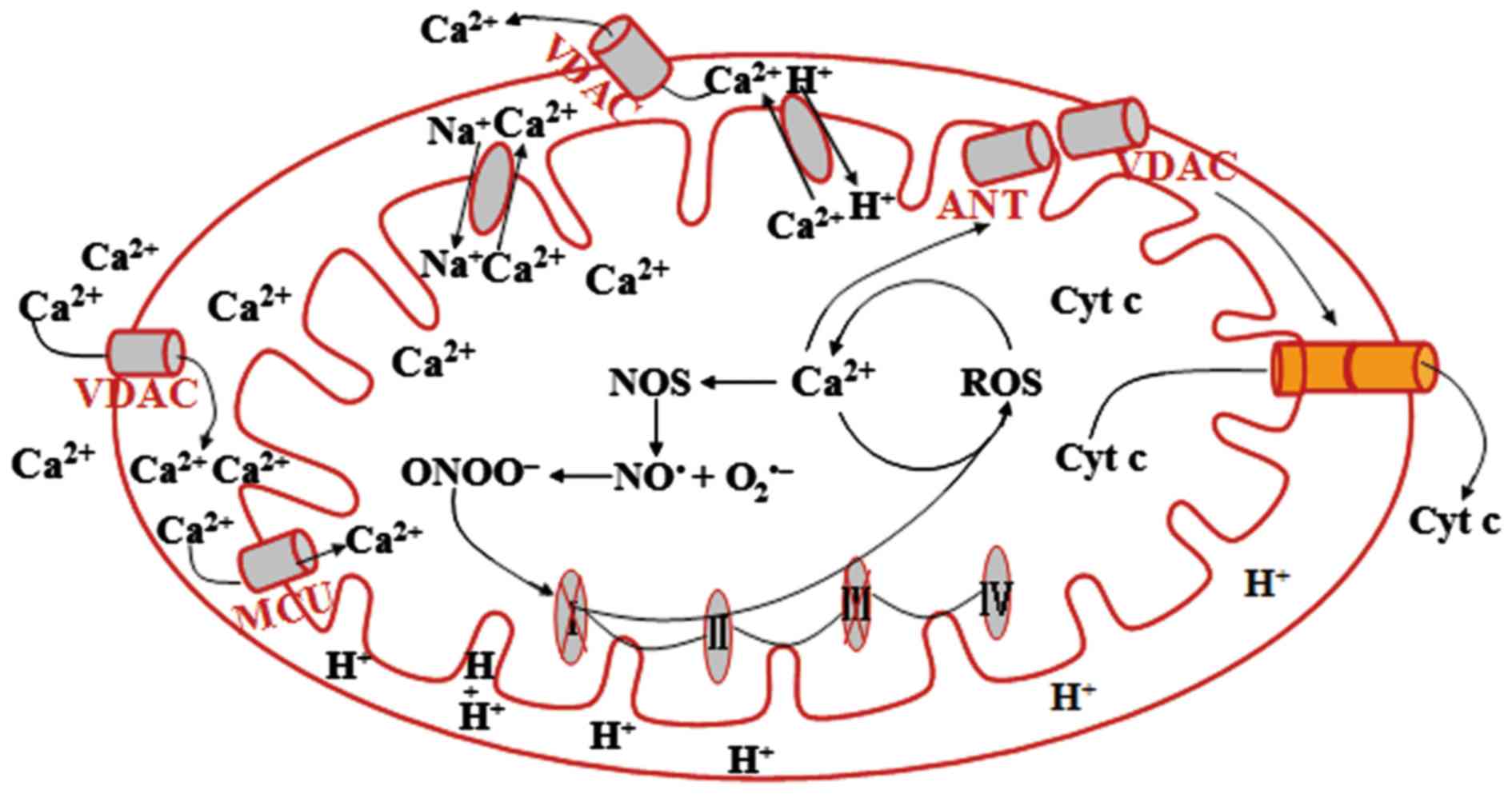
Mitochondrial Ca2+ uniporter (MCU) is a specific
transport system for Ca2+ intake across the IMM, thus
playing a role in buffering cytosolic Ca2+. The
maintenance of physiologically relevant free Ca2+ is
required for normal mitochondrial functions, but overload
contributes to the opening of the mitochondrial permeability
transition pore (mPTP) and matrix swelling, and subsequently cell
death (15). The extrusion of
mitochondrial Ca2+ depends mainly on the
Na+-dependent Na+-Ca2+ exchanger
and the H+-Ca2+ exchanger (16, 17). The Na+-H+
exchanger is a mitochondrial channel that contributes to the
maintenance of intracellular pH, which is required for
mitochondrial membrane potential formation (14).
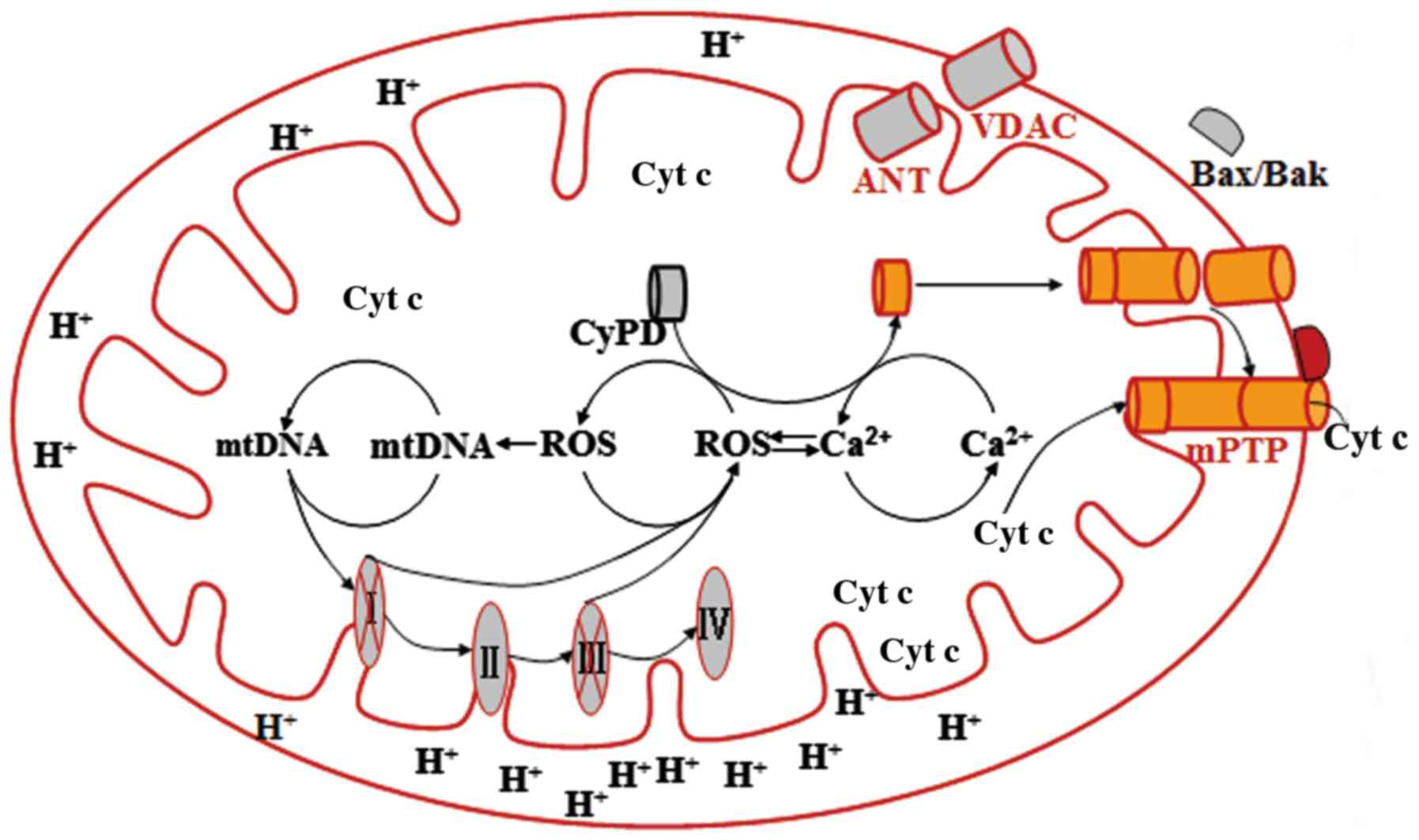
The mPTP is a transmembrane channel formed at the
contact sites between the OMM and the IMM. Although the components
of the mPTP remain controversial, the VDAC, the adenine nucleotide
translocator (ANT) and cyclophilin D (CyPD) appear to be implicated
(18,19). The mitochondrial matrix protein
CyPD mediates direct connection of the IMM protein ANT with the
VDAC by binding to a proline residue in ANT (20). The binding results in a
conformation of ANT that converts it into a non-specific pore
(21). Mitochondrial
Ca2+ overload and excessive ROS production may be key
inducers in the trans-location of CyPD from the matrix to IMM,
since each plays a crucial role in mPTP opening (13).
The ETC is a multisubunit complex that is required
for the production of ATP via oxidative phosphorylation. Synthesis
of ETC proteins depends on mitochondrial DNA (mtDNA) and nuclear
DNA. mtDNA is double stranded and encodes for 22 transfer RNAs
(tRNAs), 2 ribosomal RNAs and 13 polypeptides that are all subunits
of respiratory chain complexes (22). Energy production is the most
important function for mitochondria, and mitochondrial ATP is
generated via oxidative phosphorylation within the IMM (23). Mitochondria are a major source of
ROS, which are produced at the sites of the ETC (24,25). During oxidative phosphorylation,
the respiratory chain complexes I and III leak electrons to oxygen,
producing primarily superoxide radicals, and subsequently hydrogen
peroxide (H2O2) and hydroxyl radicals
(26,27). Overall, mitochondria participate
in energy metabolism, ROS production, calcium homeostasis, the
stress response and programmed cell death, thus determining cell
survival and death.
Mitochondria are primary intracellular source of ROS
that are generated from the interaction of unpaired electrons with
molecular O2 during oxidative phosphorylation (28,29). Respiratory chain complexes I and
III are the major sites of ROS production (26,30,31). The first generated ROS is
O2−, an amphibolic radical that cannot easily
pass through biological membranes and is converted by the
mitochondrial matrix enzyme to form H2O2 in
the mitochondrial intermembrane space and cytosol (32). H2O2 is a
stable molecule that can diffuse from the mitochondria into the
cellular cytosol and nucleus, where it can be detoxified by
glutathione peroxidase and catalase into water (33). However, when the balance of
H2O2 production and antioxidant defense is
perturbed, excessive H2O2 is accumulated and
leads to oxidative stress (34).
