Introduction
Diabetic nephropathy (DN) is the primary cause of
end-stage renal disease (ESRD), and the leading cause of morbidity
and mortality in diabetic patients (1). A number of pathways contribute to
the progression of DN. Glomerular hyperfiltration, dysregulation of
the renal vasculature, excessive oxidative stress, increased
transforming growth factor (TGF)-β, activation of protein kinase C
and the accumulation of advanced glycation end products (AGEs) may
contribute to the pathogenesis of DN (2–6).
Glomerular hypertrophy, mesangial cell proliferation, thickening of
the glomerular basement membrane, extracellular matrix expansion
and glomerular sclerosis are the pathological characteristics of
DN. However, mounting evidence indicates that renal tubular cell
injury also plays a key role in DN and may predict the declining
renal function more efficiently compared with glomerular damage
(7,8). Chronic low-grade inflammation plays
a key role in the progression and development of DN, and
inflammation in the tubulointerstitial lesion is a crucial
component of renal dysfunction (9,10).
Cell apoptosis is the process of normal cell death that activates
cell responses to change the microenvironment, albeit immoderate
apoptosis in DN may trigger pathological processes. The
hyperglycemic state is an apoptosis-facilitating microenvironment.
Apoptosis has been observed in human diabetic kidney biopsies,
particularly in tubular cells (11), as well as HK-2 cells exposed to
high glucose (HG) conditions (12,13).
p38 mitogen-activated protein kinase (p38 MAPK) is a
member of the MAPK family and is known to play a key role in
inflammation and apoptosis. Activation of p38 MAPK has been
demonstrated in renal biopsy specimens in a wide spectrum of human
glomerulonephritis, and has been correlated with renal dysfunction
and histopathological alterations (14). Previous findings demonstrated the
key role of p38 MAPK in the progression and development of DN, and
its suppression has been found to be a beneficial treatment
strategy for DN (15–17). Pentosan polysulfate (PPS) is a
semisynthetic sulfated poly-saccharide that has been used for the
treatment of interstitial cystitis (18). PPS treatment prevents the
progression of DN in mice, and the underlying mechanism may be that
PPS inhibits nuclear factor (NF)-κB activation and inflammation
stimulated by tumor necrosis factor (TNF)-α, HG and AGEs (19).
However, the protective effects of PPS against
HG-induced cell apoptosis and inflammation have not been fully
elucidated. In the present study, the anti-apoptotic and
anti-inflammatory effects of PPS on HK-2 cells were investigated,
aiming to identify its protective mechanism and assess its clinical
potential.
Materials and methods
Cell culture
HK-2 cells were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA). The cells were
cultured in Dulbecco's modified Eagle's medium (DMEM) containing
10% heat-inactivated fetal bovine serum (FBS), epidermal growth
factor, and 1% streptomycin-penicillin mixture in a 5%
CO2 incubator at 37°C. After achieving 70–85%
confluence, the cells were growth-arrested to synchronize the cell
growth for 24 h in FBS-free DMEM. The media were then changed to
serum-free media with normal glucose (NG) (5.5 mmol/l) or HG (30
mmol/l) at different time-points. PPS (200 µg/ml) and
SB203580 (10 µM) were added 2 h prior to HG.
Measurement of cell viability
The viability of HK-2 cells was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
Cell Proliferation Assay kit (American Type Culture Collection,
Manassas, VA, USA). Briefly, HK-2 cells were grown at a density of
1×104 cells/ml in 96-well culture plates in culture
media with 5.5, 15, 25, or 30 mmol/l glucose, and maintained for up
to 48 h. PPS was administered at different concentrations (50, 100,
150 and 200 µg/ml). A 10 µl MTT reagent was added to
each well and incubated at 37°C for 4 h. The culture media with MTT
reagent were then removed. The incorporated formazan crystals in
the viable cells were dissolved with 100 µl detergent
reagent and the absorbance of each well was determined using a
microplate reader (model 3550-UV; Bio-Rad, Tokyo, Japan) at a 540
nm wavelength.
Western blot analysis
Following treatment, the cells were lysed in RIPA
buffer containing protease and phosphatase inhibitors. The cell
protein was quantified with the bicinchoninic acid protein assay
method with bovine serum albumin as the standard. The protein
samples (40 µg/lane) were subsequently separated with 10–15%
SDS-PAGE and electroblotted to polyvinylidene fluoride (PVDF)
membranes (Bio-Rad, Hercules, CA, USA), which were blocked using 5%
fat-free milk for 2 h at 37°C. Subsequently, the PVDF membranes
were incubated overnight at 4°C with one of the monoclonal rabbit
anti-human primary antibodies: p38 MAPK (1:1,000; cat. no. 14451),
phospho-p38 MAPK (1:500; cat. no. 4092), caspase-3 (1:1,000; cat.
no. 14220), cleaved caspase-3 (1:500; cat. no. 9654), Bcl-2 (1:500;
cat. no. 4223), Bax (1:500; cat. no. 5023), NF-κB p65 (1:500; cat.
no. 59674) and β-actin (1:2,000; cat. no. 12620) antibodies (all
from Cell Signaling Technology, Inc., Danvers, MA, USA). Incubation
with the secondary antibody (1:5,000) (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) was performed at room temperature for 1
h. The blots were then visualized using a western blot analysis
detection system (ECL Plus; GE Healthcare, Princeton, NJ, USA).
Flow cytometric analysis of
apoptosis
The apoptotic rate of HK-2 cells was determined
using a commercially available kit (Apoptosis Detection kit; BD
Biosciences, San Jose, CA, USA) according to the manufacturer's
instructions. Briefly, following administration of the different
treatments, HK-2 cells were collected and washed twice with cold
PBS and then re-suspended in a volume of 200 µl 1X binding
buffer. Subsequently, the cells were incubated with 5 µl
propidium iodide (PI) and 5 µl fluorescein isothiocyanate
(FITC) Annexin V at room temperature for 15 min in the dark. After
the incubation period, 400 µl of 1X binding buffer were
added and results were calculated and analyzed in a FACScan flow
cytometer and Cell software (BD Biosciences).
