Introduction
Microglia have a tissue macrophage lineage and
constitute the innate immune system of the central nervous system.
The activities of microglia are critical for various neuronal
functions, including clearance of neuronal debris, tissue repair,
synaptic plasticity, and neurotrophic factor release (1,2).
Microglia also release various cytokines and growth factors to
facilitate repair of neurons affected by disordered CNS
homeostasis. However, abnormal microglial activation is
maladaptive. Many pro-neuroinflammatory cytokines and cytotoxic
mediators, including interleukin (IL)-12, IL-1β, IL-6, tumor
necrosis factor (TNF)-α and reactive oxygen species (ROS) are
induced in response to abnormal microglial activation. This
abnormal induction is injurious to neurons and results in
neurodegenerative disease (3–5).
The proper maintenance of microglial activation is important for
brain development and brain injury repair. Several studies have
characterized the neuroinflammatory response that abnormal
microglial activation induces (6,7).
Notably, this response promotes the development of
neurodegenerative diseases, such as Huntington's, Alzheimer's and
Parkinson's.
Microglia can be categorized into two types based on
distinct patterns of activation: classical M1 microglia and the
alternative M2 microglia. M1 microglia activate inflammatory
cytokines, including inducible IL-12, IL-1β, IL-6 and TNF-α, and
promote neuronal injury. In contrast, M2 microglia produce
anti-inflammatory molecules that reduce inflammation and repair
tissue injury (8–10). Microglia with an M2-like phenotype
release anti-neuroinflammatory molecules, such as nuclear factor
erythroid 2-related factor 2 (Nrf2), NADPH dehydrogenase quinone-1
(NQO-1) and heme oxygenase-1 (HO-1) (11). These multifunctional proteins are
involved in neuronal defense and repair systems. Nrf2 is a
transcription factor that is responsible for the activation of
several phase II antioxidant enzymes. Under unstimulated
conditions, the negative regulator Kelch-like ECH-associated
protein 1 (Keap1) maintains Nrf2 in the cytoplasm, which results in
its ubiquitination and proteasomal degradation. During
electrophilic stress, Keap1 is altered and Nrf2 translocates to the
nucleus, binds to the antioxidant response element (ARE) promotor
sequence, and activates the transcription of its target genes,
including HO-1 and NQO1 (12,13). Genetic and pharmacological studies
indicate that both NQO1 and HO-1 provide neuroprotection. For
example, cell culture studies have demonstrated that NQO1 and HO-1
overexpression in neuronal cells decreases oxidative damage
following exposure to amyloid βor glutamate. Consequently,
microglia from HO-1 and NQO1-overexpressing transgenic mice are
more resistant to lipopolysaccharide (LPS)-mediated and oxidative
injury (14).
AMP-activated protein kinase (AMPK) is a family of
intracellular serine/threonine protein kinases that serve roles in
context-specific metabolism in response to metabolic stress, such
as oxidative stress and neuroinflammation (15). Growing evidence suggests that AMPK
activation prevents neuronal oxidative stress and damage under
several pathological conditions. Furthermore, recent studies have
demonstrated that AMPK is a negative modulator of neuroinflammatory
responses and that AMPK has anti-neuroinflammatory properties in
LPS-treated microglia. Thus, AMPK activation indicates an
anti-neuroinflammatory response, regulates catabolism and anabolism
and improves redox balance. AMPK also promotes microglial
polarization towards the M2 phenotype (16,17). Consistent with the emerging
interplay between oxidative stress and neuroinflammation responses,
recent studies suggest the possibility of cross talk between the
AMPK and Nrf2/ARE pathways (18).
A previous study from our group reported that AMPK signaling
regulates microglia-induced Nrf2/ARE activation and that the AMPK
inhibitor compound C suppressed neuroinflammation (19). Other studies have demonstrated
that the inhibition of AMPK ameliorates the LPS-stimulated
neuroinflammatory response through the inactivation of Nrf2/ARE
signaling (20).
Natural compounds are gaining prominence as valuable
candidate substrates for the development of medications.
Bakkenolide B, the main constituent of Petasites japonicus
leaves, is generally cultured in East Asia and used as both a
vegetable and a folk remedy (21). P. japonicus has been used
to treat various diseases, including headaches, chronic cough,
fever, and asthma. A bioassay study has demonstrated that P.
japonicus extracts and compounds have antioxidant,
anti-inflammatory, and antitumor biological effects (22). P. japonicus bakkenolide B
has been previously demonstrated by our group to exhibit both
antiallergenic and anti-inflammatory effects (23). Furthermore, sesquiterpenoids from
P. japonicus have neuroprotective properties in human
neuroblastoma SH-SY5Y cells (24,25). However, the effects of bakkenolide
B on microglia-mediated neuroinflammatory activity have not been
investigated. Thus, the present study investigated the effects of
bakkenolide B on the microglial neuroinflammatory response.
Additional mechanistic experiments verified that AMPK was
associated with Nrf2/ARE activation. Taken together, the present
results indicate that bakkenolide B may a potential candidate for
treatment of abnormal neuroinflammatory-mediated neurodegenerative
diseases.
Materials and methods
Reagents
CM-H2DCFDA was obtained from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Compound C, dimethyl sulfoxide
(DMSO), MTT and other reagents were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Dulbecco's modified Eagle's
medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco
(Thermo Fisher Scientific, Inc.). Antibodies for α-tubulin [cat.
no. sc-23948, 1:10,000 dilution, mouse immunoglobulin (Ig)G],
TATA-binding protein (TBP, cat. no. sc-204, 1:500 dilution, rabbit
IgG), inducible nitric oxide synthase (iNOS, cat. no. sc-8310,
1:1,000 dilution, rabbit IgG), Nrf2 (cat. no. sc-722, 1:500
dilution, rabbit IgG), NQO1, (cat. no. sc-16464, 1:1,000 dilution,
rabbit IgG) and HO-1 (cat. no. sc-10789, 1:1,000 dilution, rabbit
IgG), as well as small interfering (si)RNAs against Nrf2 (cat. no.
sc-37049), NQO1 (cat. no. sc-37140) and HO-1 (cat. no. sc-35555),
were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Antibodies against AMPK (cat. no. 2532, 1:1,000 dilution,
rabbit IgG) and phosphorylated (p-) AMPK (cat. no. 2535, 1:1,000
dilution, rabbit IgG) were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA). FuGENE HD transfection reagent and
X-tremeGENE siRNA transfection reagent were obtained from Roche
Diagnostics (Indianapolis, IN, USA). A cytotoxicity detection kit,
measuring lactate dehydrogenase (LDH) activity, was purchased from
Roche Diagnostics (Basel, Switzerland). A mouse Quantikine ELISA
kit (for TNF-α cat. no. MTA00B; IL-1β cat. no. MLB00C; IL-6 cat. No
M6000B and IL-12 cat. no. M1270) was acquired from R&D Systems,
Inc. (Minneapolis, MN, USA). All other basic experimental supplies
and reagents were obtained from Sigma-Aldrich (Merck KGaA) and
Invitrogen (Thermo Fisher Scientific, Inc.).
Extraction, isolation, and structure
elucidation of bakkenolide B
Bakkenolide B was isolated using open column
chromatography and its structure was previously elucidated by NMR
spectroscopy (23). Briefly, the
leaves of P. japonicus (425.36 g) were ground to fine
particles using an electric mixer (HMF-3100 S; Hanil Electric,
Seoul, Korea) for extraction at room temperature with 75% EtOH. The
EtOH was then removed with a rotary evaporator and the remaining
aqueous extract was fractionated consecutively with n-hexane,
EtOAc, BuOH, and water. The acquired hexane extract (2.6728 g) was
evaporated in vacuum and the residue was used for silica gel (40
µm; J.T. Baker; Thermo Fisher Scientific, Inc.) column
chromatography (100×4.0 cm) with a step gradient of 2.5, 15, and
25% acetone in dichloromethylene and 15 and 25% MeOH in chloroform
to obtain 62 fractions. Fraction 9 (MWLSH9; 304.9 mg) was separated
on a Sephadex column (100×3.0 cm) with MeOH as the eluent to obtain
seven fractions. Fraction 3 (MWLSH9IC; 209.7 mg) was additionally
separated on a Sephadex column (100×3.0 cm) with MeOH to obtain
five fractions. Fractions 2 and 3 (MWLSH9ICIB; 202.3 mg) were
passed through a silica gel column (100×4.0 cm) with 1.5% acetone
in CH2Cl2 as the eluent to yield bakkenolide
B (173.8 mg). Pure bakkenolide B was identified using HPLC on a
Phenomenex Luna C18 column (150 4.6 mm ID; 5 µ particle
size; Phenomenex, Torrance, CA, USA) with an acetonitrile-water
reagent alcohol gradient at a flow rate of 1.0 ml per min.