Particularly in the presence of reduced metals ferrous iron
(Fe2+), via the Fenton reaction,
H2O2 can easily be converted into the highly
reactive hydroxyl radical, causing further oxidative damage
(35). It is widely accepted that
complex I inhibition is a leading cause of increased ROS formation
(34). This production of ROS
damages in turn the components of the respiratory chain,
particularly complex I, leading to its further inhibition and more
ROS production. The vicious circle formed between mitochondrial
impairment and oxidative stress has been implicated in PD
pathogenesis, and may cause the progressive degeneration of
dopaminergic neurons by triggering sequential downstream events in
neurodegenerative conditions (34,36,37). Nigral dopaminergic neurons are
frequently exposed to oxidative stress as they contain high levels
of lipids and iron, and exhibit increased dopamine metabolism.
Consequently, gradual damage to the dopaminergic neurons occurs due
to attack by more ROS (37).
Accumulating evidence supports the link of oxidative damage and
degeneration of dopaminergic neurons in PD pathogenesis (38–41). Studies in postmortem brains of
patients with PD have shown increased levels of lipid oxidation
product 4-hydroxyl-2-nonenal (HNE), DNA and RNA oxidation products
8-hydroxy-deoxyguanosine and 8-hydroxy-guanosine, and carbonyl
modifications of soluble proteins, supporting the involvement of
oxidative stress in dopaminergic neuron damage (37,42). The neurotoxins
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rote-none and
6-hydroxydopamine are well-known parkinsonism inducers that cause
oxidative stress and dopaminergic neuron degeneration in animal
models, further supporting the contribution of oxidative stress to
PD pathogenesis (43–47). ROS play a major role in causing
oxidative stress and damage to all macromolecules, including
lipids, proteins and DNA (48).
Lipids participate in membrane fluidity and
permeability, and mediate inflammatory processes and apoptotic
signals, and oxidation is the mechanism responsible for the cell
damage and death (49). The brain
contains high levels of lipids, particular polyunsaturated fatty
acids, which are the most prone to lipid peroxidation and
responsible for the susceptibility of the organ to oxidative damage
(50). The exposure of
polyunsaturated fatty acids to free radicals leads to lipid
peroxidation which causes the structural damage of membranes,
compromising their integrity and consequently cell viability
(51). HNE is one of the most
important lipid peroxidation products, and has been considered as a
inducer responsible for apoptotic cell death via activation of the
caspase cascade and induction of DNA fragmentation (52). HNE can also exacerbate oxidative
damage by decreasing the levels of glutathione, the major
non-enzymatic antioxidant in the central nervous system (53). The elevated levels of lipid
peroxidation product HNE have been detected in the SN as well as
the cerebrospinal fluid of PD patients, supporting the
reinforcement of the hypothesis that peroxidation of
polyunsaturated fatty acids leads to dopaminergic neuron damage in
oxidative conditions (40,54).
Nigral dopaminergic neurons are particularly exposed
to the increased levels of oxidative stress due to increased
dopamine metabolism (55).
