Introduction
Inflammation refers to the complex biological
response of the body to various harmful stimuli, including
pathogens, irritants, tissue necrosis, allergic reaction or cell
damage. Inflammation is induced as a protective response to a wide
range of injuries in an attempt to eliminate the cause of injury
and promote repair. However, ongoing inflammation and chronic
inflammatory processes may result in the pathogenesis of common
inflammation-associated diseases, including chronic bronchitis,
rheumatic heart disease, atherosclerosis and renal fibrosis
(1,2). Renal tubulointerstitial inflammation
is an important characteristic of chronic kidney disease (CKD).
Regardless of the initial cause, renal fibrosis is the common and
final outcome of all progressive CKDs. Renal tubulointerstitial
inflammation is generally considered an initial cause of renal
fibrosis, and is associated with the infiltration of inflammatory
cells into glomeruli and peritubular capillaries, particularly
macrophages and neutrophils, during the early events of
fibrogenesis (3). In renal
tubulointerstitial inflammation and fibrosis, the nuclear factor
(NF)-κB signaling pathways serve a critical role in CKD development
and progression. Numerous studies have indicated that inflammation
and fibrosis are closely related; therefore, a lack of inflammation
is associated with a lack of fibrosis (4–6).
Early intervention and inflammatory control in fibrosis and CKD
treatment are critical.
In renal tubulointerstitial inflammation, the renal
tubular epithelial cells are not only damaged but are also induced
to secrete numerous extracellular matrix factors and inflammatory
chemokines, which actively participate in renal interstitial
fibrosis. Proinflammatory cytokines and profibrotic factors are
secreted by renal tubular epithelial cells following injury,
thereby causing renal tubulointerstitial inflammation. In the
present study, the NRK-52E cell line was selected due to the
important role of renal tubular epithelial cells in renal
inflammation. In addition, lipopolysaccharide (LPS), which is a
product of Gram-negative bacteria, is an important inflammatory
factor that exerts host toxicity. LPS, or endotoxin, is released in
response to Gram-negative bacterial death and dissolution. Previous
studies have reported that LPS induces fever and leukocyte reaction
in renal inflammatory models when studying inflammatory renal
disease (7,8). In mouse and other animal models,
LPS-induced renal tubulointerstitial inflammation is accompanied
with a marked inflammatory response, including glomerular and
peritubular leukocyte infiltration (9,10).
In vivo mature renal inflammatory models have
been created using unilateral ureteral obstruction (UUO), which is
a classical model for the study of renal interstitial fibrosis and
inflammation. Renal obstruction caused by UUO leads to increased
urinary pressure, decreased renal blood flow, venous drainage
obstruction, infiltration of inflammatory cells and proliferation
of fibroblasts, thus leading to renal failure. This model has been
used in numerous studies regarding renal inflammation (11,12). In addition, LPS may induce a
multisystemic inflammatory response; however, the specific renal
inflammatory response was reduced in vivo LPS treatment.
These models were used in the present study to determine the
effects of left-right determination factor 1 (Lefty-1) on renal
tubulointerstitial inflammation.
Lefty is a novel member of the transforming growth
factor (TGF)-β superfamily. Lefty possesses two variants in mice:
Lefty-1 and Lefty-2, whereas Lefty-A and Lefty-B have been detected
in humans. These proteins control embryonic development, and
regulate stem cell differentiation and other functions (13). A previous study demonstrated that
Lefty may inhibit TGF-β1 signaling by reducing Smad2/3
phosphorylation, and further suppressing nuclear translocation of
the R-Smad (Smad2/3)/Smad4 complex (14). Furthermore, the Lefty protein can
regulate the cell cycle, and inhibit decidualization of the
endometrium and tumor activity (15–17). Based on these data, various
biological functions of Lefty have been revealed, and it has
received widespread attention regarding its biological regulatory
potential. However, despite the availability of information
regarding the beneficial properties of Lefty, it is currently
unknown as to whether this protein can regulate NF-κB signaling,
and its effects on renal tubulointerstitial inflammation and renal
fibrosis require elucidation. Therefore, the present study
investigated whether Lefty-1 serves a regulatory role in renal
tubulointerstitial inflammation and explored the possible
mechanisms involved in its action.
It has previously been reported that Lefty
attenuates renal tubulointerstitial injury in mice with UUO
(18). In the present study, the
protective effects of Lefty-1 on renal tubulointerstitial
inflammation were further assessed. The present study initially
investigated the effects of Lefty-1 on the expression and secretion
of LPS-induced inflammatory markers in cultured NRK-52E cells in
vitro and in a UUO model in vivo. Furthermore, the
regulatory role of the NF-κB signaling pathway in the potential
protective effects of Lefty-1 on renal interstitial inflammation
was analyzed. Through in vitro and in vivo models,
the present study aimed to evaluate the effects of Lefty-1 on renal
tubulointerstitial inflammation and to gain a preliminary
understanding of its underlying mechanism.
Materials and methods
Reagents and recombinant adenovirus
Interleukin (IL)-1β (cat. no. sc-7884; 1:100), IL-6
(cat. no. sc-7920; 1:200), tumor necrosis factor (TNF)-α (cat. no.
sc-8301; 1:100), monocyte chemotactic protein (MCP)-1 (cat. no.
sc-28879; 1:200), Smad7 (cat. no. sc-11392; 1:200), fibronectin
(cat. no. sc-9068; 1:200), collagen I (A1) (cat. no. sc-28657;
1:200), cluster of differentiation (CD)68 (cat. no. sc-9139;
1:100), F4/80 (cat. no. sc-25830; 1:200) and α-smooth muscle actin
(α-SMA; cat. no. sc-53015; 1:300) antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). NF-κB-p65 (cat.
no. 8242; 1:200), NF-κB-phosphorylated (p)-p65 (cat. no. 3033;
1:100), NF-κB inhibitor (IκB)-α (cat. no. 4812; 1:100) and p-IκB-α
(cat. no. 2859; 1:200) antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Flag (cat. no.
PAB25505; 1:200) antibodies were purchased from Abnova (Taipei,
Taiwan). Lefty-1 (cat. no. H00010637-D01P; 1:200) antibodies were
purchased from the Novus Biologicals LLC (Littleton, CO, USA). With
the exception of anti-α-SMA, which was derived from mouse, all
antibodies were monoclonal and derived from rabbit. Dulbecco’s
modified Eagle’s medium (DMEM)/high glucose medium and bovine serum
albumin (BSA) were purchased from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). LPS was obtained from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). The EZ-Cytox cell viability assay kit
(MTT) was acquired from MultiSciences Biotech Co., Ltd. (Hangzhou,
China). ELISA kits (cat. nos. EK0390, EK0412, EK0902 and EK0526)
were obtained from Wuhan Boster Biological Technology Ltd. (Wuhan,
China). The RevertAid First Strand cDNA Synthesis and quantitative
polymerase chain reaction (PCR) kits were obtained from Takara Bio,
Inc. (Otsu, Japan). Recombinant mice Lefty-1 adenovirus
(Ad-Lefty-1) was packaged and identified by Vigene Biosciences,
Inc. (Rockville, MD, USA).
Adenovirus construction
First-generation adenovirus was regulated by the
cytomegalovirus promoter using an open reading frame (ORF)
Shuttling system (Vigene Biosciences, Inc.). Briefly, mouse Lefty-1
cDNA (Vigene Biosciences, Inc.) was excised from the pENTR vector
and inserted into a pAD-ORF transfer vector. The resulting plasmids
were cotransformed into bacteria (E. coli) alongside human
adenovirus type 5 (E1 and E3 deleted). The recombinant adenoviral
constructs were transfected into 293 cells, in order to generate a
recombinant adenovirus with a high Ad-Lefty-1 titer
[1×1010 plaque-forming unit (pfu)/ml]. The doses used
were selected based on a previous report (19). Recombinant adenoviruses can
express exogenous gene fragments, and can infect dividing and
non-dividing cells in a wide range of hosts. The present study
obtained Ad-Lefty-1 through a customized adenovirus service (by
Vigene Biosciences, Inc.). In the present study, Ad-control refers
to an empty adenovirus that was used as a control in vivo
and in vitro. To detect infection efficiency, all adenovirus
vectors were labeled with flag. In addition, the effects of
adenovirus infection were verified using western blot analysis, in
order to confirm that Lefty-1 was highly expressed.
Cell culture and adenovirus
infection
NRK-52E cells were purchased from the American Type
Culture Collection (Manassas, VA, USA). The cells were cultured in
DMEM/high glucose medium supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.), 1% streptomycin and
penicillin in humidified incubator containing 95% air and 5%
CO2 at 37°C. The NRK-52E cells (70–80% confluence) were
seeded in 6-well plates, in order to determine the function of
Lefty-1 on LPS-induced inflammation. The cells were divided into
the following four groups: Control group, LPS (5 μg/ml)
treatment group, LPS + Ad-control group, and LPS + Ad-Lefty-1
group. Firstly, the cells were transfected with adenovirus at 10
pfu/cell in 1.5% serum DMEM/high glucose medium (antibiotics-free)
for 6 h, and then LPS + Ad-control and LPS + Ad-Lefty-1 groups were
washed with PBS and substitution of fresh media containing LPS was
made for 24 h. The fresh media were replaced with DMEM/high glucose
medium containing 1.5% FBS without antibiotics. The control group
received no treatment, expressions of related proteins and genes
were determined.
