Introduction
Primary open-angle glaucoma (POAG) is characterized
by degenerative changes in the optic nerve and retinal ganglion
cells (RGCs) and leads to progressive visual field loss with or
without elevated intraocular pressure (IOP) (1). Numerous studies have demonstrated
that genetic factors serve an important role in POAG. It has been
found that mutations in the optineurin (OPTN) gene may be
associated with RGC degeneration and increase susceptibility to the
development of POAG (2,3). Furthermore, studies have indicated
that OPTN is important in various physiological processes,
including the nuclear factor-κB pathway, vesicle trafficking,
protein secretion and the control of cell division (4,5).
These functions depend on the precise subcellular localization of
OPTN, its post-translational modifications and interactions with
binding partners, including Rab8, huntingtin and myosin VI
(6,7). However, how these diverse functions
of OPTN are integrated into a comprehensive network remains
unclear.
Long noncoding RNAs (lncRNAs) are a class of RNA
molecules >200 nucleotides in length that structurally resemble
mRNAs but do not encode proteins (8). Studies have shown that lncRNAs are
often interspersed between, or overlap with diverse coding and
non-coding transcripts (9). The
extensive functions of lncRNAs include precise subcellular
localization, high-order chromosomal dynamics, telomere biology and
regulation of the expression of neighboring protein-coding genes
(10–12). Highly sensitive genome tiling
arrays have demonstrated that the ability of lncRNAs to regulate
protein-coding genes may serve important roles in various human
diseases, including cardiovascular diseases, cancer and
neurological diseases (13,14). OPTN has been identified as a gene
that is associated with hereditary neurological diseases (2,15).
In addition, lncRNA as a key regulator rarely exerts a biological
function alone and may act synergistically (16,17). Thus, the analysis of
OPTN-associated lncRNAs by constructing an lncRNA-lncRNA
co-expression network for OPTN and identifying lncRNA co-expression
modules may potentially improve our understanding of the complex
molecular mechanisms associated with the pathogenesis of
OPTN-associated diseases.
To investigate the underlying role of lncRNAs in the
regulation of OPTN function, OPTN-silencing microarray data was
re-annotated in the present study to identify differentially
expressed lncRNAs and the potential functions of these lncRNAs were
explored by functional enrichment analysis of the corresponding
co-expressed genes.
Materials and methods
Microarray
The dataset GSE6819 was downloaded from the Gene
Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6819).
This microarray dataset was designed to investigate the
comprehensive molecular mechanisms by which OPTN mediates, and
identified genome-wide molecular changes upon OPTN silencing in
HeLa cells by microarray technology (18). It comprises three samples treated
with small interfering RNA (siRNA) specific for OPTN mRNA and three
samples treated with water (mock experiment) using the Human Genome
U133 (HG-U133) Plus 2.0 array (Affymetrix; Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Functional re-annotation of lncRNAs
The non-coding RNA function annotation server
(ncFANs) was used to re-annotate the probes on the HG-U133 Plus 2.0
array (19). Differentially
expressed lncRNAs were selected using the fold-change method with
an absolute log2 fold-change threshold of >1. The
fold-change values for each probe were calculated. Each probe was
then converted to its Entrez Gene ID. If one gene matched more than
one probe, its expression value was computed by averaging the
expression values of all corresponding probes.
Construction of a lncRNA-lncRNA
co-expression network and detection of co-expression modules
Pearson's correlation coefficients (PCCs) between
expressed values of each lncRNA in all samples were calculated
using the weighted correlation network analysis (WGCNA) R software
package, which is a comprehensive collection of R functions for
performing various aspects of weighted correlation network analysis
(20). The package includes
functions for network construction, module detection, gene
selection and calculations of topological properties. The
co-expressed lncRNA pairs with P<0.01 and absolute PCC >0.75
were selected. Furthermore, lncRNA-lncRNA co-expression modules
were also detected using this package. The average absolute
fold-change values of lncRNAs for all modules were then calculated.
The top three differential modules with the greatest average
absolute fold-change values were selected.
Cell culture and knockdown of OPTN
The HeLa cell line (American Type Culture
Collection, Manassas, VA, USA) was cultured in Dulbecco's modified
Eagle's medium (Hyclone, Logan, UT, USA) containing 10% fetal
bovine serum (Gibco-BRL; Thermo Fisher Scientifc, Grand Island, NY,
USA), 20 µg/ml penicillin and 20 µg/ml streptomycin
at 37°C in a humidified atmosphere with 5% CO2. The day
prior to transfection, the cells were plated in a 6-well plate
(10,000 cells/well) in an appropriate volume of growth medium
without antibiotics such that they were 30–50% confluent at the
time of transfection. siRNAs were transfected using 10
µl/well X-tremeGENE siRNA Transfection reagent (Roche
Diagnostics, Basel, Switzerland) at 10 nM final concentration, and
the cells were harvested after 48 h. The siRNA oligonucleotides
were synthesized by Guangzhou Ribobio Co., Ltd. (Guangzhou, China).
The sequences are presented in Table
I.
 | Table IsiRNA targeting sequences of
OPTN. |
Table I
siRNA targeting sequences of
OPTN.
| Genes | Sequences
(5′-3′) |
|---|
| siOPTN-1
(si-h-optineurin-1) |
GAAGCCATGAAGCTAAATA |
| siOPTN-2
(si-h-optineurin-2) |
CTGCAGCTCAAGCTGAACT |
| siOPTN-3
(si-h-optineurin-3) |
CCATCAGAGTTGAATGAAA |
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cultured HeLa cells
using a High Pure RNA Isolation kit (Roche Diagnostics).