Dopamine is an unstable molecule that can easily undergo oxidative
metabolism to form dopamine quinones and free radicals. Normally,
the released dopamine is rapidly sequestered into vesicles via
vesicular monoamine transporter 2 (VMAT2). A defect in synaptic
vesicle formation or function leads to cytoplasmic accumulation of
dopamine (56). Inhibition of
VMAT2 causes a sustained increase in the formation of dopamine
autoxidation products in the cytoplasm, which reinforces the
oxidative damage of dopaminergic neurons (56). The levels of dopamine are
regulated by monoamine oxidase (MAO)-A and MAO-B, and the latter
appears to be a predominant enzyme to metabolize dopamine in
neuronal degeneration conditions (57,58). The metabolism of dopamine mediated
by MAO-B produces H2O2 that diffuses into
neighboring dopaminergic neurons where it can react with
Fe2+ to form hydroxyl radical, leading to further damage
to the neurons (59,60). The product of dopamine quinones,
aminochrome, can also contribute to neurodegeneration by promoting
superoxide generation, α-synuclein fibrillization formation and the
neuroinflammatory response (61,62).
Iron can also promote ROS generation and trigger
neurotoxicity in neurodegenerative conditions (63,64). As a cofactor for proteins, it is
distributed widely in neuronal tissue, particularly the SN
(65). However, with aging and
degenerative processes such as PD, there is an abnormal,
progressive deposition of iron and an increased free iron
concentration in the SN pars compacta (SNpc) (66). The increased levels of iron and
hyroxyl radicals have been detected in the SN of PD animal models,
while total glutathione (glutathione and glutathione disulfide)
levels were decreased (67).
Administration of the iron chelator desferrioxamine significantly
lowers iron levels in the brain and protects against iron and
MPTP-induced neurodegeneration in PD mouse models (68). These findings suggest that iron is
involved in the process of dopaminergic neuron degeneration in PD.
Little is known regarding whether an elevated iron level is a cause
or the consequence of neuron damage. However, it is widely accepted
that iron-induced toxicity is at least partly responsible for the
neuronal cell damage associated with PD (67–69).
Several recent lines of evidence have also
implicated reactive nitrogen species in dopaminergic neuronal
damage leading to PD pathogenesis (70). Peroxynitrite (ONOO−) is
a nitrating agent that acts as a strong oxidant and can damage
numerous cellular structures and alter their function, leading to
cell death (70). High levels of
ONOO− result in oxidative damage of mitochondrial lipid
and protein, inhibition of ETC, Ca2+ overload, and
subsequent mPTP opening and mitochondria-related pro-apoptotic
mediator release (70,71).
Taken together, these results show that dopaminergic
neurons in the SNpc are frequently subjected to oxidative damage
due to their high levels of lipids and iron, as well as increased
dopamine metabolism. Damage to the mitochondrial complexes and the
subsequent increase in ROS production are considered to be central
causative events responsible for the loss of dopaminergic neurons
in PD pathogenesis under neurogenerative conditions (72,73). Overproduction of ROS causes them
to attack macromolecules such as mtDNA and components of the ETC,
and causes mtDNA strand breaks, ETC damage and mitochondrial
Ca2+ overloading, leading to further production of ROS.
The vicious circle between ROS production and Ca2+
overload favors the sustained opening of mPTP by activation of CyPD
(13). Generally, CyPD is a
mitochondrial matrix protein that can be translocated to the IMM in
response to stimuli. Once located in the IMM, this protein
interacts with ANT and changes its conformation, which results in
the binding of ANT to VDAC and subsequently mPTP opening (21). Bcl-2 family proteins such as Bax
and Bcl-2 homologous killer can also facilitate the opening of the
mPTP by translocating and oligomerizing into the OMM as a
consequence of oxidative stress (74). The opening of the mPTP causes the
collapse of mitochondrial membrane potential and the release of
pro-apoptotic mediators from the mitochondria into the cytosol, and
subsequently cell death (75)
(Fig. 1).
Damage to the ETC and subsequent ROS production form
a positive feedback circle that may be a central event driving the
progressive loss of dopaminergic neurons under neurodegenerative
conditions. Therefore, restoring the function of the ETC and
inhibition of ROS production may be a promising method for PD
treatment. Coenzyme Q10 (CoQ10), for example, functioning as an
endogenous co-enzyme of ETC proteins and an ROS scavenger, plays a
crucial role in maintaining the integrity of mitochondrial
respiration and the clearance of free radicals (76). In vitro and in vivo
studies have shown that CoQ10 inhibits paraquat-, rotenone- and
MPTP-induced mitochondrial dysfunction, and subsequently, ROS
production (77–79). As an essential cofactor in the
mitochondrial ETC and a ROS scavenger, CoQ10, is currently being
investigated in clinical trials of PD.
The most important function of the mitochondria is
the generation of ATP through the process of oxidative
phosphorylation, which depends on mtDNA-encoded proteins. mtDNA
encodes 13 proteins that are all ETC complex subunits involved in
ATP production, and 2 RNA and 22 tRNAs required for mitochondrial
protein synthesis (103). Due to
the proximity to the ETC complexes and the source of ROS release,
mtDNA is frequently exposed to oxidative stress (104). The lack of histone protein
protection results in mtDNA mutation at a high rate relative to
nuclear NDA, particularly in cells with high energy demands
(105). A number of
mitochondria-related diseases could be linked to mutations of the
mitochondrial genome (104,106–108). mtDNA deletions that are closely
associated with the deficits in normal mitochondrial activity were
previously shown to accumulate in nigral dopaminergic neurons of
aged individuals and sporadic PD subjects (106–108). Knockout of the mtDNA
transcription gene inhibits the expression of mtDNA and the
activities in the respiratory chain of dopaminergic neurons,
accompanied with progressive parkinsonism in mouse models (109). High levels of mtDNA deletions in
the brains of retenone-induced PD models were observed in the
midbrain, but not in the cortex (110). However, somatic mtDNA deletions
were shown to be slightly higher in the SN of PD patients compared
with age-matched controls (107). These observations demonstrated
that mtDNA deletions are specific to nigral neurons, which may
increase their susceptibility to oxidative stress and hence
contribute to their selective loss in PD. mtDNA is vulnerable to
oxidative damage compared with nuclear DNA, since mitochondria are
the main sites of ROS production and lack protective histones
(103,111). mtDNA damage is associated with
the instability of genes and proteins, defect in the ETC and
increased ROS production, leading to compromised bioenergetic
functions and cell damage. A number of mitochondria-related
diseases could be linked to mutations of the mitochondrial genome
(104,106–108).
