Introduction
Acute pancreatitis (AP) is a type of sudden
inflammation manifested by self-digestion, edema, hemorrhage, and
even necrosis of pancreatic tissue due to trypsin activation in the
pancreas caused by a variety of factors. AP is characterized by
trypsinogen activation, leukocyte infiltration and tissue necrosis
(1). However, the exact mechanism
underlying the signaling pathways that regulate these biological
processes in pancreatic cells has not been fully elucidated. At
present, research is mostly focused on signal transduction
mechanisms in acinar cells during AP. It is generally considered
that the involved pathways include the Ca2+ signaling
pathway (2), the
mitogen-activated protein kinase (MAPK) pathway (3), the nuclear factor (NF)-κB pathway
(4), the phosphoinositide
3-kinase (PI3K)̸AKT pathway (5)
and the NADPH pathway (6).
According to their different roles in cells, these pathways may be
divided into an abnormal trypsin secretion and activation signaling
pathway, an inflammation signaling pathway, an oxidative damage
signaling pathway, and an apoptosis and necrosis signaling pathway.
The mechanisms of these signaling pathways are complex, and one
type of cytokine may activate multiple signal transduction
pathways. Furthermore, one signal transduction pathway may be
activated by various cytokines. These pathways are regulated by
various factors that form a complex activity network in cells.
In our previous experiment, the intracellular
activation of trypsinogen was observed in rat AR42J cells
pre-treated with 200 µM/l taurolithocholic acid 3-sulfate
(TLC-S) for 20 min using (CBZ-Ile-Pro-Arg)2-rhodamine110 (BZiPAR)
(7), confirming that TLC-S
directly acts on pancreatic acinar cells to enhance the
intracellular activation of trypsinogen. Bile acid leads to
trypsinogen activation in pancreatic acinar cells through a complex
process. Additional research is required to further elucidate which
signaling pathways affect trypsinogen activation when activated.
Therefore, the changes in the whole-genome expression profile of
AR42J cells under the effect of TLC-S were investigated.
Furthermore, gene groups that may play a regulatory role were
analyzed using the modular approach of biological networks. The aim
of the present study was to provide a useful reference for
identifying new targets that regulate trypsinogen activation
mediated by the c-Jun N-terminal kinase (JNK) signaling
pathway.
Materials and methods
Detection of gene expression using a DNA
microarray
The present study was performed in accordance with
the Helsinki II Declaration, and approval for this study was
obtained from the Committee for Medical Research Ethics of the
First Affiliated Hospital of Harbin Medical University, which are
the authority of research ethics in China. Pancreatic acinar AR42J
cells were purchased from the China Center for Type Culture
Collection (Wuhan, China) and were cultured in Ham's F12K culture
medium containing 10% fetal bovine serum, 100 U/ml penicillin and
100 µg/ml streptomycin in a humidified incubator at 37°C and
5% CO2. The cells were divided into two groups, namely
the control and treatment groups. The cells in the control group
did not receive treatment, while the cells in the treatment group
were stimulated by TLC-S (200 µM/l) for 20 min. The dose of
TLC-S and the treatment duration were as previously reported by
Voronina et al (8) and
Gerasimenko et al (9).
The cells were collected and total RNA was extracted
according to the instructions for the TRIzol reagent (Invitrogen
Life Technologies; Thermo Fisher Scientific, Carlsbad, CA, USA).
Gene expression analyses were performed with the Rat 12×135K Gene
Expression Array (NimbleGen Systems, Inc., Madison, WI, USA).
Preparation of cDNA from 5 µg of total RNA, hybridizations,
washes and detection were performed in accordance with the
NimbleGen Gene Expression Analysis protocol (NimbleGen Systems,
Inc.), and the slides were scanned using the Axon GenePix 4000B
microarray scanner (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Scanned images in TIFF format were then imported
into NimbleScan software (version 2.5) for grid alignment and
expression data analysis. Expression data were normalized through
quantile normalization and the Robust Multichip Average (RMA)
algorithm included in the NimbleScan software. The Probe level
(*_norm_RMA.pair) files and Gene level
(*_RMA.calls) files were generated following
normalization. All gene level files were imported into Agilent
GeneSpring GX software (version 11.5.1) for further analysis and
were published in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63959).
Differentially expressed genes were identified through fold change
filtering.
Establishment and functional analysis of
the modules of differentially expressed genes
The protein-protein interaction (PPI) network, which
included 36,874 edges and 9,453 nodes (10), was acquired from the Human Protein
Reference Database (HPRD). HPRD is a rich resource of diverse
features of human proteins, which have been experimentally proven
(11). The differentially
expressed genes were mapped into the PPI network, and a file was
obtained on the basis of genetic interaction pairs consisting of
differentially expressed genes. The file was then imported to
Cytoscape (version 2.6.3); the interaction type was 'default
interaction'. Subsequently, 'layout', 'cytoscape layout', and
'spring embedded' was selected. Finally, a regulatory sub-network
was constructed (12).
CFinder software (http://cfinder.org/) was used to identify functional
modules in the sub-networks. Based on the clique percolation
method, CFinder (13) is a
software tool used to locate and visualize network modules and,
thus, it may be applied to detect network clusters of a specified
size.
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) was used to identify gene ontology
biological processes (GO-BP) terms from the detected modules. The
candidate gene groups were submitted to the DAVID database
(http://david.abcc.ncifcrf.gov/);
subsequently, using the complete genome of Rattus norvegicus
as the background genes and 'functional annotation tool' as the
analysis tool, the Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment results were obtained (the P-value cut-off was
0.05).
The KEGG database was used to identify pathway maps.
The candidate genes in one module were imported into the DAVID
pathway database (http://www.kegg.jp/kegg/pathway.html) as key words,
and 'map' was selected; subsequently, a thumbnail image of the
corresponding pathway was screened.
Cellular verification experiments
AR42J cells were grown to a density of 80–90%. A
total of four groups of cells growing at logarithmic phase were
treated as follows: Group A (control), AR42J cells cultured in
normal medium; group B (TLC-S treatment), AR42J cells treated with
TLC-S (200 µM) for 30 min; group C (SP600125 treatment),
AR42J cells treated with SP600125 (40 µM) for 30 min; and
group D (SP600125 + TLC-S treatment), AR42J cells pre-treated with
SP600125 (40 µM) for 30 min prior to incubation with TLC-S
(200 µM) for 30 min. Cells were harvested at the indicated
time points for subsequent experiments.