Animals and treatments
All animal studies were performed in accordance with
the Chinese Council on Animal Care Guidelines. The present study
was approved by the Animal Research Ethics Board of Wuhan
University (Wuhan, China). Male C57BL/6 mice (age, 6–7 weeks;
weight, 20–25 g) were obtained from the Wuhan University of
Laboratory Animal Research (Wuhan, China) and were divided into the
following experimental groups (n=7/group): i) Sham, in which mice
underwent a sham operation; ii) UUO (7 days); iii) UUO (7 days) +
Ad-control; and iv) UUO (7 days) + Ad-Lefty-1. Anesthesia was
induced and maintained by intraperitoneal injection of
pentobarbital sodium (50 mg/kg body weight) (20). After general anesthesia, with the
exception of the sham group, mice in the other experimental groups
were subjected to UUO. Briefly, a midline abdominal incision was
made, followed by double ligation of the upper left ureter using
4-0 silk sutures. The sham-operated mice had their ureters exposed
but not ligated. The left kidneys were then wrapped with ice-cold
saline-soaked gauze for 5 min post-injection, and 0.1 ml cold
saline containing 5.0×108 pfu Ad-control or Ad-Lefty-1
was injected into the left renal artery. The adenovirus solution,
alongside 3 units heparin, was injected using a Hamilton 800
microsyringe (Hamilton, Reno, NV, USA). The mice were maintained
under sterile conditions and were euthanized by cervical
dislocation on postoperative day 7. The left and right kidneys were
separately obtained for RNA and histological analyses.
Cytotoxicity assay
The MTT assay was performed to detect the survival
of NRK-52E cells. In the presence of an electronic-coupling
reagent, mitochondrial dehydrogenase may be reduced to form orange
formazan. Following treatment, the experimental cells were seeded
into 96-well plates at a density of 6.0×103 cells/well.
Subsequently, 10 μl MTT reagent was added to the plates for
45 min at 37°C, according to the manufacturer’s protocol.
Cytotoxicity was measured at 2, 4 and 6 h. Optical density (OD) in
each well was determined at 460 nm using a PerkinElmer VICTOR3 1420
Multilabel Counter (PerkinElmer, Inc., Waltham, MA, USA). The MTT
assay was repeated three times.
Detection of cytokine levels in cultured
supernatants
Treated cells were seeded into 6-well plates at a
density of 1.0×106 cells/well. The expression levels of
inflammatory cytokines, IL-1, IL-6, MCP-1 and TNF-α, were measured
in the supernatant using ELISA kits according to the manufacturer’s
protocol. The standards and samples were added to the wells and
were incubated at 37°C for 90 min, after which biotin-labeled
antibody was added for 60 min at 37°C. After washing with PBS three
times, avidin-peroxidase complex was added to the wells at 37°C for
30 min and the samples were then washed with PBS a further three
times. Subsequently, the chromogenic substrate TMB was added to
each well at 37°C for 25 min. Finally, wells were treated with TMB
stop solution, after which OD values were determined.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured NRK-52E cells
and tissues using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RNA was then reverse transcribed into
cDNA, which was amplified by qPCR PrimeScript One-Step RT-PCR kit
(Takara Bio, Inc.). The primer sequences are presented in Tables I and II. The reaction conditions were as
follows: Predenaturation at 95°C for 30 sec, followed by 40 cycles
at 95°C for 5 sec and 60°C for 40 sec; extension at 95°C for 5 sec
followed by 40 cycles at 60°C for 30 sec. GAPDH was used as the
reference gene. Relative quantification of gene expression was
performed using the 2−ΔΔCq method (21). The results of qPCR are presented
as the means ± standard deviation of fold-change of expression.
 | Table IMouse primers for quantitative
polymerase chain reaction. |
Table I
Mouse primers for quantitative
polymerase chain reaction.
| Gene name | Gene ID | Primer
sequences | Amplicon size
(bp) |
|---|
| α-SMA | NM_007392.3 | F:
5′-CTGGCATCGTGCTGGACTC-3′ | 291 |
| R:
5′-GCCCATCAGGCAACTCGTA-3′ | |
| Collagen I
(A1) | NM_007743.3 | F:
5′-TCCAAAGGAGAGAGCGGTAA-3′ | 310 |
| R:
5′-GACCAGGGAGACCAAACTCA-3′ | |
| Fibronectin | NM_173182.2 | F:
5′-GATGATGACCGACCAGATCCC-3′ | 88 |
| R:
5′-TGCTTCTCCGTTCACCAAGTG-3′ | |
| TNF-α | NM_013693.3 | F:
5′-CCCTCACACTCAGATCATCTTCT-3′ | 61 |
| R:
5′-GCTACGACGTGGGCTACAG-3′ | |
| IL-6 | NM_031168.2 | F:
5′-TAGTCCTTCCTACCCCAATTTCC-3′ | 76 |
| R:
5′-TTGGTCCTTAGCCACTCCTTC-3′ | |
| IL-1β | NM_008361.4 | F:
5′-CTGTGACTCATGGGATGATGATG-3′ | 75 |
| R:
5′-CGGAGCCTGTAGTGCAGTTG-3′ | |
| MCP-1 | NM_011333.3 | F:
5′-TAAAAACCTGGATCGGAACCAAA-3′ | 120 |
| R:
5′-GCATTAGCTTCAGATTTACGGGT-3′ | |
| Smad7 | NM_001042660.1 | F:
5′-GTGTTGCTGTGAATCTTACG-3′ | 228 |
| R:
5′-AGAAGAAGTTGGGAATCTGA-3′ | |
| NF-κB-p65 | NM_001357627.1 | F:
5-CCCTCGAGATGGACGATCTGTTTCCCCT-3′ | 211 |
| R:
5-CCCAAGCTTTTAGGAGCTGATCTGACTC-3′ | |
| GAPDH | NM_008084.3 | F:
5′-TGACCTCAACTACATGGTCTACA-3′ | 85 |
| R:
5′-CTTCCCATTCTCGGCCTTG-3′ | |
| Table IIRat primers for quantitative
polymerase chain reaction. |
Table II
Rat primers for quantitative
polymerase chain reaction.
| Gene name | Gene ID | Primer
sequences | Amplicon size
(bp) |
|---|
| TNF-α | NM_012675.3 | F:
5′-TGCCTCAGCCTCTTCTCATT-3′ | 180 |
| R:
5′-GGGCTTGTCACTCGAGTTTT-3′ | |
| IL-6 | NM_012589.2 | F:
5′-AGTTGCCTTCTTGGGACTGA-3′ | 218 |
| R:
5′-ACAGTGCATCATCGCTGTTC-3′ | |
| IL-1β | NM_031512.2 | F:
5′-CTGTGACTCGTGGGATGATG-3′ | 211 |
| R:
5′-AGGGATTTTGTCGTTGCTTG-3′ | |
| MCP-1 | NM_031530.1 | F:
5′-GATGCAGTTAATGCCCCACT-3′ | 168 |
| R:
5′-TTCCTTATTGGGGTCAGCAC-3′ | |
| α-SMA | NM_031004.2 | F:
5′-AACTGGTATTGTGCTGGACTCTG-3′ | 172 |
| R:
5′-CTCAGCAGTAGTCACGAAGGAATA-3′ | |
| Collagen I
(A1) | NM_053304.1 | F:
5′-CAGATTGAGAACATCCGCAGC-3′ | 310 |
| R:
5′-CGGAACCTTCGCTTCCATACTC-3′ | |
| Fibronectin | NM_019143.2 | F:
5′-GTGATCTACGAGGGACAGC-3′ | 78 |
| R:
5′-GCTGGTGGTGAAGTCAAAG-3′ | |
| GAPDH | NM_017008.4 | F:
5′-GGGTGTGAACCACGAGAAAT-3′ | 135 |
| R:
5′-ACTGTGGTCATGAGCCCTTC-3′ | |
Immunofluorescence staining
NRK-52E cells were fixed on coverslips with 4%
paraformaldehyde for 20 min at room temperature, washed three times
with PBS and permeabilized with 0.5% Triton X-100 buffer for 5 min.
Subsequently, the cells were blocked with 1% BSA in PBS for 20 min
at 4°C. The slides were then incubated overnight at 4°C with the
following primary monoclonal antibodies: Anti-NF-κB-p65 (1:100),
anti-fibronectin (1:200), anti-collagen I (1:200), anti-CD68
(1:100), anti-F4/80 (1:200) and anti-α-SMA (1:300). The cells were
then incubated with cyanine dye (Cy3)- or fluorescein
isothiocyanate (FITC)-conjugated secondary antibodies (1:100) to
visualize the primary antibodies. Finally, the nuclei were
counterstained with 4,6-diamidino-2-phenylindole. The slides were
visualized under an Olympus BX51 fluorescence upright microscope
(Olympus Corporation, Tokyo, Japan). Each experiment was repeated
three times.
Histological examination
All renal tissue specimens were examined for
morphological alterations. One half of each of the kidneys was
fixed in 4% buffered paraformaldehyde for 24 h at room temperature
for histological studies; the other half was snap-frozen in liquid
nitrogen, and stored at −80°C prior to protein or mRNA extraction.