ELISA
The secreted pro-inflammatory cytokines TNF-α,
interleukin (IL)-1β and IL-6 were analyzed in the supernatant using
commercially available ELISA kits following the manufacturer's
instructions. The absorbance at 492 nm was measured using a
microplate reader (model 3550-UV; Bio-Rad).
Statistical analysis
Experiments were performed in triplicate and
repeated three times. Values are presented as means ± standard
deviation. Analysis of variance was used for multiple comparisons
among groups. GraphPad InStat version 5.0 statistical software
(GraphPad Software, Inc., San Diego, CA, USA) was used to perform
analyses. The results were defined as statistically significant
when P<0.05.
Results
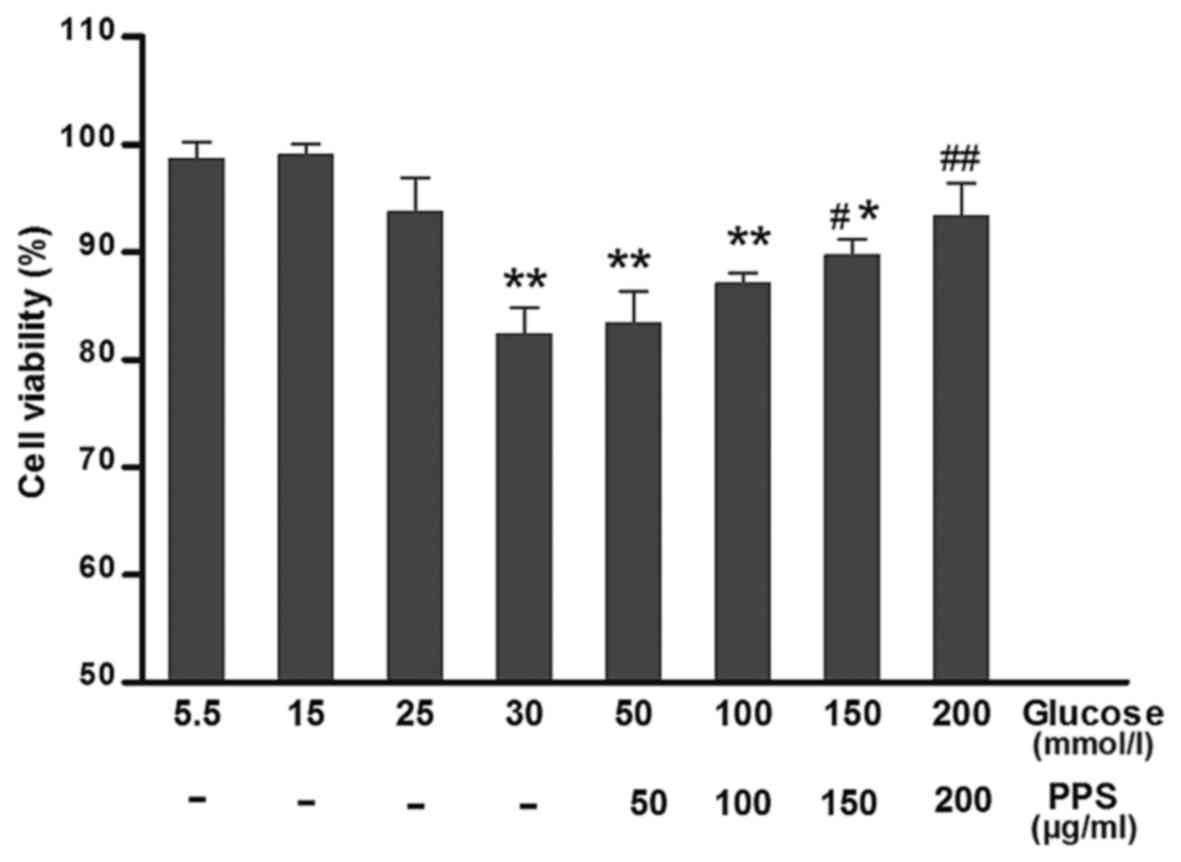
PPS enhances cell viability in HG-treated
cells
To test whether PPS treatment attenuated the
HG-induced cytotoxicity, cell viability was determined at various
levels of glucose (5–30 mmol/l) and PPS (50–200 µg/ml). The
results demonstrated that HK-2 cells incubated with 30 mmol/l
glucose exhibited evidently attenuated cell viability compared with
the cells exposed to 5 mmol/l glucose. Treatment with 200
µg/ml PPS in the HG group markedly increased cell viability
(Fig. 1). Therefore, PPS exerted
a protective effect on HK-2 cells.