Western blot analysis
Total proteins were extracted from control,
TLC-S-treated, SP600125-treated and SP600125 + TLC-S-treated
cultures at the abovementioned time points, and subjected to
western blot analysis. Total proteins were separated by 10%
SDS-polyacrylamide gel electrophoresis under reducing conditions,
and transferred onto PVDF membranes (Millipore, Billerica, MA, USA)
by electroblotting. After blocking with non-fat dry milk in
Tris-buffered saline and 500 µl Tween-20 (TBST) for 1 h at
room temperature, the membrane was incubated at 4°C overnight in a
milk solution containing the following polyclonal rabbit
antibodies: anti-p-JNK (1:1,000; cat. no. WLP1664; Wanleibio Co.,
Ltd., Shanghai, China), anti-p-JUN (1:500; cat. no. bs-3210R;
Bioss, Inc., Beijing, China), and anti-β-actin (cat. no. WL0002;
Wanleibio Co., Ltd.). After washing in TBST, the immunoreactive
proteins were visualized using horseradish peroxidase-conjugated
goat anti-rabbit IgG-HRP secondary antibodies (1:5,000; Beyotime,
Haimen, China), followed by enhanced chemiluminescence (Wanleibio
Co., Ltd.). The protein concentrations were determined using the
microassay procedure of the Gel-Pro-Analyzer. Protein expression
was determined by western blot analysis.
Trypsinogen activation assay
Following the abovementioned treatment,
2×106 cells were disrupted by sonication on ice (40 kHz,
3-5 pulses, 3 sec each time; the process was repeated after 10 min
for a total of three times). The cell homogenate was diluted with
500 µl buffer (10 mM HEPES, pH 7.5; 15% ethanol). Diluted
cell homogenate (100 µl) was then added to 24-well plates,
followed by the addition of 400 µl buffer. Finally, the
fluorescent substrate BZiPAR (Molecular Probes, Inc., Eugene, OR,
USA) was added to a final concentration of 2 µM. The
reaction was incubated in the dark for 10–15 min prior to
fluorescence intensity measurement using a multifunctional
microplate reader (excitation, 496 nm; emission, 521 nm; ELX-800;
BioTek, Winooski, VT, USA).
Statistical analysis
Data are expressed as mean ± standard deviation and
analyzed with the SPSS 10.0 software (SPSS, Inc., Chicago, IL,
USA). Analysis of variance was used to determine statistically
significant differences among the three groups, and means of every
two groups were detected with the Student-Newman-Keuls test
(q-test). P<0.05 was considered to indicate statistically
significant differences.
Results
Construction of interaction sub-networks
of differentially expressed genes
A total of 11,292 genes were detected, and 1,124
upregulated and 498 downregulated genes were identified in the
TLC-S treatment group. There were 36,875 rat PPI pairs according to
the HPRD. By mapping the differentially expressed genes onto PPI
pairs, 375 PPI pairs of differentially expressed genes were
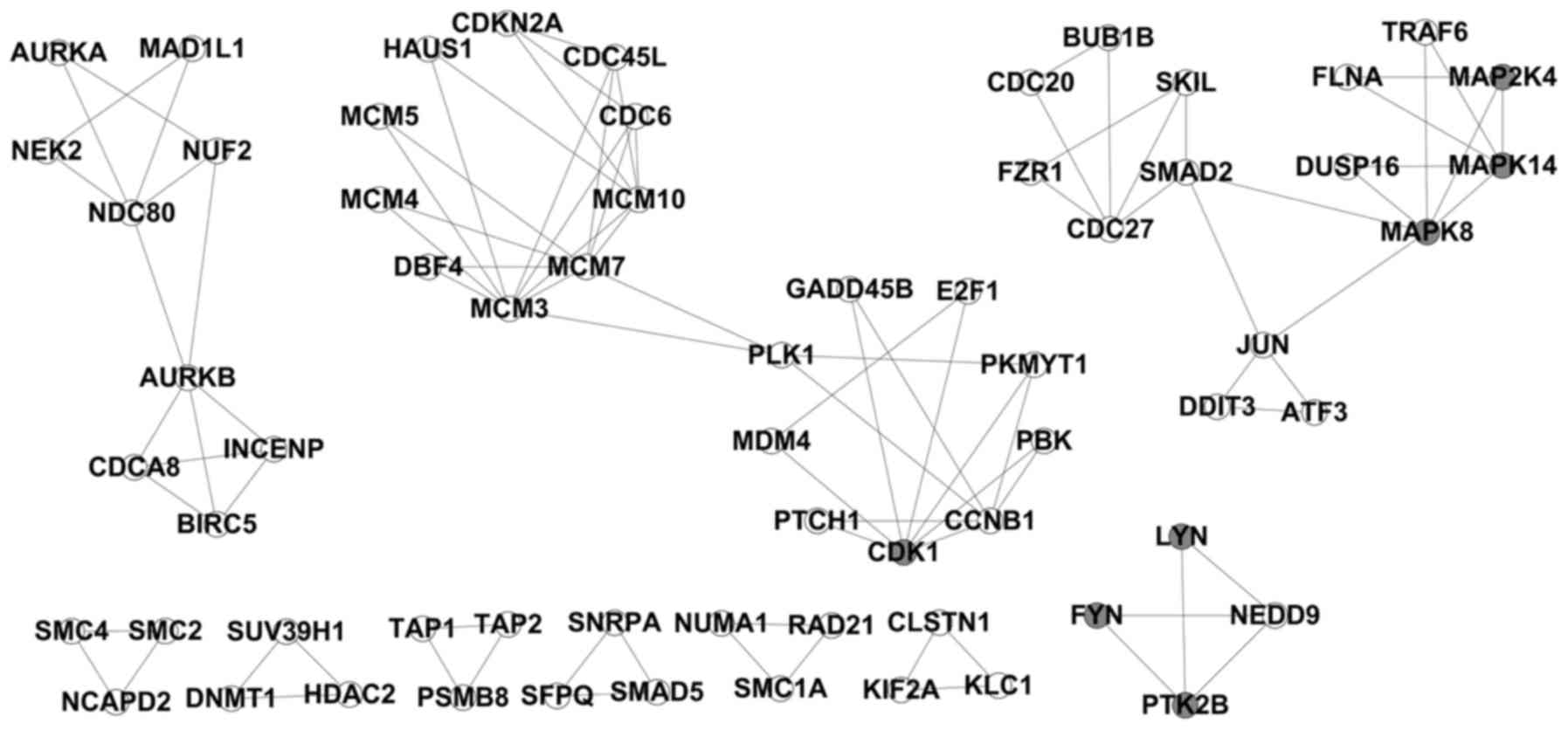
obtained, and a sub-network diagram was constructed as illustrated
in Fig. 1 using Cytoscape
software.