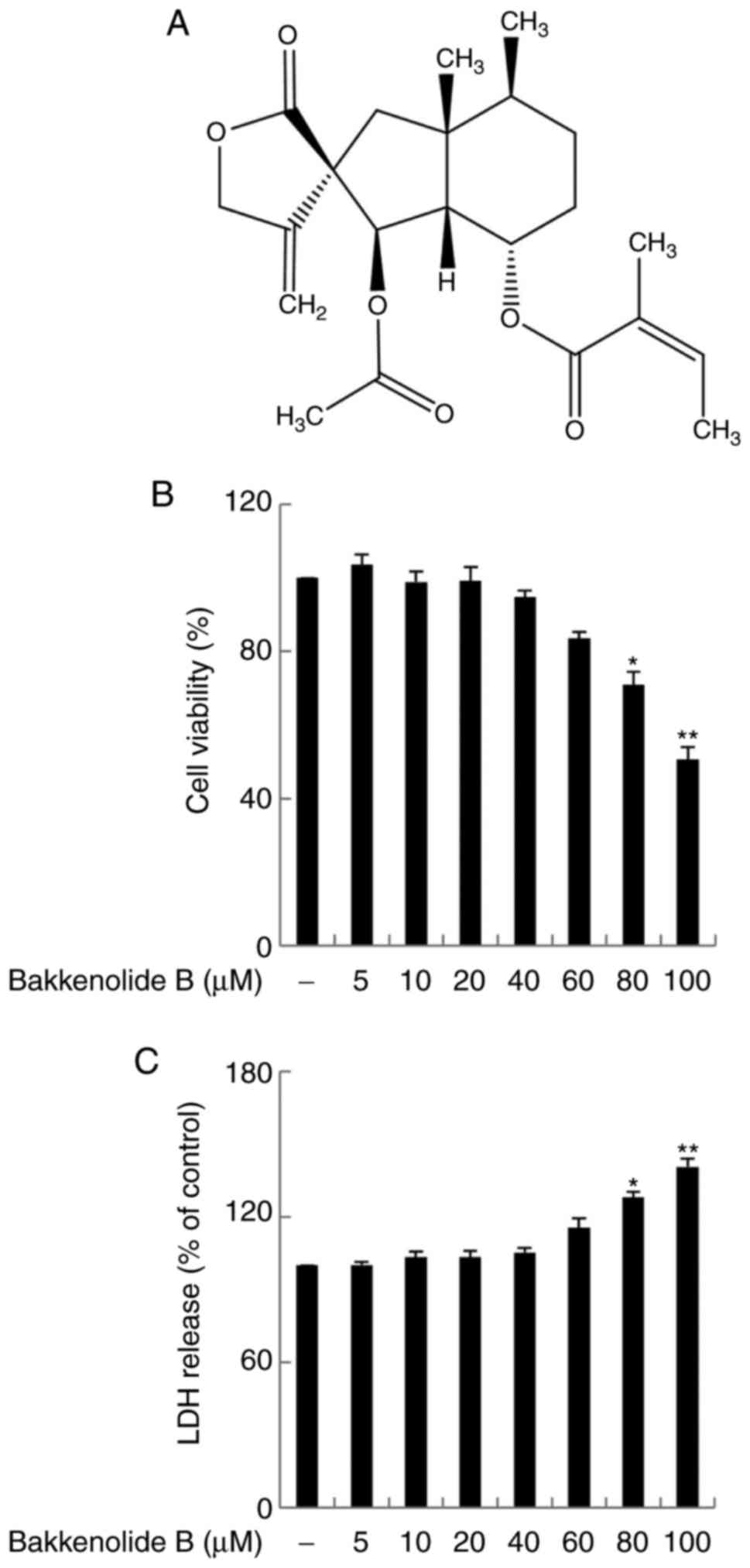
Bakkenolide B (Fig. 1A) isolated
from P. japonicus leaves was identified using 1H, 13C, and
distortionless enhancement based on polarization transfer nuclear
magnetic resonance spectroscopy in CDCl3, in contrast to previously
reported spectral data (23).
Microglia culture
Mouse BV2 microglial cells, which were a generous
gift from Professor Youn-Chul Kim at Wonkwang University (Iksan,
Korea), were cultured in DMEM supplemented with 10%
heat-inactivated fetal bovine serum at 37°C in a humidified
atmosphere containing 5% CO2. All experiments were
performed with cells between passages 15 and 25. Bakkenolide B was
solubilized with DMSO at a final concentration of 0.1% and the
stock solutions were directly added to the culture media.
Cell viability assay
The toxicity of bakkenolide B was assessed using an
LDH leakage assay and an MTT reduction assay. Briefly,
5×104 cells were seeded in 24-well plates and allowed to
grow to 80% confluence. MTT solution (50 µg/ml) was then
added to each sample. Following incubation for 6 h, the supernatant
was removed, and the formazan crystals that formed in the normal
cells were dissolved in DMSO. The absorbance of each sample was
then measured at 570 nm using a Victor3 plate reader (PerkinElmer,
Inc., Waltham, MA, USA). Extracellular LDH activity was determined
using a cytotoxicity detection kit, according to the manufacturer's
protocol. The absorbance in each well was quantified at 490 nm with
a Victor3 plate reader (PerkinElmer, Inc.).
Measurement of IL-1β, IL-6, IL-12 and
TNF-α levels
IL-1β, IL-6, IL-12 and TNF-α levels were quantified
in the culture media using an ELISA kit (TNF-α cat. no. MTA00B;
IL-1β cat. no. MLB00C; IL-6 cat. No M6000B and IL-12 cat. no.
M1270) R&D Systems, Inc.), according to the manufacturer's
protocol.
Measurement of nitric oxide (NO)
production
LPS (1 µg/ml) was added to microglia and
incubated for 24 h, following which the supernatant was obtained
and mixed with Griess reagent, as previously reported (26). The absorbance of each mixture was
then measured at 540 nm using a Victor3 plate reader (PerkinElmer,
Inc.). The concentration of NO was determined using an NO
standard.
Measurement of intracellular ROS
The intracellular ROS level, as an indicator of
general oxidative stress, was estimated using the CM-H2DCFDA
reagent. Briefly, 4×105 cells were seeded in 6-well
plates and allowed to grow to 80% confluence. The cells were then
harvested and washed thrice with phosphate buffer saline (PBS). A
total of 10 µM CM-H2DCFDA reagent was added to cells and
incubated for 30 min in a 5% CO2 at 37°C. The
fluorescence intensity was subsequently measured using a flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA). Finally, data
were quantified using the CXP version 2.0 software (Beckman
Coulter, Inc.). At least 10,000 cells were evaluated for each
condition.
Gene silencing of murine Nrf2, HO-1 and
NQO1
Knockdown of Nrf2, HO-1, and NQO1 using siRNA (10
nM) was achieved with the X-tremeGENE siRNA transfection reagent
kit, according to the manufacturer's protocols. Further experiments
were performed 24 h following transfection. Nrf2, HO-1, NQO1, or
control siRNA-transfected cells were used to measure the IL-1β,
IL-6, IL-12 and TNF-α levels.