mtDNA has an autonomously replicating genome
encoding a spectrum of mitochondrial respiratory chain proteins
(112), and their deletions
result in mitochondrial respiratory chain dysfunction (107). The ability to synthesize and
repair mitochondrial genomes (mtDNA) is required for the
mitochondria to maintain their bioactivities, which rely mainly on
mtDNA polymerase. The polymerase γ1 (POLG1) enzyme is a
nuclear-encoded gene product that plays an important role in
polymerase synthesis and mtDNA maintenance (113). Mutations in POLG1 have been
shown to be associated with severe progressive multisystem
disorders, including PD, supporting the involvement of the
defective mtDNA in dopaminergic neuron degeneration (114–116). POLG1 mutations contribute to a
gradual accumulation of secondary deletions in mtDNA, resulting in
dysfunction of the respiratory chain (117). Recently, several studies have
shown that selective increased mtDNA damage marker abasic site
levels could be detected in the SN in postmortem brains of PD
patients. Abasic sites were also shown in brain tissues from mice
treated with neuronal toxins rotenone to induce PD-like pathology
(118). Abasic sites are
segments of DNA that have lost either a purine or a pyrimidine
base, resulting in blockage of the polymerase during DNA
replication and transcription (119). These studies demonstrated that
dopaminergic neuronal injury could be ascribed to mtDNA damage,
leading to ETC inhibition and mitochondrial dysfunction in PD
pathogenesis. Therefore, the inhibition of this mtDNA damage may be
beneficial for PD in neurodegenerative conditions.
Mitochondria are dynamic organelles with constant
changes of morphology that are regulated by mitochondrial fission
and fusion. The dynamic balance between fission and fusion is
essential for the normal function of the mitochondria and plays a
vital role in cellular bioactivities (120,121). Mitochondrial fission serves as a
mechanism to facilitate the equal segregation of the mitochondria
into daughter cells in mitochondrial division, and to target
damaged segments of the mitochondria in the autophagic process.
This process is controlled by dynamin-related protein 1 (Drp1), a
cytoplasmic dynamin GTPase that translocates to the mitochondria
and locates to the OMM in response to mitochondrial dysfunction
(122,123). Cytosolic Ca2+ is a
key mediator in its translocation via the activation of calcineurin
and the dephosphorylation of Ser-637 residue in Drp1 (124). Ser-637 of Drp1 can also be
phosphorylated by calmodulin kinase Iα, leading to its
mitochondrial translocation (125). The translocated Drp1 provides a
structural device for mechanical force in physical excision of the
membrane by formatting a multimeric complex around the OMM
(126). Mitochondrial fission 1
protein (Fis1) is another important regulator protein involved in
mitochondrial fission. Overexpression of Fis1 has been described to
activate the fission process, and results in mitochondrial
fragmentation (127). This
protein can also trigger autophagy to remove damaged mitochondria,
thus contributing to the maintenance of cellular functions
(128).
Mitochondrial fusion is another mechanism to
maintain mitochondrial bioactivities by mixing the contents of
mitochondria, thus enabling its protein complementation, mtDNA
repair and equal distribution of metabolites (129). This process depends on three
GTPase proteins: Mitofusin 1 (Mfn1), Mfn2 and optic atrophy protein
1 (OPA1) (130,131). Mfn1 and Mfn2 are mitochondrial
transmembrane proteins localized to the OMM, and they mediate the
outer-membrane connection of neighboring mitochondria by forming
homo-oligomeric and hetero-oligomeric complexes via interaction
with their coiled-coil domains (132–134). OPA1 is an inner membrane protein
mediating IMM fusion. Mitochondrial membrane potential is essential
for the connection of the OMM and the IMM, and for maintenance of
the mitochondrial tubular network (135,136). Defects in the ETC complexes and
damage to the mitochondrial membrane potential contribute to
mitochondrial network disintegration, leading to increased
fragmentation and cell death (137). Recent studies have suggested
that defects in mitochondrial fission and fusion may be one of the
underlying mechanisms responsible for mitochondria-mediated
neurodegenerative disease (13).
Mitochondrial fission and fragmentation can be observed in PD
cellular models induced by the neurotoxins rotenone and
1-methyl-4-phenylpyridinium in a rat dopaminergic cell line
(138). Depolarization of the
mitochondrial membrane causes the loss of OPA1 and the MFNs,
leading to the inhibition of fusion and mitochondrial fragmentation
(139). The discharge of
membrane potential is considered to be a critical factor associated
with mitochondrial dysfunction and cell death in neurodegenerative
diseases (44,140), indicating its involvement as one
of the mechanisms responsible for the degeneration of dopaminergic
neurons in PD pathogenesis.
Recent studies have linked Parkin and the
PTEN-induced putative kinase 1 (PINK1)/Parkin pathway with the
maintenance of mitochondrial dynamics (141,142). PINK1 is a mitochondrial kinase
and can be translocated to the OMM where it is cleaved rapidly by
presenilin-associated rhomboid-like protease (143). However, when mitochondria are
impaired and deplete of membrane potential, the protease activity
is inhibited, with the subsequent accumulation of PINK1 on the
mitochondrial membrane (143,144). The elevated levels of PINK1 on
the OMM trigger the translocation of Parkin to the mitochondria by
phosphorylating the protein at Thr-175 in a kinase
activity-dependent manner (145,146). PINK1/Parkin mediate
mitochondrial fusion and fission dependent on dynamin-like GTPases,
including Mfn1, Mfn2, OPA1 and Fis1, and the potential mechanisms
of the regulation have been well documented (6). Parkin can facilitate sequestration
and elimination of damaged mitochondria via mitophagy, a protective
mechanism for maintaining the recycling of proteins and
mitochondrial homeostasis to limit cell death (147). Together, the aforementioned
studies may provide a molecular target to alleviate the severity of
mitochondria-mediated dopaminergic neuron damage in PD
pathogenesis.
PD is characterized by the progressive degeneration
and death of dopaminergic neurons in the SN. The development of
molecular and cellular mechanisms associated with dopaminergic
neuron damage has implicated the involvement of a range of events
in PD pathogenesis, through distinct and divert pathways, to cause
progressive neuron damage. Although the precise mechanisms of
neuronal damage remain unclear, abnormalities of the mitochondria
appear to be a converging point in the cell death processes.
Understanding the mechanisms underlying mitochondria-mediated
neuron death may provide a promising management solution for PD
treatment, however, this requires further elucidation.
|
1
|
Forno LS: Neuropathology of Parkinson's
disease. J Neuropathol Exp Neurol. 55:259–272. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martin LJ: Biology of mitochondria in
neurodegenerative diseases. Prog Mol Biol Transl Sci. 107:355–415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trancikova A, Tsika E and Moore DJ:
Mitochondrial dysfunction in genetic animal models of Parkinson's
disease. Antioxid Redox Signal. 16:896–919. 2012. View Article : Google Scholar :
|
|
4
|
Ryan BJ, Hoek S, Fon EA and Wade-Martins
R: Mitochondrial dysfunction and mitophagy in Parkinson's: From
familial to sporadic disease. Trends Biochem Sci. 40:200–210. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moon HE and Paek SH: Mitochondrial
dysfunction in Parkinson's disease. Exp Neurobiol. 24:103–116.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Exner N, Lutz AK, Haass C and Winklhofer
KF: Mitochondrial dysfunction in Parkinson's disease: Molecular
mechanisms and pathophysiological consequences. EMBO J.