Briefly, tissues were embedded in paraffin and serial sections were
made (4 μm). The first study were used for morphological
studies such as hematoxylin and eosin (H&E) staining, and
further analyses included immunohistochemistry, immunofluorescence,
real-time polymerase chain reaction (RT-PCR). Immunostaining was
analyzed in a blinded fashion. The antibodies used were as follows:
IL-1 (1:100), IL-6 (1:200), MCP-1 (1:200), TNF-α (1:100),
NF-κB-p-p65 (1:100), Smad7 (1:200), fibronectin (1:200), collagen I
(1:200) and α-SMA (1:300). After immunostaining with primary
antibodies (which were incubated at 4°C overnight), the sections
were developed using 3,3′-diaminobenzidine for immunohistochemical
examination. Subsequently, sections were incubated with
Cy3-conjugated goat anti-mouse (cat. no. 115-005-003; 1:200) and
FITC-conjugated goat anti-rabbit secondary antibodies (cat. no.
111-005-003; 1:200) (both from Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA) for immunohisto-fluorescence (avoid
light incubating for 1 h at 37°C). The histological images were
viewed under an Olympus BX51 upright microscope (Olympus
Corporation). Image-Pro Plus 7.0 (Media Cybernetics, Inc.,
Rockville, MD, USA) was used to semi-quantitatively analyze the
positive signals.
Western blot analysis
Total proteins were extracted using
radioimmunoprecipitation assay lysis buffer containing protease
inhibitor cocktail (Wuhan Goodbio Technology Co., Ltd., Wuhan,
China) and protein concentrations were measured using the
bicinchoninic acid assay kit (Wuhan Boster Biological Technology
Ltd.). Equal amounts of total protein (30 μg) were loaded
into each lane, separated by 10% SDS-PAGE and transferred onto
activated polyvinylidene fluoride membranes. After blocking with 5%
BSA in Tris-buffered saline at room temperature for 1 h, the
membranes were incubated overnight at 4°C with the appropriate
primary antibodies. Subsequently, the membranes were incubated with
fluorescence-labeled secondary antibodies [IRDye700 and IRDye800,
goat anti-mouse (cat. no. 925-32210; 1:1,000)/rabbit (cat. no.
925-32211; 1:1,000); both from LI-COR Biosciences, Lincoln, NE,
USA] for 1 h at 37°C, and immune complexes were detected using an
Odyssey infrared imaging system (https://www.licor.com/bio/products/imaging_systems/odyssey/;
LI-COR Biosciences).
Statistical analysis
All statistical analyses were performed using SPSS
19.0 (IBM Corp., Armonk, NY, USA). Data are expressed as the means
± standard deviation. One-way analysis of variance, followed by the
Student-Newman-Keuls test, was used for the quantitative data,
whereas the Kruskal-Wallis test was used for non-normally
distributed data. P<0.05 was considered to indicate a
statistically significant difference.
Results
Adenovirus-mediated Lefty-1
overexpression inhibits LPS-induced NRK-52E cytotoxicity
Western blot analysis and RT-qPCR were used to
examine Lefty-1 overexpression, and validate Ad-Lefty-1 infection
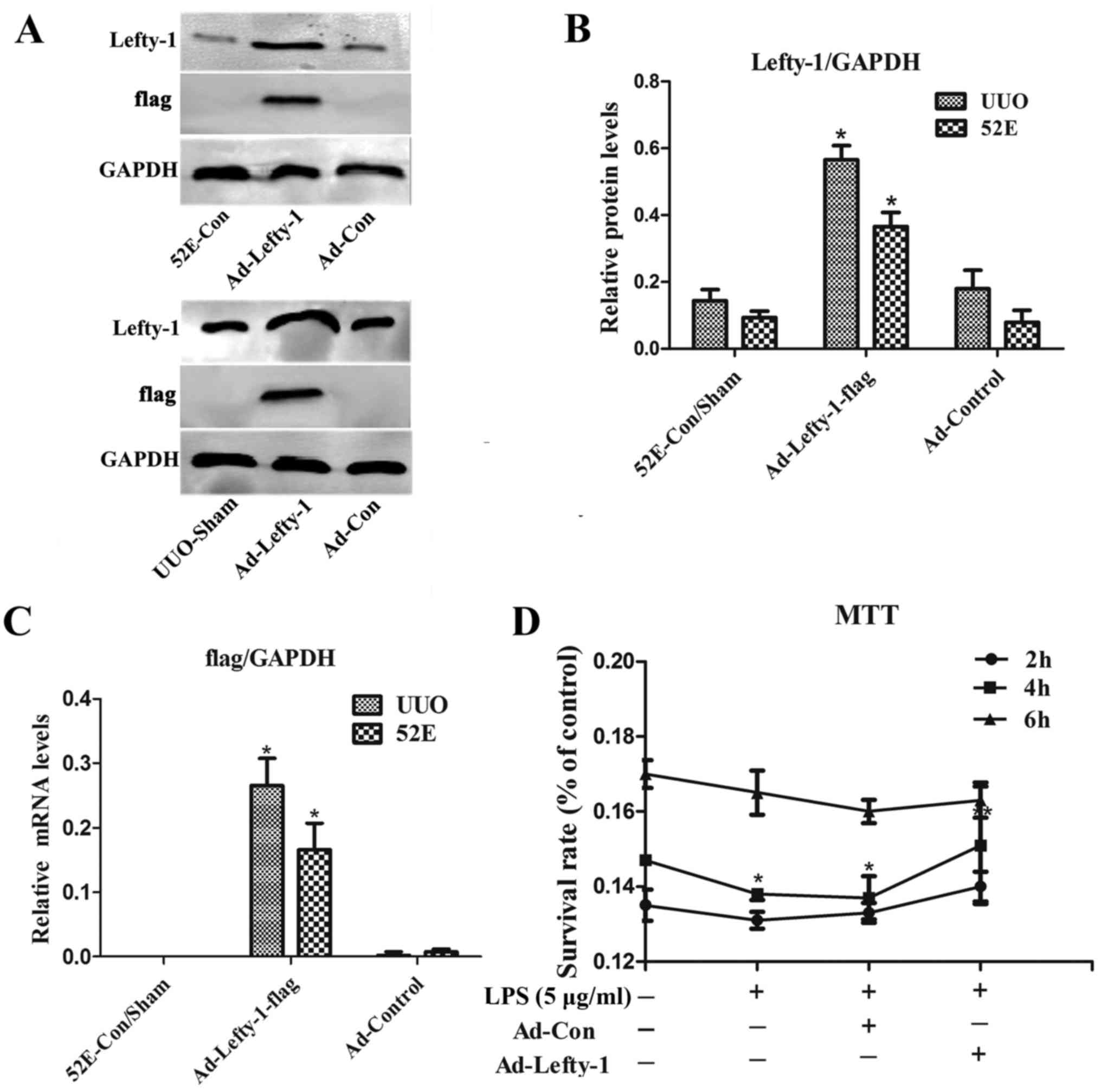
efficiency in NRK-52E cells and in UUO mice. The results indicated
that the expression levels of Lefty-1 were significantly higher in
NRK-52E cells and UUO mice stably infected with Ad-Lefty-1 compared
with in the uninfected NRK-52E cells and the sham group mice
(Fig. 1A–C). NRK-52E cytotoxicity
was evaluated using the MTT assay. Cytotoxicity was not detected in
the control groups. Conversely, LPS markedly enhanced NRK-52E
cytotoxicity, particularly 4 h after treatment, whereas Ad-Lefty-1
infection significantly suppressed LPS-induced cytotoxicity
(Fig. 1D).