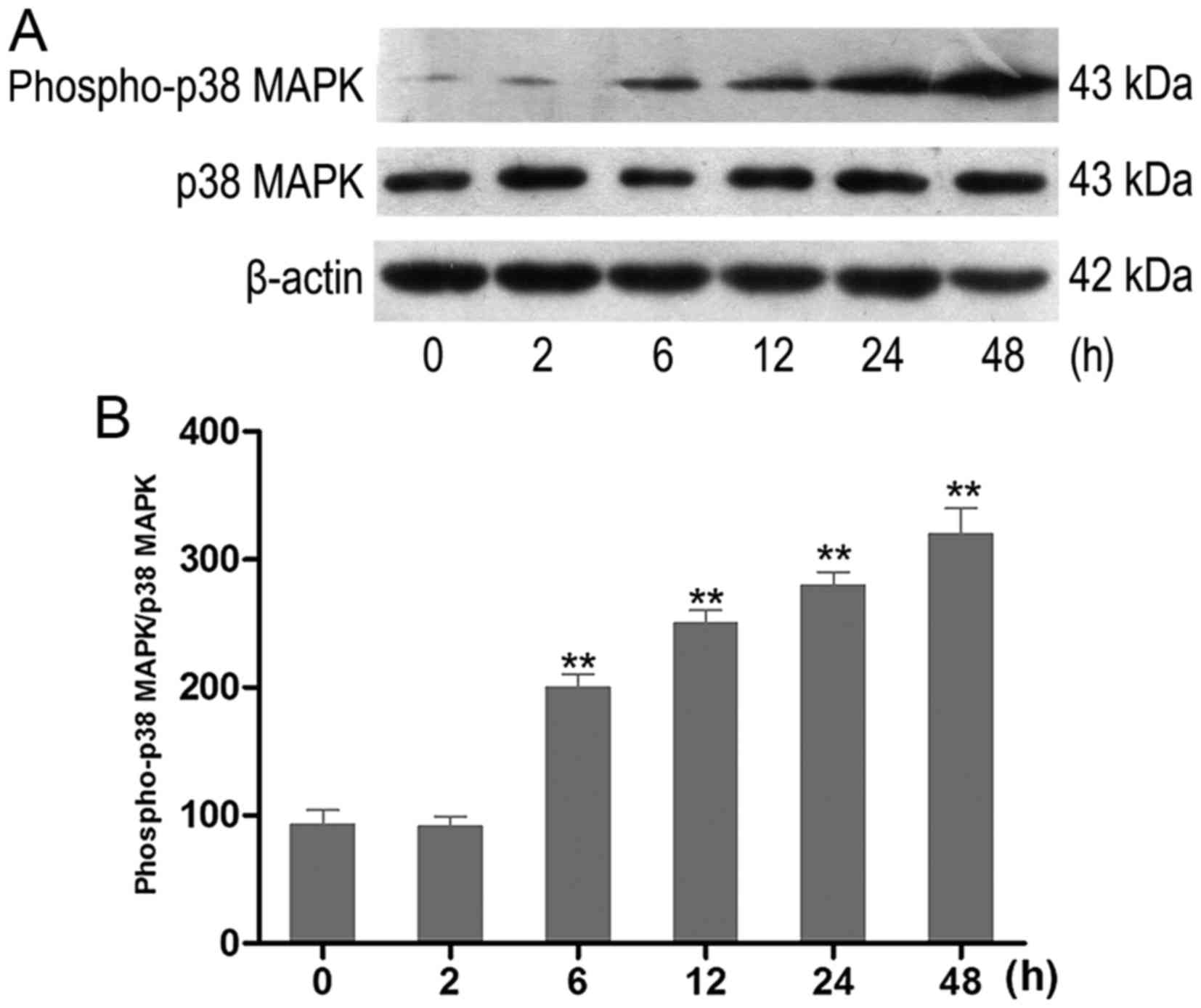
HG increases the activity of p38 MAPK in
HK-2 cells
The effect of HG on the activation of p38 MAPK in
vitro was examined. HK-2 cells were incubated with HG at
different time-points. The activity of phospho-p38 MAPK was
analyzed using western blot analysis (Fig. 2A). HK-2 cells exposed to HG
exhibited an increase of phospho-p38 MAPK in a time-dependent
manner. The increase occurred at 6 h and was most prominent at 48 h
(Fig. 2B).
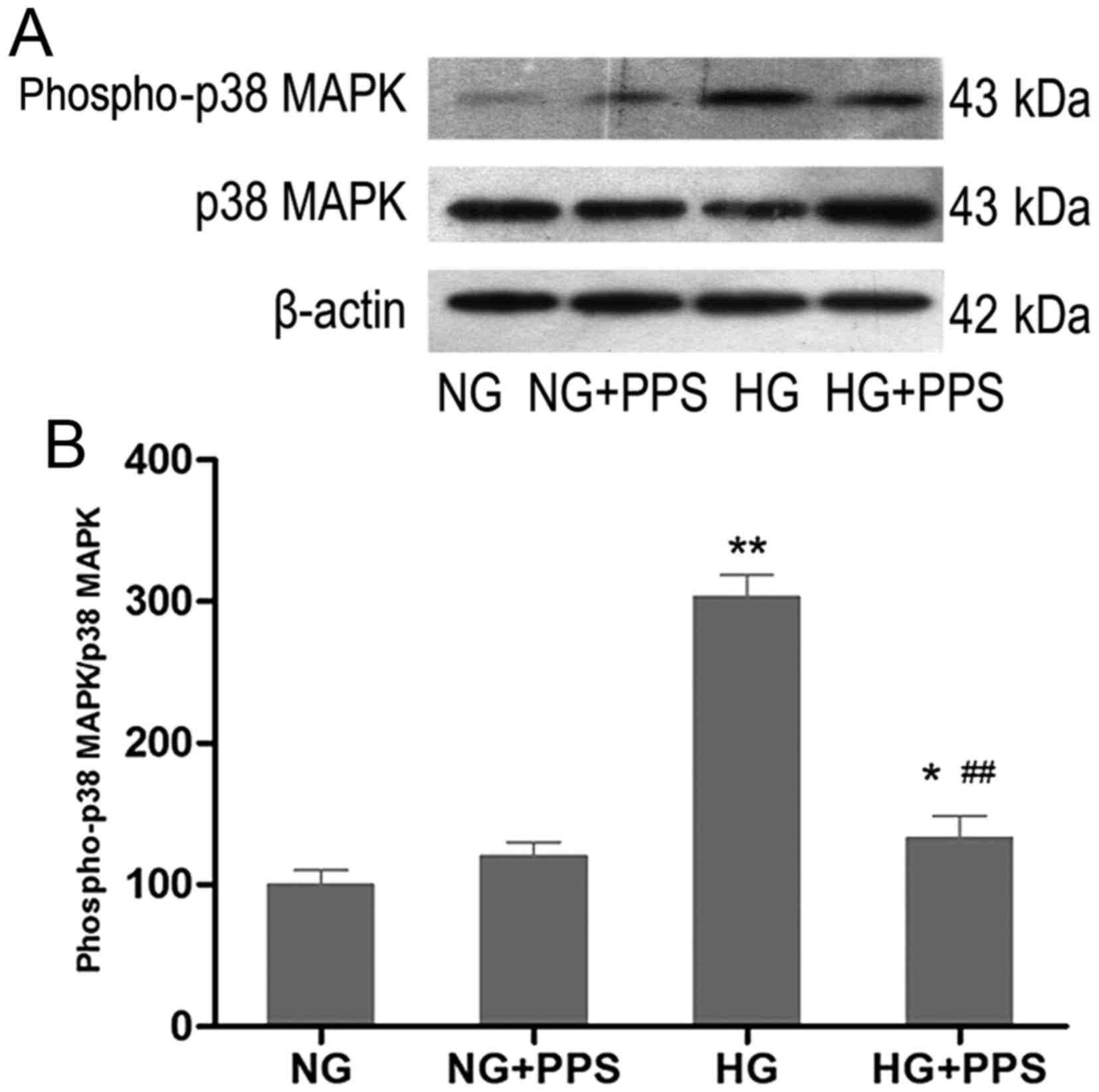
PPS inhibits HG-stimulated p38 MAPK
activation in HK-2 cells
To examine the effects of PPS on HG-stimulated p38
MAPK activation, HK-2 cells were treated with PPS. Compared with
the NG group, incubation of HK-2 cells with HG for 48 h markedly
increased the activity of p38 MAPK, which was significantly blocked
by PPS (Fig. 3A and B). There was
no evident change in p38 MAPK activation in HK-2 cells that were
incubated with NG and were treated with PPS (Fig. 3B).