31:3038–3062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mounsey RB and Teismann P: Mitochondrial
dysfunction in Parkinson's disease: Pathogenesis and
neuroprotection. Parkinsons Dis. 2010:6174722011.
|
|
8
|
Martin LJ: Mitochondrial and cell death
mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel).
3:839–915. 2010. View Article : Google Scholar
|
|
9
|
Reddy PH and Reddy TP: Mitochondria as a
therapeutic target for aging and neurodegenerative diseases. Curr
Alzheimer Res. 8:393–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reddy PH and Beal MF: Are mitochondria
critical in the pathogenesis of Alzheimer's disease? Brain Res
Brain Res Rev. 49:618–632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rostovtseva TK, Tan W and Colombini M: On
the role of VDAC in apoptosis: fact and fiction. J Bioenerg
Biomembr. 37:129–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okada SF, O'Neal WK, Huang P, Nicholas RA,
Ostrowski LE, Craigen WJ, Lazarowski ER and Boucher RC:
Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP
release and cell volume regulation in murine cells. J Gen Physiol.
124:513–526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Camara AK, Lesnefsky EJ and Stowe DF:
Potential therapeutic benefits of strategies directed to
mitochondria. Antioxid Redox Signal. 13:279–347. 2010. View Article : Google Scholar :
|
|
14
|
Bernardi P: Mitochondrial transport of
cations: Channels, exchangers, and permeability transition. Physiol
Rev. 79:1127–1155. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Teshima Y, Akao M, Jones SP and Marbán E:
Uncoupling protein-2 overexpression inhibits mitochondrial death
pathway in cardiomyocytes. Circ Res. 93:192–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Rourke B: Mitochondrial ion channels.
Annu Rev Physiol. 69:19–49. 2007. View Article : Google Scholar
|
|
17
|
Wingrove DE and Gunter TE: Kinetics of
mitochondrial calcium transport. II A kinetic description of the
sodium-dependent calcium efflux mechanism of liver mitochondria and
inhibition by ruthenium red and by tetraphenylphosphonium. J Biol
Chem. 261:15166–15171. 1986.PubMed/NCBI
|
|
18
|
Bernardi P, Krauskopf A, Basso E,
Petronilli V, Blachly-Dyson E, Di Lisa F and Forte MA: The
mitochondrial permeability transition from in vitro artifact to
disease target. FEBS J. 273:2077–2099. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leung AW and Halestrap AP: Recent progress
in elucidating the molecular mechanism of the mitochondrial
permeability transition pore. Biochim Biophys Acta. 1777:946–952.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vyssokikh MY, Katz A, Rueck A, Wuensch C,
Dörner A, Zorov DB and Brdiczka D: Adenine nucleotide translocator
isoforms 1 and 2 are differently distributed in the mitochondrial
inner membrane and have distinct affinities to cyclophilin D.
Biochem J. 358:349–358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Halestrap AP and Brenner C: The adenine
nucleotide translocase: A central component of the mitochondrial
permeability transition pore and key player in cell death. Curr Med
Chem. 10:1507–1525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ojala D, Montoya J and Attardi G: tRNA
punctuation model of RNA processing in human mitochondria. Nature.
290:470–474. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reddy PH: Amyloid precursor
protein-mediated free radicals and oxidative damage: Implications
for the development and progression of Alzheimer's disease. J
Neurochem. 96:1–13. 2006. View Article : Google Scholar
|
|
24
|
Brookes PS, Levonen AL, Shiva S, Sarti P
and Darley-Usmar VM: Mitochondria: Regulators of signal
transduction by reactive oxygen and nitrogen species. Free Radic
Biol Med. 33:755–764. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dröge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kudin AP, Bimpong-Buta NY, Vielhaber S,
Elger CE and Kunz WS: Characterization of superoxide-producing
sites in isolated brain mitochondria. J Biol Chem. 279:4127–4135.
2004. View Article : Google Scholar
|
|
27
|
Brand MD, Affourtit C, Esteves TC, Green
K, Lambert AJ, Miwa S, Pakay JL and Parker N: Mitochondrial
superoxide: Production, biological effects, and activation of
uncoupling proteins. Free Radic Biol Med. 37:755–767. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar
|
|
29
|
Cadenas E and Davies KJ: Mitochondrial
free radical generation, oxidative stress, and aging. Free Radic
Biol Med. 29:222–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kudin AP, Debska-Vielhaber G and Kunz WS:
Characterization of superoxide production sites in isolated rat
brain and skeletal muscle mitochondria. Biomed Pharmacother.
59:163–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kussmaul L and Hirst J: The mechanism of
superoxide production by NADH:ubiquinone oxidoreductase (complex I)
from bovine heart mitochondria. Proc Natl Acad Sci USA.
103:7607–7612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rush JD and Koppenol WH: Oxidizing
intermediates in the reaction of ferrous EDTA with hydrogen
peroxide. Reactions with organic molecules and ferrocytochrome c. J
Biol Chem. 261:6730–6733. 1986.PubMed/NCBI
|
|
33
|
Antunes F, Han D and Cadenas E: Relative
contributions of heart mitochondria glutathione peroxidase and
catalase to H(2)O(2) detoxification in in vivo conditions. Free
Radic Biol Med. 33:1260–1267. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morán M, Moreno-Lastres D, Marín-Buera L,
Arenas J, Martín MA and Ugalde C: Mitochondrial respiratory chain
dysfunction: Implications in neurodegeneration. Free Radic Biol
Med. 53:595–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gutteridge JM: Superoxide-dependent
formation of hydroxyl radicals from ferric-complexes and hydrogen
peroxide: An evaluation of fourteen iron chelators. Free Radic Res
Commun. 9:119–125. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hwang O: Role of oxidative stress in
Parkinson's disease. Exp Neurobiol. 22:11–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
38
|
Jenner P: Oxidative stress in Parkinson's
disease. Ann Neurol. 53(Suppl 3): S26–S38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alam ZI, Jenner A, Daniel SE, Lees AJ,
Cairns N, Marsden CD, Jenner P and Halliwell B: Oxidative DNA
damage in the parkinsonian brain: An apparent selective increase in
8-hydroxyguanine levels in substantia nigra. J Neurochem.