Complementary DNA was synthesized from 1 µg total RNA using
the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). All RT-qPCR
experiments were run in triplicate as singleplex reactions on the
7500 Fast Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using SYBR-Green (Roche Diagnostics), according
to the manufacturer's instructions. The RT-qPCR conditions involved
a denaturation step (95°C for 10 min), and amplification and
quantification were repeated 40 times (95°C for 15 sec, 56°C for 15
sec and 72°C for 30 sec, respectively). The relative gene
expression levels were quantified based on the quantification cycle
(Cq) and normalized to the reference gene glyceraldehyde
3-phosphate dehydrogenase (GAPDH). The ΔΔCq method was used for
relative quantification of gene expression, according to MIQE
guidelines (21). All primers
used in the reactions are presented in Table II.
| Table IIPrimer and probe sequences used in
reverse transcription-quantitative polymerase chain reaction. |
Table II
Primer and probe sequences used in
reverse transcription-quantitative polymerase chain reaction.
| Genes | Forward primers
(5′-3′) | Reverse primers
(5′-3′) |
|---|
| OPTN |
AAAGGCCCGGAGACTGTTG |
CATGTTCGTCATGGGTGTGAA |
| GAPDH |
GTGTGCCGGTGCAAATACAC |
CCTTTCAAGGGCCTGACACTT |
| RP1-212P9.2 |
GGCATGGACTGTGGTCATGAG |
CGTGCAGAGGCCCAGAGA |
| RP11-169D4.1 T |
GGTCAGGCTGGTCTTGAACT |
CAGGCCACGGTCACTCATATT |
Protein isolation and immunoblotting
Cells were lysed in radioimmunoprecipitation assay
buffer supplemented with protease inhibitor cocktail (Biotool LLC,
Houston, TX, USA) for 30 min on ice to extract the total protein.
Protein concentrations were determined using a bicinchoninic acid
protein assay (OriGene Technologies, Inc., Beijing, China). The
protein samples were boiled in 1X sodium dodecyl sulfate buffer for
10 min. Equal amounts of proteins (50 µg/lane) were
subjected to 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred onto polyvinylidene fluoride
membranes. The membranes were blocked with 5% skimmed milk for 1 h
at room temperature, probed with antibodies and visualized using an
enhanced chemiluminescence reagent (OriGene Technologies, Inc.).
Western blot analysis was performed to detect the OPTN protein, and
GAPDH was used as the internal reference. Primary antibodies were
anti-OPTN (1:400; sc-271549) and anti-GAPDH (1:5,000; sc-47724)
(both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
(incubation at 4°C overnight). The secondary antibody was rabbit
anti-mouse alkaline phosphatase-IgG (1:500; TA130002; OriGene
Technologies, Inc.) (incubation for 1 h at room temperature). The
protein bands were quantified using a Bio-Rad ChemiDoc™ EQ
densitometer and Bio-Rad Quantity One software 4.62 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Identification of co-expressed genes and
functional enrichment
To explore the potential functions of the
differentially expressed lncRNAs, co-expressed genes for the
lncRNAs in the selected co-expressing modules were detected. To do
this, the PCCs between each differentially expressed lncRNA and all
genes across all samples were calculated. The genes with a strict
cut-off (PCC >0.9 or <−0.9) were identified as co-expressed
genes. Gene Ontology (GO) analysis was then conducted and gene sets
were mapped to Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathways to identify the potential biological functions of the
genes co-expressed with the lncRNAs using the Database for
Annotation, Visualization and Integrated Discovery (DAVID, version
6.7) (22).
Statistical analysis
Data are presented as the mean ± standard error of
the mean, unless otherwise stated. Differences between the groups
were analyzed using analysis of variance followed by Dunnett's
test. The analysis was performed with SPSS 18.0 software (SPSS,
Inc., Chicago, IL, USA). All tests were two-sided. P<0.05 was
considered to indicate a statistically significant result.
Results
Re-annotated microarray data
A total of 3,495 lncRNAs were re-annotated. Three
lncRNAs with high fold-change values in the OPTN-silenced cells as
compared with normal cells in the microarray were identified. These
were matrix metalloprotease 12 (MMP12; upregulated), RP11-169D4.1
(upregulated) and RP1-212P9.2 (downregulated).