Transient transfection and dual
luciferase assay
Cells were transfected using the HO-1 promoter
reporter plasmid or ARE reporter plasmids (100 ng; Agilent
Technologies, Inc., Santa Clara, CA, USA) using FuGENE-HD reagent
(Roche Applied Science, Penzberg, Germany), according to the
manufacturer's protocol. A Renilla luciferase control plasmid was
co-transfected as an internal control for transfection efficiency.
Luciferase activity was examined using a dual-luciferase assay kit
(Promega Corporation, Madison, IW, USA), according to the
manufacturer's protocol. Luminescence was measured using a Victor3
plate reader (PerkinElmer, Inc.).
Western blot analysis
The cell were lysed with Radioimmunoprecipitation
assay buffer(Thermo Fisher Scientific, Inc.) supplemented with a
protease inhibitor cocktail. The nuclear extracts were prepared
using the NE-PER nuclear and cytoplasmic extraction reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Lysate protein concentrations were evaluated using Bio-Rad Protein
Assay Dye Reagent concentrate (Bio-Rad Laboratories, Hercules, CA,
USA). Proteins in each sample (30–50 µg of total protein) were
subjected to a 7.5–10% SDS-PAGE. and transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% skimmed milk in PBS with
0.1% Tween-20 for 1 h at room temperature. The membrane was
incubated with the primary antibodies (α-tubulin, TBP, iNOS, Nrf2,
NQO1 and HO-1 at room temperature for 2 h and AMPK and p-AMPK at
4°C overnight). Subsequently, the membranes were incubated with
horseadish peroxidase conjugated seceondary antibodies (goat
anti-rabbit, cat. no. sc-2004, 1:1,000; goat anto-mouse cat. no.
sc-2039, 1:1,000) at room temperature for 1 h. The membrane was
visualized using a boosted chemiluminescent immunodetection system
(Amersham; GE Healthcare, Chicago, IL, USA) and a secondary
horseradish peroxidase-conjugated antibody. The data were
quantified using an ImageQuant 350 analyzer (ImageQuant TL SecurITy
8.0 software, Amersham; GE Healthcare).
Statistical analysis
Each experiment was repeated at least three times.
Statistical analyses were conducted using the SPSS software version
18.0 (SPSS, Inc., Chicago, IL, USA). One-way analysis of variance
followed by Dunn's post-hoc test was performed to identify
differences among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Bakkenolide B ameliorates
neuroinflammatory cytokines in LPS-stimulated microglia
First, MTT and LDH assays were used to evaluate the
viability of microglia exposed to bakkenolide B (0–100 µM).
The results demonstrated that bakkenolide B treatment at
concentrations of 5–40 µM did not significantly affect
microglia viability (Fig. 1B and
C). Thus, in subsequent experiments, the microglia were
evaluated following exposure to bakkenolide B at concentrations of
5–40 µM. The anti-neuroinflammatory properties of
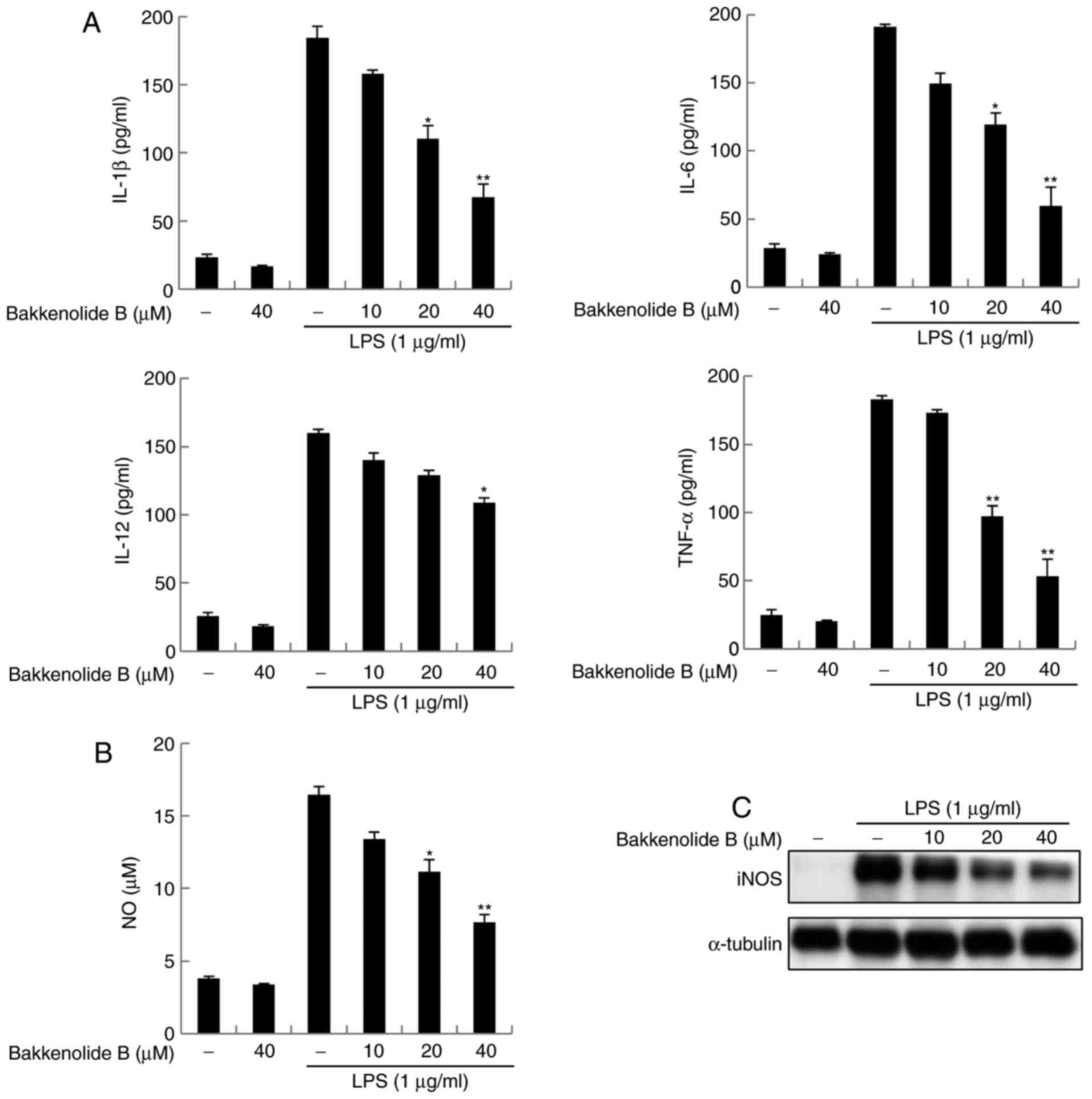
bakkenolide B were then examined in LPS-stimulated microglia.
Microglia exposed to LPS displayed increased IL-1β, IL-6, IL-12 and
TNF-α production compared with control cells exposed to vehicle.
Conversely, bakkenolide B pretreatment reversed the effects of LPS
on IL-1β, IL-6, IL-12 and TNF-α production in a dose-dependent
manner (Fig. 2A). The enzyme
activity and protein expression of iNOS were also investigated,
because this is known to be important for neuroinflammatory
responses. Treatment with bakkenolide B significantly decreased NO
production induced by LPS (Fig.