Results derived from modular detection
using CFinder software
CFinder software was applied for the modular
detection of the abovementioned obtained PPI pairs of
differentially expressed genes. Under the condition of k=3, 14
modules were identified (Fig.
2).
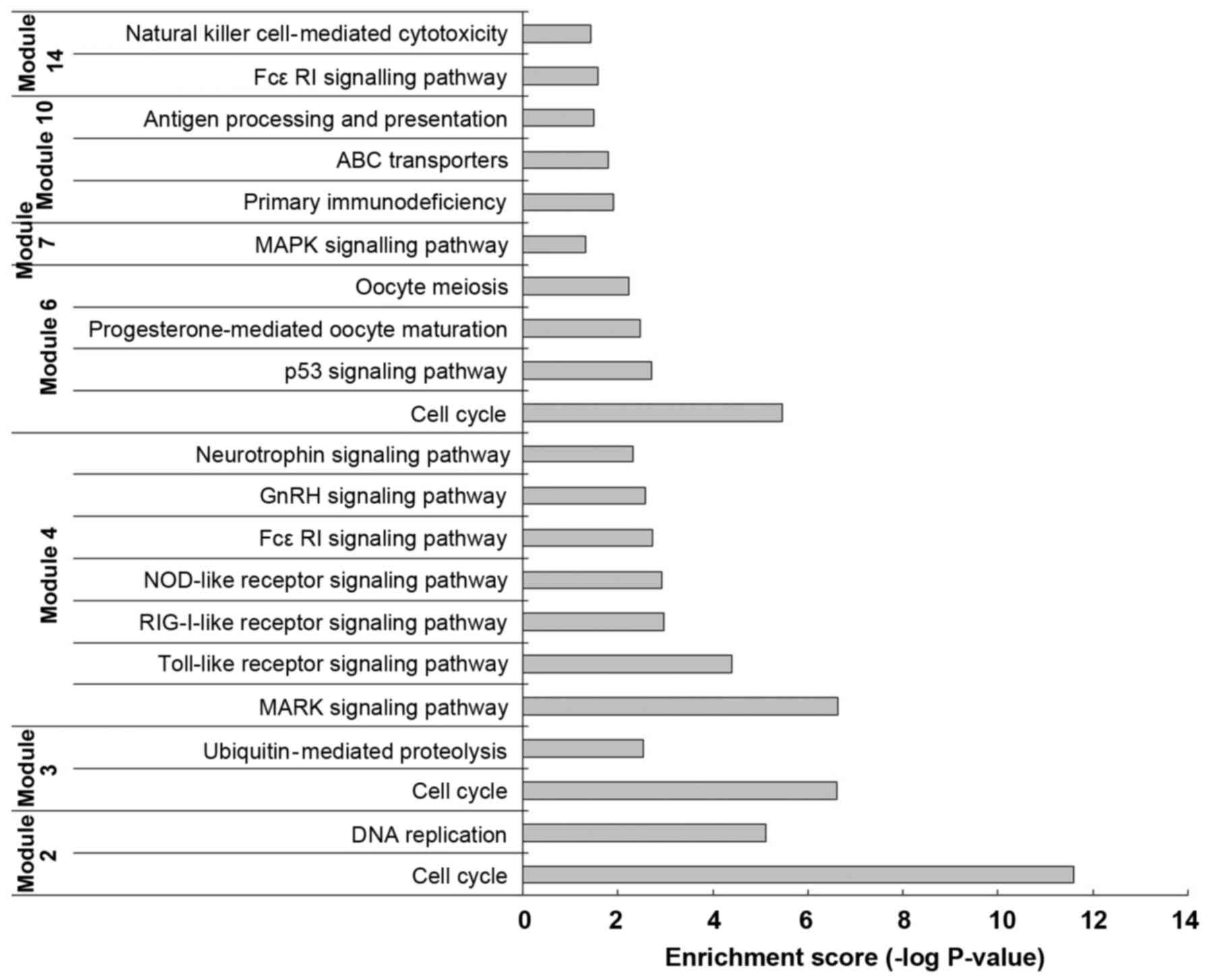
Functional analysis of each module
A GO-BP functional enrichment analysis was performed
on the 14 modules by the DAVID software. The functional analysis of
each gene group revealed that, in modules 2, 3 and 6, the
differentially expressed genes were mainly enriched in 'Cell cycle'
(rno04110) functions; in modules 4 and 7, the function of the
differentially expressed genes was mainly the 'MAPK signaling
pathway' (rno04010). Particularly in module 4, differentially
expressed genes, such as MAP2K4, MAPK14 and MAPK8, encode kinases
(Fig. 3).
