Introduction
During vascular inflammation, numerous cytokines and
adhesion molecules are overexpressed, which leads to the homing of
various immune cells into tissues (1–3).
Major events in the homing of immune cells include the migration of
leukocytes from the blood, their firm adhesion to the vascular
endothelium and transmigration across the vascular endothelium
(4). Several types of adhesion
molecule are important in the endothelium cell adhesion process.
Among these cell adhesion molecules, vascular adhesion molecule-1
(VCAM-1/CD106) is known to be increased in human endothelial cells
by pro-inflammatory cytokines, including tumor necrosis factor
(TNF)-α, interleukin (IL)-1β, IL-6 and various inflammatory
stimuli, including reactive oxygen species (ROS) and
lipopolysaccharide (LPS) (5,6).
In addition, the phosphorylation of mitogen-activated protein
kinases (MAPKs), phosphoinositide-3 kinase and the nuclear factor
(NF)-κB pathways regulates the activation of the gene expression of
VCAM-1 (3,7,8).
According to previous reports, the NF-κB signaling pathway in human
endothelial cells is important in regulating cell-to-cell adhesion
in the inflammatory response by inflammatory mediators associated
with increased expression of VCAM-1. NF-κB protein is known to be a
pivotal transcription factor for the expression of inflammatory
cytokine-mediated VCAM-1 (9,10).
The progressive accumulation of leukocyte adhesion to the
endothelium may cause vascular inflammation.
LPS is a lipoglycan and a known endotoxin present in
the outer membrane of Gram-negative bacteria, which affects
inflammation and immune cell activation in animals (11). The binding of LPS to toll-like
receptor 4, present in immune cells, affects various signal
transduction pathways, particularly the activation of inflammatory
signaling pathways involving MAPK/inhibitor of NF-κB (IκB) kinase
(IKK)/IκB/NF-κB (12). In
non-stimulated conditions, the p65 subunit of the NF-κB protein is
concealed by the IκB-α protein complex in the cytoplasm. However,
when activated by external stimuli, including inflammatory
molecules, the IKK-α/β subunit undergoes a successive
phosphorylation cascade (13).
Subsequently, the activated and released p65 subunit of NF-κB is
translocated into the nucleus and binds to DNA promoter regions,
increasing the transcription of inflammatory-mediated genes,
including cell adhesion molecules (14,15).
In ancient oriental medicine, active ingredients
isolated from natural products and herbal extracts have been used
to treat various diseases. Previous studies have shown that
Cynanchum wilfordii root extracts have been used in
traditional medicine for the therapeutic treatment and prevention
of diseases, including vascular disease, arteriosclerosis and
cancer (16–18). The active ingredient of C.
wilfordii roots, cynandione A (CA; Fig. 1A), has been isolated in a number
of studies (19,20). In addition, several studies
investigating the biological and pharmaceutical activity of CA have
revealed it exhibits protective activity against toxicity by
various stimulant agents in rat hepatocytes and cortical neurons
(21–23). Among investigations on the
inhibitory effects of CA on anti-inflammatory activity, few have
reported on the molecular mechanism underlying the anti-adhesion
effects of CA in vascular inflammation. In the present study, it
was demonstrated that CA inhibited the mRNA and protein expression
of VCAM-1 in LPS-induced human umbilical vascular endothelial cells
(HUVECs). The results confirmed that CA inhibited the expression of
pro-inflammatory cytokines, including IL-1β, IL-6, IL-8, monocyte
chemoattractant protein-1 (MCP-1) and TNF-α, in LPS-activated
HUVECs. It was also shown that CA inhibited the phosphorylation of
IKK and the transcriptional activity of NF-κB via downregulating
the phosphorylation of MAPKs. Finally, the anti-adhesion activity
of CA was confirmed using FITC-labeled immune cells. These findings
may be beneficial for the development of a useful therapeutic agent
for vascular inflammatory diseases, including atherosclerosis.