LncRNA-lncRNA co-expression network and
co-expression modules
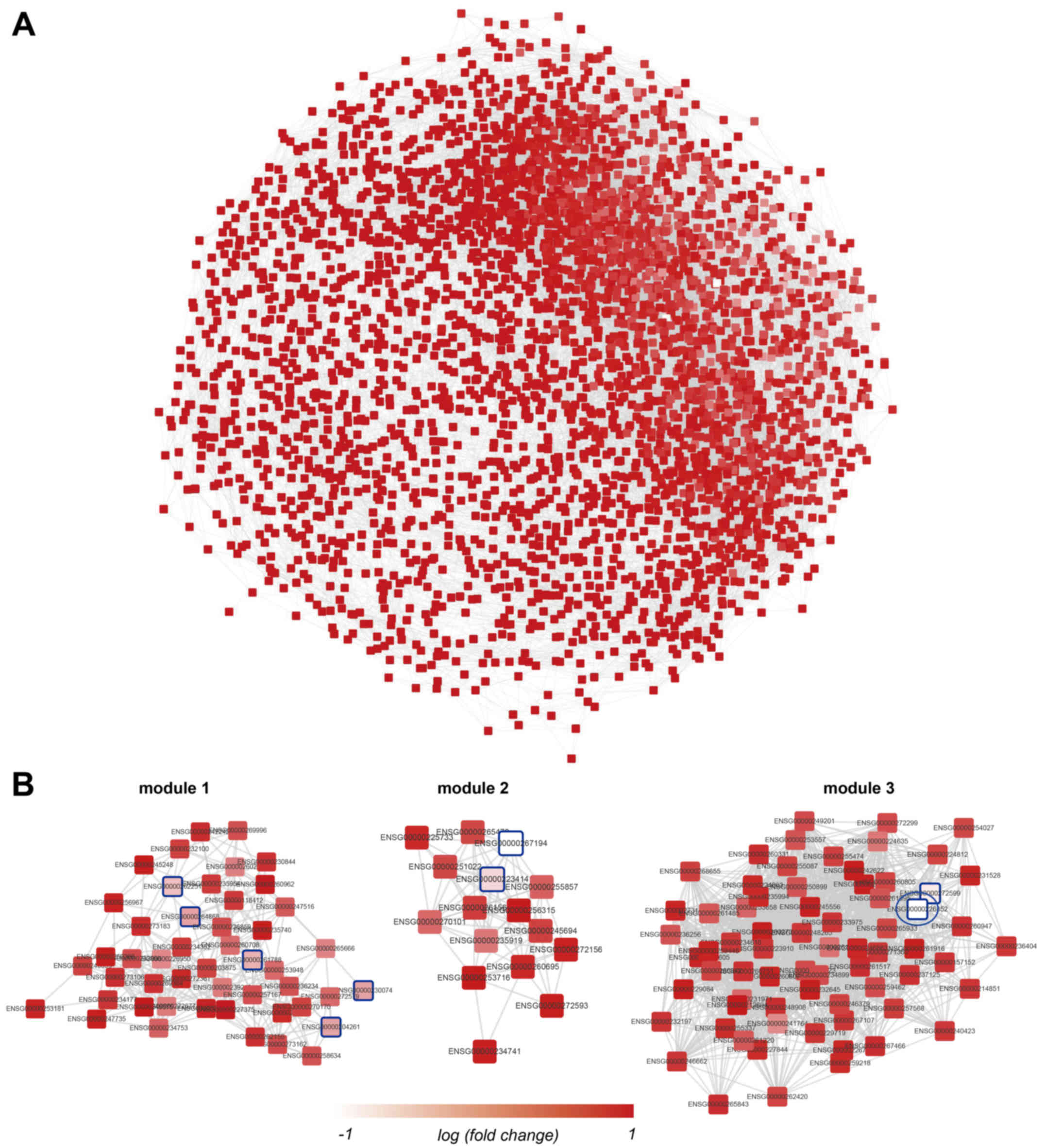
The lncRNA-lncRNA co-expression network was
constructed using the WGCNA R software package. In this network,
nodes represented lncRNAs and two nodes were connected if the PCC
between the expression values of these two lncRNAs was >0.7 or
<-0.7 and P<0.01. There were 3,495 nodes and 49,836 edges in
this network (Fig. 1A). In
addition, WGCNA analysis also detected 74 modules in this network.
The three modules with the greatest average absolute fold-change
values are presented in Fig. 1B.
In module 3, RP1-212P9.2 was the most strongly downregulated (blue
ellipse in Fig. 1B).
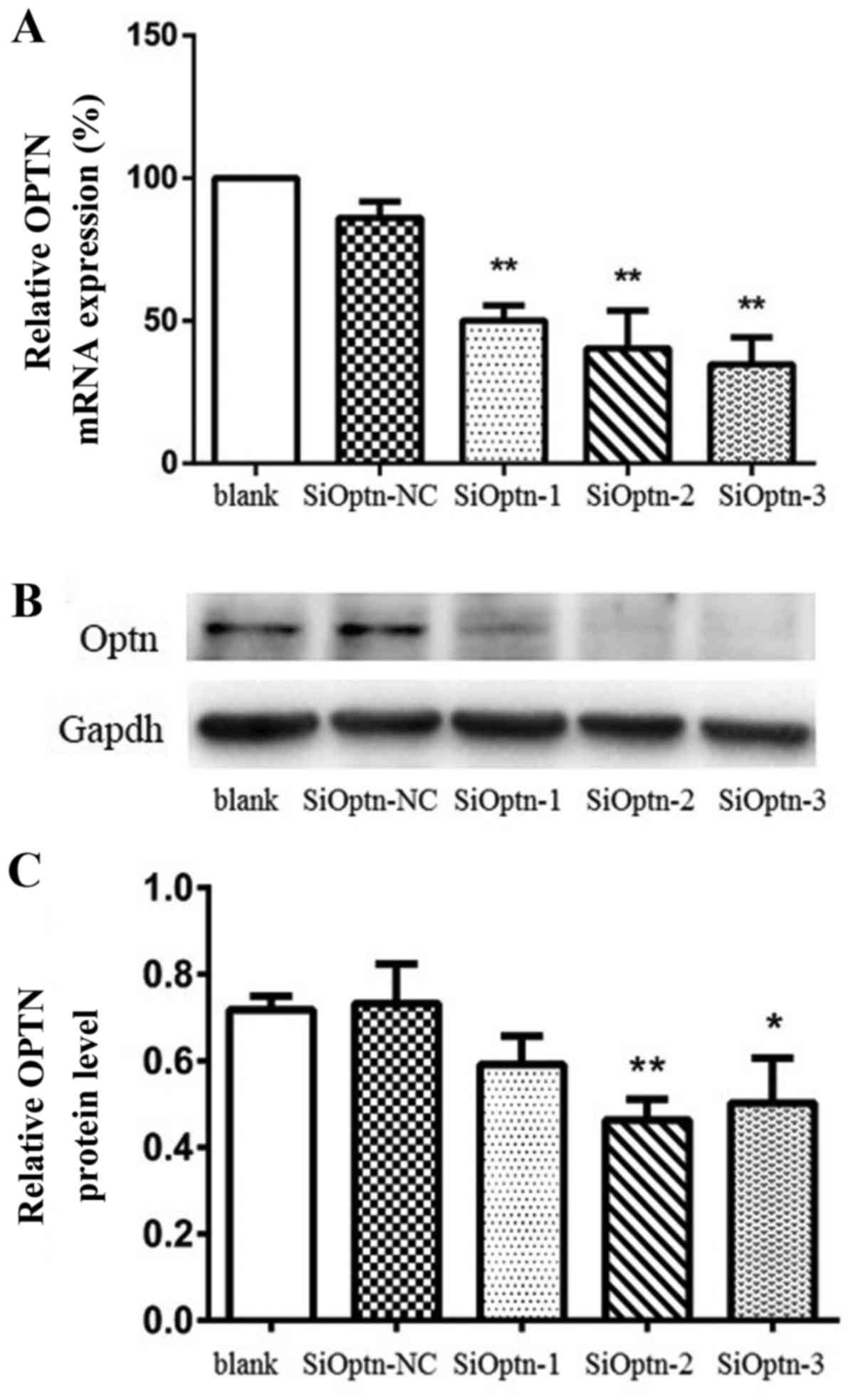
Validation of OPTN knockdown
Three OPTN siRNA-expressing sequences (siOPTN-1,
siOPTN-2 and siOPTN-3) and one negative control (siOPTN-NC) were
transiently transfected into HeLa cells. The knockdown of OPTN by
the three siRNAs was assayed using RT-qPCR. As shown in Fig. 2A, the cells transfected with
