Introduction
Bronchial asthma is a complex syndrome that presents
numerous clinical phenotypes in adults and children (1). Worldwide, 334 million people suffer
from asthma, which is also one of the most common chronic disorders
and an increasing burden on healthcare services globally (2). One of the major physiological
alterations associated with asthma is narrowing of the airways due
to exposure to bronchoconstrictors or allergens, which leads to
hyperresponsiveness, obstruction and remodeling of the airways.
Furthermore, structural alterations are associated with airway
remodeling, including goblet cell hyperplasia or metaplasia,
epithelial cell shedding, subepithelial fibrosis, smooth muscle
cell hyperplasia, angiogenesis and edema (3). Epithelial-mesenchymal transition
(EMT) has a key role in airway remodeling, which may account for
the accumulation of subepithelial mesenchymal cells, thus
contributing to airway hyperresponsiveness and the increasing mass
of contractile cells (4). During
the process of EMT, epithelial cells exhibit enhanced motility and
invasive capacity via the downregulation of epithelial markers,
including E-cadherin, and the increased expression of mesenchymal
proteins (5,6). EMT can be induced by growth factors,
including trans-forming growth factor (TGF)-β, which is secreted
from numerous cell types, such as infiltrating immune cells and
airway epithelial cells (7,8).
Although current therapies, which include long-acting β2
agonists, corticosteroids and leukotriene antagonists, remain
effective in reducing inflammation, the majority of them are
ineffective at preventing or suppressing airway remodeling
(9). Therefore, agents that
improve airway structure and reverse airway remodeling are
required.
Epigallocatechin-3-gallate (EGCG), which is a type
of polyphenol, is the most potent ingredient in green tea, and
exhibits antibacterial, antiviral, antioxidative, anticancer and
chemopreventive activities (10,11). Notably, EGCG has recently been
reported to contribute to the prevention of various degenerative
diseases, including cardiovascular diseases, arthritis and diabetes
(12,13). Specifically, the number of studies
investigating the protective effects of EGCG against asthma and
lung diseases has risen dramatically (14-16). EGCG has been reported to inhibit
nicotine-induced migration and invasion via the suppression of
angiogenesis and EMT in non-small cell lung cancer cells (17). However, the exact cellular
mechanism underlying the protective effects of EGCG against asthma
has yet to be revealed.
In the present study, ovalbumin (OVA)-challenged
asthmatic mice and TGF-β1-induced 16HBE human bronchial epithelial
cells were used as in vivo and in vitro models,
respectively. The protective effects of EGCG against TGF-β1-induced
EMT and migration of 16HBE cells, and the underlying mechanisms
associated with phosphatidylinositol 3-kinase/protein kinase B
(PI3K/AKT) signaling pathway regulation, were investigated. The
present study provides a novel insight into understanding the
protective effects of EGCG against airway remodeling in asthma, and
proposes that EGCG may be useful as an adjuvant therapy for
bronchial asthma.
Materials and methods
Ethics statement
Male Balb/c mice (6 mice each group, age, 6-8 weeks;
weight, 20-22 g) were used in the present study. Mice were procured
from Beijing Vital River Laboratory Animal Technology Co., Ltd.
(Beijing, China) and were acclimated for 1 week under standard
laboratory conditions. Mice were acclimatized for a week under
standard laboratory conditions and provided with standard
irradiated chow diet ad libitum and maintained in a specific
pathogen-free state under a strict light cycle (12:12 h) at a
temperature of 22±2°C and a relative humidity of 50±10%. All animal
experiments were performed according to the Institutional Animal
Care and Use Committees and were approved by the China Medical
University Animal Care and Use Committee (Shenyang, China).
OVA-induced asthma model
Mice were sensitized with 10 µg OVA
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) mixed with 2 mg
aluminum hydroxide in saline by intraperitoneal (i.p.) injection
once a week for 3 weeks. After 2 weeks, the mice were challenged
with aerosolized 1% OVA for 30 min three times a week for 8 weeks,
and were euthanized 24 h after the last challenge. The
non-sensitized mice were nebulized by saline in the same manner.
All mice were sacrificed at day 56 (24 h after the last OVA
treatment). The lungs were lavaged with 0.8 ml cold PBS and
bronchoalveolar lavage fluid (BALF) was processed for differential
cell counting, and determination of cytokines and chemokines. For
another set of experiments, the lung tissues were fixed with 4%
paraformaldehyde (Sinopharm Chemical Reagent Co., Ltd., Shanghai,
China) and histological examination was performed with hematoxylin
and eosin (H&E) staining.
BALF differential cell count
BALF samples were initially centrifuged at 800 × g
for 10 min and supernatants were frozen at −70°C for assessment of
inflammatory chemokines/ cytokines. The total number of cells was
counted using a cell counter and was recorded as the total number
of inflammatory cells per ml. The cell suspension was harvested for
cytokine analysis and the pellet was smeared onto slides for cell
classification and counting. The cell smear was stained with
Wright-Giemsa (Nanjing Jiangcheng Bioengineering Institute,
Nanjing, China), after which the number of leukocytes, eosinophils,
macrophages and neutrophils were recorded by counting under a light
microscopy.
Treatment with EGCG
EGCG (0.5 mg/ml; Sigma-Aldrich; Merck KGaA) was
administered in drinking water, which was provided ad
libitum 1 h after the first OVA challenge until the mice were
sacrificed. Dexamethasone (DEX, 1 mg/kg; Dalian Meilun Biotech Co.,
Ltd., Dalian, China), which was used as a positive control, was
injected (i.p.) 1 h prior to every nebulization.
Cell culture
The 16HBE human bronchial epithelial cell line
(FMGBio Co., Ltd., Shanghai, China) was cultured in Dulbecco's
modified Eagle's medium containing 100 U penicillin, 100 mg/ml
streptomycin (all Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and 10% fetal bovine serum (FBS; Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) in a humidified incubator containing
5% CO2 at 37°C. Cells were initially pretreated with 5
ng/ml TGF-β1 (PeproTech, Inc., Rocky Hill, NJ, USA) at 37°C for 24
h and were then cotreated with 50 µM EGCG (Sigma-Aldrich;
Merck KGaA) at 37°C for a further 24 h. In order to investigate the
effects of the PI3K pathway, cells were pretreated with LY294002
(Sigma-Aldrich; Merck KGaA) at 37°C for 1 h prior to treatment with
TGF-β.
ELISA
Samples from mice treated with or without OVA and/or
EGCG were harvested and washed twice in cold PBS. The
concentrations of interleukin (IL)-4, IL-5 and TGF-β1 in the BALF
or whole lung tissues were determined using mouse IL-4/-5 and mouse
TGF-β1 detection kits (EK0405, EK0408 and EK0515; Wuhan Boster
Biological Technology, Ltd., Wuhan, China) according to the
manufacturer's protocols. Protein concentrations were determined
using a bicinchoninic acid (BCA) protein assay kit (Beyotime
Institute of Biotechnology, Shanghai, China) according to the
manufacturer's protocol. All of the samples, and the standards
diluted to different concentrations, were added to the wells at a
volume of 100 µl. The absorbance was measured using a
microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) at
450 nm, and the concentration levels of IL-4, IL-5 and TGF-β1 were
calculated from the standard curve.
Western blot analysis
Proteins were harvested from the lung tissues of
asthmatic mice treated with or without EGCG, and from cells
pretreated with 5 ng/ml TGF-β1 for 24 h and then coincubated with
50 µM EGCG for a further 24 h. Tissue or cell extracts were
prepared in ice-cold radioimmunoprecipitation buffer plus
phenylmethylsulfonyl fluoride (both Beyotime Institute of
Biotechnology) for 30 min on ice. The lysates were centrifuged at
20,000 × g for 10 min at 4̊C. Subsequently, protein concentration
was determined using a BCA protein assay kit according to the
manufacturer's protocol. Total proteins (40 µg) were
separated by 5-10% SDS-PAGE for 2.5 h, after which the separated
proteins were transferred to polyvinylidene difluoride membranes
(EMD Millipore, Billerica, MA, USA) for 1.5 h at 80 V. After that,
the PVDF membranes were blocked with TTBS buffer sharking for 5
min, then the membranes were transferred into blocking buffer (95%
TTBS buffer and 5% skimmed milk powder) sharking for 1 h at room
temperature. Each membrane was then incubated with the following
primary antibodies: Anti-phosphorylated (p)-AKT (cat. no.
sc-135651) and anti-AKT (cat. no. sc-8312) (1:200; both Santa Cruz
Biotechnology, Inc., Dallas, TX, USA); anti-phosphatase and tensin
homolog (PTEN; cat. no. ab32199, 1:1,000; Abcam, Cambridge, MA,
USA); anti-PI3K (cat. no. BA1352, 1:400; Wuhan Boster Biological
Technology, Ltd.); anti-p-PI3K (cat. no. bs-5538R, 1:500; BIOSS,
Beijing, China); anti-E-cadherin (cat. no. WL00941), anti-α-smooth
muscle actin (α-SMA; cat. no. WL0002a) and anti-β-actin (cat. no.