69:1196–1203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yoritaka A, Hattori N, Uchida K, Tanaka M,
Stadtman ER and Mizuno Y: Immunohistochemical detection of
4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl
Acad Sci USA. 93:2696–2701. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li DW, Yao M, Dong YH, Tang MN, Chen W, Li
GR and Sun BQ: Guanosine exerts neuroprotective effects by
reversing mitochondrial dysfunction in a cellular model of
Parkinson's disease. Int J Mol Med. 34:1358–1364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM,
Chong WL, Looi WF, Huang SH, Wang H and Chan YH: Oxidative damage
in Parkinson disease: Measurement using accurate biomarkers. Free
Radic Biol Med. 48:560–566. 2010. View Article : Google Scholar
|
|
43
|
Callio J, Oury TD and Chu CT: Manganese
superoxide dismutase protects against 6-hydroxydopamine injury in
mouse brains. J Biol Chem. 280:18536–18542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vila M and Przedborski S: Targeting
programmed cell death in neurodegenerative diseases. Nat Rev
Neurosci. 4:365–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Perier C, Bové J, Vila M and Przedborski
S: The rotenone model of Parkinson's disease. Trends Neurosci.
26:345–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun SY, An CN and Pu XP: DJ-1 protein
protects dopaminergic neurons against 6-OHDA/MG-132-induced
neurotoxicity in rats. Brain Res Bull. 88:609–616. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Heikkila RE, Hess A and Duvoisin RC:
Dopaminergic neurotoxicity of
1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science.
224:1451–1453. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Barzilai A and Yamamoto K: DNA damage
responses to oxidative stress. DNA Repair (Amst). 3:1109–1115.
2004. View Article : Google Scholar
|
|
49
|
Ruipérez V, Darios F and Davletov B:
Alpha-synuclein, lipids and Parkinson's disease. Prog Lipid Res.
49:420–428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mariani E, Polidori MC, Cherubini A and
Mecocci P: Oxidative stress in brain aging, neurodegenerative and
vascular diseases: An overview. J Chromatogr B Analyt Technol
Biomed Life Sci. 827:65–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Montine TJ, Neely MD, Quinn JF, Beal MF,
Markesbery WR, Roberts LJ II and Morrow JD: Lipid peroxidation in
aging brain and Alzheimer's disease. Free Radic Biol Med.
33:620–626. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu W, Kato M, Akhand AA, Hayakawa A,
Suzuki H, Miyata T, Kurokawa K, Hotta Y, Ishikawa N and Nakashima
I: 4-hydroxynonenal induces a cellular redox status-related
activation of the caspase cascade for apoptotic cell death. J Cell
Sci. 113:635–641. 2000.PubMed/NCBI
|
|
53
|
Schmidt H, Grune T, Müller R, Siems WG and
Wauer RR: Increased levels of lipid peroxidation products
malondialdehyde and 4-hydroxynonenal after perinatal hypoxia.
Pediatr Res. 40:15–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Montine KS, Quinn JF, Zhang J, Fessel JP,
Roberts LJ II, Morrow JD and Montine TJ: Isoprostanes and related
products of lipid peroxidation in neurodegenerative diseases. Chem
Phys Lipids. 128:117–124. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lotharius J and Brundin P: Pathogenesis of
Parkinson's disease: Dopamine, vesicles and alpha-synuclein. Nat
Rev Neurosci. 3:932–942. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fornstedt B and Carlsson A: A marked rise
in 5-S-cysteinyl-dopamine levels in guinea-pig striatum following
reserpine treatment. J Neural Transm. 76:155–161. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Youdim MB, Edmondson D and Tipton KF: The
therapeutic potential of monoamine oxidase inhibitors. Nat Rev
Neurosci. 7:295–309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fowler JS, Volkow ND, Wang GJ, Logan J,
Pappas N, Shea C and MacGregor R: Age-related increases in brain
monoamine oxidase B in living healthy human subjects. Neurobiol
Aging. 18:431–435. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nagatsu T and Sawada M: Molecular
mechanism of the relation of monoamine oxidase B and its inhibitors
to Parkinson's disease: Possible implications of glial cells. J
Neural Transm Suppl. 71:53–65. 2006. View Article : Google Scholar
|
|
60
|
Kumar MJ and Andersen JK: Perspectives on
MAO-B in aging and neurological disease: Where do we go from here?
Mol Neurobiol. 30:77–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Norris EH, Giasson BI, Hodara R, Xu S,
Trojanowski JQ, Ischiropoulos H and Lee VM: Reversible inhibition
of alpha-synuclein fibrillization by dopaminochrome-mediated
conformational alterations. J Biol Chem. 280:21212–21219. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zecca L, Wilms H, Geick S, Claasen JH,
Brandenburg LO, Holzknecht C, Panizza ML, Zucca FA, Deuschl G,
Sievers J, et al: Human neuromelanin induces neuroinflammation and
neurodegeneration in the rat substantia nigra: Implications for
Parkinson's disease. Acta Neuropathol. 116:47–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jomova K and Valko M: Advances in
metal-induced oxidative stress and human disease. Toxicology.
283:65–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Núñez MT, Urrutia P, Mena N, Aguirre P,
Tapia V and Salazar J: Iron toxicity in neurodegeneration.
Biometals. 25:761–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sadrzadeh SM and Saffari Y: Iron and brain
disorders. Am J Clin Pathol. 121(Suppl): S64–S70. 2004.PubMed/NCBI
|
|
66
|
Sian-Hülsmann J, Mandel S, Youdim MB and
Riederer P: The relevance of iron in the pathogenesis of
Parkinson's disease. J Neurochem. 118:939–957. 2011. View Article : Google Scholar
|
|
67
|
Sziráki I, Mohanakumar KP, Rauhala P, Kim
HG, Yeh KJ and Chiueh CC: Manganese: A transition metal protects
nigrostriatal neurons from oxidative stress in the iron-induced
animal model of parkinsonism. Neuroscience. 85:1101–1111. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lan J and Jiang DH: Desferrioxamine and
vitamin E protect against iron and MPTP-induced neurodegeneration
in mice. J Neural Transm Vienna. 104:469–481. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Faucheux BA, Martin ME, Beaumont C, Hauw
JJ, Agid Y and Hirsch EC: Neuromelanin associated redox-active iron
is increased in the substantia nigra of patients with Parkinson's
disease. J Neurochem. 86:1142–1148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yokoyama H, Kuroiwa H, Yano R and Araki T:
Targeting reactive oxygen species, reactive nitrogen species and
inflammation in MPTP neurotoxicity and Parkinson's disease. Neurol
Sci. 29:293–301. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Duchen MR: Mitochondria in health and
disease: Perspectives on a new mitochondrial biology. Mol Aspects
Med. 25:365–451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Henchcliffe C and Beal MF: Mitochondrial
biology and oxidative stress in Parkinson disease pathogenesis. Nat
Clin Pract Neurol. 4:600–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mythri RB, Jagatha B, Pradhan N, Andersen
J and Bharath MM: Mitochondrial complex I inhibition in Parkinson's
disease: How can curcumin protect mitochondria? Antioxid Redox
Signal. 9:399–408. 2007. View Article : Google Scholar
|
|
74
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Crompton M: The mitochondrial permeability
transition pore and its role in cell death. Biochem J. 341:233–249.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Burchell VS, Gandhi S, Deas E, Wood NW,
Abramov AY and Plun-Favreau H: Targeting mitochondrial dysfunction
in neurodegenerative disease: Part I. Expert Opin Ther Targets.