2B). In addition, pretreatment with bakkenolide B resulted in
markedly decreased protein expression levels of iNOS (Fig. 2C). CM-H2DCFDA staining, which is a
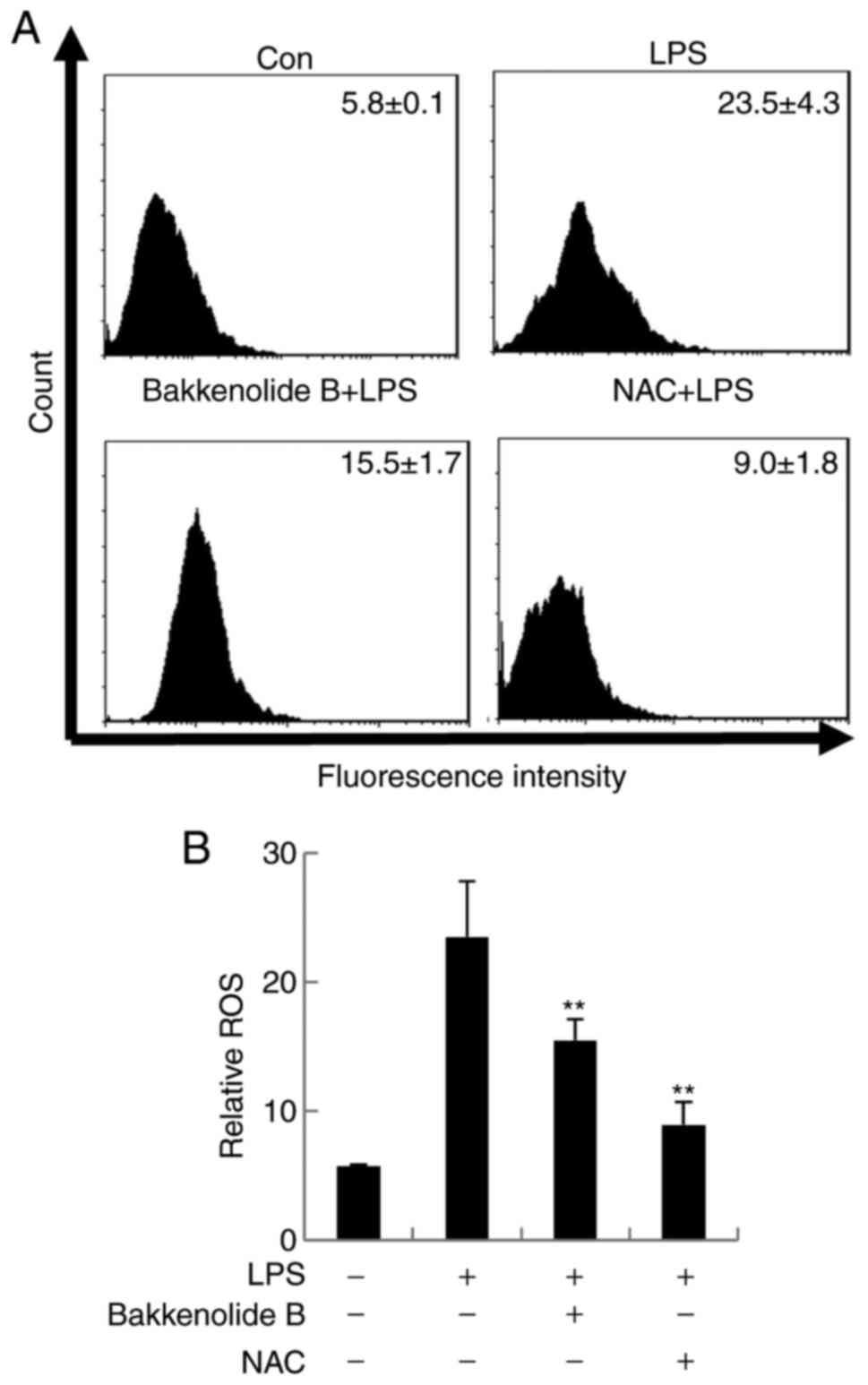
general oxidative stress indicator, was used to assess the impact
of bakkenolide B pretreatment on LPS-stimulated microglia. The
results demonstrated that LPS exposure increased ROS production to
23.5±4.3% in LPS-stimulated microglia compared to 5.8±0.1% in
control untreated cells. Conversely, bakkenolide B pretreatment
significantly decreased the LPS-induced ROS production (15.5±1.7%;
Fig. 3). As expected,
N-acetyl-L-cysteine (NAC; a ROS scavenger) significantly attenuated
the LPS-induced ROS (Fig. 3).
Taken together, these results suggest that bakkenolide B can
inhibit the production of pro-neuroinflammatory cytokines and
mediators in LPS-stimulated microglia.
Bakkenolide B activates Nrf2/ARE
signaling and reduces neuroinflammation in LPS-stimulated
microglia
To better understand the molecular pathway through
which bakkenolide B inhibits the LPS-induced neuroinflammatory
response, we evaluated whether Nrf2/ARE signaling is induced in
bakkenolide B-treated microglia. First, the levels of nuclear Nrf2
accumulation activation were examined using western blotting and a
luciferase promoter assay following treatment of microglia with
bakkenolide B. As presented in Fig.
4A, nuclear Nrf2 accumulation rapidly increased 1 h following
bakkenolide B treatment. Furthermore, bakkenolide B
dose-dependently increased nuclear Nrf2 accumulation (Fig. 4A). Nuclear extracts were analyzed
for the α-tubulin cytoplasmic marker and minimal cytoplasmic
contamination was observed (Fig.
4A). Consistent with this result, bakkenolide B significantly
induced the expression of HO-1 and NQO1 in microglia compared with
controls, in a time- and dose-dependent manner (Fig. 4B). Bakkenolide B-treated microglia
lysates were prepared to measure ARE promoter activity (based on
luciferase activity normalized to renilla luciferase activity), and
the results indicated that bakkenolide B dose-dependently increased
ARE-promoter activity (Fig. 4C).
Consistent with this finding, bakkenolide B also significantly
increased the transcriptional activity of the HO-1 promoter in a
dose-dependent manner (Fig. 4D).
To test the hypothesis that the anti-neuroinflammatory properties
of bakkenolide B are diminished following knockdown of Nrf2, HO-1
and NQO1, TNF-α levels were evaluated using a Quantikine ELISA kit
following exposure of microglia to LPS. The results revealed that
TNF-α production was markedly increased in LPS-stimulated cells
transfected with the control siRNA, while exposure to bakkenolide B
and the control siRNA significantly decreased TNF-α production
compared with the LPS-only group (Fig. 4E). Notably, transfection of siRNA
for Nrf2, HO-1 or NQO1 partially reversed the bakkenolide
B-mediated inhibition of TNF-α production in LPS-stimulated
microglia (Fig. 4E). These
findings suggest that bakkenolide B may have significant
anti-neuroinflammatory properties mediated by the Nrf2/ARE
pathway.