PPS prevents HG-stimulated apoptosis in
HK-2 cells by inhibiting the activation of p38 MAPK
Previous findings have demonstrated that HG may
increase apoptosis via the activation of p38 MAPK in HK-2 cells
(20–22). Whether the p38 MAPK pathway is
involved in the protective effects of PPS against HG-induced HK-2
cell apoptosis was also investigated. Cell apoptosis was examined
after 48 h of treatment with HG. Incubation with HG caused a marked
increase of the Bax/Bcl-2 ratio and the cleaved caspase-3/caspase-3
ratio in HK-2 cells, which was significantly attenuated by PPS and
SB203580 (Fig. 4A and B). Flow
cytometry was used to analyze the apoptosis of HK-2 cells. Higher
HK-2 cell apoptosis was induced by HG compared with NG, which was
improved by PPS or SB203580, as verified by Annexin V-FITC and PI
staining (Fig. 4C and D). In
addition, it was determined whether the combination of PPS and
SB20358 exerted a synergistic effect on HG-stimulated apoptosis in
HK-2 cells. The results demonstrated that this combination resulted
in a mild increase of the Bax/Bcl-2 and cleaved caspase-3/caspase-3
ratios compared with PPS or SB203580 alone, although the difference
was not statistically significant (P<0.05; Fig. 4E and F). Therefore, suppression of
the p38 MAPK pathway blocked HG-stimulated apoptosis in HK-2
cells.
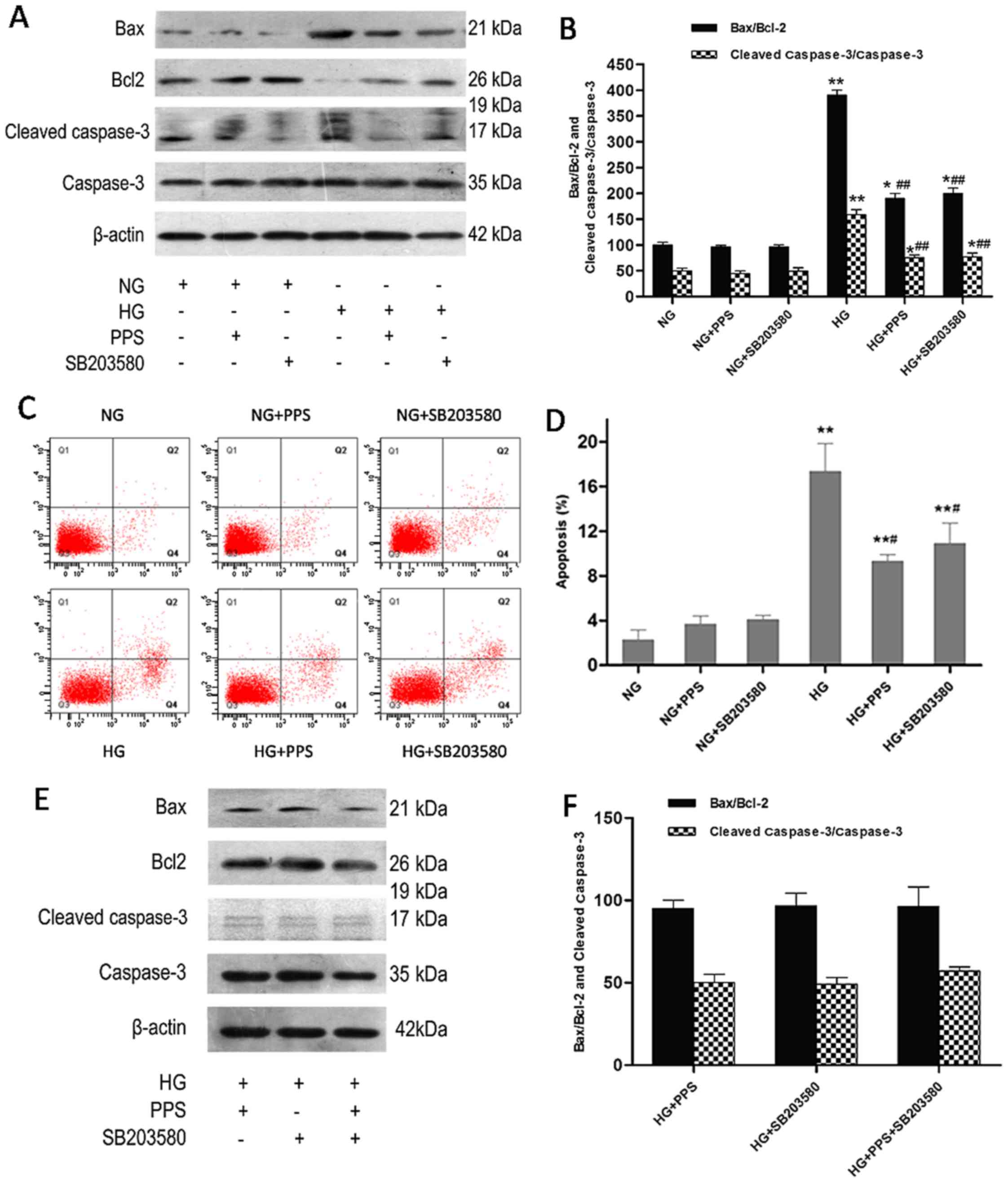 | Figure 4Effect of PPS or SB2035 on HG-induced
apoptosis in HK-2 cells. (A) HK-2 cells were grown in HG and NG in
the absence or presence of PPS or SB203580 for 48 h. The expression
levels of caspase-3, cleaved caspase-3, Bax, Bcl-2 and β-actin were
determined using western blot analysis. (B) Densitometric analysis
for western blot analysis. (C) Apoptosis of HK-2 cells was
determined by flow cytometry. (D) Analysis of apoptotic rate using
flow cytometry. (E) Effects of PPS and SB203580 on the expression
of caspase-3, cleaved caspase-3, Bax, Bcl-2 and β-actin were
determined using western blot analysis. (F) Densitometric analysis
for western blotting. NG: 5.5 mmol/l D-glucose; NG + PPS: 5.5
mmol/l D-glucose plus 200 µg/ml PPS; NG + SB203580: 5.5
mmol/l D-glucose plus 10 µM SB203580; HG: 30 mmol/l
D-glucose; HG + PPS: 30 mmol/l D-glucose plus 200 µg/ml PPS;