WL0001) (1:1,000; Wanleibio Co., Ltd., Shenyang, China) overnight
at 4̊C. Subsequently, the membranes were washed with TBST buffer
(150 mM NaCl, 10 mM Tris-HCl and 1% Tween-20) for 30 min and were
incubated with horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G (cat. no. A0208, 1:5,000; Beyotime Institute of
Biotechnology) at room temperature for 1 h. Finally, the blots were
developed using an enhanced chemiluminescence detection (ECL) kit
(ECL detection reagent; 7Sea Biotech, Shanghai, China). β-actin was
used as an internal positive control. The relative amounts of the
transferred proteins were semi-quantified by scanning the
autoradiographic films using a gel densitometer and normalizing the
levels to those of β-actin. Semi-quantitative analysis was
conducted using Gel-Pro Analyzer software (version 4.0; Media
Cybernetics, Inc., Rockville, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from cells pretreated with 5 ng/ml
TGF-β1 for 24 h then coincubated with 50 µM EGCG for a
further 24 h. Total RNA was extracted using a high purity total RNA
extraction kit (BioTeke Corporation, Beijing, China) according to
the manufacturer's protocol. mRNA expression levels were then
examined by RT-qPCR using total RNA. cDNA was synthesized from
total RNA using the Super M-MLV RT kit at 25°C for 10 min, at 42°C
for 50 min, at 95°C for 5 min (BioTeke Corporation). cDNA was then
amplified by qPCR. Primers used for RT-qPCR were as follows:
E-cadherin forward, 5′-ATGCCGCCATCGCTTACAC-3′ and reverse,
5′-CGACGTTAGCCTCGTTCTCA-3′; α-SMA forward,
5′-CCTGAAGAGCATCCCACCCT-3′ and reverse,
5′-ACCATCTCCAGAGTCCAGCACG-3′; PTEN forward,
5′-AAGGAAGTGAATCTGTATTGGGGT-3′ and reverse,
5′-TTGTTGCTGTGTTTCTTACCTATG-3′; and β-actin forward,
5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT-3′. Each RT-qPCR experiment consisted
of a predenaturation cycle at 95°C for 10 min; subsequently, the
cDNA was amplified for 40 cycles, including a denaturation step at
95°C for 10 sec, a primer annealing step at 60°C for 20 sec and an
extension step at 72°C for 30 sec; finally, the samples were
maintained at 4°C for 5 min. The PCR results were verified by
varying the number of PCR cycles for each cDNA and set of primers.
qPCR was performed using an Exicycler™ 96 Real-Time Quantitative
Thermal block (Bioneer Corporation, Daejeon, Korea) with β-actin as
a control. RT-qPCR was performed at least in quadruplicate. The
2−ΔΔCt method was used to determine the relative mRNA
folding changes (18). Data are
presented as the mean of the results from at least three
experiments.
Transwell assay
Transwell chambers (Corning Inc., Corning, NY, USA)
were used to measure cell migration. Cells that were pretreated
with 5 ng/ml TGF-β1 for 24 h or LY294002 for 1 h, then coincubated
with 50 µM EGCG for a further 24 h, were seeded into the
upper chamber at a density of 2×104 in 200 µl
fresh medium without FBS. In the lower chamber, 800 µl
medium supplemented with 20% FBS was added. Three duplicate wells
were set up for each group. Following 24 h incubation at 37°C,
cells infiltrating through the filter were fixed with 4%
paraformaldehyde for 20 min and were stained with 0.5% crystal
violet (Amresco, LLC, Solon, OH, USA) for 5 min. Cell numbers on
the lower membrane were counted under high power lens (×200
magnification) in five random visual fields using an inverted
microscope (Motic Incorporation, Ltd., Hong Kong, China).
Wound healing assay
Cells were seeded onto 6-well plates until they
reached 80-90% confluence. Subsequently, the cells were pretreated
with 5 ng/ml TGF-β1 for 24 h or LY294002 for 1 h, and were then
coincubated with 50 µM EGCG for a further 24 h. The medium
was then discarded and a straight scratch was made to the cell
layer using a 200-µl pipette tip, to simulate a wound. The
plates were washed with serum-free medium, and the cells were
observed and images were captured under a inverted phase contrast
microscope (AE31; Motic Incorporation, Ltd.) to ensure there were
enough cells in the leading edge of the wound. Cell culture
continued in serum-free medium at 37°C for 12 and 24 h, after which
images were captured. The rate of migration of the cells was
calculated by measuring the distance traveled toward the center of
the wound.
Histological analysis
The lung tissues collected from asthmatic mice
treated with or without EGCG were fixed with 4% paraformaldehyde
for 30 min, embedded in paraffin wax and then sectioned at room
temperature into 5-µm slices. The sections were stained with
hematoxylin (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) and eosin (Sinopharm Chemical Reagent Co., Ltd.).
Morph ological features of the sections were observed under a light
microscope (DP73; Olympus Corporation, Tokyo, Japan).
Immunofluorescence
Immunofluorescent staining was perfo- rmed on
paraffin-embedded lung tissue sections obtained from asthmatic
mice. Samples from lung tissues were isolated, fixed with 4%
paraformaldehyde at room temperature for 20 min and embedded in
paraffin wax. Microwave antigen retrieval was conducted in citrate
buffer for 10 min, followed by 15 min of cooling at room
temperature. Slides and tissue sections were blocked and
permeabilized in 2% goat serum (OriGene Technologies, Inc.,
Beijing, China) at room temperature for 30 min, and were then
incubated with anti-E-cadherin (cat. no. sc-8426, 1:50; Santa Cruz
Biotechnology, Inc.) and anti-α-SMA (cat. no. 14395-1-AP, 1:200;
ProteinTech Group, Inc., Chicago, IL, USA) antibodies at 4°C
overnight. Sections were then incubated with a secondary antibody
[FITC-labeled goat anti-mouse IgG (H+L), cat. no. A0568; and
Cy3-labeled goat anti-rabbit IgG (H+L), cat. no. A0516; Beyotime
Institute of Biotechnology] in blocking reagent at room temperature
for 90 min. Both antibodies were diluted in 1% bull serum albumin
in PBS. Samples were counterstained with DAPI (Beyotime Institute
of Biotechnology) and observed under an inverted fluorescence
microscope (Olympus Corporation); all images were taken at ×400
magnification. Semi-quantitative analysis of immunofluorescent
staining was conducted using Image-Pro Plus 6.0 (Media Cybernetics,
Inc.) software and the results are presented as average optical
density (AOD) values.
Statistical analysis
All data are expressed as the mean ± standard
deviation from at least three experiments. Data were analyzed using
the one-way analysis of variance followed by Bonferroni's multiple
comparisons, where appropriate. P<0.05 was considered to
indicate a statistically significant difference. Statistical
analyses were performed using Prism version 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA).
Results
EGCG inhibits inflammation and
inflammatory cell infiltration into the lungs of OVA-challenged
asthmatic mice
During the pathological process of chronic asthma,
antigen-induced airway inflammation is characterized by an
increased number of inflammatory cells into the lung subepithelial
spaces or the airways (19).
Therefore, the effects of EGCG on the accumulation of inflammatory
cells in the BALF and their infiltration into the lungs were
examined following an OVA challenge. As shown in Fig. 1A, there was a significant increase
in the number of total leukocytes in the BALF from OVA-sensitized
and challenged mice (from 6.25±1.08 to
95.5±19.60×104/ml, P<0.01). Conversely, the number of
total leukocytes was significantly decreased in the BALF of
OVA-challenged mice treated with EGCG (from 95.5±19.60 to
52.8±12.1×104/ml, P<0.01). Furthermore, the number of
macrophages was significantly decreased (from
32.7±8.20×104/ml in the OVA group to
19.91±6.79×104/ml in the OVA + EGCG group, P<0.01),
whereas the number of eosinophils (from
44.58±12.04×104/ml in the OVA group to
20.0±2.70×104/ml in the OVA + EGCG group, P<0.01) and
neutrophils (from 951.44±231.26×104/ml in the OVA group
to 620.67±205.36 104/ml in the OVA + EGCG group,
P<0.01) were decreased by 40-50% in the BALF of EGCG-treated
OVA-challenged mice compared with in OVA-challenged mice. In
addition, the effects of EGCG on asthmatic mice were comparable to
those of the standard drug DEX.