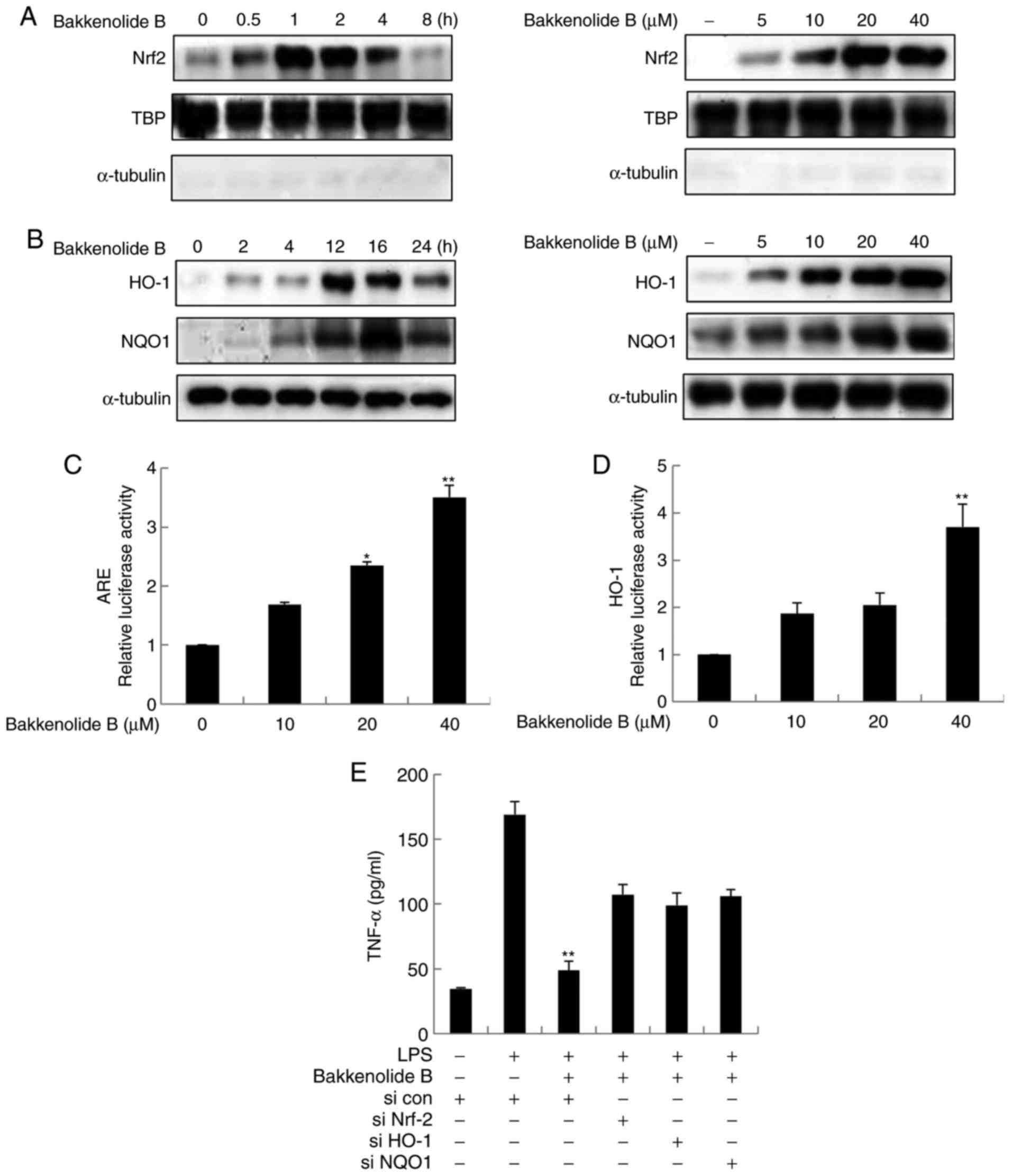 | Figure 4Bakkenolide B pretreatment suppresses
the LPS-induced neuroinflammatory response via activation of
Nrf2/ARE signaling. (A) The effects of bakkenolide B on Nrf2
nuclear accumulation were determined by western blotting. Microglia
were incubated with 40 µM bakkenolide B for different time
periods (0, 0.5, 1, 2, 4 and 8 h). Alternatively, cells were
treated with different concentrations of bakkenolide B for 4 h.
Nuclear lysates were then collected, and the purity of nuclear
extracts was confirmed by the absence of cytoplasmic α-tubulin,
while TBP was used as the internal loading control for nuclear
protein. (B) Microglia were treated with bakkenolide B (40
µM) for 4, 8, 12, 16 and 24 h. Alternatively, the cells were
treated with bakkenolide B at the indicated concentrations for 4 h.
Then the protein levels of HO-1 and NQO1 were measured by western
blotting. α-tubulin was used as an internal loading control. (C and
D) Microglia were transfected with the ARE or HO-1 luciferase
reporter plasmid, then incubated with bakkenolide B for 8 h. Equal
amounts of cell extract were assayed for dual-luciferase activity.
*P<0.05 and **P<0.01 compared with
control. (E) Microglia were transiently transfected with Nrf2, HO-1
or NQO1 siRNA or control siRNA and pretreated with bakkenolide B
(40 µM) for 1 h, then exposed to LPS for 24 h, and then
analyzed using a TNF-α Quantikine ELISA kit. *P<0.05
and **P<0.01 compared with LPS+control siRNA group.
All data are presented as the means ± standard error of the mean
(n=3). LPS, lipopolysaccharide; Nrf2, nuclear factor erythroid
2-related factor 2; ARE, antioxidant response element; TBP, TATA
binding protein; HO-1, heme oxygenase-1; NQO1, NADPH dehydrogenase
quinone-1; si, small interfering; TNF, tumor necrosis factor. |
Bakkenolide B-induced AMPK-mediated
Nrf2/ARE activation is vital for the suppression of LPS-induced
neuroinflammation
To determine if AMPK signaling mediates bakkenolide
B-induced Nrf2/ARE activation, the AMPK activation level were
determined following bakkenolide B treatment in microglia using
western blotting and antibodies targeting total and phosphorylated
(at Thr 172) AMPK. As expected, bakkenolide B induced AMPK
phosphorylation in a time- and dose-dependent manner (Fig. 5A and B). To determine if AMPK
inhibition decreases Nrf2/ARE activation, nuclear Nrf2 accumulation
and protein expression of HO-1 and NQO1 were evaluated by western
blot analysis. As expected, the AMPK inhibitor compound C
significantly inhibited bakkenolide B-induced AMPK phosphorylation
(Fig. 5C). Notably, AMPK
inhibition decreased bakkenolide B-induced nuclear Nrf2
accumulation and protein expression of HO-1 and NQO1 (Fig. 5C). To confirm that AMPK-mediated
Nrf2/ARE signaling regulated the anti-neuroinflammatory properties
of bakkenolide B protection against LPS-induced neuroinflammation,
microglia were pretreated with compound C prior to exposure to
bakkenolide B. Following treatment with bakkenolide B, microglia
were exposed to LPS for 24 h. As illustrated in Fig. 5D, compound C pretreatment
partially reversed the bakkenolide B-mediated inhibition of TNF-α
production. Taken together, these results suggest that AMPK
activity is required for bakkenolide B-induced Nrf2 activation and
subsequent anti-neuroinflammatory activity.
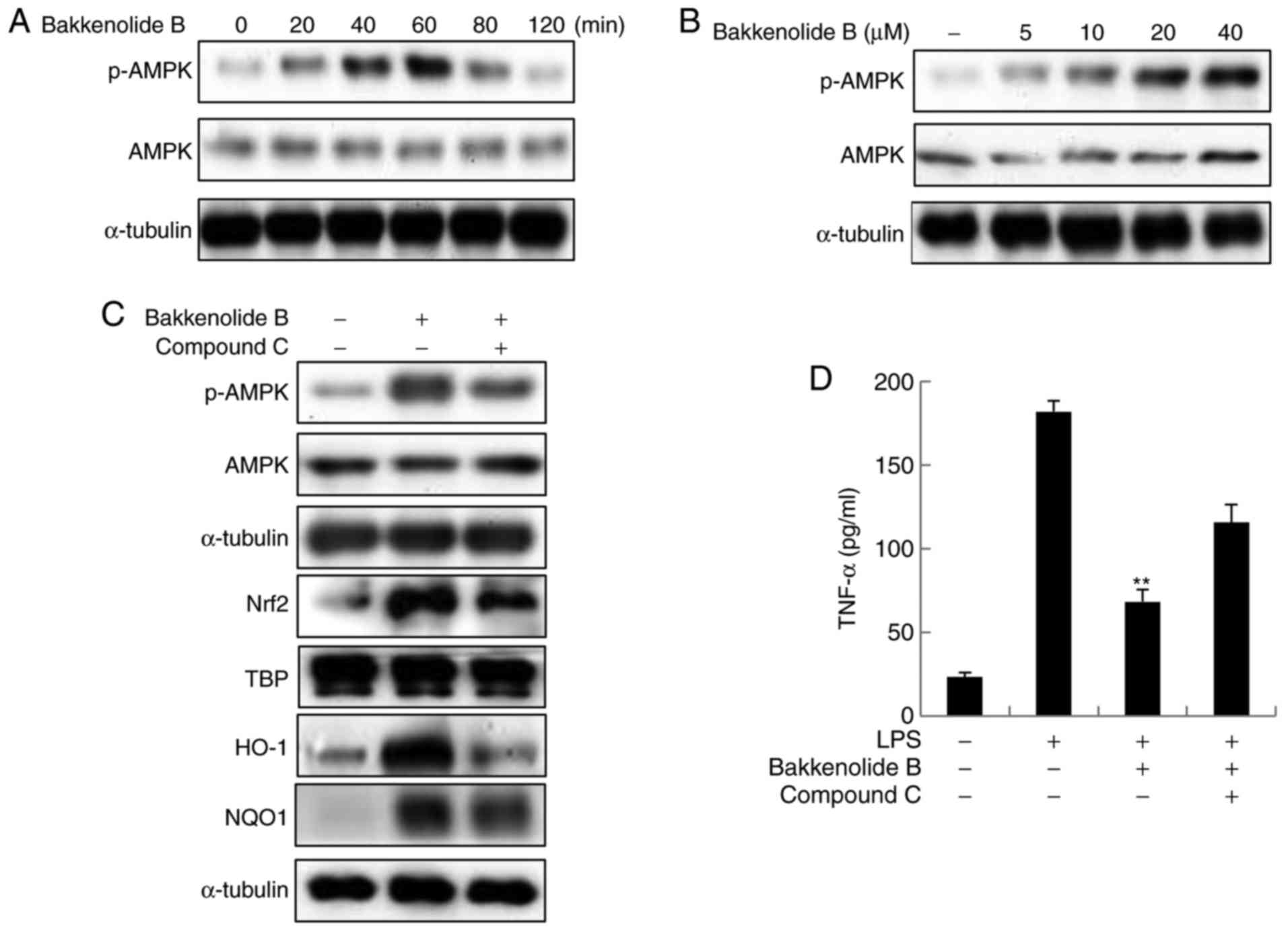 | Figure 5Anti-neuroinflammatory properties of
bakkenolide B are regulated by AMPK. (A) Cells were treated with
bakkenolide B (40 µM) for the indicated times, following
which the cell extracts were prepared for western blotting. (B)
Cells were incubated with the indicated doses of bakkenolide B for
1 h, following which the cell extracts were prepared for western
blotting. (C) Cells were pretreated with 20 µM compound C
for 1 h, then treated with bakkenolide B (40 µM) for 1 h.