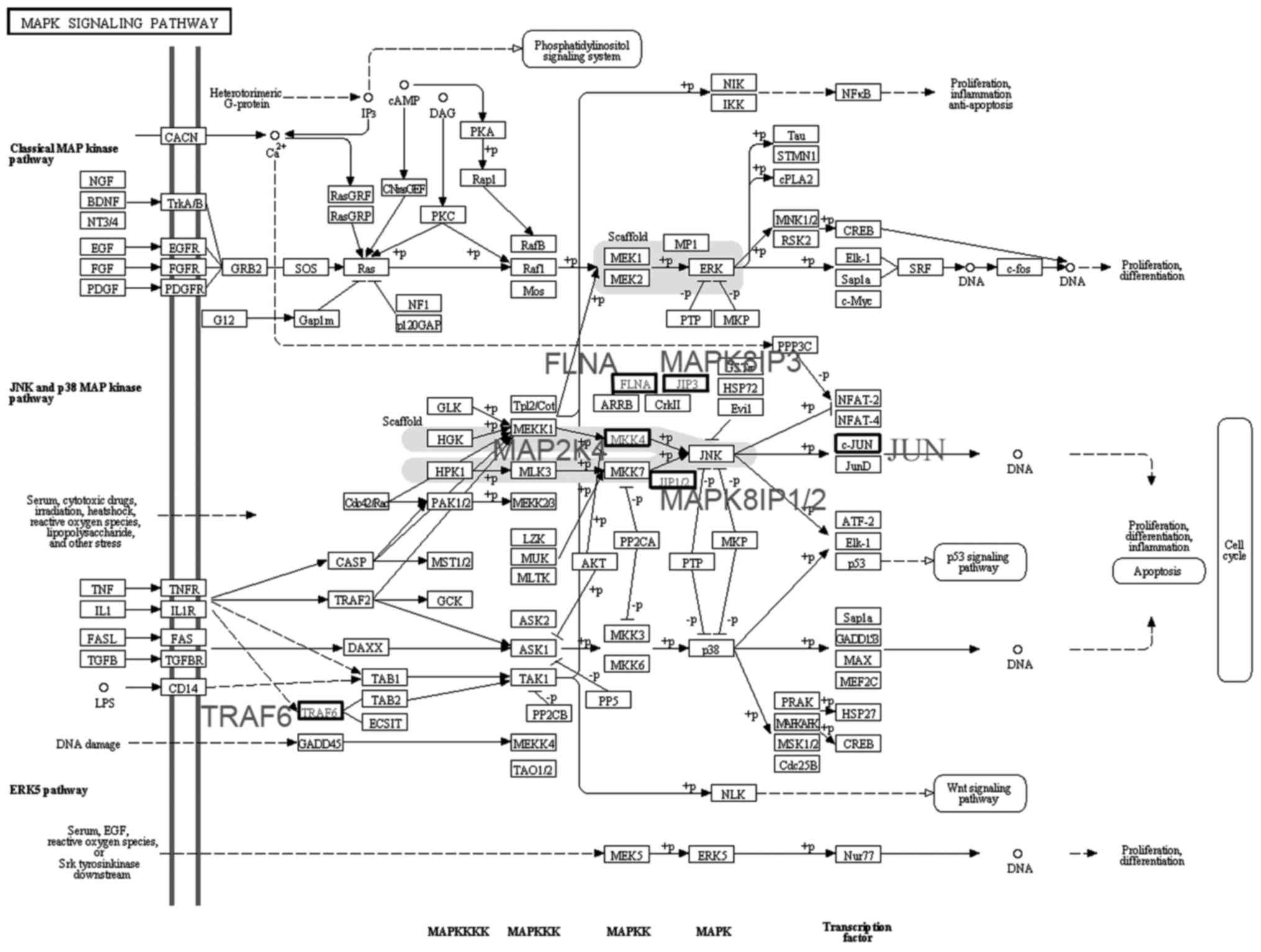
A total of six differentially expressed genes in
module 4 were imported into the KEGG map. It was observed that
MAP2K4, MAPK8 and FLNA are part of the JNK pathway in the 'MAPK
signaling pathway' (map04010) (Fig.
4). MAP2K4 is a MAPK kinase (MAPKK) and phosphorylates the
downstream MAPK MAPK8 (JNK1).
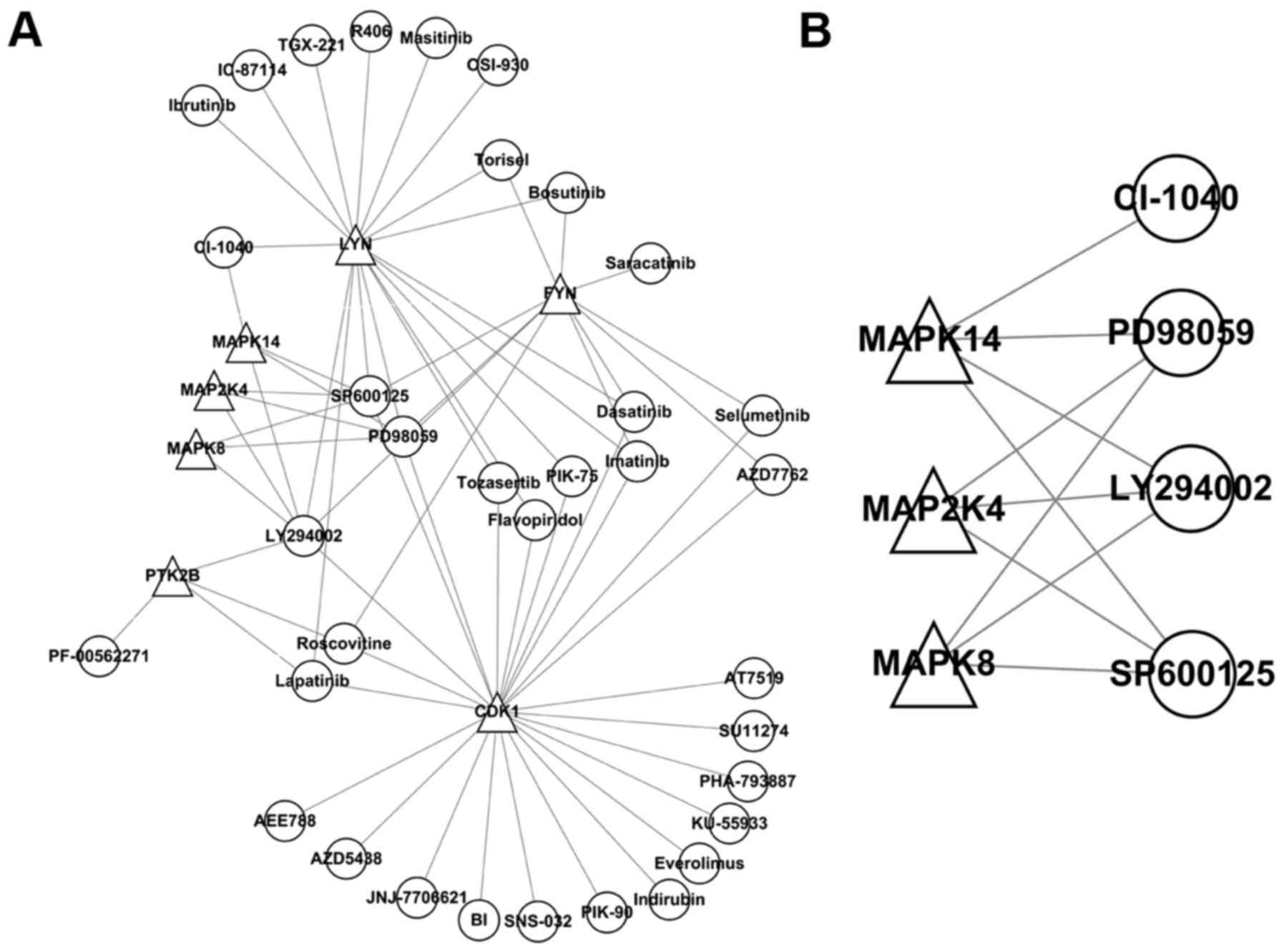
A regulatory network (Fig. 5A) of seven kinase inhibitors was
constructed and arranged by the number of kinases regulated:
LY294002 (n=6); SP600125 and PD98059 (n=5); and lapatinib,
roscovitine, CI-1040, bosutinib, Torisel, selumetinib, imatinib,
AZD7762, PIK-75, tozasertib and flavopiridol (n=2). A regulatory
network (Fig. 5B) constructed
with three kinases from module 4 contained LY294002 (PI3K
inhibitor), SP600125 (JNK inhibitor) and PD98059 (MEK inhibitor).