14:369–385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Moon Y, Lee KH, Park JH, Geum D and Kim K:
Mitochondrial membrane depolarization and the selective death of
dopaminergic neurons by rotenone: Protective effect of coenzyme
Q10. J Neurochem. 93:1199–1208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
McCarthy S, Somayajulu M, Sikorska M,
Borowy-Borowski H and Pandey S: Paraquat induces oxidative stress
and neuronal cell death; neuroprotection by water-soluble Coenzyme
Q10. Toxicol Appl Pharmacol. 201:21–31. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cleren C, Yang L, Lorenzo B, Calingasan
NY, Schomer A, Sireci A, Wille EJ and Beal MF: Therapeutic effects
of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of
Parkinsonism. J Neurochem. 104:1613–1621. 2008. View Article : Google Scholar
|
|
80
|
Dubois C, Prevarskaya N and Vanden Abeele
F: The calcium-signaling toolkit: Updates needed. Biochim Biophys
Acta. 1863:1337–1343. 2016. View Article : Google Scholar
|
|
81
|
Santo-Domingo J, Wiederkehr A and De
Marchi U: Modulation of the matrix redox signaling by mitochondrial
Ca(2). World J Biol Chem. 6:310–323. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nicholls DG: Mitochondrial function and
dysfunction in the cell: Its relevance to aging and aging-related
disease. Int J Biochem Cell Biol. 34:1372–1381. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kirichok Y, Krapivinsky G and Clapham DE:
The mitochondrial calcium uniporter is a highly selective ion
channel. Nature. 427:360–364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gincel D, Zaid H and Shoshan-Barmatz V:
Calcium binding and translocation by the voltage-dependent anion
channel: A possible regulatory mechanism in mitochondrial function.
Biochem J. 358:147–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Takeuchi A, Kim B and Matsuoka S: The
destiny of Ca(2+) released by mitochondria. J Physiol Sci.
65:11–24. 2015. View Article : Google Scholar
|
|
86
|
Plovanich M, Bogorad RL, Sancak Y, Kamer
KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J,
Speciner L, et al: MICU2, a paralog of MICU1, resides within the
mitochondrial uniporter complex to regulate calcium handling. PLoS
One. 8:e557852013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Perocchi F, Gohil VM, Girgis HS, Bao XR,
McCombs JE, Palmer AE and Mootha VK: MICU1 encodes a mitochondrial
EF hand protein required for Ca(2+) uptake. Nature. 467:291–296.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
McCormack JG and Denton RM: Mitochondrial
Ca2+ transport and the role of intramitochondrial
Ca2+ in the regulation of energy metabolism. Dev
Neurosci. 15:165–173. 1993. View Article : Google Scholar
|
|
89
|
Balaban RS: Cardiac energy metabolism
homeostasis: Role of cytosolic calcium. J Mol Cell Cardiol.
34:1259–1271. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Alderton WK, Cooper CE and Knowles RG:
Nitric oxide synthases: Structure, function and inhibition. Biochem
J. 357:593–615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Jekabsone A, Ivanoviene L, Brown GC and
Borutaite V: Nitric oxide and calcium together inactivate
mitochondrial complex I and induce cytochrome c release. J Mol Cell
Cardiol. 35:803–809. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Muravchick S and Levy RJ: Clinical
implications of mitochondrial dysfunction. Anesthesiology.
105:819–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
O'Rourke B: Pathophysiological and
protective roles of mitochondrial ion channels. J Physiol.
529:23–36. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Di Lisa F and Bernardi P: A CaPful of
mechanisms regulating the mitochondrial permeability transition. J
Mol Cell Cardiol. 46:775–780. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Jones SP, Teshima Y, Akao M and Marbán E:
Simvastatin attenuates oxidant-induced mitochondrial dysfunction in
cardiac myocytes. Circ Res. 93:697–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Celardo I, Martins LM and Gandhi S:
Unravelling mitochondrial pathways to Parkinson's disease. Br J
Pharmacol. 171:1943–1957. 2014. View Article : Google Scholar :
|
|
98
|
Surmeier DJ, Guzman JN, Sanchez-Padilla J
and Goldberg JA: The origins of oxidant stress in Parkinson's
disease and therapeutic strategies. Antioxid Redox Signal.
14:1289–1301. 2011. View Article : Google Scholar :
|
|
99
|
Perier C, Tieu K, Guégan C, Caspersen C,
Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S
and Vila M: Complex I deficiency primes Bax-dependent neuronal
apoptosis through mitochondrial oxidative damage. Proc Natl Acad
Sci USA. 102:19126–19131. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar
|
|
102
|
Javadov S, Choi A, Rajapurohitam V, Zeidan
A, Basnakian AG and Karmazyn M: NHE-1 inhibition-induced
cardioprotection against ischaemia/reperfusion is associated with
attenuation of the mitochondrial permeability transition.
Cardiovasc Res. 77:416–424. 2008. View Article : Google Scholar
|
|
103
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Yang JL, Weissman L, Bohr VA and Mattson
MP: Mitochondrial DNA damage and repair in neurodegenerative
disorders. DNA Repair (Amst). 7:1110–1120. 2008. View Article : Google Scholar
|
|
105
|
Levy RJ and Deutschman CS: Deficient
mitochondrial biogenesis in critical illness: Cause, effect, or
epiphenomenon? Crit Care. 11:1582007. View
Article : Google Scholar : PubMed/NCBI
|
|
106
|
Elstner M, Müller SK, Leidolt L, Laub C,
Krieg L, Schlaudraff F, Liss B, Morris C, Turnbull DM, Masliah E,
et al: Neuromelanin, neurotransmitter status and brainstem location
determine the differential vulnerability of catecholaminergic
neurons to mitochondrial DNA deletions. Mol Brain. 4:432011.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kraytsberg Y, Kudryavtseva E, McKee AC,
Geula C, Kowall NW and Khrapko K: Mitochondrial DNA deletions are
abundant and cause functional impairment in aged human substantia
nigra neurons. Nat Genet. 38:518–520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bender A, Krishnan KJ, Morris CM, Taylor
GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock
T, et al: High levels of mitochondrial DNA deletions in substantia
nigra neurons in aging and Parkinson disease. Nat Genet.