Total cell extracts were subjected to immunoblotting using
anti-AMPK, Nrf2, HO-1 and NQO1 antibodies. (D) Cells were
pretreated with 20 µM compound C for 1 h, then treated with
bakkenolide B (40 µM) for 1 h, then exposed to LPS for 24 h,
and then analyzed using a TNF-α Quantikine ELISA kit. All data are
presented as the means ± standard error of the mean (n=3).
**P<0.01 compared with LPS alone group. AMPK,
AMP-activated protein kinase; Nrf2, nuclear factor erythroid
2-related factor 2; HO-1, heme oxygenase-1; NQO1, NADPH
dehydrogenase quinone-1; TNF, tumor necrosis factor; p,
phosphorylated; TBP, TATA binding protein. |
Discussion
The prevalence of neurodegenerative disorders has
increased nowadays and has become one of the most serious health
issues worldwide. Neurodegenerative disorders are associated with a
higher risk of neurodegenerative diseases, including Huntington's,
Alzheimer's and Parkinson's (27,28). Abnormal microglia activation has a
crucial role in the development of neurodegenerative disorders,
although the exact mechanisms have not been fully elucidated.
Limiting the production of neuroinflammatory mediators is crucial
for neuronal protection and repair. Abnormal activation of the
microglia and upregulation of pro-neuroinflammatory mediators are
pathological features of neurodegenerative disorders (29–31). Furthermore, abnormal microglial
accumulation and release of neuroinflammatory mediators is
deleterious to neighboring neurons and may be related to
neurodegenerative disease (32).
The current study presents several major findings. First,
bakkenolide B was demonstrated to have the potential for use as a
treatment to ameliorate abnormal neuroinflammatory responses.
Second, AMPK/Nrf2/ARE signaling was demonstrated to have a
significant role in the anti-neuroinflammatory activities of
bakkenolide B. The present study is the first to demonstrate the
anti-neuroinflammatory mechanisms that regulate the effects of
bakkenolide B in LPS-stimulated microglia.
Neuronal injury results from ongoing abnormal
microglia activation or deficient/suppressed neuron recovery.
Abnormal microglia activation contributes to the development of
neurodegenerative disorder, and recent evidence has highlighted the
anti-neuroinflammatory effects in microglia, providing novel
potential avenues for treatment of neurodegenerative diseases
(33). These proinflammatory
cytokines, including TNF-α, IL-1β, IL-6 and IL-12, are secreted
from microglia and initiate, amplify, and perpetuate the
neuroinflammatory response in the brain. These cytokines are the
primary endogenous mediators of the neuroinflammatory reaction,
causing not only abnormal microglia activation, but also triggering
damage to adjacent neurons (34,35). The present study demonstrated that
following LPS exposure, IL-1β, IL-6, IL-12 and TNF-α in microglia
are significantly increased. However, pretreatment with bakkenolide
B significantly reduced the levels of IL-1β, IL-6, IL-12 and TNF-α
in microglia. ROS were also induced via inflammatory mediators
during the neuroinflammatory response in microglia, but this
LPS-induced production of ROS was diminished following bakkenolide
B pretreatment. These results demonstrated that the
anti-neuroinflammatory properties of bakkenolide B in microglia may
be attributed to the inhibition of IL-1β, IL-6, IL-12 and TNF-α,
and ROS production.
Nrf2/ARE signaling in microglia is essential to
reducing neuronal cell death and preserving cognitive function.
Studies of microglia have reported that HO-1 and NQO1
overexpression, mediated by Nrf2 activation, has a strong
anti-neuroinflammatory effect (11). This finding has been observed in
both cell culture and animal models treated with different natural
compounds that activate Nrf2/ARE to reduce the LPS-induced
pro-inflammatory response and inhibit oxidative damage. Natural
compounds exhibit both antioxidant and anti-inflammatory effects
depending on the Nrf2/ARE signal that prevents neurodegeneration.
Several studies have reported that phase II antioxidant enzymes may
contribute to neural deterioration in neurodegenerative disorders
and diseases. Remarkably, Nrf-2 signaling activates phase II
antioxidant enzymes, such as HO-1 and NQO-1 (12,13). These enzymes serve crucial roles
in neurodegenerative disorders via abnormal neuroinflammatory
responses. The present results also indicate that bakkenolide
B-induced Nrf2 activation upregulated HO-1 and NOQ1 expression and
consequently diminished LPS-mediated neuroinflammatory
responses.
AMPK is an essential factor in the protein kinase
cascade that has a vital role in regulation of energy metabolism,
which is important for the regulation of cell homeostasis. A
previous study has reported that the molecular mechanisms and
signaling pathways regulating AMPK signal activation that mediate
its neuroprotective effects are driven by its
anti-neuroinflammatory activities (36). Furthermore, the neuroprotective
action of AMPK may not only be mediated by its antioxidant and
anti-inflammatory action, but also due to the enhancement of
Nrf2/ARE signaling. Indeed, studies have demonstrated the
involvement of molecules downstream of AMPK in the modulation of
Nrf2 activation and HO-1 and NQO-1 expression in neurons and
microglia (18,19). More recently, the role of AMPK in
the induction of neuroprotective factors has been confirmed in mice
that show improved recovery following stroke in response to
AMPK-dependent Nrf2/ARE signaling activation in the hippocampus
(37). Thus, Nrf2/ARE activation
has been demonstrated to be neuroprotective. Consistent with this
finding, the present study demonstrated that the
anti-neuroinflammatory properties of bakkenolide B were attributed
to the activation of AMPK/Nrf2/ARE signaling. Bakkenolide
B-mediated AMPK signaling was critical in Nrf2/ARE activation,
which subsequently suppressed LPS-mediated neuroinflammatory
responses. These results suggest that bakkenolide B may reduce
LPS-stimulated neuroinflammatory responses by inducing the
AMPK/Nrf2/ARE signaling pathway.