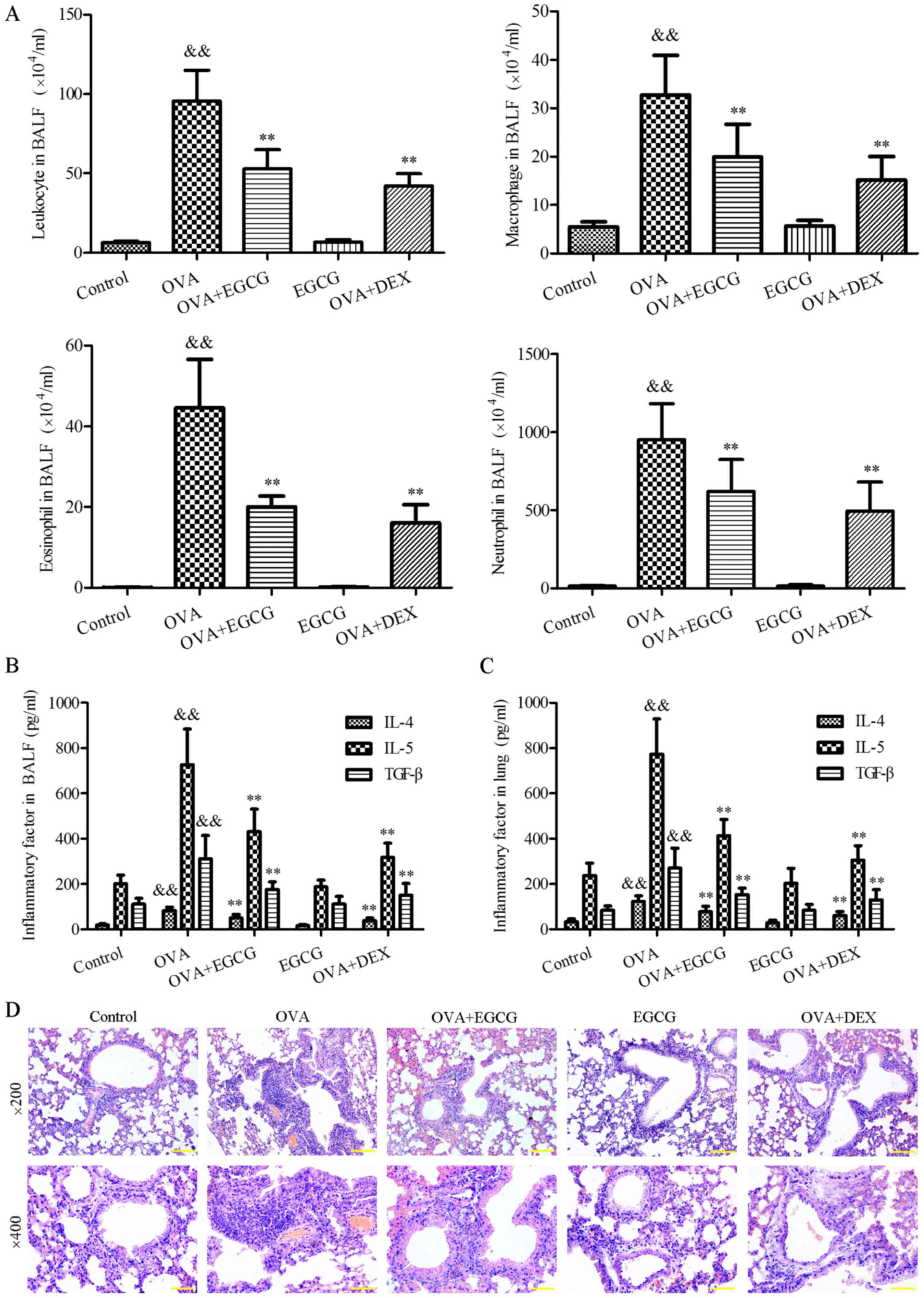 | Figure 1EGCG inhibits lung inflammation and
inflammatory cell infiltration into the lungs of OVA-challenged
asthmatic mice. (A) EGCG inhibited leukocyte accumulation in the
BALF of OVA-challenged asthmatic mice. The total, as well as
differential, cell counts were measured in the BALF collected
following the final OVA challenge. The number of total cells,
macrophages, eosinophils and neutrophils per ml of BALF is
presented as the mean ± standard deviation (n=6). EGCG inhibited
the expression of IL-4, IL-5 and TGF-β in (B) BALF and (C) lung
tissues following an OVA challenge. IL-4, IL-5 and TGF-β levels
were measured by ELISA. (D) H&E staining. Fixed lungs from the
different experimental groups were sectioned, stained with H&E
and examined under a light microscope. A representative image from
each group is shown (n=6, ×200 and ×400 magnification). DEX was
used as a positive control. Data are are presented as the mean ±
standard deviation. &&P<0.01 vs. the control
group; **P<0.01 vs. the OVA group. BALF,
bronchoalveolar lavage fluid; DEX, dexamethasone; EGCG,
epigallocatechin-3-gallate; H&E, hematoxylin and eosin; IL,
interleukin; OVA, ovalbumin; TGF-β, transforming growth
factor-β. |
Protein levels of IL-4, IL-5 and TGF-β1 were
detected in the BALF (Fig. 1B)
and lung tissues (Fig. 1C) of
mice following an OVA challenge using ELISA. As shown in Fig. 1B, OVA induced a significant
increase in these cytokines in the BALF compared with in the
control group. The concentrations of IL-4, IL-5 and TGF-β1 in the
control group vs. the OVA group were: 33.32±12.72 vs. 122.94±25.22
pg/ml, 237.80±54.10 vs. 772.11±156.86 pg/ml and 83.41±19.36 vs.
270.24±88.77 pg/ml, respectively (P<0.01, Fig. 1B). Administration of EGCG during
OVA challenge normalized the elevated cytokine levels, with potent
effects on IL-4, IL-5 and TGF-β1 in the OVA + EGCG group. The
concentrations of the cytokines were as follows: IL-4, 122.94±25.22
pg/ml in the OVA group vs. 77.02±24.95 pg/ml in the OVA + EGCG
group; IL-5, 772.11±156.86 pg/ml in the OVA group vs. 413.11±73.11
pg/ml in the OVA + EGCG group; and TGF-β1, 270.24±88.77 pg/ml in
the OVA group vs. 152.92±29.23 pg/ml in the OVA + EGCG group
(P<0.01; Fig. 1B). In
addition, the levels of IL-4, IL-5 and TGF-β1 in the lung tissues
from OVA-challenged mice exhibited similar trends to those in the
BALF. The results demonstrated that OVA increased the IL-4 levels
from 18.88±5.33 pg/ml in the control group to 82.02±15.70 pg/ml in
the OVA group. In addition, IL-5 levels were increased from
201.37±37.18 pg/ml in the control group to 726.11±157.35 pg/ml in
the OVA group, whereas TGF-β1 were increased from 110.86±25.70
pg/ml in the control group vs. 311.44±102.33 pg/ml in the OVA group
(P<0.01, Fig. 1C). However,
administration of EGCG inhibited these increased cytokine levels
(IL-4, from 82.02±15.70 to 49.85±15.92 pg/ml; IL-5, from
726.11±157.35 to 431.13±99.40 pg/ml; and TGF-β1 from 311.44±102.33
to 176.25±33.67 pg/ml, P<0.01, Fig. 1C), whereas administration of EGCG
alone (EGCG group) had no effect on cytokine response in the BALF
and lung tissues compared with in the control group. In addition,
the effects of EGCG were compared with those of DEX; no significant
differences were detected between the two groups.
H&E staining revealed that inflammatory cell
infiltration, particularly of eosinophils (red staining) as well as
other inflammatory cells (blue staining), into the peribronchial
and perivascular spaces of OVA-challenged mice was increased
(Fig. 1D). Conversely, in
EGCG-treated asthmatic mice, infiltration of eosinophils and other
inflammatory cells into the lung tissue was markedly decreased
compared with in OVA-challenged mice (Fig. 1D). These results indicated that
EGCG inhibits lung inflammation in OVA-challenged asthmatic mice
with the same effect as DEX, which is an anti-inflammatory and
anti-immune disease drug.