HG + SB203580: 30 mmol/l D-glucose plus 10 µM SB203580; and
HG + PPS + SB203580: 30 mmol/l M D-glucose plus 200 µg/ml
PPS plus 10 µM SB203580. Experiments were repeated at least
three times, and the results are presented as the mean ± standard
deviation. *P<0.05 and **P<0.01 vs. NG;
#P<0.01 vs. HG; ##P<0.01 vs. HG. PPS,
pentosan polysulfate; HG, high glucose; NG, normal glucose. |
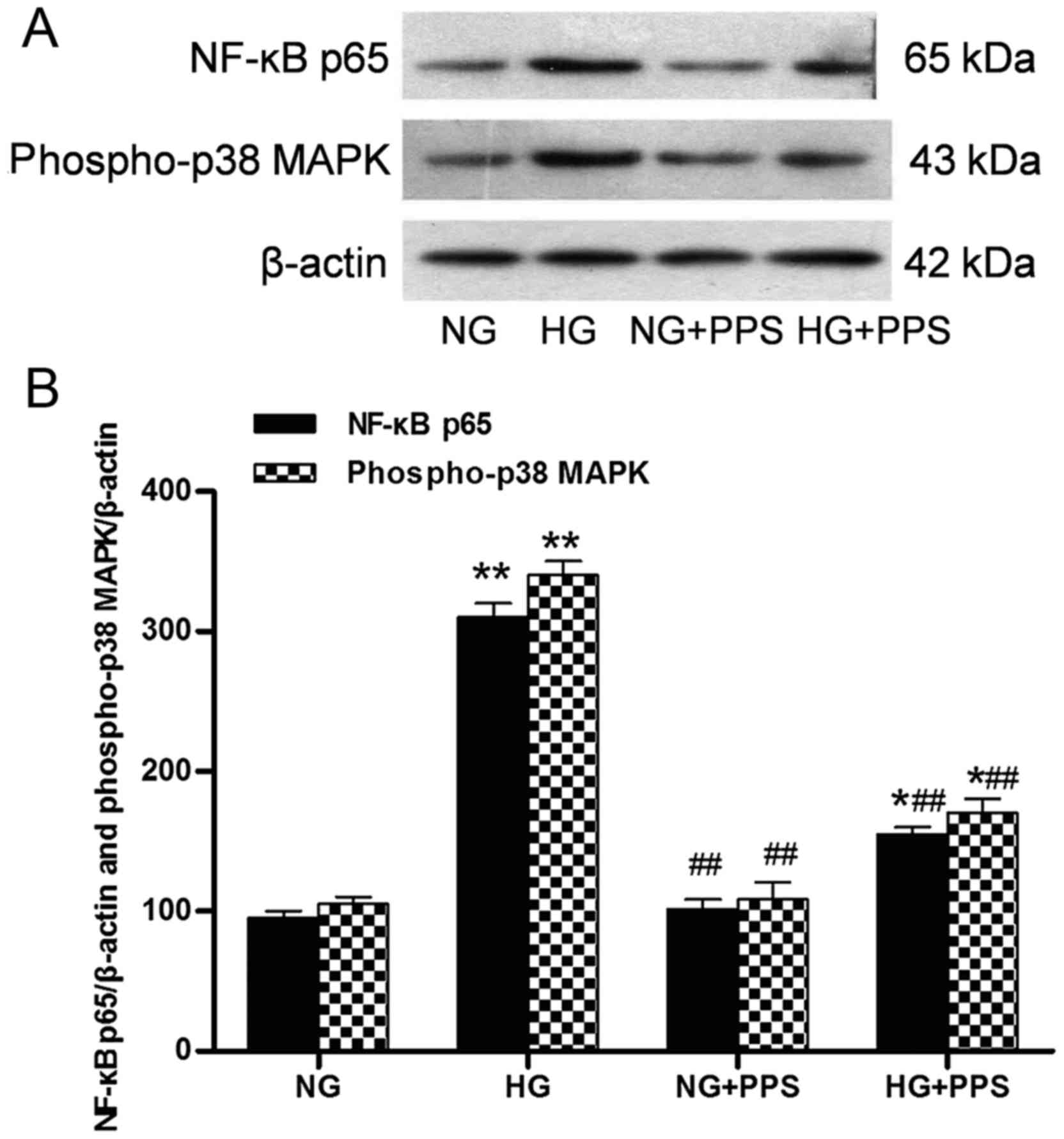
PPS inhibits HG-induced inflammatory
response in HK-2 cells
Our previous results demonstrated that PPS directly
affects the inflammatory action of TNF-α in proximal tubular cells
(19). The action of PPS was also
assessed. Treatment with HG stimulated a marked increase in the
activation of the inflammatory response, p38 MAPK and NF-κB, as
indicated by the increased levels of phospho-p38 MAPK and NF-κB p65
in the HG vs. the NG group. Compared with the HG group, PPS
treatment significantly inhibited this HG-stimulated NF-κB and p38
MAPK activation in the HG + PPS group (Fig. 5A and B). However, no significant
change was observed in the activation of NF-κB and p38 MAPK between
the NG and NG + PPS groups (Fig
5B), suggesting that PPS does not exert any toxic effect on
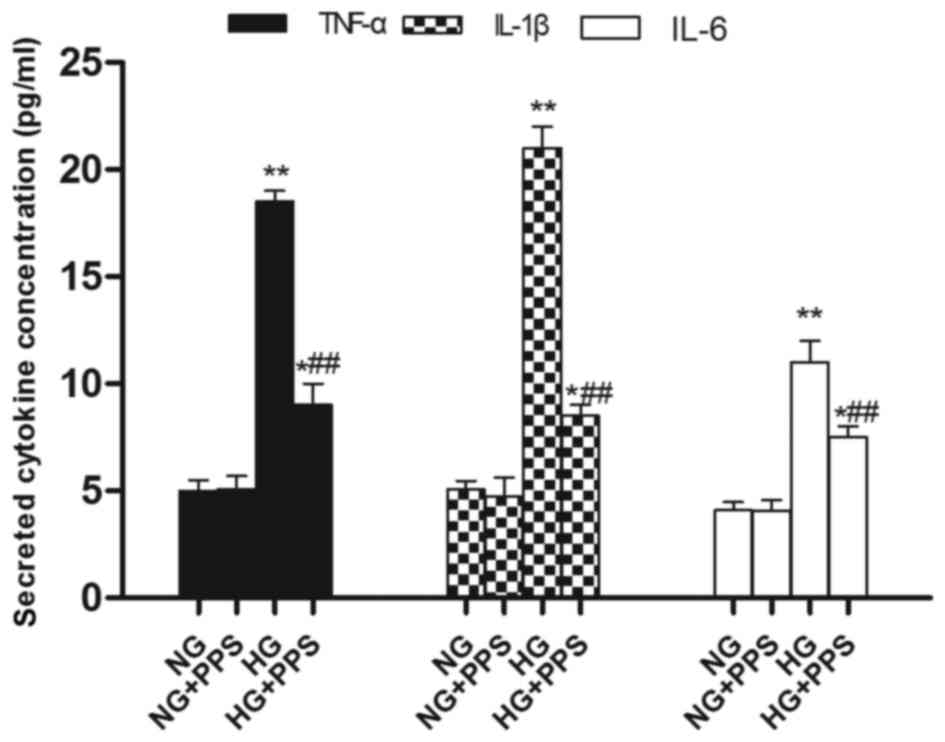
normal HK-2 cells. Compared with the NG group, the secretion of the
pro-inflammatory cytokines TNF-α, IL-1β and IL-6 was evidently
increased in the HG group, which was in agreement with the
activation of NF-κB and p38 MAPK (Fig. 6). Compared with the HG group, PPS
treatment markedly reduced HG-stimulated pro-inflammatory cytokine
production in the HG + PPS group (Fig. 6).
Discussion
In the present study, the anti-apoptotic effects and
anti-inflammatory properties of PPS were evaluated using HG-treated
HK-2 cells. The results revealed that PPS treatment ameliorated
apoptosis and inflammation by inhibiting the activation of p38 MAPK
stimulated by HG. Whether PPS treatment attenuated the HG-induced
cytotoxicity was also investigated by assessing the
nephroprotective effect of PPS in terms of cell viability. The
results demonstrated that PPS markedly enhanced cell viability,
suggesting that PPS protects against HG-induced cytotoxicity.