siOPTN-1, siOPTN-2 and siOPTN-3 exhibited 50.12, 40.37 and 34.74%
OPTN expression relative to the untransfected cells, respectively,
all of which were found to be significant reductions as compared
with the control (P<0.01; n=3). OPTN levels in the cells
transfected with siOPTN-NC were not observed to be significantly
different from those in the untransfected cells (86.01%; P>0.05;
n=3), indicating that the transfection itself did not affect OPTN
expression. Western blot analysis was used to validate the RT-qPCR
results (Fig. 2B and C). As shown
in Fig. 2C, siOPTN-2 and -3
significantly inhibited OPTN protein expression (P<0.05; n=3).
Therefore, siOPTN-2 and -3 were used to construct stably
transfected cell lines, which were used in the following
experiments.
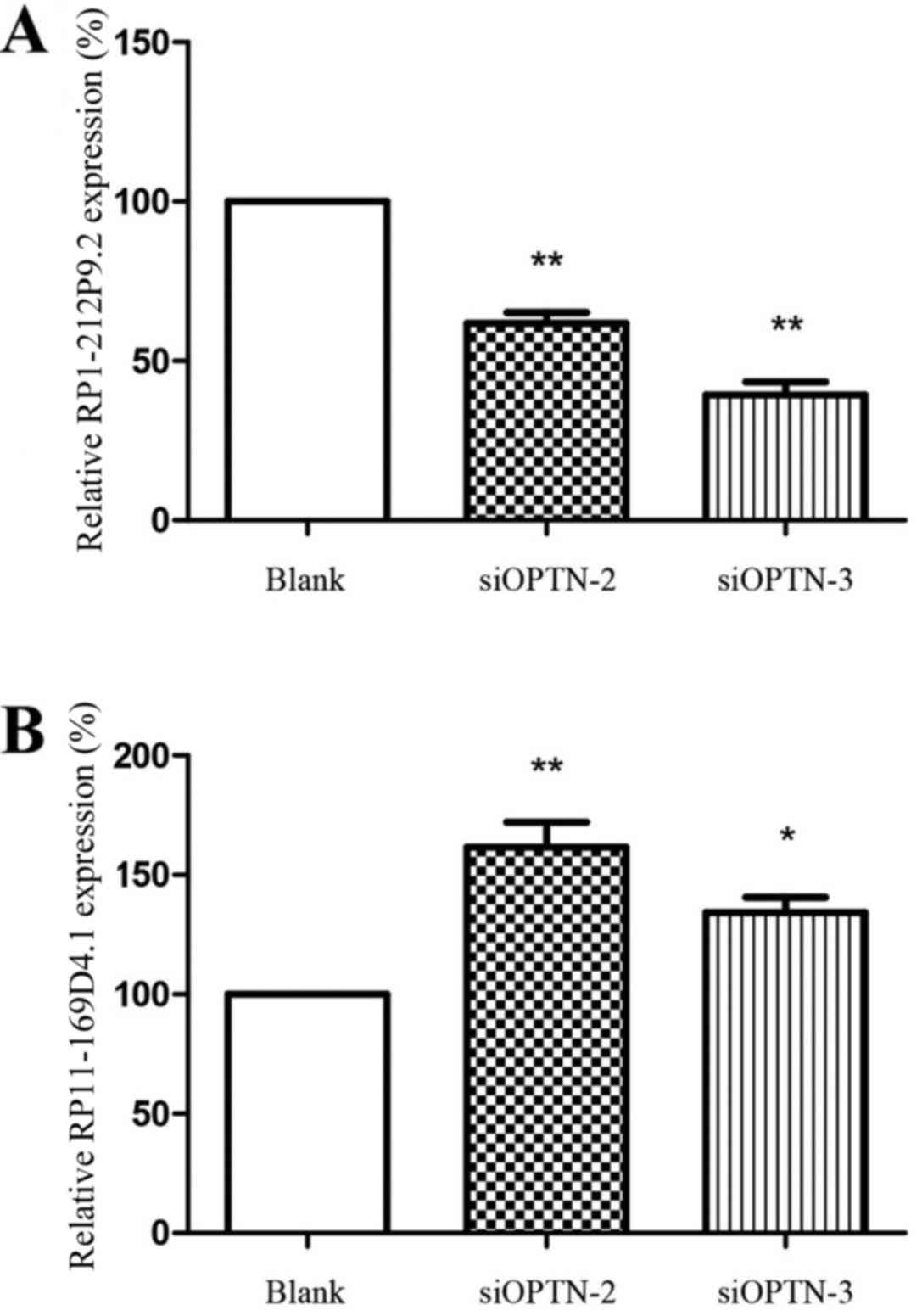
Validation of lncRNAs with RT-qPCR
To verify the results obtained with the re-annotated
data, RT-qPCR assays were performed to detect the differences of
lncRNA expression between cells transfected with siOPTN-2 and -3
and untransfected cells (Fig. 3).