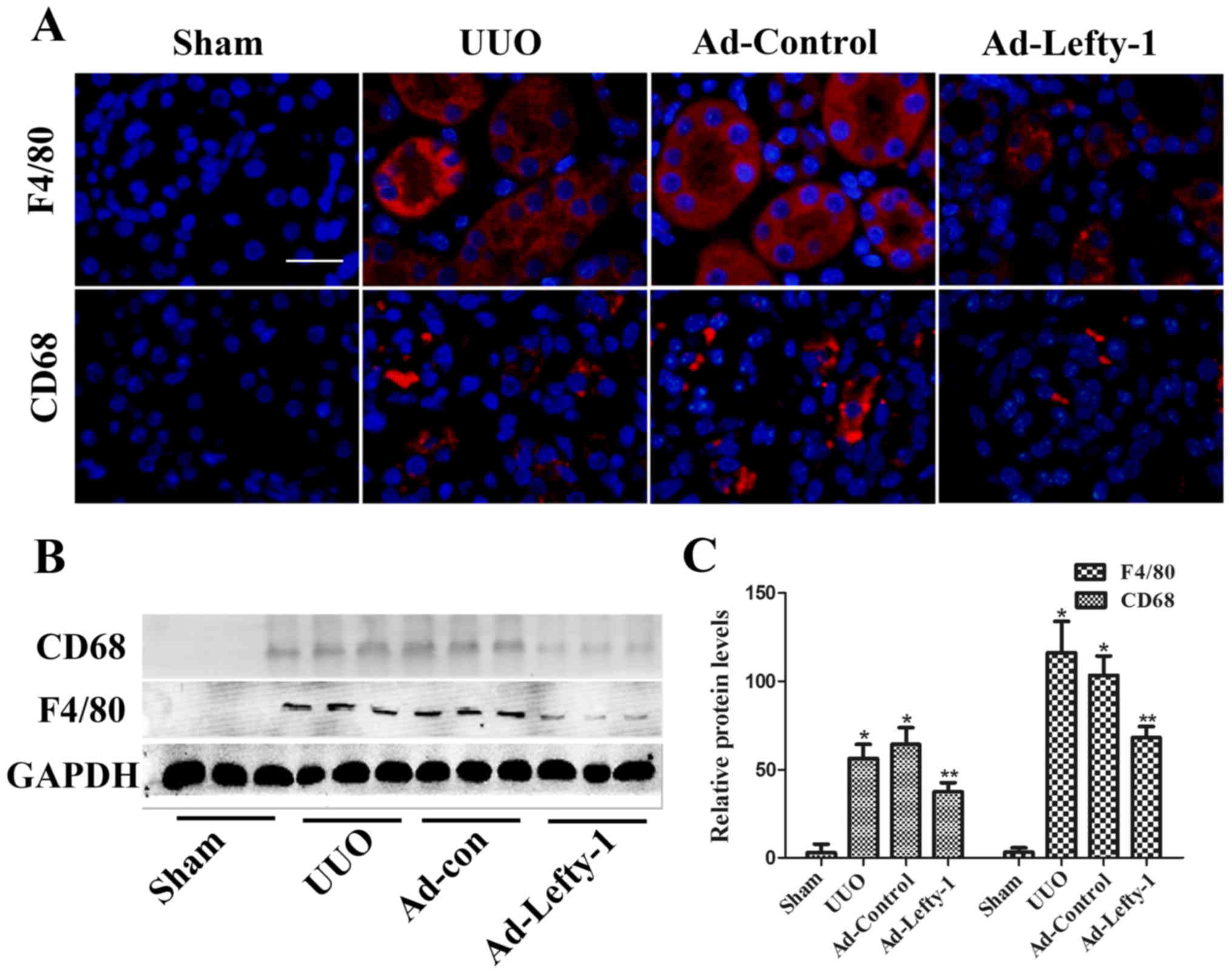
Lefty-1 overexpression suppresses the
release of inflammatory mediators and macrophage infiltration
The present study aimed to determine the protective
function of Lefty-1 on the suppression of proinflammatory mediators
and macrophage infiltration in vivo via histological
examination and RT-qPCR. As shown in Fig. 2, mice exhibited progressive
tubulointerstitial damage, which was characterized by tubular
dilation with flattened epithelium, and tubular and glomerular
atrophy, on day 7 following UUO. In addition, expansion of the
renal interstitial space, and inflammatory cytokine (IL-1β, IL-6
and TNF-α) and chemokine (MCP-1) expression were increased
alongside progressive inflammation. Furthermore, immunofluorescence
staining indicated that CD68 and F4/80 expression, and positive
macrophage infiltration, were increased in UUO mice compared with
in sham-operated mice (Fig. 3).
These increases were not significantly inhibited by Ad-control.
Conversely, Ad-Lefty-1 infection suppressed macrophage
infiltration, and inflammatory cytokine and chemokine expression.
These findings suggested that Ad-Lefty-1-induced improvement of
histological damage was associated with a reduction in
inflammation.
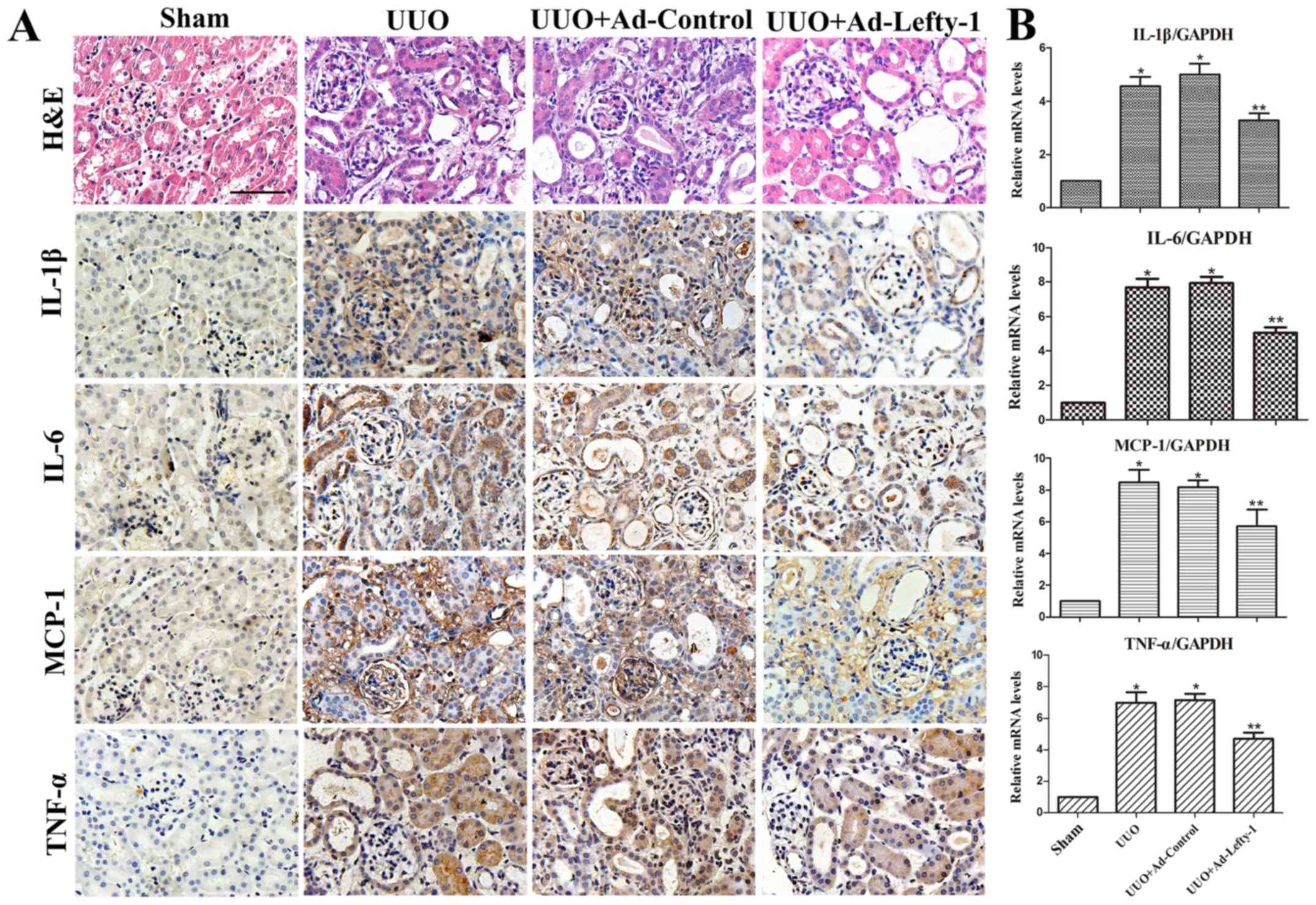 | Figure 2Effects of Lefty-1 on UUO-induced
renal tubulointerstitial inflammation. (A) Overexpression of
Lefty-1 suppressed inflammatory cytokine (IL-1β, IL-6 and TNF-α)
and chemokine (MCP-1) expression 7 days after UUO, as determined by
immunohistochemistry. Magnification, ×200; scale bar, 20 μm
(B) Reverse transcription-quantitative polymerase chain reaction.