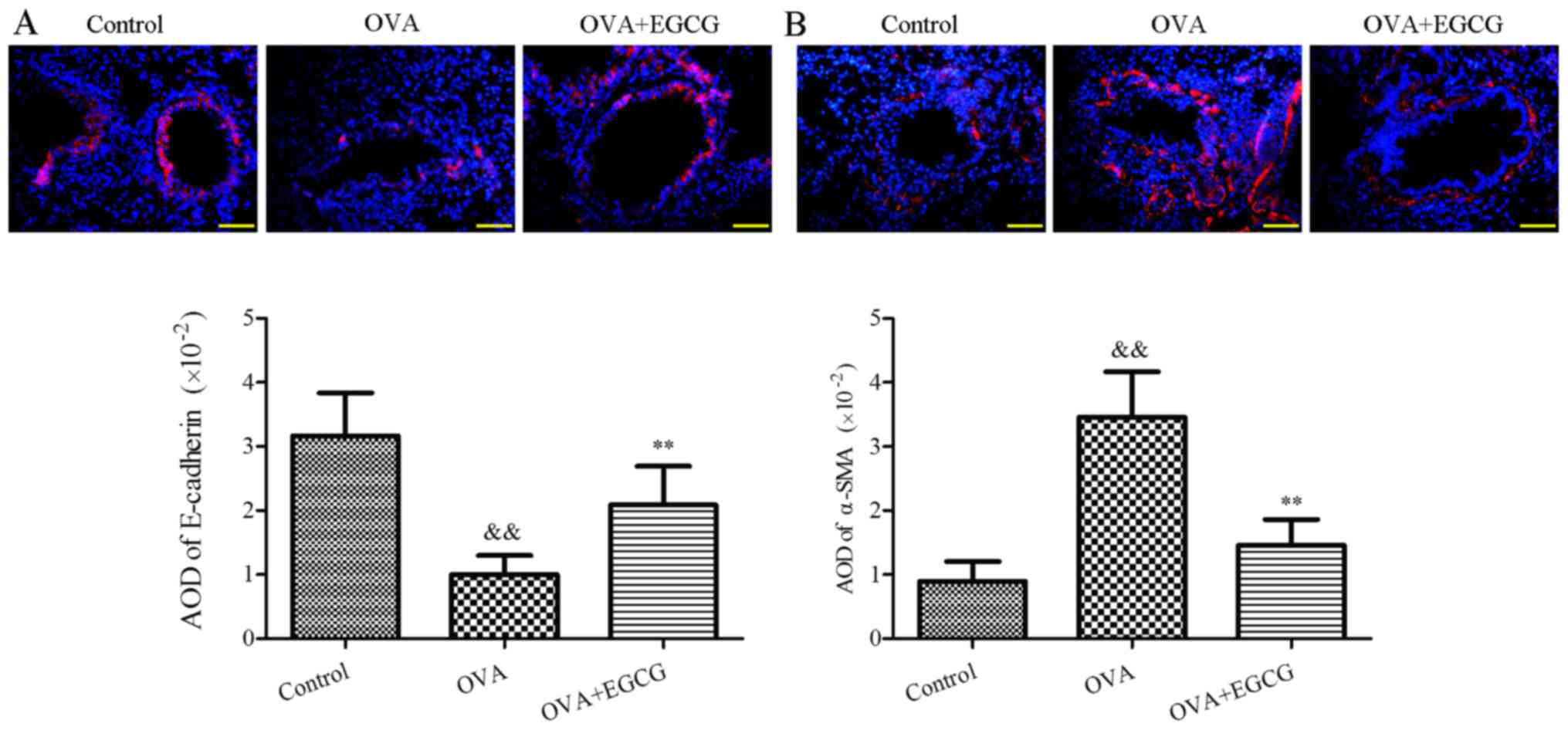
EGCG suppresses airway EMT in
OVA-challenged asthmatic mice
The present study demonstrated that the expression
levels of TGF-β1 were markedly enhanced in OVA-challenged mice
(Fig. 1); the increased levels of
TGF-β1 may initiate signaling cascades in epithelial cells that
lead to EMT. To deter-mine the effects of EGCG on OVA-challenged
asthmatic mice, the expression levels of the epithelial marker
E-cadherin and the mesenchymal marker α-SMA were detected in the
airways of asthmatic mice with or without EGCG treatment by
immunofluorescence analysis. The results indicated that the airways
of mice challenged with OVA exhibited reduced E-cadherin expression
and increased α-SMA expression compared with in the healthy control
mice; however, EGCG increased the expression of E-cadherin and
decreased the expression of α-SMA (Fig. 2A and B). The AOD values of
E-cadherin and α-SMA were as follows: E-cadherin expression was
reduced from 3.17±0.67 (×10−2) in the control group to
1.00±0.30 (×10−2) in the OVA group, and α-SMA expression
was increased from 0.90±0.30 (×10−2) in the control
group to 3.46±0.71 (×10−2) in the OVA group (P<0.01).
In the OVA + EGCG group, the AOD values of E-cadherin were
increased from 1.00±0.30 to 2.09±0.60 (×10−2), whereas
the AOD values of α-SMA were decreased from 3.46±0.71 to 1.46±0.40
(×10−2) (Fig. 2A and
B). These results suggested that EGCG may attenuate EMT via
increasing the levels of E-cadherin and suppressing α-SMA
expression in OVA-challenged asthmatic mice.
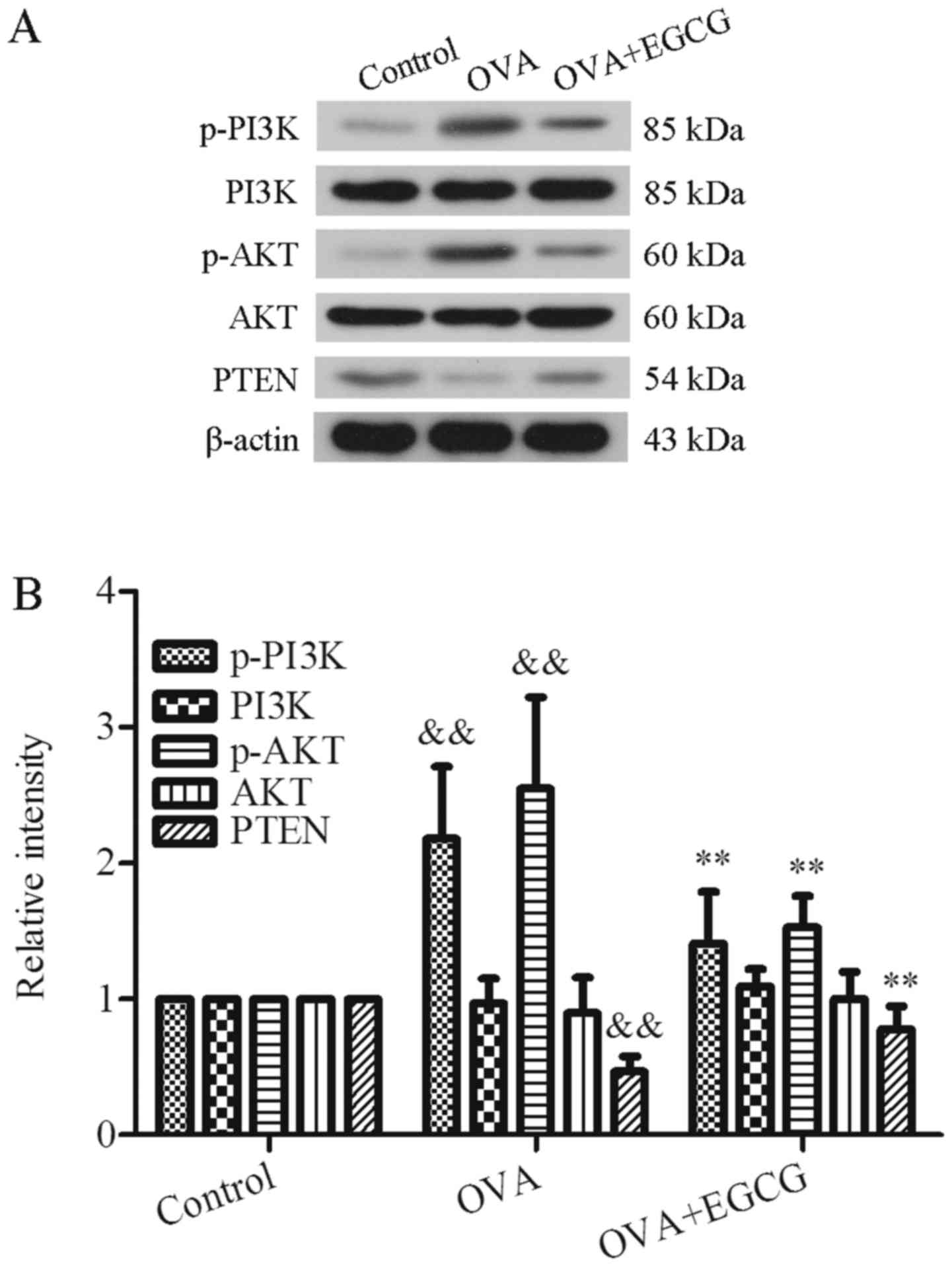
EGCG inhibits the PI3K/AKT signal pathway
via upregu- lation of PTEN in OVA-challenged asthmatic mice
To determine the effects of EGCG on the PI3K/AKT
signaling pathway, the expression levels of PI3K, p-PI3K, AKT and
p-AKT were detected in OVA-challenged lung tissue with or without
EGCG treatment by western blot analysis (Fig. 3). As shown in Fig. 3A, compared with in the control
group, the expression levels of PI3K and AKT were not changed,
whereas the expression levels of p-PI3K and p-AKT were upregulated
in the OVA group. Conversely, the expression levels of p-PI3K and
p-AKT were significantly reduced by EGCG treatment.