RP1-212P9.2 was significantly downregulated (P<0.01) and
RP11-169D4.1 was significantly upregulated (P<0.01), which was
consistent with the re-annotated data.
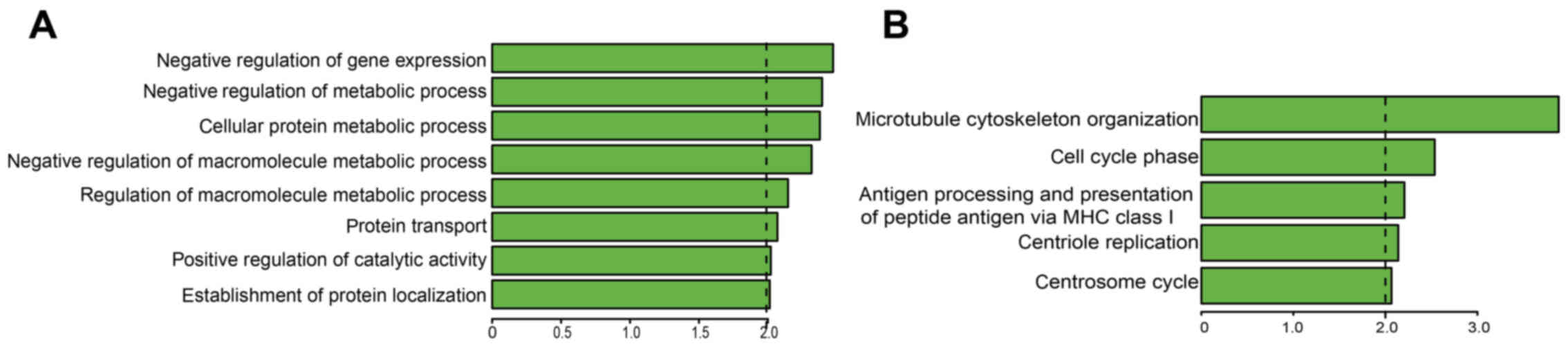
Detection of lncRNA functions
Gene enrichment provides a means of evaluating gene
and gene product enrichment according to function, to facilitate
the identification of GOs with particular relevance (23,24). In the present study, it was
identified that the RP1-212P9.2 and RP11-169D4.1 co-expressed mRNAs
were most significantly associated with 'negative regulation of
gene expression' and 'microtubule cytoskeleton organization'
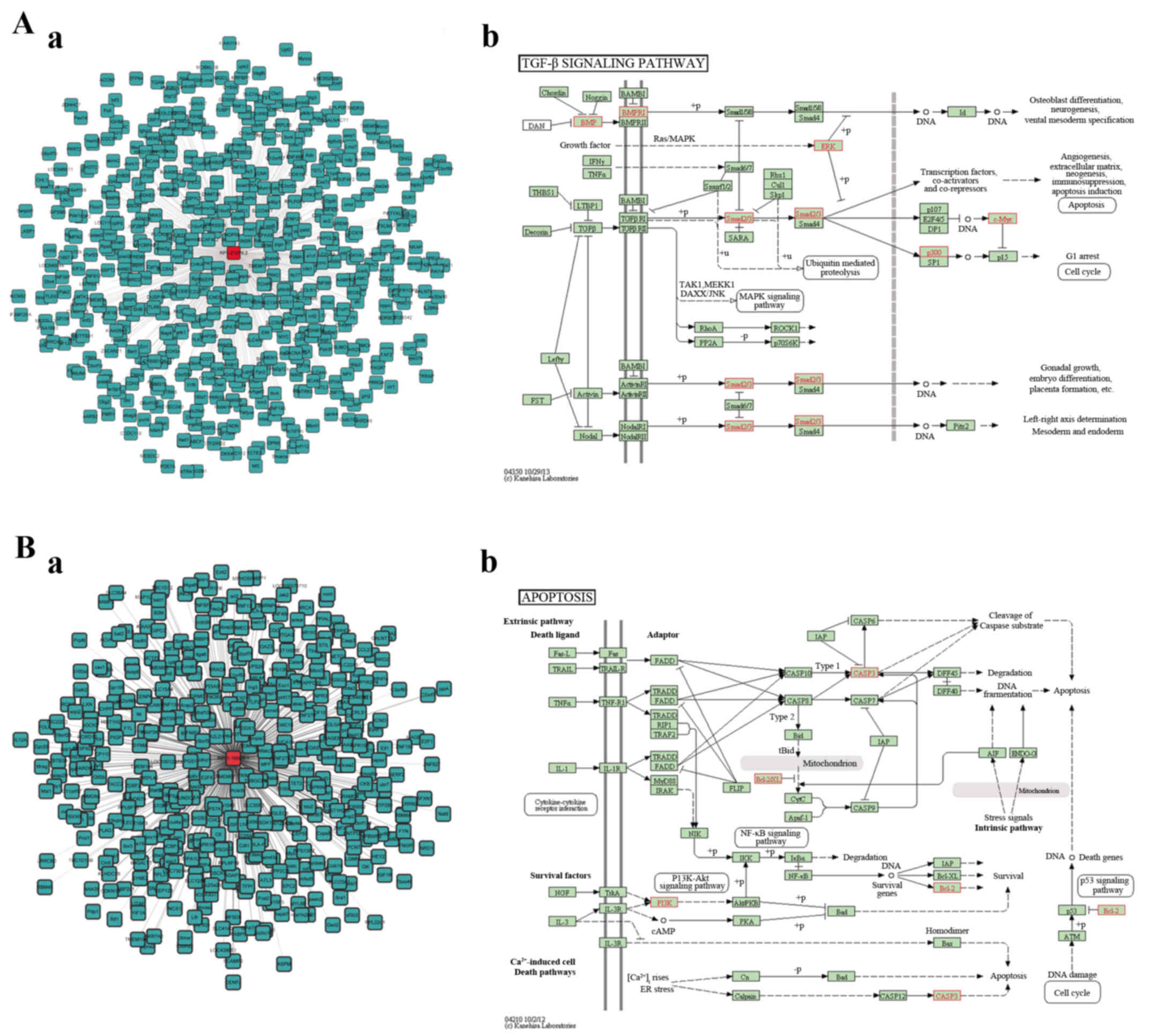
(ontology, biological process), respectively (Fig. 4). To illustrate the potential
biological functions of lncRNAs in the modules, a co-expression
network was constructed for the co-expressed coding genes of
RP1-212P9.2 and RP11-169D4.1 and these coding genes were mapped to
KEGG pathways using KEGG data mapping tools. The pathways comprised
apoptosis (path, 04210), oxidative phosphorylation (path, 00190),
axon guidance (path, 04360), ubiquitin-mediated proteolysis (path,
04120), cell cycle (path, 04110), lysosome (path, 04142),
endocytosis (path, 04144), transforming growth factor (TGF)-β
signaling pathway (path, 04350) and neurotropin signaling pathway
(path, 04722). Among these pathways, the TGF-β signaling pathway
and apoptosis pathway are associated with basic biological
processes of neural functions. The red genes represent co-expressed
genes of the annotated lncRNAs (Fig.
5A-b and B-b).
Discussion
As the sensitivity of genome tiling arrays has
increased, the concept of the functional genome has been revised to
encompass an abundance of newly discovered lncRNA transcripts.
lncRNAs are widely expressed in the mammalian nervous system and
numerous lncRNAs are likely to serve critical roles in neuronal
development and activity (13).
However, the changes of lncRNA expression associated with ocular
disorders are just beginning to be elucidated (25).
Previous studies have shown that disturbing the
homeostasis of OPTN by overexpression or knockdown results in
adverse consequences on cellular function, leading to the
progression of neurodegenerative diseases including amyloid lateral
sclerosis, Paget's disease of bone and POAG (2,15,26). Glaucoma is the second most common