Data are expressed as the means ± standard deviation of at least
three independent experiments. n=6. *P<0.001 vs. the
sham control group; **P<0.05 vs. the UUO or UUO +
Ad-control-treated groups. Ad, adenovirus; H&E, hematoxylin and
eosin; IL, interleukin; Lefty-1, left-right determination factor 1;
MCP-1, monocyte chemotactic protein-1; TNF-α, tumor necrosis
factor-α; UUO, unilateral ureteral obstruction. |
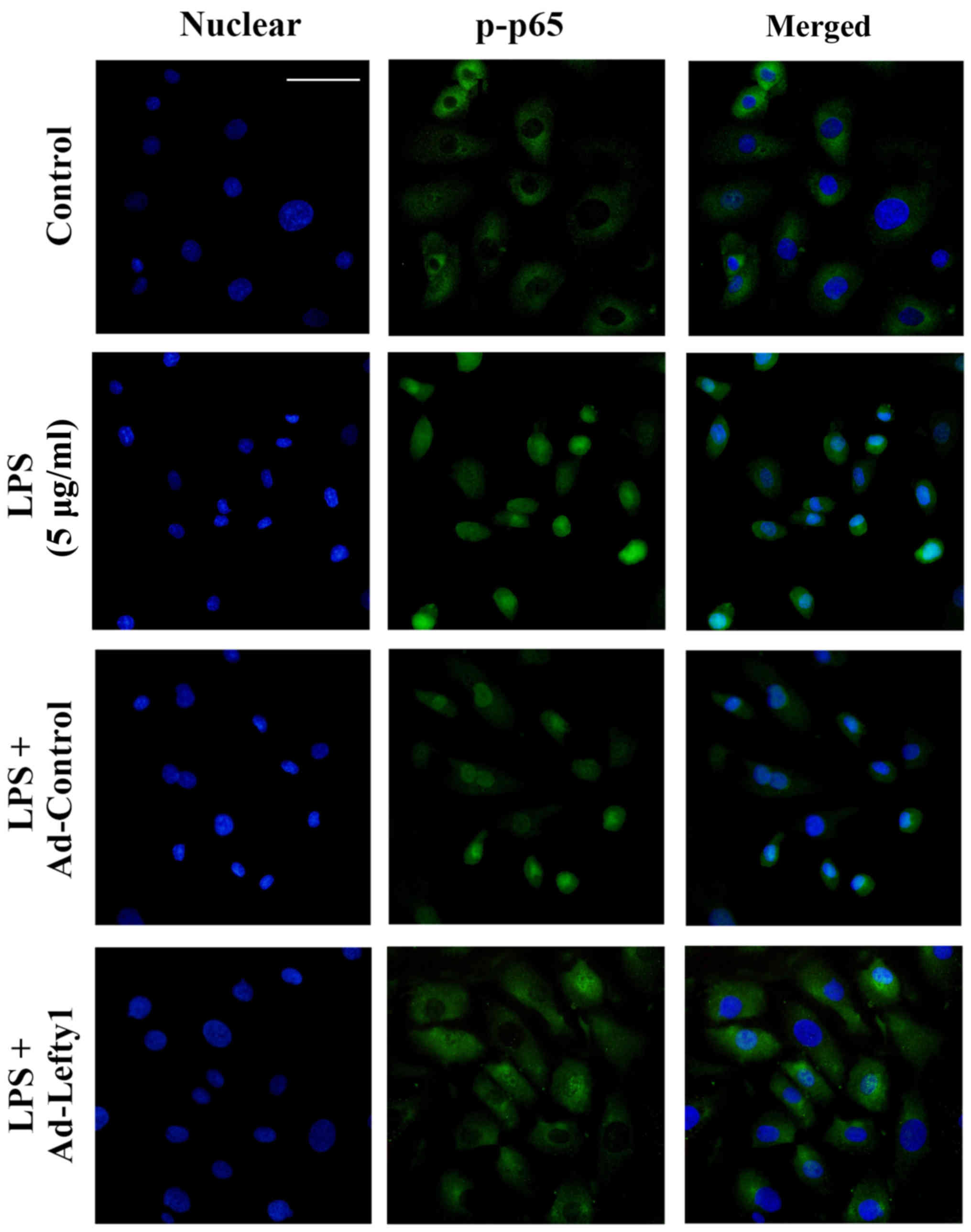
Lefty-1 overexpression suppresses
LPS-induced NF-κB-p-p65 nuclear translocation in vitro
The present study performed immunofluorescence
staining to examine the distribution of NF-κB-p-p65 and to confirm
that Lefty-1 inhibited LPS-induced nuclear translocation of
NF-κB-p-p65 in NRK-52E cells. The results indicated that
NF-κB-p-p65 protein was predominately detected in the cytoplasm of
normal cells (Fig. 4). Following
LPS stimulation for 1 h, increased NF-κB-p-p65 staining was
observed in the nucleus. Conversely, Ad-Lefty-1 could decrease
NF-κB-p-p65 nuclear staining, thus suggesting that Lefty-1
inhibited NF-κB-p-p65 nuclear translocation. However, Ad-control
infection had no affect on NF-κB-p-p65 nuclear staining.
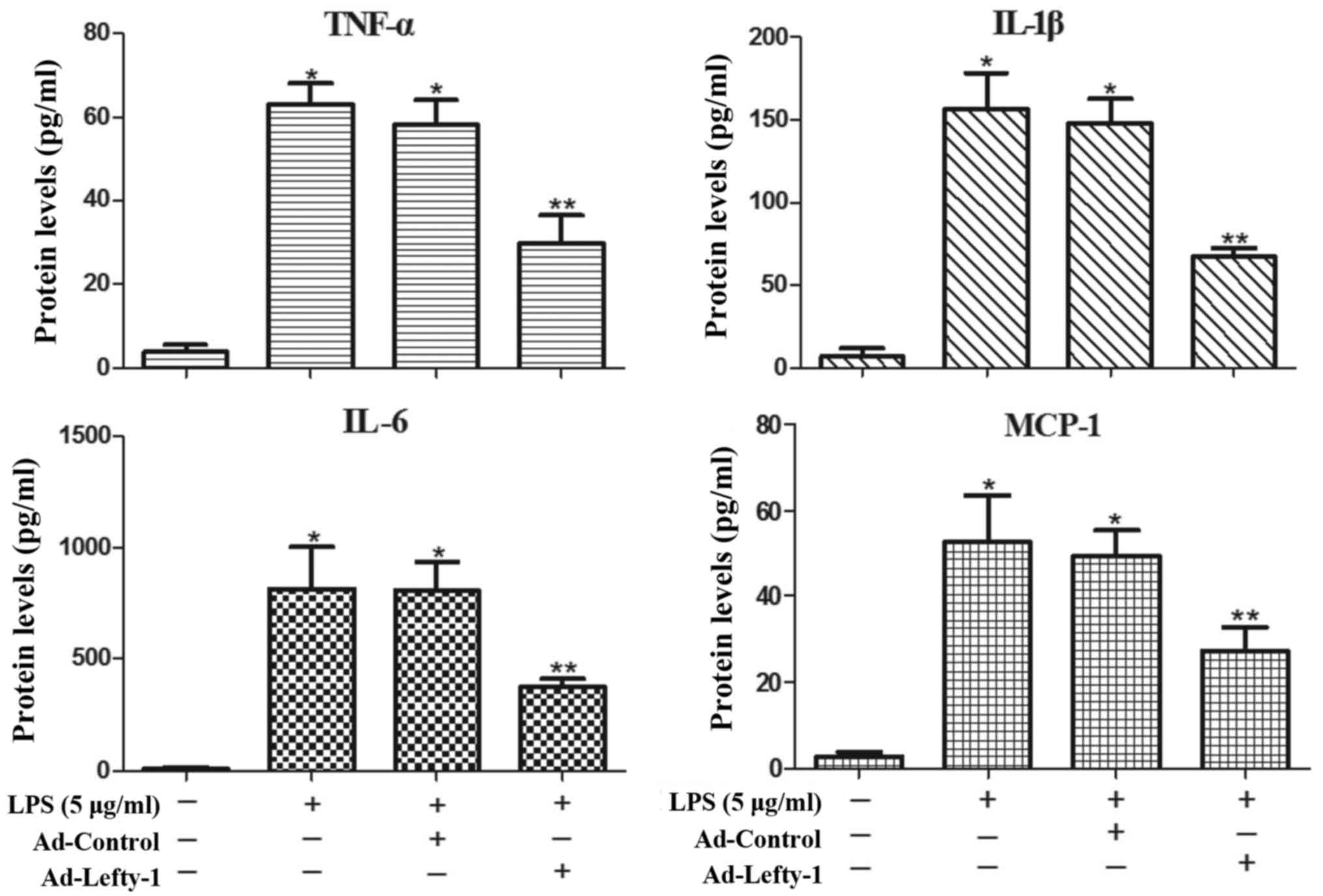
Lefty-1 overexpression attenuates
LPS-induced inflammatory responses in NRK-52E cells
The present study also aimed to determine whether
Lefty-1 affects the synthesis of proinflammatory cytokines (IL-1β,
IL-6 and TNF-α) and chemokines (MCP-1) in LPS-treated cells. Based
on the results of ELISA analyses (Fig. 5), the expression levels of these
inflammatory proteins were significantly increased in
LPS-challenged NRK-52E cells. However, infection with Ad-Lefty-1
significantly decreased cytokine and chemokine expression. These
in vitro results were similar to those observed in the in
vivo analyses. These findings indicated that Lefty-1 may
mediate the inhibition of proinflammatory cytokines and
chemokines.
Lefty-1 overexpression inhibits renal
fibrosis in vivo and in vitro
Inflammation is a protective response in numerous
types of kidney injury; however, unresolved inflammation promotes
progressive renal fibrosis (4).
Therefore, the present study assessed the expression of fibrotic
proteins, in order to verify the association between fibrosis and
inflammation.
The effects of Lefty-1 were determined on fibrosis
in vivo and in vitro (Fig. 6). Immunofluorescence staining and
RT-qPCR analysis indicated that the expression levels of
fibronectin, collagen I and α-SMA were increased in kidneys from
UUO mice compared with in the sham-operated kidneys (Fig. 6A and C). This increase was
significantly inhibited by Ad-Lefty-1, but not by Ad-control. The
expression levels of fibrotic markers exhibited similar trends in
NRK-52E cells; LPS-induced alterations were significantly inhibited
by Ad-Lefty-1, but not by Ad-control (Fig. 6B and C).