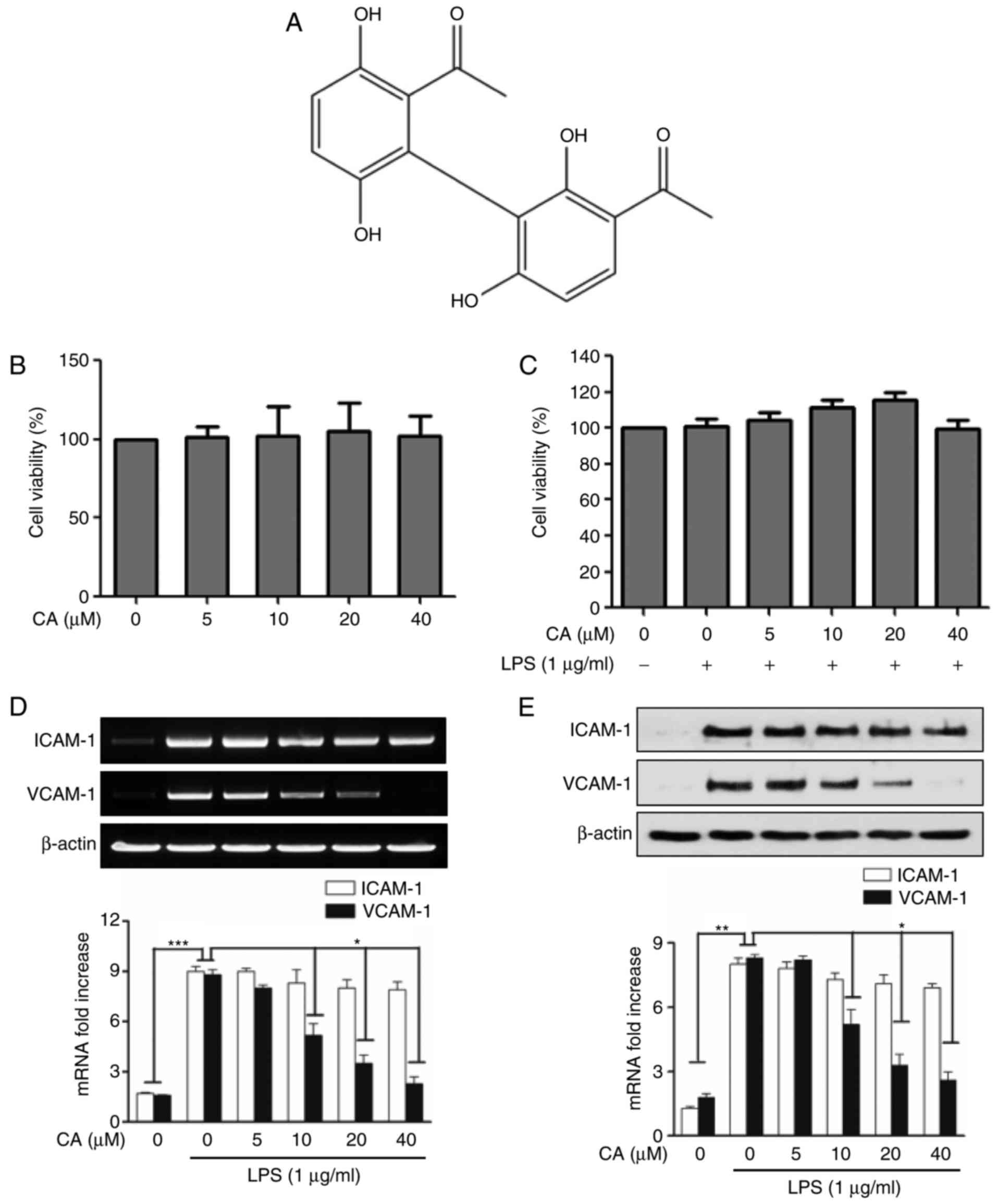 | Figure 1Effects of CA on mRNA and protein
expression of cell adhesion molecules in LPS-stimulated human
umbilical vascular endothelial cells. (A) Chemical structure of CA.
Cell viability assessment of (B) CA-treated cells and (C) cells
treated with CA+LPS was evaluated using a
3-(4,5-dimetnythiazol-2-yl)-2,5-di-phenyl-thetazolium bromide
assay. The cells were cultured for 24 h at each indicated CA
concentration and the results were measured by optical density
value. Results are shown as a percentage of the negative control,
which was treated with medium alone. Each value is presented as the
mean ± standard deviation. (D) Reverse transcription-quantitative
polymerase chain reaction experiments were performed to compare
mRNA levels of cell adhesion molecules. The values for intensity
are presented as the mean ± standard deviation of three independent
experiments. ***P<0.001, compared with treatment with
medium alone; *P<0.05, compared with treatment with
LPS alone. (E) Western blot analyses were performed to compare the
expression levels of cell adhesion molecules. The intensity of each
band was compared and data are presented as a graph. The values for
intensity are presented the mean ± standard deviation from three
independent experiments. **P<0.01, compared with
treatment with medium alone; *P<0.05, compared with
treatment with LPS alone. CA, cynandione A; LPS,
lipopolysaccharide; ICAM-1, intercellular adhesion molecule-1;
VCAM-1, vascular adhesion molecule-1. |
Materials and methods
Materials and cell culture
Human endothelial cells were cultured in medium 199
containing fetal bovine serum (FBS) and antibiotics
(penicillin/streptomycin) prepared by Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Anti-human VCAM-1 (1:200; cat
no. SC-13160) and anti-b-actin (1:200; cat no. SC-47778) antibodies
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The antibodies targeting phosphorylated (p-)c-Jun N-terminal
kinase (JNK; 46 kDa, 1:1,000; cat no. MAB1205), p-extracellular
signal-regulated kinase (ERK)1/2 (44,42 kDa, 1:1,000; cat no.