cause of blindness in humans, and the most common form of this
disease is POAG, which is caused by the irreversible progressive
degeneration of RGCs (27). To
investigate the molecular mechanisms and signal pathways of OPTN in
POAG, Weisschuh et al (18) used RNA interference technology to
silence the expression of OPTN in HeLa cells, and found a series of
differentially expressed genes using microarray technology. Based
on this, the OPTN-silencing microarray data were re-annotated,
differentially expressed lncRNAs were identified and an
lncRNA-lncRNA co-expression network was constructed in the present
study. As a result, three lncRNAs, namely MMP12, RP11-169D4.1 and
RP1-212P9.2 with fold-change values >2 or <0.5 were
identified. Among them, the first two lncRNAs were upregulated,
while the third was downregulated. RT-qPCR assays were performed to
verify the results of the re-annotated data and these demonstrated
that RP1-212P9.2 was downregulated (P<0.01), while RP11-169D4.1
was upregulated (P<0.01), consistent with the re-annotated
data.
It is difficult to predict the functions of lncRNAs
on the basis of their nucleotide sequences only, since the majority
of them are poorly conserved in mammals, as compared with
protein-encoding genes (28).
Therefore, to explore the potential functions of the lncRNAs in
OPTN-silenced cells, a lncRNA/mRNA co-expression network was
constructed based on the correlation analysis. This indicated that
RP1-212P9.2 and RP11-169D4.1 co-expressed mRNAs are targeted to
'negative regulation of gene expression' and 'microtubule
cytoskeleton organization' (ontology, biological process),
respectively. These biological processes may be involved with the
regulation of gene products, and neuronal migration, polarity and
differentiation (29). KEGG
pathway analysis indicated that the mRNAs co-expressed with the
lncRNAs were involved in biological functions including apoptosis,
oxidative phosphorylation, axon guidance, ubiquitin-mediated
proteolysis, the cell cycle, lysosome, endocytosis, the TGF-β
signaling pathway and the neurotropin signaling pathway. These
signaling pathways are closely associated with pathological
processes such as neurodegeneration (30,31), neuronal cell death, survival and
migration (32), suggesting that
the lncRNA-mediated network may serve an extensive role in the
pathogenesis of OPTN-associated diseases.