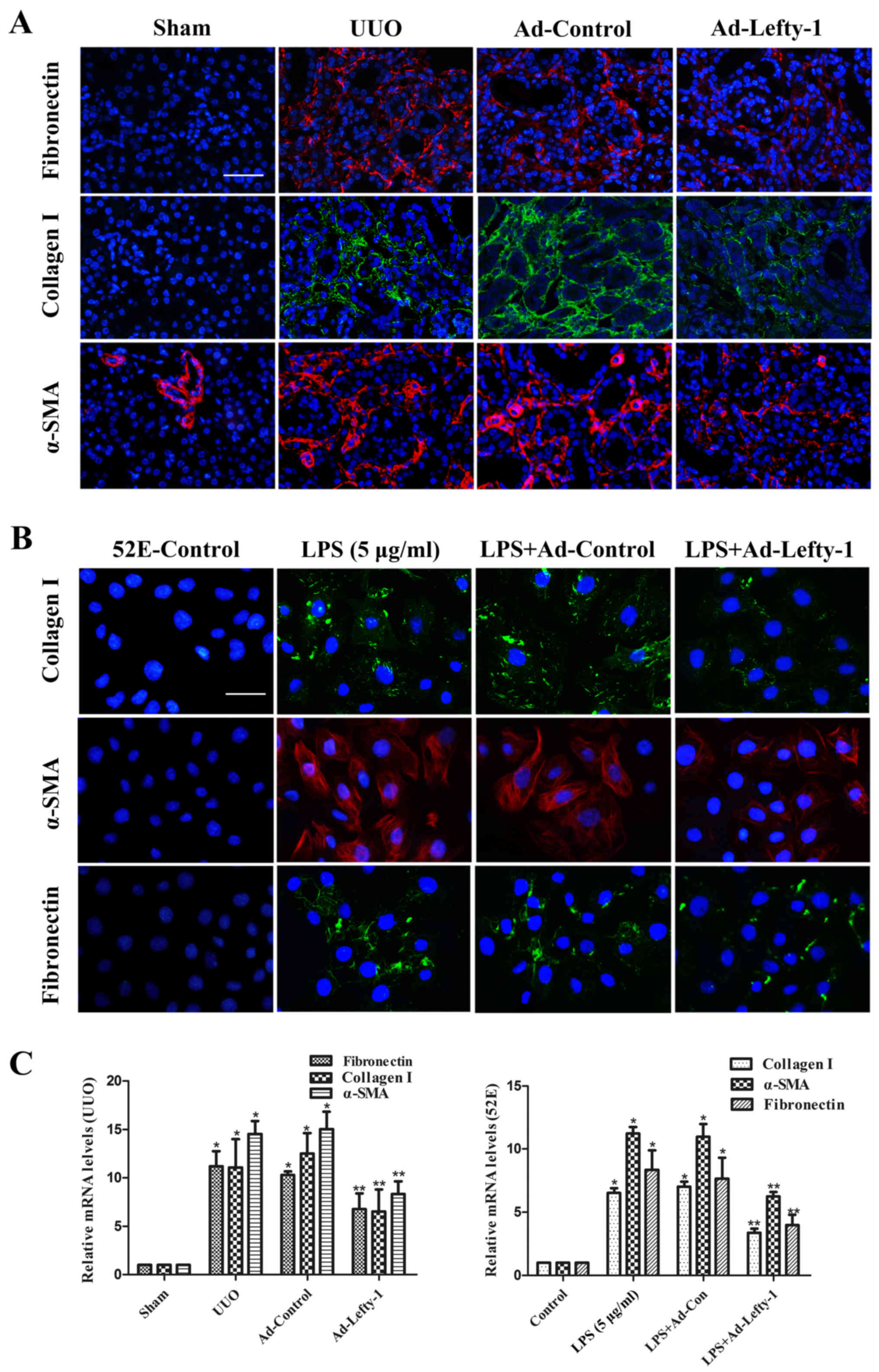 | Figure 6Immunofluorescence staining of
fibronectin, collagen and α-SMA in kidney tissues and NRK-52E
cells. (A and B) Compared with in the UUO mice and LPS-treated
NRK-52E cells, the expression levels of fibronectin, collagen I and
α-SMA were decreased in Ad-Lefty-1-infected groups. Ad-control had
no significant effects. Magnification: (A) ×200, scale bar, 20
μm; (B) ×400, scale bar, 40 μm. (C) Reverse
transcription-quantitative polymerase chain reaction analyses. n=5,
*P<0.001 vs. the sham or control groups;
**P<0.05 vs. the UUO, LPS (5 μg/ml), UUO +
Ad-control or LPS + Ad-control-treated groups. α-SMA, α-smooth
muscle actin; Ad, adenovirus; Lefty-1, left-right determination
factor 1; LPS, lipopolysaccharide; UUO, unilateral ureteral
obstruction. |
Lefty-1 overexpression regulates NF-κB
signaling pathways
Numerous reports have suggested that NF-κB signaling
pathways are important for the regulation of macrophage
infiltration and inflammatory cytokine expression (22). Therefore, the present study
examined whether Lefty-1 may suppress NF-κB pathways in
vitro and in vivo. The results indicated that
NF-κB-p-p65 protein expression was significantly inhibited by
Ad-Lefty-1, whereas NF-κB-p65 protein was not altered. In addition,
IκB-α expression and its phosphorylation were detected. The
expression levels of p-IκB-α were increased in the LPS-stimulated
group and were decreased in the Ad-Lefty-1 group, whereas IκB-α
protein expression was not altered. In addition, LPS-induced
downregulation of Smad7 expression was reversed following
Ad-Lefty-1 administration (Fig. 7A
and B). Furthermore, NF-κB-p65 and Smad7 mRNA expression levels
were examined in UUO mice with or without Ad-Lefty-1 treatment
(Fig. 7C and D). The expression
levels of NF-κB-p65 were not significantly altered in these groups.
However, Smad7 expression levels were significantly downregulated
in the UUO group and were significantly upregulated in the
Ad-Lefty-1 group. These findings suggested that administering
Ad-Lefty-1 could reverse UUO-induced downregulation of Smad7, which
was similar to the previous observations in vitro. These
data suggested that Lefty-1 may modulate the inflammatory response,
probably via the Smad7-regulated NF-κB signaling pathway.
Smad7-mediated inhibition of NF-κB activation via IκB-α induction
may be considered the central mechanism by which Lefty-1 prevents
renal inflammation.
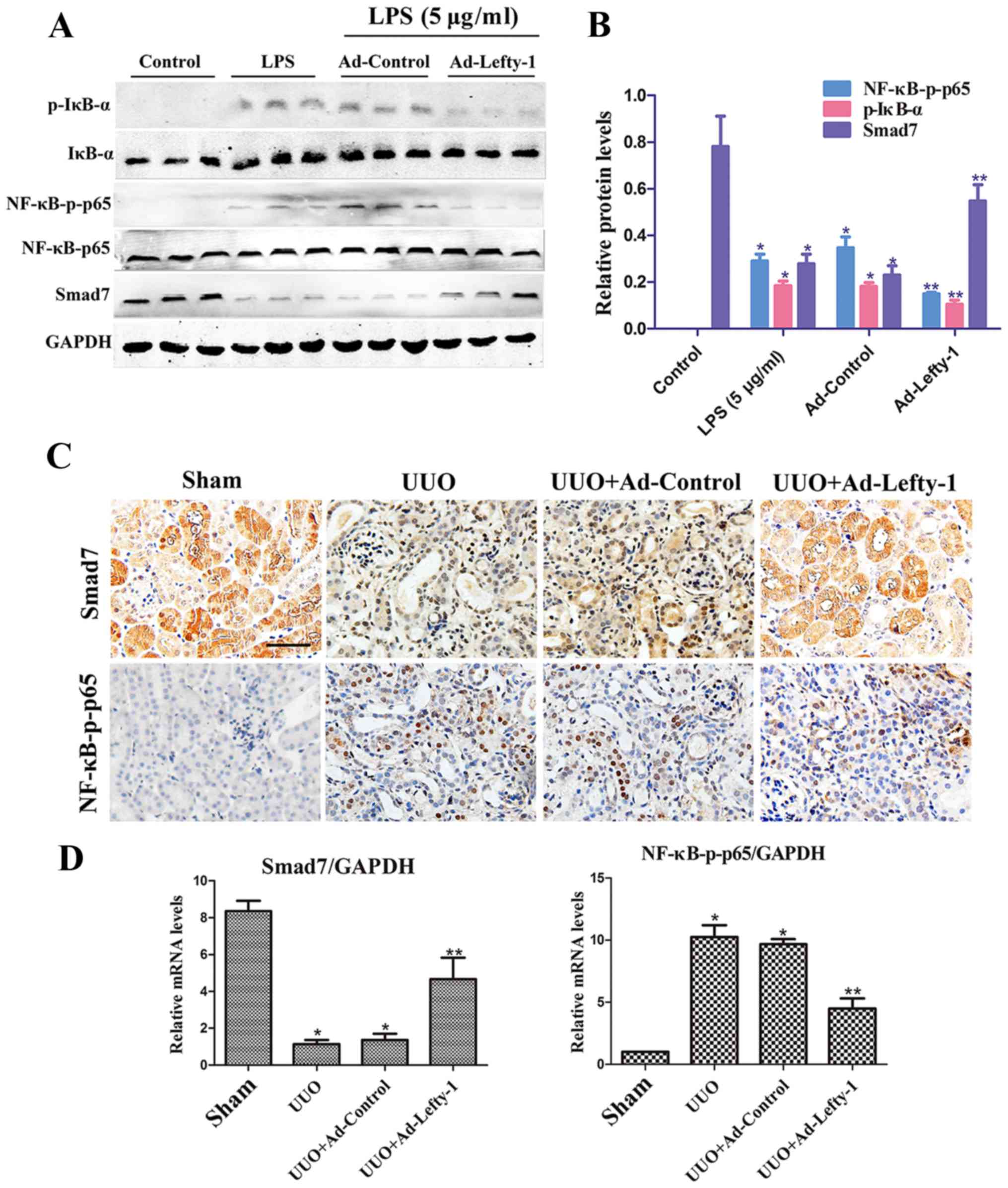 | Figure 7Overexpression of Lefty-1 modulates
NF-κB/Smad7 signaling in renal tubulointerstitial inflammation. (A)
Western blot analysis was conducted to determine the effects of
Lefty-1 on LPS-treated NRK-52E cells. Ad-Lefty-1 significantly
inhibited NF-κB-p-p65 and p-IκB-α expression, and enhanced Smad7
expression. (B) Semi-quantitative analysis of western blotting. (C)
Histological examination validated the in vitro results.