MAB18251), p-p38 (38 kDa, 1:500; cat no. MAB8691), JNK, ERK and p38
antibodies were from R&D Systems, Inc. (Minneapolis, MN, USA).
Antibodies against p-IKK α/β (87, 85 kDa, 1:1,000; cat no. #2697)
p-IκB-α (40 kDa, 1:2,000; cat no. #9246), anti-p65 (65 kDa,
1:1,000; cat no. #8242) and all non-phosphorylated antibodies were
prepared and acquired from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Signal inhibitors AG490 (a Janus kinase 2
inhibitor), U0126 (an MAPK kinase/ERK inhibitor) and SB203580 (a
p38 inhibitor) were purchased from Cell Signaling Technology, Inc.,
and PDTC (an NF-κB inhibitor) was from EMD Millipore (Billerica,
MA, USA). The primers used for polymerase chain reaction (PCR)
analysis were prepared from Bioneer Corporation (Daejeon, Korea).
Other chemicals were purchased commercially from Sigma-Aldrich;
Merck Millipore (Darmstadt, Germany).
The HUVECs (BMS, Seoul, Korea) were grown over three
to six passages in cell culture medium at 37°C under a humidified
95 and 5% (v/v) mixture of air and CO2 supplemented with
10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 3
ng/ml bFGF and 5 U/ml heparin. The U937 human monocyte-like cell
line (American Type Culture Collection, Manassas, VA, USA;
CRL-1593.2™) was also cultured in complete RPMI-1640 cell culture
medium under the same conditions as the HUVECs.
Cell viability assay
The HUVECs (1×104 cells/well) were
removed from the cell growth medium and incubated in serum-free
medium for 18 h. These cells were then incubated with or without
LPS (1 µg/ml) in the presence of each indicated CA
concentration (0, 5, 10, 20 and 40 µM) for 48 h at 37°C Cell
viability was measured by the addition of 50 µg/ml
3-(4,5-dimetnythiazol-2-yl)-2,5-diphenyl-thetazolium bromide (MTT).
Following incubation for 2 h, purple formazan had formed, the
supernatants were removed from each well and 100 µl DMSO was
added to completely dissolve the formazan crystals. Data was
quantified by measuring the absorbance at 540 nm using a
spectrophotometric multi-well microplate reader (Multiskan MS;
Thermo Fisher Scientific, Inc.).
Reverse transcriptase-polymerase chain
reaction (RT-PCR) analysis
Total RNA from HUVECs was isolated according to the
manufacturer's protocol using an RNA-Bee isolation kit (Tel-Test,
Inc., Friendswood, TX, USA). The cDNA was prepared by reverse
transcription using M-MuLV reverse transcriptase (Fermentas; Thermo
Fisher Scientific, Inc.) and PCR amplification was performed
according to the manufacturer's protocol using an AccuPower PCR
PreMix (Bioneer Coporation; Daejeon, Korea). Using 250 ng of cDNA
and 10 pmole/µl of forward and reverse primers, PCR was
performed as follows: Intercellular adhesion molecule (ICAM)-1 (556
bp, annealing temp. 60°C, 25 cycles), forward 5′-CAG TGA CCA TCT
ACA GCT TTC CGG-3′ and reverse 5′-GCT GCT ACC ACA GTG ATG ATG ACA
A-3′; VCAM-1 (742 bp, annealing temp. 62°C, 32 cycles), forward
5′-AAT TTA TGT GTG TGA AGG AG-3′ and reverse 5′-GCA TGT CAT ATT CAC
AGA A-3′; IL-1β (264 bp, annealing temp. 62°C, 33 cycles), forward
5′-GGA TAT GGA GCA ACA ACT GG-3′ and reverse 5′-ATG TAC CAG TTG GGG
AAC TG-3′; IL-6 (497 bp, annealing temp. 54°C, 30 cycles), forward
5′-TGA CAA ACA AAT TCG GTA CAT CC-3′ and reverse 5′-ATC TGA GGT GCC
CAT GCT AC-3′; IL-8 (292 bp, annealing temp. 63°C, 28 cycles),
forward 5′-ATG ACT TCC AAG CTG GCC GTG GCT-3′ and reverse 5′-TCT
CAG CCC TCT TCA AAA ACT TCT C-3′; MCP-1 (261 bp, annealing temp.