Notably, the TGF-β signaling pathway and apoptosis
pathway are associated with basic biological processes of neural
diseases (33–35). The TGF-β signaling pathway
regulates diverse cellular processes, including cell proliferation,
differentiation, plasticity and migration (36). In addition, it is involved in the
development of the nervous system and exhibits neuroprotective
functions (33). Furthermore,
TGF-β is a vital growth factor in the pathogenesis of glaucoma
(37). It stimulates the
proliferation of human Tenon's fibroblasts and alters the
cytoskeleton, which causes ocular hypertension and fibronectin
deposition in the trabecular meshwork (TM) (38). A previous study indicates that
bone morphogenetic proteins (BMPs) antagonize TGF-β signaling
(39). Furthermore, in
glaucomatous TM cells and tissues, a BMP antagonist directly
elevated IOP through the canonical TGF-β/SMAD pathway (40). In accordance with these studies,
the TGF-β signaling pathway gained the highest count score during
KEGG analysis in the present study.
The apoptosis pathway is a genetically controlled
mechanism of cell death involved in the regulation of tissue
homeostasis. KEGG analysis in the present study indicated that
RP11-169D4.1 is enriched in apoptosis. This implies that apoptosis
is a crucial event in OPTN-silenced cells. POAG is characterized by
progressive degeneration induced by the apoptosis of RGCs in optic
nerves (41). It has been
reported that RGC degeneration is partly initiated via apoptotic
pathways; among the involved pathways, the phosphoinositide
3-kinase/Akt pathway, Bcl-2 family and caspase family were
suggested to be the most important (42). In summary, differentially
expressed lncRNAs in OPTN-silenced cells may be crucial in
regulating the molecular mechanisms of glaucoma. However, in the
future, further exploration of this hypothesis is necessary.
Although the detailed mechanisms of OPTN-associated
diseases have not been fully elucidated, the differentially
expressed lncRNAs may serve important roles in their corresponding
signaling networks, and facilitate the regulation of coding genes.
lncRNAs possibly regulate gene expression through directing
methylation complexes, initiating chromatin remodeling, targeting
transcription factors and blocking nearby transcription (43). The present understanding of the
regulatory effect of lncRNA in OPTN mutation is limited. In the
future, alternative techniques may be used to determine the
biological functions of the lncRNAs, such as lncRNA silencing and
structure disruption (44).
In conclusion, the present study demonstrated the
ability of functional re-annotation to identify differentially
expressed lncRNAs in the microarray dataset. This approach has been
successful in the identification of altered lncRNA expression in
previous studies (45,46). The present study provides new
insights into the involvement of lncRNAs in the pathogenesis of
OPTN-associated diseases. However, further study is required to
understand the biological functions and molecular mechanisms of the
distinct lncRNAs implicated in OPTN-associated diseases.
References
|
1
|
Chi ZL, Akahori M, Obazawa M, Minami M,
Noda T, Nakaya N, Tomarev S, Kawase K, Yamamoto T, Noda S, et al:
Overexpression of optineurin E50K disrupts Rab8 interaction and
leads to a progressive retinal degeneration in mice. Hum Mol Genet.
19:2606–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rezaie T, Child A, Hitchings R, Brice G,
Miller L, Coca-Prados M, Héon E, Krupin T, Ritch R, Kreutzer D, et
al: Adult-onset primary open-angle glaucoma caused by mutations in
optineurin. Science. 295:1077–1079. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Allingham RR, Liu Y and Rhee DJ: The
genetics of primary open-angle glaucoma: a review. Exp Eye Res.
88:837–844. 2009. View Article : Google Scholar
|
|
4
|
Gleason CE, Ordureau A, Gourlay R, Arthur
JSC and Cohen P: Polyubiquitin binding to optineurin is required
for optimal activation of TANK-binding kinase 1 and production of
interferon β. J Biol Chem. 286:35663–35674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hattula K and Peränen J: FIP-2, a
coiled-coil protein, links huntingtin to Rab8 and modulates
cellular morphogenesis. Curr Biol. 10:1603–1606. 2000. View Article : Google Scholar
|
|
6
|
del Toro D, Alberch J, Lázaro-Diéguez F,
Martín-Ibáñez R, Xifró X, Egea G and Canals JM: Mutant huntingtin
impairs post-Golgi trafficking to lysosomes by delocalizing
optineurin/Rab8 complex from the Golgi apparatus. Mol Biol Cell.
20:1478–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagabhushana A, Chalasani ML, Jain N,
Radha V, Rangaraj N, Balasubramanian D and Swarup G: Regulation of
endocytic trafficking of transferrin receptor by optineurin and its
impairment by a glaucoma-associated mutant. BMC Cell Biol.
11:42010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kapranov P, Cheng J, Dike S, Nix DA,
Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J,
Hofacker IL, et al: RNA maps reveal new RNA classes and a possible
function for pervasive transcription. Science. 316:1484–1488. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carninci P, Kasukawa T, Katayama S, Gough
J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al
RIKEN Genome Exploration Research Group and Genome Science Group
(Genome Network Project Core Group): The transcriptional landscape
of the mammalian genome. Science. 309:1559–1563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mercer TR, Dinger ME, Sunkin SM, Mehler MF
and Mattick JS: Specific expression of long noncoding RNAs in the
mouse brain. Proc Natl Acad Sci USA. 105:716–721. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amaral PP and Mattick JS: Noncoding RNA in
development. Mamm Genome. 19:454–492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ng SY, Lin L, Soh BS and Stanton LW: Long
noncoding RNAs in development and disease of the central nervous
system. Trends Genet. 29:461–468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson R: Long non-coding RNAs in
Huntington's disease neurodegeneration. Neurobiol Dis. 46:245–254.
2012. View Article : Google Scholar
|
|
14
|
Zhu J, Liu S, Ye F, Shen Y, Tie Y, Zhu J,
Jin Y, Zheng X, Wu Y and Fu H: The long noncoding RNA expression
profile of hepatocellular carcinoma identified by microarray
analysis. PLoS One. 9:e1017072014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maruyama H, Morino H, Ito H, Izumi Y, Kato
H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al:
Mutations of optineurin in amyotrophic lateral sclerosis. Nature.