Thus, SP600125 (JNK inhibitor) was selected for the following
experiments.
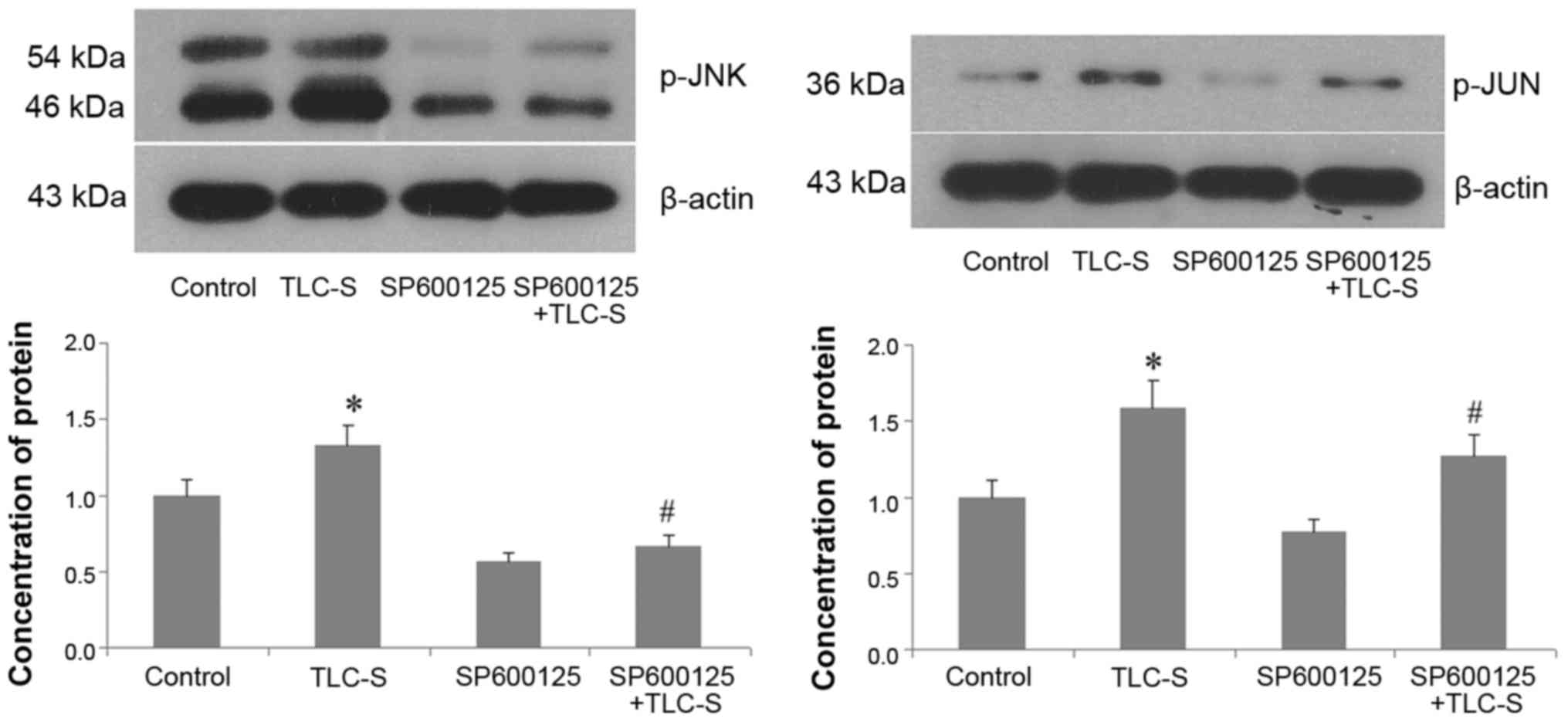
Western blot results
Western blots of the control and TLC-S groups
revealed that, during trypsinogen activation, the p-JNK levels were
markedly increased in AR42J cells following TLC-S treatment. In the
TLC-S and SP600125 + TLC-S groups, the p-JNK levels following TLC-S
stimulation were significantly reduced by pre-treatment with
SP600125 (JNK inhibitor). p-JUN levels trended with p-JNK levels
(Fig. 6).
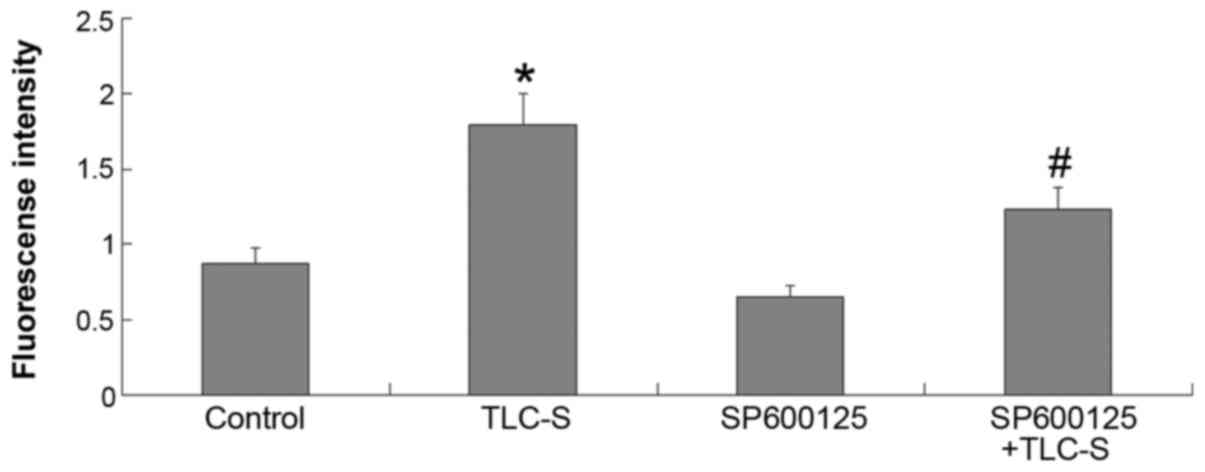
Results of trypsinogen activation
detection
Based on the results of the control and TLC-S
groups, the extent of trypsinogen activation was found to be
markedly increased in AR42J cells following TLC-S treatment. Based
on the results of the TLC-S and SP600125 + TLC-S groups, the amount
of trypsinogen activation following TLC-S stimulation was found to
be markedly reduced by pretreatment with SP6001255 (JNK inhibitor)
(Fig. 7).