Magnification, ×200; scale bar, 20 μm (D) Reverse
transcription-quantitative polymerase chain reaction for NF-κB-p65
and Smad7 expression. n=6, *P<0.001 vs. the sham or
control groups; **P<0.05 vs. the UUO, LPS (5
μg/ml), UUO + Ad-control or LPS + Ad-control-treated groups.
Ad, adenovirus; IκB-α, NF-κB inhibitor-α; Lefty-1, left-right
determination factor 1; LPS, lipopolysaccharide; NF-κB, nuclear
factor-κB; p-, phosphorylated; UUO, unilateral ureteral
obstruction. |
Discussion
The pathological process of inflammation, which is
associated with acute and chronic renal injury, has an important
role in structural and functional damage in various types of renal
disease. Various inflammatory processes induce renal fibrotic
damage, crescent formation and mesangial cell apoptosis. Renal
tubulointerstitial inflammation culminates in fibrosis, which
contributes to progressive renal disease. Emerging evidence has
indicated that renal tubulointerstitial inflammation is closely
associated with renal fibrosis. Therefore, inflammatory processes
are considered to be involved in the pathogenesis of numerous renal
diseases, including CKD and end-stage renal disease (23–25). These findings indicated that early
renal inflammatory intervention is important. The pathogenesis of
inflammation has been detected in numerous types of renal disease
and has become a common target for novel drug development.
The present study provided credible evidence to
suggest the importance of Lefty-1 as a mediator of renal
inflammation and fibrosis in vitro and in vivo. Based
on the results of the present study, Lefty-1 may effectively
inhibit expression of the NF-κB signaling pathway, interfere with
IκB-α hydrolysis and nuclear translocation of the NF-κB-p-p65
protein, and serve an anti-inflammatory role. Furthermore, Lefty-1
was able to recover Smad7 expression, an important inhibitory Smad
protein. A previous study reported that Smad7 can regulate numerous
signal transduction pathways associated with inflammation (26). In addition, Lefty-1 is an
important regulator of TGF-β signaling, which possesses various
biological properties that are involved in mediating stem cell
differentiation and embryonic development (27,28). Based on these findings, the
present study investigated the role of Lefty-1 in UUO- and
LPS-induced renal expression of IL-1β, IL-6, TNF-α and MCP-1 using
in vivo and in vitro models, respectively. The
present results demonstrated that Lefty-1 significantly inhibited
proinflammatory cytokine and chemokine expression, and macrophage
infiltration, in vivo and in vitro. Furthermore,
protective effects of Lefty-1 were observed against renal
inflammation, which may be due to the suppression of NF-κB
signaling activation, which may be mediated by Smad7. These
findings were consistent with the previous findings that Lefty is a
novel tumor suppressor with anti-inflammatory potential (29).
The inflammatory process in the kidney is complex,
and is closely associated with numerous types of cell signaling
pathways and cytokines, which involve various cells,
cytokines/chemokines, adhesion molecules and growth factors
(30). Previous studies have
focused on TGF-β and epithelial or endothelial cells for
mesenchymal transition in myofibroblast transformation, which leads
to fibrosis (3). Inflammation is
involved in the multi-stage process of fibrosis and is closely
associated with scarring, where persistent and excessive tissue
repair results in the replacement of normal parenchymal cells with
fibrous tissue (31,32). However, the exact mechanism
underlying how renal tubulointerstitial inflammation leads to
fibrosis has yet to be elucidated. Furthermore, current treatment
of renal inflammatory diseases focuses on reducing inflammatory
cytokines, and the use of nonspecific immunosuppressants,
anticoagulants and anti-platelet drugs; however, such treatment is
less effective (33). Enhancement
of renal interstitial inflammation appears to be the main factor
for renal fibrosis, particularly in obstructive nephropathy.
Therefore, a better understanding of the renal interstitial
inflammation-associated mechanism may promote the development of
early treatment strategies for renal interstitial inflammation, and
even for renal fibrosis and its associated ailments. Among all
normal renal cells, renal tubular epithelial cells have an
important role in renal inflammation (34,35). Impaired renal tubular epithelial
cells, which are able to secrete proinflammatory cytokines and a
series of profibrotic factors, induce interstitial inflammation and
vascular contraction that aggravates renal ischemia. This
phenomenon leads to the apoptosis of renal tubular epithelial
cells, renal tubular atrophy and renal interstitial fibrosis.
Activated NF-κB signaling is the main signaling
mechanism that activates and promotes macrophage infiltration and
interstitial inflammation. Previous studies have reported that UUO-
or LPS-mediated renal interstitial inflammation is associated with
various intracellular signaling pathways, including NF-κB,
mitogen-activated protein kinase and other cascades (36–38). Therefore, NF-κB signaling is
considered important for renal interstitial inflammation, as
numerous molecules, including IL-1β, IL-6, TNF-α and MCP-1, are
involved in the early stage of the immune response, and each stage
of inflammation is affected by NF-κB regulation. In quiescent
cells, NF-κB and IκB form a complex, which is inactive in the
cytoplasm. When the cells are stimulated by extracellular signals,
IκB kinase activation leads to IκB phosphorylation, and NF-κB
nuclear localization sites are exposed. The rapid translocation of
dissociated NF-κB to the nucleus, in combination with the specific
κB sequence, induces gene transcription. Therefore, NF-κB-targeted
therapeutic strategies may be effective in the treatment of renal
inflammatory diseases; numerous anti-inflammatory agents achieve
therapeutic effects by suppressing NF-κB signaling (39,40). Smad7 is an inhibitory Smad that
suppresses NF-κB-driven inflammatory responses in renal
inflammation and fibrosis, and has garnered attention in recent
years. Smad7 overexpression can induce IκB-α, thereby inhibiting
NF-κB-induced renal inflammation. Therefore, Smad7 may have a
potential therapeutic role in renal interstitial inflammation
(41–43). In agreement with these
observations, the present results indicated that the
anti-inflammatory effects of Lefty-1 were partially involved with
inhibition of NF-κB activation, by blocking nuclear translocation
of the NF-κB/p65 protein and IκB-α degradation. In addition,
Lefty-1 was able to recover Smad7 levels by inhibiting NF-κB
activation. It is well accepted that TGF-β may induce inhibition of
NF-κB-mediated renal inflammation via induction of Smad7-dependent
IκB-α expression (44). Notably,
Smad7 expression is reduced in diseased kidneys, which causes an
imbalance within the TGF-β/Smads and NF-κB signaling pathways,
resulting in the development of renal inflammation and fibrosis. A
previous study, and the present study, indicated that Lefty-1 is a
crucial mediator of the NF-κB and TGF-β/Smads pathways; these
results revealed the numerous regulatory and balancing functions of
Lefty-1 (45).
In conclusion, the association between renal
interstitial inflammation and fibrosis is complex and significant
in CKD pathogenesis. In particular, inflammation is often the
initiating factor of fibrosis. Therefore, early intervention and
control of renal interstitial inflammation are crucial in the
treatment of renal fibrosis and CKD. The present findings provided
clear evidence to suggest that Lefty-1 attenuated renal fibrosis by
inhibiting renal interstitial inflammation. Further research
revealed that Lefty-1 effectively inhibited NF-κB-p65 nuclear
translocation, IκB-α degradation and Smad7 degradation at the early
stage of renal interstitial inflammation. These findings provide a
novel theoretical basis for further development and utilization of
Lefty-1 in the treatment of renal interstitial inflammation.
Abbreviations:
|
CKD
|
chronic kidney disease
|
|
LPS
|
lipopolysaccharide
|
|
UUO
|
unilateral ureteral obstruction
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
References
|
1
|
Ozaki E, Campbell M and Doyle SL:
Targeting the NLRP3 inflammasome in chronic inflammatory diseases:
Current perspectives. J Inflamm Res. 8:15–27. 2015.PubMed/NCBI
|
|
2
|
Chen J, Zhao Y and Liu Y: The role of
nucleotides and purinergic signaling in apoptotic cell clearance -
implications for chronic inflammatory diseases. Front Immunol.
5:6562014. View Article : Google Scholar
|
|
3
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
Inflammatory processes in renal fibrosis. Nat Rev Nephrol.
10:493–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee SB and Kalluri R: Mechanistic
connection between inflammation and fibrosis. Kidney Int Suppl.
78:S22–S26. 2010. View Article : Google Scholar
|
|
6
|
Rodríguez-Iturbe B and García García G:
The role of tubulointerstitial inflammation in the progression of
chronic renal failure. Nephron Clin Pract. 116:c81–c88. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mayeux PR: Pathobiology of
lipopolysaccharide. J Toxicol Environ Health. 51:415–435. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bosshart H and Heinzelmann M: Targeting
bacterial endotoxin: Two sides of a coin. Ann N Y Acad Sci.