465:223–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moran VA, Perera RJ and Khalil AM:
Emerging functional and mechanistic paradigms of mammalian long
non-coding RNAs. Nucleic Acids Res. 40:6391–6400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weisschuh N, Alavi MV, Bonin M and
Wissinger B: Identification of genes that are linked with
optineurin expression using a combined RNAi - microarray approach.
Exp Eye Res. 85:450–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao Q, Xiao H, Bu D, Xie C, Miao R, Luo
H, Zhao G, Yu K, Zhao H, Skogerbø G, et al: ncFANs: A web server
for functional annotation of long non-coding RNAs. Nucleic Acids
Res. 39:W118–W124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
23
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schlitt T, Palin K, Rung J, Dietmann S,
Lappe M, Ukkonen E and Brazma A: From gene networks to gene
function. Genome Res. 13:2568–2576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yan B, Tao ZF, Li XM, Zhang H, Yao J and
Jiang Q: Aberrant expression of long noncoding RNAs in early
diabetic retinopathy. Invest Ophthalmol Vis Sci. 55:941–951. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Albagha OM, Visconti MR, Alonso N,
Langston AL, Cundy T, Dargie R, Dunlop MG, Fraser WD, Hooper MJ,
Isaia G, et al: Genome-wide association study identifies variants
at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's
disease of bone. Nat Genet. 42:520–524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soto I, Oglesby E, Buckingham BP, Son JL,
Roberson ED, Steele MR, Inman DM, Vetter ML, Horner PJ and
Marsh-Armstrong N: Retinal ganglion cells downregulate gene
expression and lose their axons within the optic nerve head in a
mouse glaucoma model. J Neurosci. 28:548–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chodroff RA, Goodstadt L, Sirey TM, Oliver
PL, Davies KE, Green ED, Molnár Z and Ponting CP: Long noncoding
RNA genes: conservation of sequence and brain expression among
diverse amniotes. Genome Biol. 11:R722010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kapitein LC and Hoogenraad CC: Building
the neuronal microtubule cytoskeleton. Neuron. 87:492–506. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Espinet C, Gonzalo H, Fleitas C, Menal MJ
and Egea J: Oxidative stress and neurodegenerative diseases: A
neurotrophic approach. Curr Drug Targets. 16:20–30. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wakatsuki S, Furuno A, Ohshima M and Araki
T: Oxidative stress-dependent phosphorylation activates ZNRF1 to
induce neuronal/axonal degeneration. J Cell Biol. 211:881–896.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herrup K, Neve R, Ackerman SL and Copani
A: Divide and die: Cell cycle events as triggers of nerve cell
death. J Neurosci. 24:9232–9239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dobolyi A, Vincze C, Pál G and Lovas G:
The neuroprotective functions of transforming growth factor beta
proteins. Int J Mol Sci. 13:8219–8258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang SP, Wang ZH, Peng DY, Li SM, Wang H
and Wang XH: Therapeutic effect of mesenchymal stem cells in rats
with intracerebral hemorrhage: Reduced apoptosis and enhanced
neuroprotection. Mol Med Rep. 6:848–854. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu G, Wang X, Shao G and Liu Q:
Genetically modified Schwann cells producing glial cell
line-derived neurotrophic factor inhibit neuronal apoptosis in rat
spinal cord injury. Mol Med Rep. 9:1305–1312. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao B and Chen YG: Regulation of TGF-β
signal transduction. Scientifica (Cairo). 2014:8740652014.
|
|
37
|
Wordinger RJ, Clark AF, Agarwal R, Lambert
W and Wilson SE: Expression of alternatively spliced growth factor
receptor isoforms in the human trabecular meshwork. Invest
Ophthalmol Vis Sci. 40:242–247. 1999.PubMed/NCBI
|
|
38
|
McDowell CM, Tebow HE, Wordinger RJ and
Clark AF: Smad3 is necessary for transforming growth factor-beta2
induced ocular hypertension in mice. Exp Eye Res. 116:419–423.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zode GS, Clark AF and Wordinger RJ: Bone
morphogenetic protein 4 inhibits TGF-β2 stimulation of
extracellular matrix proteins in optic nerve head cells: role of
gremlin in ECM modulation. Glia. 57:755–766. 2009. View Article : Google Scholar
|
|
40
|
Sethi A, Wordinger RJ and Clark AF:
Gremlin utilizes canonical and non-canonical TGFβ signaling to
induce lysyl oxidase (LOX) genes in human trabecular meshwork
cells. Exp Eye Res. 113:117–127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kuehn MH, Fingert JH and Kwon YH: Retinal
ganglion cell death in glaucoma: mechanisms and neuroprotective
strategies. Development. 1:32005.
|
|
42
|
Levkovitch-Verbin H: Retinal ganglion cell
apoptotic pathway in glaucoma: Initiating and downstream
mechanisms. Prog Brain Res. 220:37–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sánchez Y and Huarte M: Long non-coding
RNAs: Challenges for diagnosis and therapies. Nucleic Acid Ther.
23:15–20. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gao W, Chan JY and Wong TS: Differential
expression of long noncoding RNA in primary and recurrent
nasopharyngeal carcinoma. Biomed Res Int. 2014:4045672014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Michelhaugh SK, Lipovich L, Blythe J, Jia
H, Kapatos G and Bannon MJ: Mining Affymetrix microarray data for
long non-coding RNAs: Altered expression in the nucleus accumbens
of heroin abusers. J Neurochem. 116:459–466. 2011. View Article : Google Scholar
|