Accumulating evidence indicates that renal tubular
injury plays a critical role in the pathogenesis of human and
animal DN (7,23). Apoptosis is associated with the
progression of human DN (11,24). The apoptosis-associated marker
cleaved caspase-3/caspase-3 and Bax/Bcl-2 protein levels were
investigated, and PPS was found to modulate these protein levels,
indicating that PPS treatment suppressed HG-stimulated apoptosis in
HK-2 cells. The present study confirmed that the anti-apoptotic
properties of PPS required inhibition of the p38 MAPK pathways. Our
results are consistent with those of previous studies whereby HG
and hyperglycemia induced renal tubular cell apoptosis primarily
through regulating the Bcl-2/caspase/poly(ADP-ribose) polymerase
pathway (12,13,20,25). p38 MAPK is a member of the MAPK
superfamily, including JNK, ERK1/2 and ERK5, and it plays a key
role in renal cell death and the development of DN. It has been
demonstrated that phosphorylated p38 MAPK is expressed mainly in
diabetic tubules, which is associated with tubular lesions
(26), and coincides with the
localization of TGF-β (27),
indicating the role of p38 MAPK in TGF-β-mediated renal fibrosis.
Similarly, previous findings demonstrated the expression of
activated p38 MAPK and the production of fibrotic and inflammatory
markers, such as TGF-β, collagen Ⅲ and TNF-α, in diabetic mouse
kidneys (21). Accumulating
evidence indicates that activation of the p38 MAPK signaling
pathway induces renal tubular cell apoptosis, whereas suppressing
it significantly ameliorates tubular cell apoptosis and renal
dysfunction (20,28). In the present study, PPS, which
exerted its effects through downregulating p38 MAPK activation, may
provide a therapeutic alternative to prevent p38 MAPK-mediated DN.
In the present study, the p38 MAPK inhibitor SB203580 was used as
the positive control to investigate the role of PPS in
HG-stimulated apoptosis of HK-2 cells. The findings revealed that,
while administration of PPS or SB203580 alone attenuated HG-induced
HK-2 cell apoptosis, combination treatment resulted in a mild but
not statistically significant increase, indicating a dose-dependent
cytotoxic action of the inhibitor.