38:515–517. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ekstrand MI, Terzioglu M, Galter D, Zhu S,
Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS,
Trifunovic A, et al: Progressive parkinsonism in mice with
respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci
USA. 104:1325–1330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tanner CM, Kamel F, Ross GW, Hoppin JA,
Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR,
et al: Rotenone, paraquat, and Parkinson's disease. Environ Health
Perspect. 119:866–872. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Halliwell B: Role of free radicals in the
neurodegenerative diseases: Therapeutic implications for
antioxidant treatment. Drugs Aging. 18:685–716. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Reeve AK, Krishnan KJ and Turnbull D:
Mitochondrial DNA mutations in disease, aging, and
neurodegeneration. Ann NY Acad Sci. 1147:21–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ropp PA and Copeland WC: Cloning and
characterization of the human mitochondrial DNA polymerase, DNA
polymerase gamma. Genomics. 36:449–458. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Luoma P, Melberg A, Rinne JO, Kaukonen JA,
Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L,
Majamaa K, et al: Parkinsonism, premature menopause, and
mitochondrial DNA polymerase gamma mutations: Clinical and
molecular genetic study. Lancet. 364:875–882. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wong LJ, Naviaux RK, Brunetti-Pierri N,
Zhang Q, Schmitt ES, Truong C, Milone M, Cohen BH, Wical B, Ganesh
J, et al: Molecular and clinical genetics of mitochondrial diseases
due to POLG mutations. Hum Mutat. 29:E150–E172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Gui YX, Xu ZP, Lv W, Zhao JJ and Hu XY:
Evidence for polymerase gamma, POLG1 variation in reduced
mitochondrial DNA copy number in Parkinson's disease. Parkinsonism
Relat Disord. 21:282–286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hudson G and Chinnery PF: Mitochondrial
DNA polymerase-gamma and human disease. Hum Mol Genet.
15:R244–R252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Sanders LH, McCoy J, Hu X, Mastroberardino
PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B and Greenamyre JT:
Mitochondrial DNA damage: Molecular marker of vulnerable nigral
neurons in Parkinson's disease. Neurobiol Dis. 70:214–223. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wilson DM III and Barsky D: The major
human abasic endonuclease: Formation, consequences and repair of
abasic lesions in DNA. Mutat Res. 485:283–307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Benard G and Karbowski M: Mitochondrial
fusion and division: Regulation and role in cell viability. Semin
Cell Dev Biol. 20:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Soubannier V and McBride HM: Positioning
mitochondrial plasticity within cellular signaling cascades.
Biochim Biophys Acta. 1793:154–170. 2009. View Article : Google Scholar
|
|
122
|
Schrader M: Shared components of
mitochondrial and peroxisomal division. Biochim Biophys Acta.
1763:531–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ishihara N, Jofuku A, Eura Y and Mihara K:
Regulation of mitochondrial morphology by membrane potential, and
DRP1-dependent division and FZO1-dependent fusion reaction in
mammalian cells. Biochem Biophys Res Commun. 301:891–898. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Cereghetti GM, Stangherlin A, Martins de
Brito O, Chang CR, Blackstone C, Bernardi P and Scorrano L:
Dephosphorylation by calcineurin regulates translocation of Drp1 to
mitochondria. Proc Natl Acad Sci USA. 105:15803–15808. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y,
Tomizawa K, Nairn AC, Takei K, Matsui H and Matsushita M: CaM
kinase I alpha-induced phosphorylation of Drp1 regulates
mitochondrial morphology. J Cell Biol. 182:573–585. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Reddy PH, Reddy TP, Manczak M, Calkins MJ,
Shirendeb U and Mao P: Dynamin-related protein 1 and mitochondrial
fragmentation in neurodegenerative diseases. Brain Res Brain Res
Rev. 67:103–118. 2011. View Article : Google Scholar
|
|
127
|
James DI, Parone PA, Mattenberger Y and
Martinou JC: hFis1, a novel component of the mammalian
mitochondrial fission machinery. J Biol Chem. 278:36373–36379.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gomes LC and Scorrano L: High levels of
Fis1, a pro-fission mitochondrial protein, trigger autophagy.
Biochim Biophys Acta. 1777:860–866. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Santos D and Cardoso SM: Mitochondrial
dynamics and neuronal fate in Parkinson's disease. Mitochondrion.
12:428–437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Reddy PH: Amyloid beta, mitochondrial
structural and functional dynamics in Alzheimer's disease. Exp
Neurol. 218:286–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Reddy PH: Mitochondrial dysfunction in
aging and Alzheimer's disease: Strategies to protect neurons.
Antioxid Redox Signal. 9:1647–1658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chen H, Chomyn A and Chan DC: Disruption
of fusion results in mitochondrial heterogeneity and dysfunction. J
Biol Chem. 280:26185–26192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ishihara N, Eura Y and Mihara K: Mitofusin
1 and 2 play distinct roles in mitochondrial fusion reactions via
GTPase activity. J Cell Sci. 117:6535–6546. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Züchner S, Mersiyanova IV, Muglia M,
Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E,
Patitucci A, Senderek J, et al: Mutations in the mitochondrial
GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A.
Nat Genet. 36:449–451. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
135
|
Armstrong JS: Mitochondria-directed
therapeutics. Antioxid Redox Signal. 10:575–578. 2008. View Article : Google Scholar
|
|
136
|
Chan DC: Mitochondria: Dynamic organelles
in disease, aging, and development. Cell. 125:1241–1252. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: More than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Barsoum MJ, Yuan H, Gerencser AA, Liot G,
Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, et al:
Nitric oxide-induced mitochondrial fission is regulated by
dynamin-related GTPases in neurons. EMBO J. 25:3900–3911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Head B, Griparic L, Amiri M, Gandre-Babbe
S and van der Bliek AM: Inducible proteolytic inactivation of OPA1
mediated by the OMA1 protease in mammalian cells. J Cell Biol.
187:959–966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Abou-Sleiman PM, Muqit MM and Wood NW:
Expanding insights of mitochondrial dysfunction in Parkinson's
disease. Nat Rev Neurosci. 7:207–219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang
Y, Wang JW, Yang L, Beal MF, Vogel H and Lu B: Mitochondrial
pathology and muscle and dopaminergic neuron degeneration caused by
inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl
Acad Sci USA. 103:10793–10798. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Park J, Lee SB, Lee S, Kim Y, Song S, Kim
S, Bae E, Kim J, Shong M, Kim JM and Chung J: Mitochondrial
dysfunction in Drosophila PINK1 mutants is complemented by parkin.
Nature. 441:1157–1161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Jin SM, Lazarou M, Wang C, Kane LA,
Narendra DP and Youle RJ: Mitochondrial membrane potential
regulates PINK1 import and proteolytic destabilization by PARL. J
Cell Biol. 191:933–942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kim Y, Park J, Kim S, Song S, Kwon SK, Lee
SH, Kitada T, Kim JM and Chung J: PINK1 controls mitochondrial
localization of Parkin through direct phosphorylation. Biochem
Biophys Res Commun. 377:975–980. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Sha D, Chin LS and Li L: Phosphorylation
of parkin by Parkinson disease-linked kinase PINK1 activates parkin
E3 ligase function and NF-kappaB signaling. Hum Mol Genet.
19:352–363. 2010. View Article : Google Scholar
|
|
147
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010. View Article : Google Scholar : PubMed/NCBI
|