62°C, 30 cycles), forward 5′-TCT GTG CCT GCT GCT CAT AG-3′ and
reverse 5′-TTT GCT TGT CCA GGT GGT CC-3′; and TNF-α (444 bp,
annealing temp. 62°C, 32 cycles), forward 5′-GAC TGA CAA GCC TGT
AGC CCA TGT TGT TGT AGC A-3′ and reverse 5′-GCA ATG ATC CCA AAG TAG
ACC TGC CCA GAC-3′; β-actin (482 bp, annealing temp. 58°C, 28
cycles), forward 5′-TGA GAC CTT CAA CAC CCC AG-3′ and reverse
5′-CAC TGT GTT GGC GTA CAG GT-3′. Each band intensity was detected
for all bands and standardization relative β-actin and measured
using ImageJ software (Ver.1.51e; National Institutes of Health,
Bethesda, MD, USA).
Western blot analysis
The HUVECs were treated with cell lysis buffer to
obtain cell extracts. Protein quantity was determined using a
Bradford assay kit according to the manufacturer's protocol (Thermo
Fisher Scientific, Inc.) and equal quantities of protein (30
µg) were separated by performing electrophoresis on 6-15%
SDS-PAGE gels. The proteins were then transferred onto a
nitrocellulose membrane, which was gently agitated and washed with
3–5% non-fat dry milk in Tris-buffered saline-Tween-20 buffer,
containing 0.5 mol/l Tris-HCl (pH 7.5), 0.15 mol/l NaCl and 1 g/l
Tween-20. The membrane was incubated with each primary antibody
overnight at 4°C according to manufacturer's protocol followed by
the secondary antibodies (1:2,000) 2 h at room temperature which
was then visualized through enhanced chemiluminescence and exposure
to X-ray films. The intensity of each band was measured as
aforementioned using ImageJ software.
Luciferase reporter assay
VCAM-1 promoter region information was obtained by
searching human VCAM-1 gene sequences (www.ncbi.nlm.nih.gov/), following which the
pVCAM-1/Luc plasmid construct, containing the −1,350 to +45 bp
promoter region in the pGL3-basic vector (Promega, Madison, WI,
USA), was constructed. The HUVECs were transiently transfected with
empty vector (pGL3-basic), constructed pVCAM-1/Luc or pNF-κB/Luc
(Stratagene; Agilent Technologies, Inc., Santa Clara, CA, USA)
using Lipofectamine™ 2000 reagent (Stratagene; Agilent
Technologies, Inc.). Following 24 h of transfection, cell extracts
were prepared and luciferase activity was detected using the
Renilla luciferase assay kit according to the manufacturer's
protocol (Promega). Data was quantified by measuring the
fluorescence intensity at 480 nm (excitation) and 560 nm (emission)
using a fluorescence multi-well microplate reader (Wallac 1420
Victor 2; PerkinElmer, Inc., Waltham, MA, USA).
Fluorescence microscopy analysis
Growth medium was removed from the HUVECs and
serum-free medium containing CA was stimulated with or without LPS
(1 µg/ml) for 2 h. The HUVECs were incubated overnight at
4°C with the FITC-labeled NF-κB p65 antibody [(F-6) Alexa
FluorR488; 65 kDa; 1:100; cat. no. SC-8008; Santa Cruz
Biotechnology, Inc.], and then treated with 3.7% paraformaldehyde
solution to preserve the cell state and then washed in PBS. The
fluorescence intensities were analyzed for the distribution and
translocation of p65 protein. The fixed cells were stained with
mounting solution containing 4′–6-diamidino-2-phenylindole to
indicate the location of nuclei in the cells.