Although numerous factors contribute to the
pathogenesis of DN, it is increasingly considered as an
inflammatory disease (9,10). Infiltration by macrophages and
lymphocytes and the overproduction of pro-inflammatory chemokines
and cytokines are observed in renal diabetic tissues, which promote
further inflammation in the kidney (29-33). Monocyte chemoattractant protein-1
(MCP-1) promotes the recruitment of macrophages and upregulates the
production of pro-inflammatory cytokines (34). Diabetic db/db mice and
streptozotocin-stimulated diabetic mice, which lack the MCP-1 gene,
are resistant to DN (35,36). Intercellular adhesion molecule-1
(ICAM-1) facilitates the infiltration of leukocytes into diabetic
kidneys (37). Diabetic
ICAM-1-deficient mice exhibit amelioration of urinary albumin
excretion, glomerular hypertrophy and renal fibrosis, which are
correlated with a reduction in macrophage accumulation (38,39). NF-κB is one of the crucial
transcription factors involved in the progression of DN (40). In response to inflammatory and
stressful stimuli, p38 MAPK is activated by upstream kinases and
MAPK3/6, and is then trans-located to the nucleus, modulating the
activation and expression of various genes, including NF-κB
(15,17,37), pro-inflammatory cytokines (TNF-α,
IL-6 and IL-1β) (41), chemokines
(chemokine ligand 2), cell adhesion molecules (ICAM-1) and growth
factors (vascular endothelial growth factor, TGF-β and cytoplasmic
transmembrane growth factor) (5,42,43). Our findings confirmed that PPS
treatment significantly prevented NF-κB and p38 MAPK activation in
HG-treated HK-2 cells. Furthermore, the production of
pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β) was markedly
alleviated by PPS treatment. The results are consistent with those
of our previous findings, which demonstrated that PPS inhibited the
inflammation mediated by TNF-α, HG and AGEs (19). It is hypothesized that PPS blocked
the activation of NF-κB and reduced the expression of
pro-inflammatory cytokines by suppressing the p38 MAPK pathway. It
has been reported that apoptosis may be induced by HG through a
mechanism associated with inflammation (44). Collectively, based on the present
findings and previous results, the anti-inflammatory properties of
PPS are considered to be another possible mechanism underlying its
anti-apoptotic effects.
In conclusion, the present findings demonstrated
that PPS treatment attenuates HG-stimulated apoptosis and
inflammation in HK-2 cells through inhibition of p38 MAPK
activation, thereby preventing DN. The protective effect of PPS in
HK-2 cells is correlated with suppression of the p38 MAPK signaling
pathway. The present study provides experimental evidence to
investigate the possible use of PPS as a promising therapeutic
agent against DN.
Acknowledgments
The present study was supported by the Xiamen
Municipal Bureau of Science and Technology (grant no.
3502Z20134015), the Natural Science Foundation of Fujian Province
(grant no. 2015J01532) and the projects of the Medical and Health
Technology Program in Zhejiang Province (grant no. 2018KY679).
References
|
1
|
Gross JL, de Azevedo MJ, Silveiro SP,
Canani LH, Caramori ML and Zelmanovitz T: Diabetic nephropathy:
Diagnosis, prevention, and treatment. Diabetes Care. 28:164–176.
2005. View Article : Google Scholar
|
|
2
|
Tan AL, Forbes JM and Cooper ME: AGE,
RAGE, and ROS in diabetic nephropathy. Semin Nephrol. 27:130–143.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Magee GM, Bilous RW, Cardwell CR, Hunter
SJ, Kee F and Fogarty DG: Is hyperfiltration associated with the
future risk of developing diabetic nephropathy? A meta-analysis.
Diabetologia. 52:691–697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishikawa T, Edelstein D, Du XL, Yamagishi
S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP,
et al: Normalizing mitochondrial superoxide production blocks three
pathways of hyperglycaemic damage. Nature. 404:787–790. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Navarro-González JF and Mora-Fernández C:
The role of inflammatory cytokines in diabetic nephropathy. J Am
Soc Nephrol. 19:433–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Noh H and King GL: The role of protein
kinase C activation in diabetic nephropathy. Kidney Int Suppl.
72(Suppl): S49–S53. 2007. View Article : Google Scholar
|
|
7
|
Magri CJ and Fava S: The role of tubular
injury in diabetic nephropathy. Eur J Intern Med. 20:551–555. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gilbert RE and Cooper ME: The
tubulointerstitium in progressive diabetic kidney disease: More
than an aftermath of glomerular injury. Kidney Int. 56:1627–1637.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mora C and Navarro JF: Inflammation and
diabetic nephropathy. Curr Diab Rep. 6:463–468. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tuttle KR: Linking metabolism and
immunology: Diabetic nephropathy is an inflammatory disease. J Am
Soc Nephrol. 16:1537–1538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar D, Robertson S and Burns KD:
Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem.
259:67–70. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Verzola D, Bertolotto MB, Villaggio B,
Ottonello L, Dallegri F, Salvatore F, Berruti V, Gandolfo MT,
Garibotto G and Deferrari G: Oxidative stress mediates apoptotic
changes induced by hyperglycemia in human tubular kidney cells. J
Am Soc Nephrol. 15(Suppl 1): S85–S87. 2004. View Article : Google Scholar
|
|
13
|
Verzola D, Bertolotto MB, Villaggio B,
Ottonello L, Dallegri F, Frumento G, Berruti V, Gandolfo MT,
Garibotto G and Deferran G: Taurine prevents apoptosis induced by
high ambient glucose in human tubule renal cells. J Investig Med.
50:443–451. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stambe C, Nikolic-Paterson DJ, Hill PA,
Dowling J and Atkins RC: p38 mitogen-activated protein kinase
activation and cell localization in human glomerulonephritis:
Correlation with renal injury. J Am Soc Nephrol. 15:326–336. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adhikary L, Chow F, Nikolic-Paterson DJ,
Stambe C, Dowling J, Atkins RC and Tesch GH: Abnormal p38
mitogen-activated protein kinase signalling in human and
experimental diabetic nephropathy. Diabetologia. 47:1210–1222.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen H, Brahmbhatt S, Gupta A and Sharma
AC: Duration of streptozotocin-induced diabetes differentially
affects p38-mitogen-activated protein kinase (MAPK) phosphorylation
in renal and vascular dysfunction. Cardiovasc Diabetol. 4:32005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du Y, Tang J, Li G, Berti-Mattera L, Lee
CA, Bartkowski D, Gale D, Monahan J, Niesman MR, Alton G, et al:
Effects of p38 MAPK inhibition on early stages of diabetic
retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci.
51:2158–2164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nickel JC, Barkin J, Forrest J, Mosbaugh
PG, Hernandez-Graulau J, Kaufman D, Lloyd K, Evans RJ, Parsons CL
and Atkinson LE; Elmiron Study Group: Randomized, double-blind,
dose-ranging study of pentosan polysulfate sodium for interstitial
cystitis. Urology. 65:654–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu J, Guan TJ, Zheng S, Grosjean F, Liu W,
Xiong H, Gordon R, Vlassara H, Striker GE and Zheng F: Inhibition
of inflammation by pentosan polysulfate impedes the development and
progression of severe diabetic nephropathy in aging C57B6 mice. Lab
Invest. 91:1459–1471. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu H, Shi Y, Deng X, Su Y, Du C, Wei J,
Ren Y, Wu M, Hou Y and Duan H: Inhibition of c-Src/p38 MAPK pathway
ameliorates renal tubular epithelial cells apoptosis in db/db mice.