Semi-quantitative analysis of p-PI3K and p-AKT expression indicated
that p-PI3K expression was decreased from 2.18±0.53 in the OVA
group to 1.41±0.38 in the OVA + EGCG group, and p-AKT expression
was decreased from 2.55±0.67 in the OVA group to 1.53±0.23 in the
OVA + EGCG group (P<0.01, Fig.
3B). Therefore, treatment with EGCG may decrease the
phosphorylation of PI3K and AKT. Furthermore, it has been reported
that upregulation of PI3K is likely due to downregulation of PTEN
(20,21). The present study also examined the
protein expression levels of PTEN in OVA-challenged asthmatic mice
with or without EGCG treatment by western blot analysis. The
results demonstrated that PTEN was down-regulated from 1.00±0.00 in
the control group to 0.47±0.11 in the OVA group (P<0.01),
whereas EGCG recovered the decreased levels of PTEN to similar
levels as in the control group (from 0.47±0.11 in the OVA group to
0.78±0.17 in the OVA + EGCG group; P<0.01). Therefore, it may be
suggested that EGCG upregulates PTEN expression, and downregulates
the expression of p-PI3K and p-AKT, to modulate the PI3K/AKT
signaling pathway in OVA-challenged asthmatic mice.
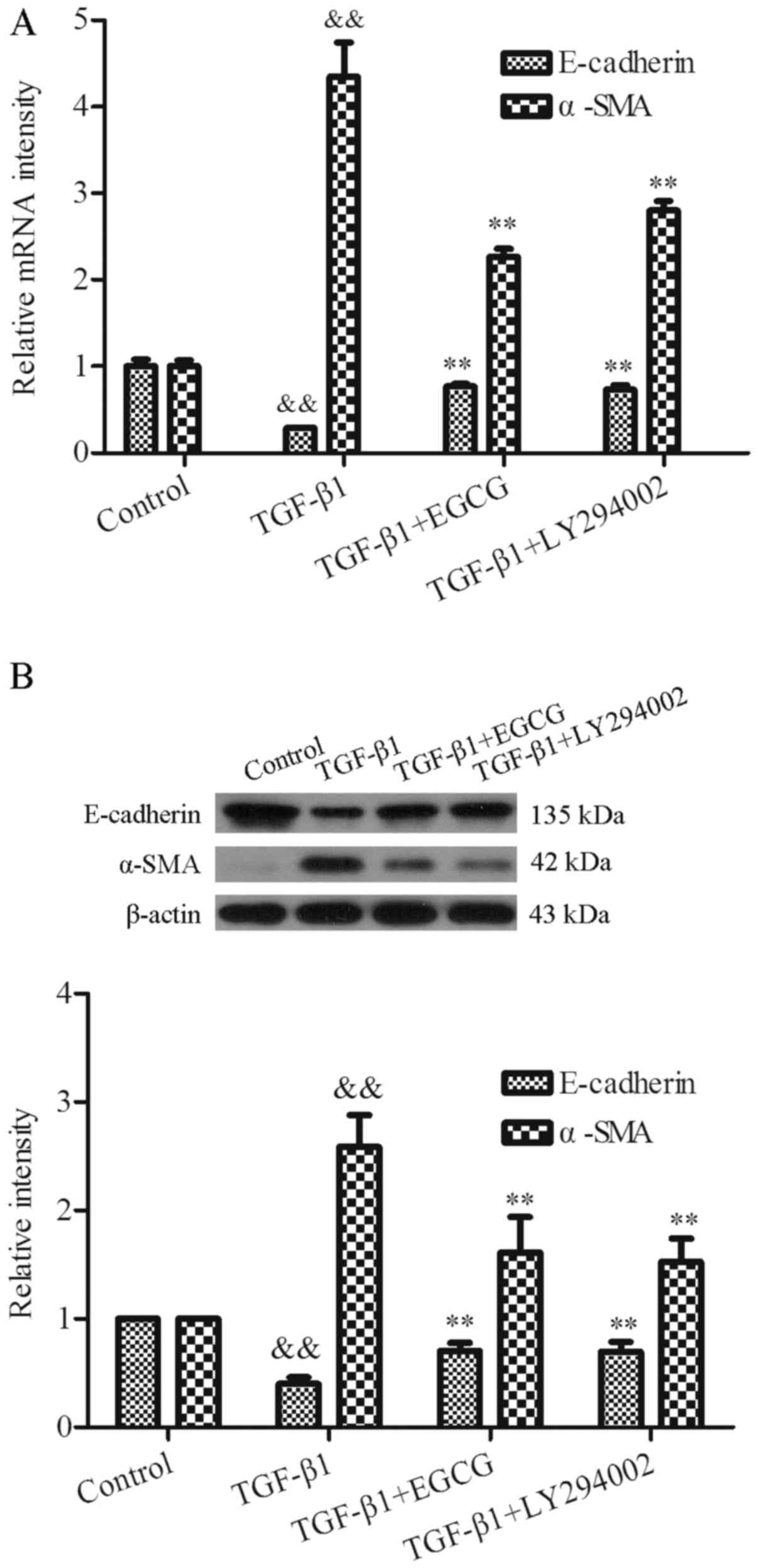
EGCG suppresses TGF-β1-induced EMT in
16HBE cells
To further study the mechanism underlying how EGCG
inhibits EMT in asthma, the 16HBE human bronchial epithelial cell
line was used in subsequent experiments. It has previously been
reported that the PI3K/AKT signaling pathway is involved in
EGCG-induced apoptosis of human pancreatic carcinoma cells
(22). In the present study,
LY294002, which is a selective inhibitor of PI3K, was used as a
positive control to explore the potential mechanism underlying the
effects of EGCG on EMT in TGF-β1-induced 16HBE cells. As expected,
RT-qPCR analysis indicated that the mRNA expression levels of
E-cadherin were decreased, whereas the mRNA expression levels of
α-SMA were significantly increased in response to TGF-β1, which
indicated that TGF-β1 may induce EMT in 16HBE cells. However, EGCG
increased the mRNA expression levels of E-cadherin from 0.29±0.00
to 0.77±0.03, and decreased the levels of α-SMA from 4.35±0.39 to
2.27±0.09 (P<0.01); EGCG had a similar effect to LY294002 in
TGF-β1-induced 16HBE cells (Fig.
4A). Similarly, the western blot analysis indicated that the
expression levels of E-cadherin were downregulated, whereas the
expression levels of α-SMA were markedly upregulated by TGF-β1 in
16HBE cells. Conversely, EGCG upregulated the expression of
E-cadherin and inhibited the expression of α-SMA in TGF-β1-induced
16HBE cells, similar to LY294002 (Fig. 4B). Semi-quantitative analysis of
E-cadherin and α-SMA expression demonstrated that E-cadherin
expression was increased from 0.40±0.06 to 0.71±0.07 in the TGF-β1
+ EGCG group, whereas α-SMA was decreased from 2.59±0.29 to
1.61±0.33 in the TGF-β1 + EGCG group (P<0.01, Fig. 4B). Therefore, it may be
hypothesized that EGCG inhibits TGF-β1-induced EMT in 16HBE cells,
which is likely associated with the PI3K/AKT signaling pathway.
EGCG inhibits the PI3K/AKT signal pathway
via upregula- tion of PTEN expression in TGF-β1-induced 16HBE
cells
The expression levels of PI3K, p-PI3K, AKT and p-AKT
were detected in TGF-β1-induced 16HBE cells with or without EGCG
treatment by western blot analysis. The results indicated that the
expression levels of PI3K and AKT were not altered by TGF-β1
compared with in the control group, whereas the expression levels
of p-PI3K and p-AKT were upregulated in 16HBE cells treated with
TGF-β1. However, the expression levels of these proteins were
significantly downregulated by EGCG treatment. In addition, in the
TGF-β1 + LY294002 group, the expression levels of p-PI3K and p-AKT
were inhibited (Fig. 5A). p-PI3K
expression was decreased from 2.72±0.35 in the TGF-β1 group to
1.98±0.18 in the TGF-β1 + EGCG group, and p-AKT expression was
decreased from 2.34±0.36 in the TGF-β1 group to 1.71±0.25 in the
TGF-β1 + EGCG group (P<0.01). The PI3K/AKT pathway may be
regulated via PTEN; therefore, the present study explored whether
EGCG regulates PTEN expression at the mRNA and protein levels
(Fig. 5B and C). The results
demonstrated that the mRNA and protein expression levels of PTEN
were decreased by TGF-β1, whereas EGCG increased the expression
levels to a large extent. PTEN mRNA expression was increased from
0.12±0.01 in the TGF-β1 group to 0.43±0.00 in the TGF-β1 + EGCG
group (Fig. 5B, P<0.01), and
PTEN protein expression was increased from 0.41±0.04 in the TGF-β1
group to 0.57±0.07 in the TGF-β1 + EGCG group (Fig. 5C, P<0.01). These results
clearly indicated that EGCG may inhibit EMT in asthma via the
PTEN/PI3K/AKT signaling pathway.