Discussion
In the post-genomics era, elucidating the mechanisms
that underlie a large number of biological processes has become one
of the most important research goals; however, this process is
complex and challenging. Therefore, studies focusing on only a few
biological molecules (genes or proteins) cannot systematically
reveal the mechanisms underlying biological processes. A
comprehensive system consisting of a large number of molecules
requires a multi-dimensional systemic analysis. If only a complete
complex network is taken into account, it is very difficult to
screen and identify suitable biological target molecules or
relevant pathways. In recent years, researchers have noticed the
modularization of genetic functions. Using the network module
information, the search scope may be greatly reduced. For example,
Liu et al (14) used
genetic network characteristics to classify cancer and analyze the
involved pathways. There is a close association between biological
network modules and functions; each module corresponds to a
different biological function (15). Wagner and Fell (16) defined metabolic pathway
sub-networks with high local connectivity as 'modules'. Segal et
al (17) defined a group of
genes that have the same regulatory programs, functions and
expression patterns as a 'regulatory module'. Several other
scholars believe that a genetic functional module is a group of
genes with high functional relevance; the module must respond to an
experimental stimulus consistently, and all component genes must
exhibit consistent expression responses. Through investigating the
functional modularization of multiple genes, we may be able to
understand the regulatory mechanisms of regulatory pathways more
definitively and accurately.
The present study attempted to deepen our
understanding of the changes in TLC-S-stimulated AR42J cells
through a genetic functional modular analysis. Whole-genome
expression profile chip arrays were applied to detect genes that
were differentially expressed in pancreatic acinar AR42J cells
treated with TLC-S for 20 min. Based on the HPRD database, a PPI
network was obtained, which was then processed by CFinder software
to derive 14 modules. Among these 14 modules, the GO-BP enrichment
analysis identified two as modules of interest.
On the basis of GO-BP analysis, it was observed that
in modules 4 and 7, the function of differentially expressed genes
is mainly enriched in biological processes such as the 'MAPK
signaling pathway'. This result suggests that this pathway plays an
important role in TLC-S stimulation of AR42J cells to activate
trypsin. The MAPK signal transduction pathway is an important
intracellular signaling pathway (18). MAPKs are a class of
serine/threonine protein kinases that are widespread in mammalian
cells and mainly include MAPKK kinases (MAPKKKs), MAPKKs and MAPKs.
MAPKs are expressed in all eukaryotic cells, and activated MAPKs
convey extracellular stimuli into the cell; moreover, MAPKs
phosphorylate transcription factors and various protein kinases at
the substrate level, regulate related gene transcription and
participate in physiological processes, such as cell proliferation,
differentiation, transformation and apoptosis (19). MAPKs are also associated with the
pathogenesis of inflammation, cancer and a variety of other
diseases. Currently, there are at least four MAPK family members,
namely ERK1/2, JNK, p38 and ERK5, which interact with other
proteins to regulate cellular functions (20). The existing literature confirms
that the MAPK signaling pathway plays a crucial role in trypsinogen
activation (21). Dabrowski et
al (22) and Grady et
al (23) demonstrated that
activation of the MAPK pathway is an early event in AP and leads to
further development of acute pancreatic disease.
In module 4, differentially expressed genes that
encode the kinases MAP2K4, MAPK14 and MAPK8 were found. KEGG map
analysis revealed that MAP2K4, MAPK8 and FLNA are part of the JNK
pathway. JNK was initially identified as a protein kinase that
specifically phosphorylates the transcription factor c-JUN in the
nucleus (24,25). From the 'MAPK signaling pathway'
map (map04010), it may be observed that a MAPKKK activates MAP2K4
through phosphorylation as part of the JNK signaling pathway.
MAP2K4 is a MAPKK and may phosphorylate downstream MAPK8 (JNK1), a
MAPK (26). JNK is activated by
an upstream signal and further phosphorylates the 63rd and 73rd
serine residues at the amino terminus of the nuclear transcription
factor c-JUN, thereby activating c-JUN and enhancing its
transcriptional activity. Stress may activate the JNK signaling
pathway, leading to the transcriptional activation of c-JUN by
phosphorylation (27). The JNK
signaling pathway is part of the MAPK signaling pathway and acts as
an important messenger to convey extracellular signals from the
cell surface into the nucleus (28). The JNK gene has three subtypes,
JNK1 (MAPK8), JNK2 and JNK3; JNK1 (MAPK8) and JNK2 are present in a
wide array of tissue types (29).
The activation of JNK is closely associated with inflammation,
nerve cell degeneration (30) and
apoptosis (31). JNK is activated
by different stimuli, such as inflammatory cytokines and stress
factors (32). Research has
revealed that the JNK signaling pathway plays an important role in
the pathogenesis of AP. In 2000, Schafer reported that the JNK
signaling pathway may produce inflammatory cells, thus playing a
key role in the pathogenesis of AP (33). Thereafter, the importance of the
JNK signaling pathway in the pathogenesis of AP was demonstrated by
a series of research findings (34,35). A specific inhibitor of the JNK
signaling pathway exerted a therapeutic effect in a rat model of AP
(35,36). In a study of an acute edematous
pancreatitis model, Grady et al (23) also found that JNK activation is an
early event in the pathogenesis of AP. In the present study, it was
observed that the JNK pathway was activated in AR42J cells
following TLC-S stimulation, and that this pathway plays an
important role in trypsinogen activation.