1096:1–17. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abdel-Bakky MS, Hammad MA, Walker LA and
Ashfaq MK: Silencing of tissue factor by antisense
deoxyoligonucleotide prevents monocrotaline/LPS renal injury in
mice. Arch Toxicol. 85:1245–1256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhong F, Chen H, Jin Y, Guo S, Wang W and
Chen N: Analysis of the gene expression profile of curcumin-treated
kidney on endotoxin-induced renal inflammation. Inflammation.
36:80–93. 2013. View Article : Google Scholar
|
|
11
|
Zhang J, Zheng L, Yuan X, Liu C, Yuan Q,
Xie F, Qiu S, Peng Z, Tang Y, Meng J, et al: Mefunidone ameliorates
renal inflammation and tubulointerstitial fibrosis via suppression
of IKKβ phosphorylation. Int J Biochem Cell Biol. 80:109–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou X, Sun X, Gong X, Yang Y, Chen C,
Shan G and Yao Q: Astragaloside IV from Astragalus membranaceus
ameliorates renal interstitial fibrosis by inhibiting inflammation
via TLR4/NF-кB in vivo and in vitro. Int Immunopharmacol. 42:18–24.
2017. View Article : Google Scholar
|
|
13
|
Tabibzadeh S and Hemmati-Brivanlou A:
Lefty at the crossroads of ‘stemness’ and differentiative events.
Stem Cells. 24:1998–2006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ulloa L and Tabibzadeh S: Lefty inhibits
receptor-regulated Smad phosphorylation induced by the activated
transforming growth factor-beta receptor. J Biol Chem.
276:21397–21404. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H, Li H, Bai L and Yu H: Lefty inhibits
in vitro decidualization by regulating P57 and cyclin D1
expressions. Cell Biochem Funct. 32:657–664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cavallari C, Fonsato V, Herrera MB, Bruno
S, Tetta C and Camussi G: Role of Lefty in the anti tumor activity
of human adult liver stem cells. Oncogene. 32:819–826. 2013.
View Article : Google Scholar
|
|
17
|
Tang M, Naidu D, Hearing P, Handwerger S
and Tabibzadeh S: LEFTY, a member of the transforming growth
factor-beta superfamily, inhibits uterine stromal cell
differentiation: A novel autocrine role. Endocrinology.
151:1320–1330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu C, Xu M, Wang W and Zhang J: Lefty1
alleviates renal tubulointerstitial injury in mice with unilateral
ureteral obstruction. Mol Med Rep. 13:901–908. 2016. View Article : Google Scholar
|
|
19
|
Ghayur A, Liu L, Kolb M, Chawla A, Lambe
S, Kapoor A and Margetts PJ: Adenovirus-mediated gene transfer of
TGF-β1 to the renal glomeruli leads to proteinuria. Am J Pathol.
180:940–951. 2012. View Article : Google Scholar
|
|
20
|
Georgi MK, Vigilance J, Dewar AM and Frame
MD: Terminal arteriolar network structure/function and plasma
cytokine levels in db/db and ob/ob mouse skeletal muscle.
Microcirculation. 18:238–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Liu W, Zhu H and Fang H: Propofol
potentiates sevoflurane-induced inhibition of nuclear
factor-κB-mediated inflammatory responses and regulation of
mitogen-activated protein kinases pathways via Toll-like receptor 4
signaling in lipopolysaccharide-induced acute lung injury in mice.
Am J Med Sci. 354:493–505. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anders HJ and Ryu M: Renal
microenvironments and macrophage phenotypes determine progression
or resolution of renal inflammation and fibrosis. Kidney Int.
80:915–925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lan HY: Diverse roles of TGF-β/Smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067. 2011.
View Article : Google Scholar :
|
|
25
|
Brown NJ: Contribution of aldosterone to
cardiovascular and renal inflammation and fibrosis. Nat Rev
Nephrol. 9:459–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu L, Chen S and Chen Y: Unraveling the
biological functions of Smad7 with mouse models. Cell Biosci.
1:442011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosa A, Papaioannou MD, Krzyspiak JE and
Brivanlou AH: miR-373 is regulated by TGFβ signaling and promotes
mesendoderm differentiation in human Embryonic Stem Cells. Dev
Biol. 391:81–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu H, Deng F, Liu Y, Chen M, Zhang X, Sun
X, Dong Z, Liu X and Ge J: Characterization and retinal neuron
differentiation of WERI-Rb1 cancer stem cells. Mol Vis.
18:2388–2397. 2012.PubMed/NCBI
|
|
29
|
Miyata N, Azuma T, Hozawa S, Higuchi H,
Yokoyama A, Kabashima A, Igarashi T, Saeki K and Hibi T:
Transforming growth factor β and Ras/MEK/ERK signaling regulate the
expression level of a novel tumor suppressor Lefty. Pancreas.
41:745–752. 2012.PubMed/NCBI
|
|
30
|
Manresa MC, Godson C and Taylor CT:
Hypoxia-sensitive pathways in inflammation-driven fibrosis. Am J
Physiol Regul Integr Comp Physiol. 307:R1369–R1380. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Torres IB, Moreso F, Sarró E, Meseguer A
and Serón D: The Interplay between inflammation and fibrosis in
kidney transplantation. BioMed Res Int. 2014:7506022014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Manucha W and Vallés PG: Apoptosis
modulated by oxidative stress and inflammation during obstructive
nephropathy. Inflamm Allergy Drug Targets. 11:303–312. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Declèves AE and Sharma K: Novel targets of
antifibrotic and anti-inflammatory treatment in CKD. Nat Rev
Nephrol. 10:257–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bolisetty S, Zarjou A, Hull TD, Traylor
AM, Perianayagam A, Joseph R, Kamal AI, Arosio P, Soares MP, Jeney
V, et al: Macrophage and epithelial cell H-ferritin expression
regulates renal inflammation. Kidney Int. 88:95–108. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oh SW, Lee YM, Kim S, Chin HJ, Chae DW and
Na KY: Cobalt chloride attenuates oxidative stress and inflammation
through NF-κB inhibition in human renal proximal tubular epithelial
cells. J Korean Med Sci. 29(Suppl 2): S139–S145. 2014. View Article : Google Scholar :
|
|
36
|
Zhong F, Chen H, Han L, Jin Y and Wang W:
Curcumin attenuates lipopolysaccharide-induced renal inflammation.
Biol Pharm Bull. 34:226–232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu F, Liang W, Ren Z, Wang G and Ding G:
Surfactant protein D inhibits lipopolysaccharide-induced monocyte
chemoattractant protein-1 expression in human renal tubular
epithelial cells: Implication for tubulointerstitial fibrosis. Clin
Exp Immunol. 167:514–522. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Choi YH, Kim GY and Lee HH:
Anti-inflammatory effects of cordycepin in
lipopolysaccharide-stimulated RAW 264.7 macrophages through
Toll-like receptor 4-mediated suppression of mitogen-activated
protein kinases and NF-κB signaling pathways. Drug Des Devel Ther.
8:1941–1953. 2014. View Article : Google Scholar :
|
|
39
|
Oh YC, Jeong YH, Ha JH, Cho WK and Ma JY:
Oryeongsan inhibits LPS-induced production of inflammatory
mediators via blockade of the NF-kappaB, MAPK pathways and leads to
HO-1 induction in macrophage cells. BMC Complement Altern Med.
14:2422014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suh SH, Lee KE, Kim IJ, Kim O, Kim CS,
Choi JS, Choi HI, Bae EH, Ma SK, Lee JU, et al: Alpha-lipoic acid
attenuates lipopolysaccharide-induced kidney injury. Clin Exp
Nephrol. 19:82–91. 2015. View Article : Google Scholar
|
|
41
|
Yan X and Chen YG: Smad7: Not only a
regulator, but also a cross-talk mediator of TGF-β signalling.
Biochem J. 434:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Meng XM, Huang XR, Xiao J, Chung AC, Qin
W, Chen HY and Lan HY: Disruption of Smad4 impairs TGF-β/Smad3 and
Smad7 transcriptional regulation during renal inflammation and
fibrosis in vivo and in vitro. Kidney Int. 81:266–279. 2012.
View Article : Google Scholar
|
|
43
|
Liu GX, Li YQ, Huang XR, Wei L, Chen HY,
Shi YJ, Heuchel RL and Lan HY: Disruption of Smad7 promotes ANG
II-mediated renal inflammation and fibrosis via Sp1-TGF-β/Smad3-NF.
κB-dependent mechanisms in mice. PLoS One. 8:e535732013. View Article : Google Scholar
|
|
44
|
Ng YY, Hou CC, Wang W, Huang XR and Lan
HY: Blockade of NFkappaB activation and renal inflammation by
ultrasound-mediated gene transfer of Smad7 in rat remnant kidney.
Kidney Int Suppl. 67:S83–S91. 2005. View Article : Google Scholar
|
|
45
|
Zhang L, Zhang J, Xu C, Zhou X, Wang W,
Zheng R, Hu W and Wu P: Lefty-1 alleviates TGF-β1-induced
fibroblast-myofibroblast transdifferentiation in NRK-49F cells.
Drug Des Devel Ther. 9:4669–4678. 2015. View Article : Google Scholar :
|