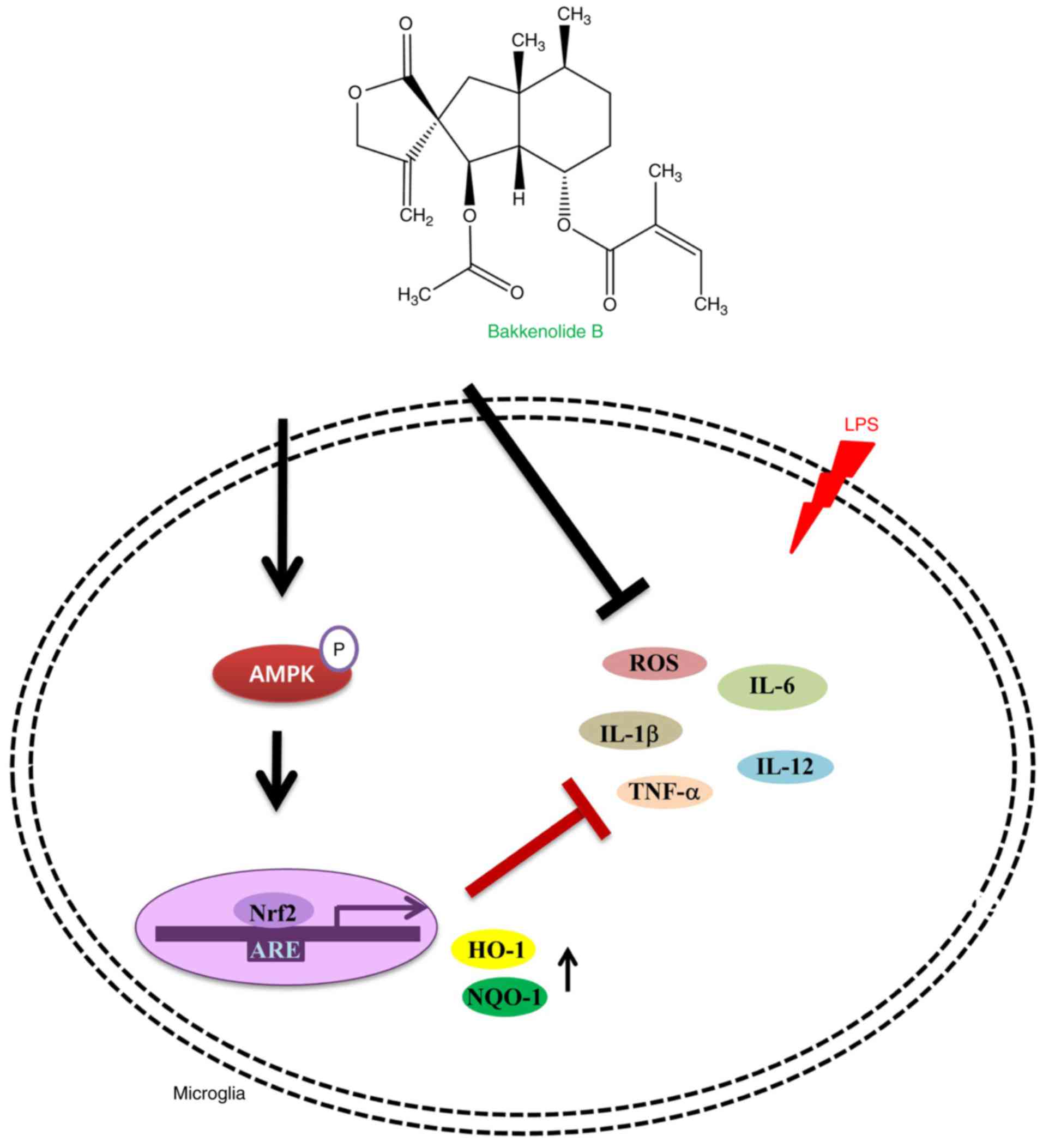
In conclusion, this is the first study linking
bakkenolide B, isolated from P. japonicus, with AMPK
signaling. These findings are especially important because AMPK
mediates activation of Nrf2, HO-1 and NQO1. Bakkenolide B mediated
the activation of AMPK, subsequently induced nuclear Nrf2
translocation, and promoted Nrf-2-mediated HO-1 and NQO1
expression, leading to attenuation of LPS-stimulated
neuroinflammatory response (Fig.
6). Overall, these results suggest that bakkenolide B exposure
may be a promising therapeutic strategy to protect against abnormal
microglia activation in neurodegenerative diseases.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant nos.
NRF-2015R1D1A1A01059450 and NRF-2016R1D1A3B03934083).
References
|
1
|
Letiembre M, Liu Y, Walter S, Hao W,
Pfander T, Wrede A, Schulz-Schaeffer W and Fassbender K: Screening
of innate immune receptors in neurodegenerative diseases: A similar
pattern. Neurobiol Aging. 30:759–768. 2009. View Article : Google Scholar
|
|
2
|
Frank-Cannon TC, Alto LT, McAlpine FE and
Tansey MG: Does neuroinflammation fan the flame in
neurodegenerative diseases? Mol Neurodegener. 4:472009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mueller AM, Yoon BH and Sadiq SA:
Inhibition of hyaluronan synthesis protects against central nervous
system (CNS) autoimmunity and increases CXCL12 expression in the
inflamed CNS. J Biol Chem. 289:22888–22899. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Slusarczyk J, Trojan E, Glombik K,
Piotrowska A, Budziszewska B, Kubera M, Popiolek-Barczyk K, Lason
W, Mika J and Basta-Kaim A: Anti-inflammatory properties of
tianeptine on lipopolysaccharide-induced changes in microglial
cells involve toll-like receptor-related pathways. J Neurochem.
136:958–970. 2016. View Article : Google Scholar
|
|
5
|
Tao L, Zhang F, Hao L, Wu J, Jia J, Liu
JY, Zheng LT and Zhen X: 1-O-tigloyl-1-O-deacetyl-nimbolinin B
inhibits LPS-stimulated inflammatory responses by suppressing
NF-kappaB and JNK activation in microglia cells. J Pharmacol Sci.
125:364–374. 2014. View Article : Google Scholar
|
|
6
|
Ransohoff RM, Schafer D, Vincent A,
Blachère NE and Bar-Or A: Neuroinflammation: Ways in which the
immune system affects the brain. Neurotherapeutics. 12:896–909.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jebelli J, Hooper C and Pocock JM:
Microglial p53 activation is detrimental to neuronal synapses
during activation-induced inflammation: Implications for
neurodegeneration. Neurosci Lett. 583:92–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia CY, Zhang S, Gao Y, Wang ZZ and Chen
NH: Selective modulation of microglia polarization to M2 phenotype
for stroke treatment. Int Immunopharmacol. 25:377–382. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaire BP, Kwon OW, Park SH, Chun KH, Kim
SY, Shin DY and Choi JW: Neuroprotective effect of 6-paradol in
focal cerebral ischemia involves the attenuation of
neuroinflammatory responses in activated microglia. PLoS One.
10:e01202032015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JA, Kim JH, Woo SY, Son HJ, Han SH,
Jang BK, Choi JW, Kim DJ, Park KD and Hwang O: A novel compound
VSC2 has anti-inflammatory and antioxidant properties in microglia
and in parkinson's disease animal model. Br J Pharmacol.
172:1087–100. 2015. View Article : Google Scholar :
|
|
11
|
Mazzuferi M, Kumar G, van Eyll J, Danis B,
Foerch P and Kaminski RM: Nrf2 defense pathway: Experimental
evidence for its protective role in epilepsy. Ann Neurol.
74:560–568. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Onasanwo SA, Velagapudi R, El-Bakoush A
and Olajide OA: Inhibition of neuroinflammation in BV2 microglia by
the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant
protective mechanism. Mol Cell Biochem. 414:23–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jayasooriya RG, Lee KT, Choi YH, Moon SK,
Kim WJ and Kim GY: Antagonistic effects of acetylshikonin on
LPS-induced NO and PGE2 production in BV2 microglial cells via
inhibition of ROS/PI3K/akt-mediated NF-kappaB signaling and
activation of Nrf2-dependent HO-1. In Vitro Cell Dev Biol Anim.