Next, a regulatory network of seven kinase
inhibitors was constructed and arranged by the number of kinases
regulated: LY294002 (n=6); SP600125 and PD98059 (n=5); and
lapatinib, roscovitine, CI-1040, bosutinib, Torisel, selumentinib,
imatinib, AZD7762, PIK-75, tozasertib and flavopiridol (n=2). In
the regulatory network constructed containing three kinases of
module 4, there were LY294002 (PI3K inhibitor), SP600125 (JNK
inhibitor) and PD98059 (MEK inhibitor). Thus, SP600125 (JNK
inhibitor) was selected for subsequent experiments. This JNK
signaling pathway inhibitor (SP600125) is a broad-spectrum
inhibitor of serine/threonine kinases capable of inhibiting JNK
activation. SP600125 is an ATP-competitive, efficient, selective
and reversible inhibitor of JNK (37) that significantly inhibits
JNK-mediated MAPK activation. The selectivity of SP600125 for JNK
is 300 times that for ERK1 and p38 (37). Irrera et al (38) demonstrated that SP600125 improves
AP. Nishi et al (39)
proposed that SP600125 is one of the most effective inhibitors for
the treatment of inflammatory diseases involving the MAPK signaling
pathway, such as AP.
The results were verified by four sets of
experiments. From western blots, it was observed that the
phosphorylation of JNK and JUN was significantly increased in AR42J
cells following TLC-S treatment. However, pre-treatment with the
inhibitor SP600125 led to a significant reduction in
phosphorylation in AR42J cells following TLC-S treatment. The same
trend was observed by trypsinogen activation assays, further
indicating that the JNK signaling pathway plays an important role
in trypsinogen activation following TLC-S stimulation of AR42J
cells; treatment with the inhibitor SP600125 blocked the JNK
signaling pathway and, thus, reduced the amount of trypsinogen
activation. This finding further indicates that trypsinogen
activation is mediated by the JNK signaling pathway in the
pathogenesis of AP.
From the abovementioned experiments and analysis, it
may be concluded that the biological process of kinase
phosphorylation plays a key role in activating trypsinogen in AR42J
cells following TLC-S stimulation. Phosphorylation is an important
and universal post-translational modification and is also the most
important regulatory modification in prokaryotic and eukaryotic
organisms (40). Phosphorylation
provides a dynamic means of regulating protein activity (41). During trypsinogen activation,
phosphorylation is required for the activation of MAP2K4, MAPK8,
MAPK14 and other kinases. MAPK8 activation requires the
phosphorylation of threonine and tyrosine residues (26), whereas MAPK14 activation requires
the phosphorylation of its serine/tyrosine residues (42). Phosphorylation is required for the
activation of the abovementioned kinases, and the kinases are
closely associated with trypsinogen activation. Therefore,
phosphorylation of kinases participating in the JNK signaling
pathway is considered to be necessary for trypsinogen
activation.
In conclusion, the JNK signaling pathway is involved
in regulating trypsinogen activation in rat pancreatic AR42J cells.
The present study provides a useful reference for better
understanding the pathogenesis of AP and identifying new targets to
regulate trypsinogen activation. Moreover, it provides valuable
information for the treatment of AP.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81370566). The
authors would like to thank Fenghe Inc. (Shanghai, China) for their
asistance with the bioinformatics analysis.
Glossary
Abbreviations
Abbreviations:
|
AP
|
acute pancreatitis
|
|
TLC-S
|
taurolithocholic acid 3-sulfate
|
|
RMA
|
robust multichip average
|
|
PPI
|
protein-protein interaction
|
|
HPRD
|
human protein reference database
|
|
DAVID
|
database for annotation visualization
and integrated discovery
|
|
GO-BP
|
gene ontology biological processes
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
References
|
1
|
Awla D, Hartman H, Abdulla A, Zhang S,
Rahman M, Regnér S and Thorlacius H: Rho-kinase signalling
regulates trypsinogen activation and tissue damage in severe acute
pancreatitis. Br J Pharmacol. 162:648–658. 2011. View Article : Google Scholar :
|
|
2
|
Li J, Zhou R, Zhang J and Li ZF: Calcium
signaling of pancreatic acinar cells in the pathogenesis of
pancreatitis. World J Gastroenterol. 20:16146–16152. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao MH, Xu J, Cai HD, Lv ZW, Feng YJ, Li
K, Chen CQ and Li YY: p38 MAPK inhibition alleviates experimental
acute pancreatitis in mice. Hepatobiliary Pancreat Dis Int.
14:101–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu G, Wan R, Hu Y, Ni J, Yin G, Xing M,
Shen J, Tang M, Chen C, Fan Y, et al: Pancreatic acinar
cells-derived cyclophilin A promotes pancreatic damage by
activating NF-κB pathway in experimental pancreatitis. Biochem
Biophys Res Commun. 444:75–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu P, Wang J, Yang ZW, Lou XL and Chen C:
Regulatory roles of the PI3K/Akt signaling pathway in rats with
severe acute pancreatitis. PLoS One. 8:e817672013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao WL, Xiang XH, Chen K, Xu W and Xia SH:
Potential role of NADPH oxidase in pathogenesis of pancreatitis.
World J Gastrointest Pathophysiol. 5:169–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Z, Lu M, Chu J, Qiao X, Meng X, Sun B,
Zhang W and Xue D: Early proteome analysis of rat pancreatic acinar
AR42J cells treated with taurolithocholic acid 3-sulfate.
Pancreatology. 12:248–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Voronina SG, Gryshchenko OV, Gerasimenko
OV, Green AK, Petersen OH and Tepikin AV: Bile acids induce a
cationic current, depolarizing pancreatic acinar cells and
increasing the intracellular Na+ concentration. J Biol Chem.
280:1764–1770. 2005. View Article : Google Scholar
|
|
9
|
Gerasimenko JV, Flowerdew SE, Voronina SG,
Sukhomlin TK, Tepikin AV, Petersen OH and Gerasimenko OV: Bile
acids induce Ca2+ release from both the endoplasmic reticulum and
acidic intracellular calcium stores through activation of inositol
trisphosphate receptors and ryanodine receptors. J Biol Chem.
281:40154–40163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Keshava Prasad TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human Protein Reference
Database-2009 update. Nucleic Acids Res. 37:D767–D772. 2009.