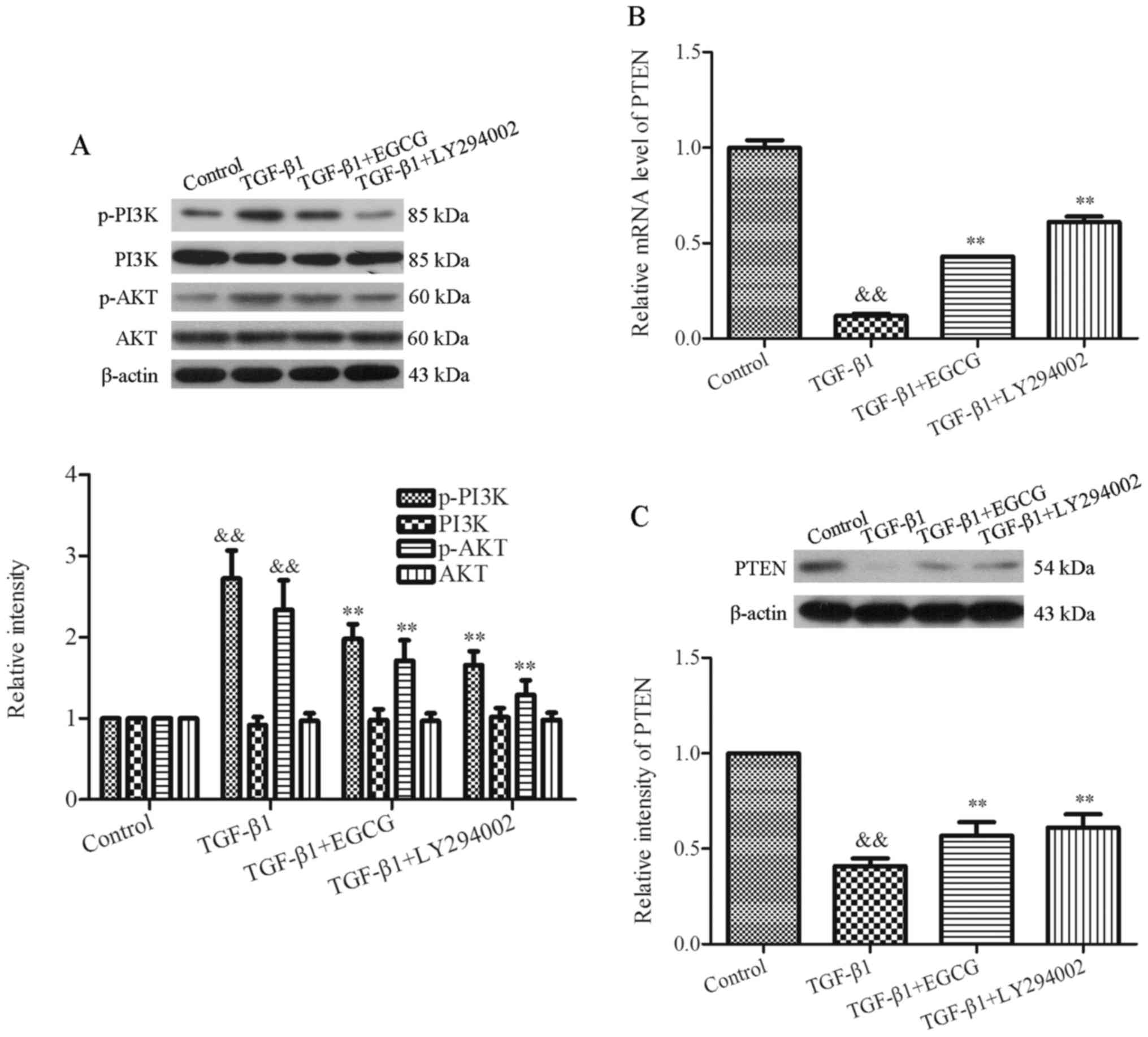 | Figure 5EGCG inhibits the PI3K/AKT signal
pathway via upregulation of PTEN in TGF-β1-induced 16HBE cells. (A)
Protein expression levels of p-PI3K, PI3K, p-AKT and AKT following
treatment of TGF-β1-induced 16HBE cells with or without EGCG were
determined by western blot analysis. Blots underwent
semi-quantitative analysis of gray intensity. (B) Relative mRNA
expression levels of PTEN in TGF-β1-induced 16HBE cells, as
determined by reverse transncription-quantitative polymerase chain
reaction analysis. (C) Protein expression levels of PTEN following
treatment of TGF-β1-induced 16HBE cells with or without EGCG, as
determined by western blot analysis. Blots underwent
semi-quantitative analysis of gray intensity. Data are presented as
the mean ± standard deviation. &&P<0.01 vs.
the control group; **P<0.01 vs. the TGF-β1 group.
AKT, protein kinase B; EGCG, epigallocatechin-3-gallate; LY294002,
inhibitor of PI3K activity; p-, phosphorylated; PI3K,
phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin
homolog; TGF-β1, transforming growth factor-β1. |
EGCG inhibits the migration of
TGF-β1-induced 16HBE cells
During asthma, EMT is reported not only to initiate
phenotypic alterations in airway epithelial cells, but also to
facilitate migration and invasion of these cells into subepithelial
regions of the airway wall, where they contribute to fibrosis
(23). It has also been
demonstrated that TGF-β1-induced EMT results in enhancement of cell
migration through the PI3K/AKT pathway (24). In the present study, the effects
of EGCG on TGF-β1-induced migration of 16HBE cells were
investigated using wound healing and Transwell assays (Fig. 6). The results demonstrated that
TGF-β1 markedly increased migration of 16HBE cells, particularly
after 24 h. However, cells treated with EGCG exhibited decreased
migratory ability after 12 and 24 h (Fig. 6A). The distance of migration was
markedly decreased in the TGF-β1 + EGCG group, and was similar to
that in the TGF-β1 + LY294002 group. The migration rates in the
TGF-β1, TGF-β1 + EGCG and TGF-β1 + LY294002 groups were 42.75±4.34,
34.02±3.73 and 32.15±3.64% after 12 h, and 66.87±7.33, 49.97±6.62
and 41.64±5.34% after 24 h, respectively (P<0.01, Fig. 6B).
A Transwell assay was also conducted to investigate
EGCG-induced suppression of migration. Compared with in the control
group, TGF-β1 increased the migratory ability of 16HBE cells,
whereas EGCG suppressed the increased migration (Fig. 6C). Furthermore, the results
indicated that the number of cells adherent to the lower membranes
of the Transwell chamber were markedly increased from 40.00±4.09%
in the control group to 103.00±10.65% in the TGF-β1 group
(P<0.01). However, EGCG decreased the numbers of cell from
103.00±10.65 to 69.60±7.30%; the number of migratory cells was
almost two times lower in the TGF-β1 + EGCG group than in the
TGF-β1 group (P<0.01). A similar effect was detected in the
TGF-β1 + LY294002 group (62.80±7.12%) (Fig. 6D). These results suggested that
EGCG may suppress TGF-β1-induced migration of 16HBE cells.
Discussion
Structural alterations in the airway epithelium
result in abnormal lung function, and airway hyperresponsiveness
and remodeling, which involves differentiation of airway epithelial
cells into myofibroblasts via EMT, which consequently intensifies
the degree of subepithelial fibrosis. Physical alterations in the
asthmatic epithelium include a decrease in E-cadherin (25) and an increase in the mesenchymal
marker α-SMA (12). Damage to the
airway epithelium from allergens or other environmental factors can
lead to the release of inflammatory factors, which promote
epithelial cell migration to allow for wound repair (26). Furthermore, although EMT is a
normal biological process that occurs during wound healing, this
process becomes detrimental to the airway allowing transient
properties to become permanent during allergic responses, including
asthma. EGCG has been reported to attenuate TGF-β1-induced EMT in
renal tubular epithelial cells (12,27). Kim et al reported that EGCG
may be useful as an adjuvant therapy for bronchial asthma (28). However, the cellular and molecular
mechanisms underlying the effects of EGCG on the pathogenesis
and/or treatment of asthma remain to be fully understood. The
present study demonstrated that EGCG improves lung function through
inhibiting inflammation and EMT induced by an OVA challenge, and
suppressing activation of the PI3K/AKT signal pathway via
upregulation of PTEN in OVA-challenged asthmatic mice in
vivo. In vitro studies confirmed the inhibitory effects
of EGCG on TGF-β1-induced EMT in 16HBE human bronchial epithelial
cells. Furthermore, the results revealed that EGCG inhibited
TGF-β1-induced migration through suppression of the PI3K/AKT signal
pathway via increasing the expression of PTEN in 16HBE cells. These
observations provide a novel insight into understanding the
protective effects of EGCG on airway remodeling in asthma, and
reveal the potential mechanisms underlying the effects of EGCG on
bronchial asthma in vivo and in vitro.