Mol Cell Endocrinol. 417:27–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rane MJ, Song Y, Jin S, Barati MT, Wu R,
Kausar H, Tan Y, Wang Y, Zhou G, Klein JB, et al: Interplay between
Akt and p38 MAPK pathways in the regulation of renal tubular cell
apoptosis associated with diabetic nephropathy. Am J Physiol Renal
Physiol. 298:F49–F61. 2010. View Article : Google Scholar :
|
|
22
|
Iwayama H and Ueda N: Role of
mitochondrial Bax, caspases, and MAPKs for ceramide-induced
apoptosis in renal proximal tubular cells. Mol Cell Biochem.
379:37–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blantz RC and Singh P: Glomerular and
tubular function in the diabetic kidney. Adv Chronic Kidney Dis.
21:297–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baba K, Minatoguchi S, Sano H, Kagawa T,
Murata I, Takemura G, Hirano T, Ohashi H, Takemura M, Fujiwara T,
et al: Involvement of apoptosis in patients with diabetic
nephropathy: A study on plasma soluble Fas levels and pathological
findings. Nephrology (Carlton). 9:94–99. 2004. View Article : Google Scholar
|
|
25
|
Habib SL: Diabetes and renal tubular cell
apoptosis. World J Diabetes. 4:27–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sakai N, Wada T, Furuichi K, Iwata Y,
Yoshimoto K, Kitagawa K, Kokubo S, Kobayashi M, Hara A, Yamahana J,
et al: Involvement of extracellular signal-regulated kinase and p38
in human diabetic nephropathy. Am J Kidney Dis. 45:54–65. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fujita H, Omori S, Ishikura K, Hida M and
Awazu M: ERK and p38 mediate high-glucose-induced hypertrophy and
TGF-beta expression in renal tubular cells. Am J Physiol Renal
Physiol. 286:F120–F126. 2004. View Article : Google Scholar
|
|
28
|
Bocanegra V, Gil Lorenzo AF, Cacciamani V,
Benardón ME, Costantino VV and Vallés PG: RhoA and MAPK signal
transduction pathways regulate NHE1-dependent proximal tubule cell
apoptosis after mechanical stretch. Am J Physiol Renal Physiol.
307:F881–F889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galkina E and Ley K: Leukocyte recruitment
and vascular injury in diabetic nephropathy. J Am Soc Nephrol.
17:368–377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chow F, Ozols E, Nikolic-Paterson DJ,
Atkins RC and Tesch GH: Macrophages in mouse type 2 diabetic
nephropathy: Correlation with diabetic state and progressive renal
injury. Kidney Int. 65:116–128. 2004. View Article : Google Scholar
|
|
31
|
Sassy-Prigent C, Heudes D, Mandet C,
Bélair MF, Michel O, Perdereau B, Bariéty J and Bruneval P: Early
glomerular macrophage recruitment in streptozotocin-induced
diabetic rats. Diabetes. 49:466–475. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Furuta T, Saito T, Ootaka T, Soma J, Obara
K, Abe K and Yoshinaga K: The role of macrophages in diabetic
glomerulosclerosis. Am J Kidney Dis. 21:480–485. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bohle A, Wehrmann M, Bogenschütz O, Batz
C, Müller CA and Müller GA: The pathogenesis of chronic renal
failure in diabetic nephropathy. Investigation of 488 cases of
diabetic glomerulosclerosis. Pathol Res Pract. 187:251–259. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Viedt C and Orth SR: Monocyte
chemoattractant protein-1 (MCP-1) in the kidney: Does it more than
simply attract monocytes. Nephrol Dial Transplant. 17:2043–2047.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chow FY, Nikolic-Paterson DJ, Ozols E,
Atkins RC, Rollin BJ and Tesch GH: Monocyte chemoattractant
protein-1 promotes the development of diabetic renal injury in
streptozotocin-treated mice. Kidney Int. 69:73–80. 2006. View Article : Google Scholar
|
|
36
|
Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols
E, Rollins BJ and Tesch GH: Monocyte chemoattractant
protein-1-induced tissue inflammation is critical for the
development of renal injury but not type 2 diabetes in obese db/db
mice. Diabetologia. 50:471–480. 2007. View Article : Google Scholar
|
|
37
|
Lim AK and Tesch GH: Inflammation in
diabetic nephropathy. Mediators Inflamm. 146154:2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chow FY, Nikolic-Paterson DJ, Ozols E,
Atkins RC and Tesch GH: Intercellular adhesion molecule-1
deficiency is protective against nephropathy in type 2 diabetic
db/db mice. J Am Soc Nephrol. 16:1711–1722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Okada S, Shikata K, Matsuda M, Ogawa D,
Usui H, Kido Y, Nagase R, Wada J, Shikata Y and Makino H:
Intercellular adhesion molecule-1-deficient mice are resistant
against renal injury after induction of diabetes. Diabetes.
52:2586–2593. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sanchez AP and Sharma K: Transcription
factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol
Med. 11:e132009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saklatvala J: The p38 MAP kinase pathway
as a therapeutic target in inflammatory disease. Curr Opin
Pharmacol. 4:372–377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mezzano S, Aros C, Droguett A, Burgos ME,
Ardiles L, Flores C, Schneider H, Ruiz-Ortega M and Egido J:
NF-kappaB activation and overexpression of regulated genes in human
diabetic nephropathy. Nephrol Dial Transplant. 19:2505–2512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang B, Hodgkinson A, Oates PJ, Millward
BA and Demaine AG: High glucose induction of DNA-binding activity
of the transcription factor NFkappaB in patients with diabetic
nephropathy. Biochim Biophys Acta. 1782:295–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lewko B and Stepinski J: Hyperglycemia and
mechanical stress: Targeting the renal podocyte. J Cell Physiol.
221:288–295. 2009. View Article : Google Scholar : PubMed/NCBI
|