View Article : Google Scholar
|
|
11
|
Goel R, Harsha HC, Pandey A and Prasad TS:
Human Protein Reference Database and Human Proteinpedia as
resources for phosphoproteome analysis. Mol Biosyst. 8:453–463.
2012. View Article : Google Scholar
|
|
12
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar :
|
|
13
|
Adamcsek B, Palla G, Farkas IJ, Derényi I
and Vicsek T: CFinder: Locating cliques and overlapping modules in
biological networks. Bioinformatics. 22:1021–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu CC, Chen WS, Lin CC, Liu HC, Chen HY,
Yang PC, Chang PC and Chen JJ: Topology-based cancer classification
and related pathway mining using microarray data. Nucleic Acids
Res. 34:4069–4080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barabási AL and Oltvai ZN: Network
biology: Understanding the cell's functional organization. Nat Rev
Genet. 5:101–113. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wagner A and Fell DA: The small world
inside large metabolic networks. Proc Biol Sci. 268:1803–1810.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Segal E, Shapira M, Regev A, Pe'er D,
Botstein D, Koller D and Friedman N: Module networks: Identifying
regulatory modules and their condition-specific regulators from
gene expression data. Nat Genet. 34:166–176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
King LA, Toledo AH, Rivera-Chavez FA and
Toledo-Pereyra LH: Role of p38 and JNK in liver ischemia and
reperfusion. J Hepatobiliary Pancreat Surg. 16:763–770. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tanaka T and Iino M: Sec8 regulates
cytokeratin8 phosphorylation and cell migration by controlling the
ERK and p38 MAPK signalling pathways. Cell Signal. 27:1110–1119.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Finch AR, Caunt CJ, Perrett RM,
Tsaneva-Atanasova K and McArdle CA: Dual specificity phosphatases
10 and 16 are positive regulators of EGF-stimulated ERK activity:
indirect regulation of ERK signals by JNK/p38 selective MAPK
phosphatases. Cell Signal. 24:1002–1011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma B, Wu L, Lu M, Gao B, Qiao X, Sun B,
Xue D and Zhang W: Differentially expressed kinase genes associated
with trypsinogen activation in rat pancreatic acinar cells treated
with taurolithocholic acid 3-sulfate. Mol Med Rep. 7:1591–1596.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dabrowski A, Grady T, Logsdon CD and
Williams JA: Jun kinases are rapidly activated by cholecystokinin
in rat pancreas both in vitro and in vivo. J Biol Chem.
271:5686–5690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grady T, Dabrowski A, Williams JA and
Logsdon CD: Stress-activated protein kinase activation is the
earliest direct correlate to the induction of secretagogue-induced
pancreatitis in rats. Biochem Biophys Res Commun. 227:1–7. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: Conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith JL, Schaffner AE, Hofmeister JK,
Hartman M, Wei G, Forsthoefel D, Hume DA and Ostrowski MC: ets-2 is
a target for an akt (Protein kinase B)/jun N-terminal kinase
signaling pathway in macrophages of motheatenviable mutant mice.
Mol Cell Biol. 20:8026–8034. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hickson JA, Huo D, Vander Griend DJ, Lin
A, Rinker-Schaeffer CW and Yamada SD: The p38 kinases MKK4 and MKK6
suppress metastatic colonization in human ovarian carcinoma. Cancer
Res. 66:2264–2270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang SH, Sharrocks AD and Whitmarsh AJ:
MAP kinase signalling cascades and transcriptional regulation.
Gene. 513:1–13. 2013. View Article : Google Scholar
|
|
28
|
Goolsby TV and Lombardo FA: Extravasation
of chemotherapeutic agents: Prevention and treatment. Semin Oncol.
33:139–143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ji RR, Gereau RW IV, Malcangio M and
Strichartz GR: MAP kinase and pain. Brain Res Rev. 60:135–148.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brnjic S, Olofsson MH, Havelka AM and
Linder S: Chemical biology suggests a role for calcium signaling in
mediating sustained JNK activation during apoptosis. Mol Biosyst.
6:767–774. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carmen JC, Southard RC and Sinai AP: The
complexity of signaling in host-pathogen interactions revealed by
the Toxoplasma gondii-dependent modulation of JNK phosphorylation.
Exp Cell Res. 314:3724–3736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Zhao H, Liu T, Wan C, Liu X, Gao
Z, Hou X, Jiang L and Liu F: Activation of transcription factor
AP-1 in response to thermal injury in rat small intestine and IEC-6
cells. BMC Gastroenterol. 15:832015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schäfer C and Williams JA: Stress kinases
and heat shock proteins in the pancreas: possible roles in normal
function and disease. J Gastroenterol. 35:1–9. 2000.PubMed/NCBI
|
|
34
|
Samuel I, Zaheer S, Fisher RA and Zaheer
A: Cholinergic receptor induction and JNK activation in acute
pancreatitis. Am J Surg. 186:569–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wagner AC, Mazzucchelli L, Miller M,
Camoratto AM and Göke B: CEP-1347 inhibits caerulein-induced rat
pancreatic JNK activation and ameliorates caerulein pancreatitis.
Am J Physiol Gastrointest Liver Physiol. 278:G165–G172. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minutoli L, Altavilla D, Marini H,
Passaniti M, Bitto A, Seminara P, Venuti FS, Famulari C, Macrì A,
Versaci A and Squadrito F: Protective effects of SP600125 a new
inhibitor of c-jun N-terminal kinase (JNK) and
extracellular-regulated kinase (ERK1/2) in an experimental model of
cerulein-induced pancreatitis. Life Sci. 75:2853–2866. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shin M, Yan C and Boyd D: An inhibitor of
c-jun aminoterminal kinase (SP600125) represses c-Jun activation,
DNA-binding and PMA-inducible 92-kDa type IV collagenase
expression. Biochim Biophys Acta. 1589:311–316. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Irrera N, Bitto A, Interdonato M,
Squadrito F and Altavilla D: Evidence for a role of
mitogen-activated protein kinases in the treatment of experimental
acute pancreatitis. World J Gastroenterol. 20:16535–16543. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nishi H, Fong JH, Chang C, Teichmann SA
and Panchenko AR: Regulation of protein-protein binding by coupling
between phosphorylation and intrinsic disorder: Analysis of human
protein complexes. Mol Biosyst. 9:1620–1626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hunter T: Signaling-2000 and beyond. Cell.
100:113–127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|