Allergen challenges are known to cause persistent
inflammation in the airway, which leads to tissue damage. To handle
this oxidative insult, the lungs possess inherent mechanisms to
repair tissue damage. However, incessant and excessive oxidative
insult leads to the release of excessive amounts of inflammatory
mediators, including cytokines, interleukins and growth factors,
causing chronic inflammation (29). Persistent inflammation in airway
tissues may result in structural alterations, including airway
remodeling and obstruction, which are not fully reversible and
result in the progressive loss of lung function. It is generally
accepted that airway remodeling involves thickening of the basement
membrane, goblet cell metaplasia, deposition of extracellular
matrix proteins and mucus hypersecretion, thus resulting in
reversible airflow limitations, airway hyperresponsiveness and a
decline in lung function (30,31). In the present study,
OVA-sensitized mice were examined and the effects of EGCG on the
parameters of airway remodeling were analyzed. The results
demonstrated that besides increased infiltration of inflammatory
cells into the lungs and their accumulation in the BALF, an
increased production of IL-4, IL-5 and TGF-β1 was detected in the
lungs and BALF, which corresponded to increased inflammation in
response to OVA (Fig. 1). In
addition, the results revealed that EGCG significantly inhibited
accumulation of inflammatory cells, and prevented infiltration of
eosinophils and other inflammatory cells into lung tissues and BALF
in OVA-sensitized mice (Fig. 1).
It has previously been reported that EGCG protects against toluene
diisocyanate-induced airway inflammation in a murine model of
asthma (28). Choi et al
also demonstrated that EGCG reduces mucin expression in asthmatic
mice and nasal epithelial cells from patients with allergic
inflammation (32). Therefore,
the present in vivo results suggested that administration of
EGCG to OVA-sensitized mice may inhibit airway inflammation in
chronic asthmatic lungs. However, the molecular mechanisms
underlying how EGCG prevents these events require further
clarification.
Chronic inflammation of the airways usually results
in airway remodeling, which is frequently associated with severe
chronic asthma, which poorly responds to conventional
anti-inflammatory therapies (33). EMT of airway epithelial cells has
been reported to be an important inducer of airway remodeling,
since it increases the number of mesenchymal cells migrating to the
subepithelial connective tissues where they enhance the production
of extracellular matrix proteins leading to bronchial wall fibrosis
(34). In addition, it has
previously been demonstrated that inflammatory cells are able to
promote EMT. Lange-Sperandio et al reported that monocytes
or macrophages induce EMT of renal epithelial cells during
unilateral ureteral obstruction (35). Minshall et al also
demonstrated that eosinophils are involved in EMT of esophageal
mucosal epithelial cells (36).
In the present study, inflammatory cells, including macrophages,
eosinophils and neutrophils, were increased in response to an OVA
challenge, both in the lung tissue and BALF of asthmatic mice,
whereas EGCG significantly decreased infiltration of these cells
(Fig. 1A). Furthermore, α-SMA
expression is a marker of myofibroblasts, a cell type that suggests
an advanced phase of EMT (37).
Conversely, E-cadherin is an epithelial cell transmembrane protein
in the extracellular domain, which serves an essential role in
maintaining the structural integrity of bronchial epithelia and in
epithelial polarization; it has previously been demonstrated that a
decrease in E-cadherin expression alone could induce EMT in
carcinoma cells (38). In
OVA-sensitized asthmatic mice, the downregulation of E-cadherin was
reversed, whereas the upregulation of α-SMA expression was
decreased, by EGCG treatment (Fig.
2), thus suggesting that EGCG exerts inhibitory effects on
OVA-challenged EMT in vivo.
EGCG also inhibited the increase of cytokines,
including TGF-β1, which is a major inducer of EMT and therefore is
considered the most active player in the process of airway
remodeling (33). TGF-β1 is a
potent contributor to extracellular matrix formation and has been
known as the key mediator of subepithelial fibrosis in asthma
(39). In epithelial cells,
TGF-β1 induces biochemical and morphological alterations toward the
mesenchymal phenotype. Therefore, TGF-β1-induced 16HBE human
bronchial epithelial cells were used to analyze the effects of EGCG
on EMT in vitro. As expected, TGF-β1 decreased the protein
and mRNA expression levels of E-cadherin and induced the
upregulation of α-SMA in 16HBE cells, as determined by RT-qPCR and
western blot analysis. Conversely, EGCG increased the expression of
E-cadherin and inhibited the upregulation of α-SMA in TGF-β1
induced-16HBE cells (Fig. 4).
Therefore, it may be concluded that EGCG inhibits EMT in
vivo and in vitro.
It has previously been demonstrated that the
PI3K/AKT signal pathway is required for early alterations during
EMT (40,41). Various studies have reported that
EGCG acts via the PI3K/AKT signaling to exert antioxidant,
anti-athero-sclerotic and antitumor activities (42-44). In addition, EGCG has been
identified as a PI3K inhibitor using a scintillation proximity
assay (13). The present in
vivo study demonstrated that EMT was inhibited in
OVA-challenged asthmatic mice by EGCG through suppression of
PI3K/AKT pathway activation (Fig.
3). A previous study revealed that TGF-β1 may induce a
reversible mesenchymal transition in mammary epithelial NMuMG cells
via the PI3K/AKT pathway (45).
The PI3K/AKT signal pathway is one of the most commonly activated
signal pathways in numerous types of human cancer (46). In the present study,
TGF-β1-induced activation of PI3K and AKT, and downregulation of
PTEN, were significantly reversed by EGCG treatment in vitro
(Fig. 5). In addition, PTEN has
been reported to inhibit cell migration and proliferation, and
promote cell apoptosis via its lipid phosphatase activity, which
uses PI3K as a physiological substrate (47). PTEN can also block activation of
PI3K by dephosphorylating the signaling lipid
phosphatidylinositol-3,4,5-triphosphate (48). Therefore, it may be hypothesized
that EGCG inhibits EMT through the PI3K/AKT pathway via
upregulating the expression of PTEN in vivo and in
vitro.
Activation of EMT signaling usually causes
epithelial cells to differentiate into myofibroblasts, enabling
migration and invasion within the epithelial layer, thereby
initiating subepithelial fibrosis in airway remodeling.
Furthermore, PI3K has been implicated in the regulation of cell
migration of human neutrophils (49). The present results suggested that
TGF-β1 enhanced the basal migration of 16HBE cells, whereas
blockade of PI3K with LY294002 reduced basal and TGF-β1-stimulated
cell migration (Fig. 6).
Furthermore, EGCG exhibited a similar inhibitory effect as LY294002
with regards to TGF-β1-stimulated migration of 16HBE cells. These
data are in agreement with a previous study, which reported that
EGCG may serve a critical role in cell motility and migration
(50). Therefore, the present
study indicated that EGCG may inhibit migration induced by TGF-β1
via the PI3K/AKT signal pathway in 16HBE cells.
In conclusion, the present study demonstrated that
EGCG inhibits inflammation and EMT through the PI3K/AKT signaling
pathway via upregulating the expression of PTEN in asthma in
vivo and in vitro. In addition, EGCG may exert
antimigratory effects on TGF-β1-induced 16HBE cells, thus
suggesting it may be able to reduce airway remodeling in the
epithelium. The present study provides a novel insight supporting a
role for EGCG in the prevention of airway inflammation and
remodeling in asthma, and proposes that EGCG may be useful as an
adjuvant therapy for bronchial asthma.
References
|
1
|
Busse WW and Lemanske RF Jr: Asthma. N
Engl J Med. 344:350–362. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Global Asthma Network: The global asthma
report. 2014, http://www.globalasthmareport.org/resources/Global_Asthma_Report_2014.pdf.
Accessed Dec 22, 2016.
|
|
3
|
Lemanske RF Jr and Busse WW: 6. Asthma. J
Allergy Clin Immunol. 111(Suppl 2): S502–S519. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fischer KD, Hall SC and Agrawal DK:
Vitamin D supplem entation reduces induction of
epithelial-mesenchymal transition in allergen sensitized and
challenged mice. PLoS One. 11. pp. e01491802016, View Article : Google Scholar
|
|
5
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Willis BC and Borok Z: TGF-beta-induced
EMT: mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kelly MM, O'Connor TM, Leigh R, Otis J,
Gwozd C, Gauvreau GM, Gauldie J and O'Byrne PM: Effects of
budesonide and formoterol on allergen-induced airway responses,
inflammation, and airway remodeling in asthma. J Allergy Clin
Immunol. 125:349–356. 2010. View Article : Google Scholar
|
|
10
|
Khan N and Mukhtar H: Multitargeted
therapy of cancer by green tea polyphenols. Cancer Lett.