Cell adhesion assay
The U937 cell line has the monocyte phenotype and
can therefore be used in monocyte adhesion experiments, which were
performed as described in a previous study (24). The HUVECs were cultured to
confluence in 6-well plates and pre-treated with each signal
inhibitor (10 µM) with CA at the indicated concentrations
for 12 h. The U937 cells were cultured in 1% FBS and 5 µg/ml
calcein-AM (Invitrogen; Thermo Fisher Scientific, Inc.) RPMI medium
at 37°C for 30 min for fluorescent labeling. The
fluorescence-labeled U937 cells (1×107) were
re-suspended in HUVEC culture medium and then incubated for 30 min
with the pre-cultured HUVECs. The non-adherent fluorescent U937
cells were washed with PBS and the fluorescence intensity of
attached cells was quantitated using a Wallace Victor2 1420
Multi-label counter (PerkinElmer, Inc.).
Statistical analysis
Unless otherwise stated, all experiments were
performed with triplicate samples and repeated at least three
times. The data are presented as the mean ± standard deviation and
statistical comparisons between groups were performed using one-way
analysis of variance followed by Student's t-tests using SigmaPlot
software (version 11; Systat Software Inc., San Jose, CA, USA).
P<0.05 was considered to indicate a statistically significant
differences.
Results
CA inhibits the LPS-induced mRNA and
protein expression of VCAM-1 in HUVECs
The present study examined cell viability using an
MTT assay to determine the in vitro experimental
concentration of CA. As shown in Fig.
1B and C, the cells were treated with CA for 24 h in the
presence or absence of LPS. It was confirmed that treatment of the
HUVECs with CA was not toxic up to a concentration of 40 µM
CA. Based on this information, the mRNA expression levels of ICAM-1
and VCAM-1 were examined in the LPS-induced HUVECs at the indicated
concentrations of CA. The results showed that CA did not affect the
mRNA expression of ICAM-1, however, the mRNA expression of VCAM-1
was decreased in a concentration-dependent manner in the
LPS-induced HUVECs (Fig. 1D).
Subsequently, the results of western blot analysis showed that the
protein expression levels of ICAM-1 and VCAM-1 were significantly
increased by LPS stimulation in the HUVECs. Similar to the mRNA
expression levels, CA markedly inhibited the protein expression of
VCAM-1 and had no effect on the protein expression level of ICAM-1
in the LPS-stimulated HUVECs (Fig.
1E).
CA inhibits the pro-inflammatory cytokine
expression in LPS-stimulated HUVECs