51:975–986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen J, Yin W, Tu Y, Wang S, Yang X, Chen
Q, Zhang X, Han Y and Pi R: L-F001, a novel multifunctional ROCK
inhibitor, suppresses neuroinflammation in vitro and in vivo:
Involvement of NF-κB inhibition and Nrf2 pathway activation. Eur J
Pharmacol. 806:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ismaiel AA, Espinosa-Oliva AM, Santiago M,
García-Quintanilla A, Oliva-Martín MJ, Herrera AJ, Venero JL and de
Pablos RM: Metformin, besides exhibiting strong in vivo
anti-inflammatory properties, increases mptp-induced damage to the
nigrostriatal dopaminergic system. Toxicol Appl Pharmacol.
298:19–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Syed Hussein SS, Kamarudin MN and Kadir
HA: (+)-Catechin attenuates NF-kappaB activation through regulation
of akt, MAPK, and AMPK signaling pathways in LPS-induced BV-2
microglial cells. Am J Chin Med. 43:927–952. 2015. View Article : Google Scholar
|
|
17
|
Xu Y, Xu Y, Wang Y, Wang Y, He L, Jiang Z,
Huang Z, Liao H, Li J, Saavedra JM, et al: Telmisartan prevention
of LPS-induced microglia activation involves M2 microglia
polarization via CaMKKβ-dependent AMPK activation. Brain Behav
Immun. 50:298–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee YY, Park JS, Lee EJ, Lee SY, Kim DH,
Kang JL and Kim HS: Anti-inflammatory mechanism of ginseng saponin
metabolite Rh3 in lipopolysaccharide-stimulated microglia: Critical
role of 5′-adenosine monophosphate-activated protein kinase
signaling pathway. J Agric Food Chem. 63:3472–3480. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park SY, Jin ML, Wang Z, Park G and Choi
YW: 2,3,4′,5-tetrahyd roxystilbene-2-O-β-d-glucoside exerts
anti-inflammatory effects on lipopolysaccharide-stimulated
microglia by inhibiting NF-κB and activating AMPK/Nrf2 pathways.
Food Chem Toxicol. 97:159–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Cao Y, Ao G, Hu L, Liu H, Wu J,
Wang X, Jin M, Zheng S, Zhen X, et al: CaMKKβ-dependent activation
of AMP-activated protein kinase is critical to suppressive effects
of hydrogen sulfide on neuroinflammation. Antioxid Redox Signal.
21:1741–1758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong XW, Li RJ, Gao X and Row KH:
Bakkenolides from petasites tatewakianus. Fitoterapia. 81:153–156.
2010. View Article : Google Scholar
|
|
22
|
Zhang FJ, Wang Q, Wang Y and Guo ML:
Anti-allergic effects of total bakkenolides from petasites
tricholobus in ovalbumin-sensitized rats. Phytother Res.
25:116–121. 2011. View
Article : Google Scholar
|
|
23
|
Lee KP, Kang S, Park SJ, Choi YW, Lee YG
and Im DS: Anti-allergic and anti-inflammatory effects of
bakkenolide B isolated from petasites japonicus leaves. J
Ethnopharmacol. 148:890–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Jin DQ, Xie C, Wang H, Wang M, Xu
J and Guo Y: Isolation, characterization, and neuroprotective
activities of sesquiterpenes from petasites japonicus. Food Chem.
141:2075–2082. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu J, Yang B, Guo Y, Jin DQ, Guo P, Liu C,
Hou W, Zhang T, Gui L and Sun Z: Neuroprotective bakkenolides from
the roots of valeriana jatamansi. Fitoterapia. 82:849–853. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee YJ, Park SY, Kim SG, Park DJ, Kang JS,
Lee SJ, Yoon S, Kim YH, Bae YS and Choi YW: Identification of a
novel compound that inhibits iNOS and COX-2 expression in
LPS-stimulated macrophages from schisandra chinensis. Biochem
Biophys Res Commun. 391:1687–1692. 2010. View Article : Google Scholar
|
|
27
|
Fan H, Wu PF, Zhang L, Hu ZL, Wang W, Guan
XL, Luo H, Ni M, Yang JW, Li MX, et al: Methionine sulfoxide
reductase A negatively controls microglia-mediated
neuroinflammation via inhibiting ROS/MAPKs/NF-κB signaling pathways
through a catalytic antioxidant function. Antioxid Redox Signal.
22:832–847. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song SY, Jung YY, Hwang CJ, Lee HP, Sok
CH, Kim JH, Lee SM, Seo HO, Hyun BK, Choi DY, et al: Inhibitory
effect of ent-sauchinone on amyloidogenesis via inhibition of
STAT3-mediated NF-κB activation in cultured astrocytes and
microglial BV-2 cells. J Neuroinflammation. 11:1182014. View Article : Google Scholar
|
|
29
|
Wu LH, Lin C, Lin HY, Liu YS, Wu CY, Tsai
CF, Chang PC, Yeh WL and Lu DY: Naringenin suppresses
neuroinflammatory responses through inducing suppressor of cytokine
signaling 3 expression. Mol Neurobiol. 53:1080–1091. 2016.
View Article : Google Scholar
|
|
30
|
Kang SM, More SV, Park JY, Kim BW, In PJ,
Yoon SH and Choi DK: A novel synthetic HTB derivative, BECT
inhibits lipopolysaccharide-mediated inflammatory response by
suppressing the p38 MAPK/JNK and NF-κB activation pathways.
Pharmacol Rep. 66:471–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Santa-Cecilia FV, Socias B, Ouidja MO,
Sepulveda-Diaz JE, Acuña L, Silva RL, Michel PP, Del-Bel E, Cunha
TM and Raisman-Vozari R: Doxycycline suppresses microglial
activation by inhibiting the p38 MAPK and NF-κB signaling pathways.
Neurotox Res. 29:447–459. 2016. View Article : Google Scholar
|
|
32
|
Vinoth Kumar R, Oh TW and Park YK:
Anti-inflammatory effects of ginsenoside-Rh2 inhibits LPS-induced
activation of microglia and overproduction of inflammatory
mediators via modulation of TGF-β1/smad pathway. Neurochem Res.
41:951–957. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jeong YH, Kim Y, Song H, Chung YS, Park SB
and Kim HS: Anti-inflammatory effects of α-galactosylceramide
analogs in activated microglia: Involvement of the p38 MAPK
signaling pathway. PLoS One. 9:e870302014. View Article : Google Scholar
|
|
34
|
Shu Z, Yang B, Zhao H, Xu B, Jiao W, Wang
Q, Wang Z and Kuang H: Tangeretin exerts anti-neuroinflammatory
effects via NF-κB modulation in lipopolysaccharide-stimulated
microglial cells. Int Immunopharmacol. 19:275–282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tanaka T, Kai S, Matsuyama T, Adachi T,
Fukuda K and Hirota K: General anesthetics inhibit LPS-induced
IL-1β expression in glial cells. PLoS One. 8:e829302013. View Article : Google Scholar
|
|
36
|
Lin HY, Huang BR, Yeh WL, Lee CH, Huang
SS, Lai CH, Lin H and Lu DY: Antineuroinflammatory effects of
lycopene via activation of adenosine monophosphate-activated
protein kinase-alpha1/heme oxygenase-1 pathways. Neurobiol Aging.
35:191–202. 2014. View Article : Google Scholar
|
|
37
|
Ashabi G, Khalaj L, Khodagholi F,
Goudarzvand M and Sarkaki A: Pre-treatment with metformin activates
Nrf2 antioxidant pathways and inhibits inflammatory responses
through induction of AMPK after transient global cerebral ischemia.
Metab Brain Dis. 30:747–754. 2015. View Article : Google Scholar
|