269:269–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suganuma M, Okabe S, Sueoka N, Sueoka E,
Matsuyama S, Imai K, Nakachi K and Fujiki H: Green tea and cancer
chemo-prevention. Mutat Res. 428:339–344. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Liu N, Su X, Zhou G, Sun G, Du F,
Bian X and Wang B: Epigallocatechin-3-gallate attenuates
transforming growth factor-beta1 induced epithelial-mesenchymal
transition via Nrf2 regulation in renal tubular epithelial cells.
Biomed Pharmacother. 70:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van Aller GS, Carson JD, Tang W, Peng H,
Zhao L, Copeland RA, Tummino PJ and Luo L: Epigallocatechin gallate
(EGCG), a major component of green tea, is a dual
phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res
Commun. 406:194–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu SY, Silverberg JI, Joks R, Durkin HG
and Smith-Norowitz TA: Green tea (Camelia sinensis) mediated
suppression of IgE production by peripheral blood mononuclear cells
of allergic asthmatic humans. Scand J Immunol. 76:306–310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bani D, Giannini L, Ciampa A, Masini E,
Suzuki Y, Menegazzi M, Nistri S and Suzuki H:
Epigallocatechin-3-gallate reduces allergen-induced asthma-like
reaction in sensitized guinea pigs. J Pharmacol Exp Ther.
317:1002–1011. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kimata H: Effect of viewing a humorous vs.
nonhumorous film on bronchial responsiveness in patients with
bronchial asthma. Physiol Behav. 81:681–684. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi J, Liu F, Zhang W, Liu X, Lin B and
Tang X: Epigallo-catechin-3-gallate inhibits nicotine-induced
migration and invasion by the suppression of angiogenesis and
epithelial-mesenchymal transition in non-small cell lung cancer
cells. Oncol Rep. 33:2972–2980. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen S, Xu Y, Chen Y, Li X, Mou W, Wang L,
Liu Y, Reisfeld RA, Xiang R, Lv D and Li N: SOX2 gene regulates the
transcriptional network of oncogenes and affects tumorigenesis of
human lung cancer cells. PLoS One. 7:e363262012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yadav UC, Naura AS, Aguilera-Aguirre L,
Ramana KV, Boldogh I, Sur S, Boulares HA and Srivastava SK: Aldose
redu-ctase inhibition suppresses the expression of Th2 cytokines
and airway inflammation in ovalbumin-induced asthma in mice. J
Immunol. 183:4723–4732. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Quah SY, Dong JM, Manser E, Tang
JP and Zeng Q: PRL-3 down-regulates PTEN expression and signals
through PI3K to promote epithelial-mesenchymal transition. Cancer
Res. 67:2922–2926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SR, Lee KS, Park SJ, Min KH, Lee KY,
Choe YH, Lee YR, Kim JS, Hong SJ and Lee YC: PTEN down-regulates
IL-17 expression in a murine model of toluene diisocyanate-induced
airway disease. J Immunol. 179:6820–6829. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu S, Wang XJ, Liu Y and Cui YF:
PI3K/AKT/mTOR signaling is involved in
(-)-epigallocatechin-3-gallate-induced apoptosis of human
pancreatic carcinoma cells. Am J Chin Med. 41:629–642. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Johnson JR, Roos A, Berg T, Nord M and
Fuxe J: Chronic respiratory aeroallergen exposure in mice induces
epithelial-mesenchymal transition in the large airways. PLoS One.
6. pp. e161752011, View Article : Google Scholar
|
|
24
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor beta-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hackett TL, de Bruin HG, Shaheen F, van
den Berge M, van Oosterhout AJ, Postma DS and Heijink IH:
Caveolin-1 controls airway epithelial barrier function.
Implications for asthma. Am J Respir Cell Mol Biol. 49:662–671.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fischer KD and Agrawal DK: Vitamin D
regulating TGF-β induced epithelial-mesenchymal transition. Respir
Res. 15:1462014. View Article : Google Scholar
|
|
27
|
Chang JZ, Yang WH, Deng YT, Chen HM and
Kuo MY: EGCG blocks TGFβ1-induced CCN2 by suppressing JNK and p38
in buccal fibroblasts. Clin Oral Investig. 17:455–461. 2013.
View Article : Google Scholar
|
|
28
|
Kim SH, Park HJ, Lee CM, Choi IW, Moon DO,
Roh HJ, Lee HK and Park YM: Epigallocatechin-3-gallate protects
toluene diisocyanate-induced airway inflammation in a murine model
of asthma. FEBS Lett. 580:1883–1890. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pascual RM and Peters SP: Airway
remodeling contributes to the progressive loss of lung function in
asthma: an overview. J Allergy Clin Immunol. 116:477–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tagaya E and Tamaoki J: Mechanisms of
airway remodeling in asthma. Allergol Int. 56:331–340. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sumi Y and Hamid Q: Airway remodeling in
asthma. Allergol Int. 56:341–348. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choi YS, Bae CH, Song SY and Kim YD: The
effect of epigallocatechin-3-gallate in allergic airway
inflammation. Rhinology. 52:406–412. 2014.PubMed/NCBI
|
|
33
|
Cho JY: Recent advances in mechanisms and
treatments of airway remodeling in asthma: a message from the bench
side to the clinic. Korean J Intern Med. 26:367–383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Royce SG, Cheng V, Samuel CS and Tang ML:
The regulation of fibrosis in airway remodeling in asthma. Mol Cell
Endocrinol. 351:167–175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lange-Sperandio B, Trautmann A, Eickelberg
O, Jayachandran A, Oberle S, Schmidutz F, Rodenbeck B, Hömme M,
Horuk R and Schaefer F: Leukocytes induce epithelial to mesenchymal
transition after unilateral ureteral obstruction in neonatal mice.
Am J Pathol. 171:861–871. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minshall EM, Leung DY, Martin RJ, Song YL,
Cameron L, Ernst P and Hamid Q: Eosinophil-associated TGF-beta1
mRNA expression and airways fibrosis in bronchial asthma. Am J
Respir Cell Mol Biol. 17:326–333. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boukhalfa G, Desmoulière A, Rondeau E,
Gabbiani G and Sraer JD: Relationship between alpha-smooth muscle
actin expression and fibrotic changes in human kidney. Exp Nephrol.
4:241–247. 1996.PubMed/NCBI
|
|
38
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Makinde T, Murphy RF and Agrawal DK: The
regulatory role of TGF-beta in airway remodeling in asthma. Immunol
Cell Biol. 85:348–356. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Higaki M and Shimokado K:
Phosphatidylinositol 3-kinase is required for growth factor-induced
amino acid uptake by vascular smooth muscle cells. Arterioscler
Thromb Vasc Biol. 19:2127–2132. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Krymskaya VP, Hoffman R, Eszterhas A,
Ciocca V and Panettieri RA Jr: TGF-beta 1 modulates EGF-stimulated
phosphatidylinositol 3-kinase activity in human airway smooth
muscle cells. Am J Physiol. 273:L1220–L1227. 1997.
|
|
42
|
Zhang Y, He Q, Dong J, Jia Z, Hao F and
Shan C: Effects of epigallocatechin-3-gallate on proliferation and
differentiation of mouse cochlear neural stem cells: involvement of
PI3K/Akt signaling pathway. Eur J Pharm Sci. 88:267–273. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xuan F and Jian J: Epigallocatechin
gallate exerts protective effects against myocardial
ischemia/reperfusion injury through the PI3K/Akt pathway-mediated
inhibition of apoptosis and the restoration of the autophagic flux.
Int J Mol Med. 38:328–336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu S, Xu ZL, Sun L, Liu Y, Li CC, Li HM,
Zhang W, Li CJ and Qin W: (-)-Epigallocatechin-3-gallate induces
apoptosis in human pancreatic cancer cells via PTEN. Mol Med Rep.
14:599–605. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Piek E, Moustakas A, Kurisaki A, Heldin CH
and ten Dijke P: TGF-(beta) type I receptor/ALK-5 and Smad proteins
mediate epithelial to mesenchymal transdifferentiation in NMuMG
breast epithelial cells. J Cell Sci. 112:4557–4568. 1999.PubMed/NCBI
|
|
46
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
progress pitfalls, and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kwak YG, Song CH, Yi HK, Hwang PH, Kim JS,
Lee KS and Lee YC: Involvement of PTEN in airway
hyperresponsiveness and inflammation in bronchial asthma. J Clin
Invest. 111:1083–1092. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hirsch E, Katanaev VL, Garlanda C,
Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F
and Wymann MP: Central role for G protein-coupled phosphoinositide
3-kinase gamma in inflammation. Science. 287:1049–1053. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Barenys M, Gassmann K, Baksmeier C, Heinz
S, Reverte I, Schmuck M, Temme T, Bendt F, Zschauer TC, Rockel TD,
et al: Epigallocatechin gallate (EGCG) inhibits adhesion and
migration of neural progenitor cells in vitro. Arch Toxicol.
91:827–837. 2017. View Article : Google Scholar
|