The present study subsequently examined the activity
of CA on the mRNA expression levels of IL-1β, IL-6, IL-8, MCP-1 and
TNF-α in HUVECs. In the LPS-stimulated HUVECs, the gene expression
levels of the pro-inflammatory cytokines IL-1β, IL-6, IL-8, MCP-1
and TNF-α increased ~6–9-fold, compared with those in the untreated
control. In the CA-treated HUVECs, the mRNA level of each
pro-inflammatory cytokine was significantly decreased in a CA
concentration-dependent manner (Fig.
2).
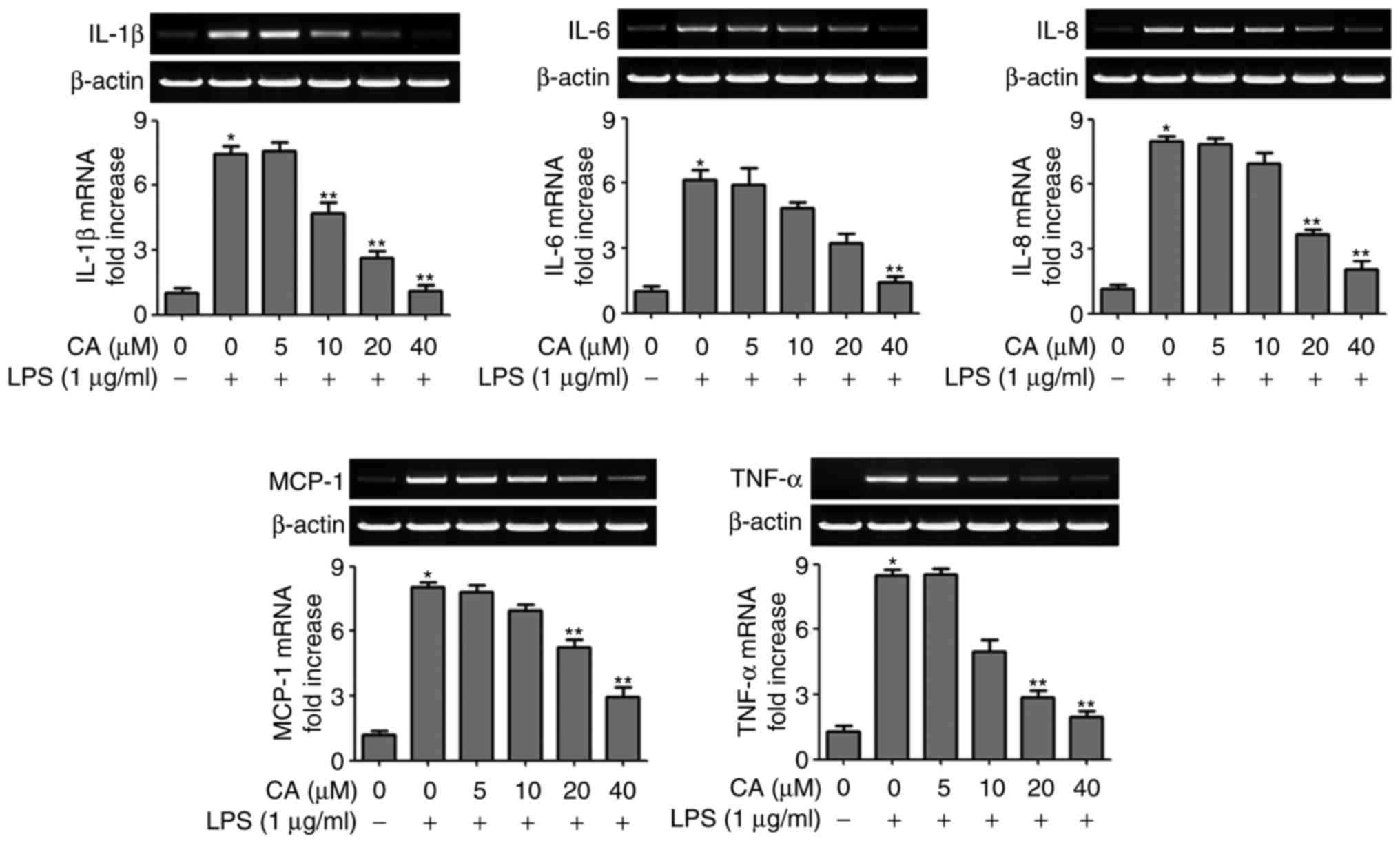 | Figure 2Inhibitory effect of CA on expression
of LPS-induced pro-inflammatory cytokines and chemoattractant
cytokines in HUVECs. Reverse transcription polymerase chain
reaction analyses were performed to compare the mRNA expression
levels of IL-1β, IL-6, IL-8, MCP-1 and TNF-α. HUVECs were
pretreated with CA at the indicated concentration for 1 h and then
treated with LPS. The values for intensity are presented as the
mean ± standard deviation of three independent experiments.
*P<0.01, compared with treatment with medium alone;
**P<0.05, compared with treatment with LPS alone.
HUVECs, human umbilical vascular endothelial cells; CA, cynandione
A; LPS, lipopolysaccharide; IL, interleukin; MCP-1, monocyte
chemoattractant protein-1; TNF-α, tumor necrosis factor-α. |
Role of MAPKs in the inhibition of VCAM-1
by CA in LPS-induced HUVECs
To reveal the molecular mechanism for the inhibitory
activity of CA on the expression of VCAM-1 and pro-inflammatory
cytokines, the present study analyzed the phosphorylation of MAPKs
in the LPS-induced HUVECs with or without CA exposure. The effects
of CA and commercial signal inhibitors on the mRNA expression
levels of VCAM-1 and pro-inflammatory cytokines were examined in
the LPS-activated HUVECs. The HUVECs were treated with AG490 (a
Janus kinase inhibitor), U0126 (an MEK/ERK inhibitor), SB203580 (a
p38 inhibitor), PDTC (an NF-κB inhibitor) and at the indicated
concentration of CA. After 1 h, the incubated HUVECs in the
commercial signal inhibitors and CA were stimulated with LPS. Our
data showed that, with the exception of AG490, all the commercial
signal inhibitors inhibited the mRNA and protein levels of VCAM-1
in LPS-induced HUVECs. CA inhibited the mRNA and protein expression
levels of VCAM-1 to levels similar to those in the U0126, SB203580
and PDTC treatment groups (Fig.
3A). This result indicated that key signal inhibitors,
including U0126, SB203580 and PDTC, inhibited the expression of
VCAM-1 and the upstream signaling molecules of VCAM-1 in the
LPS-stimulated HUVECs.
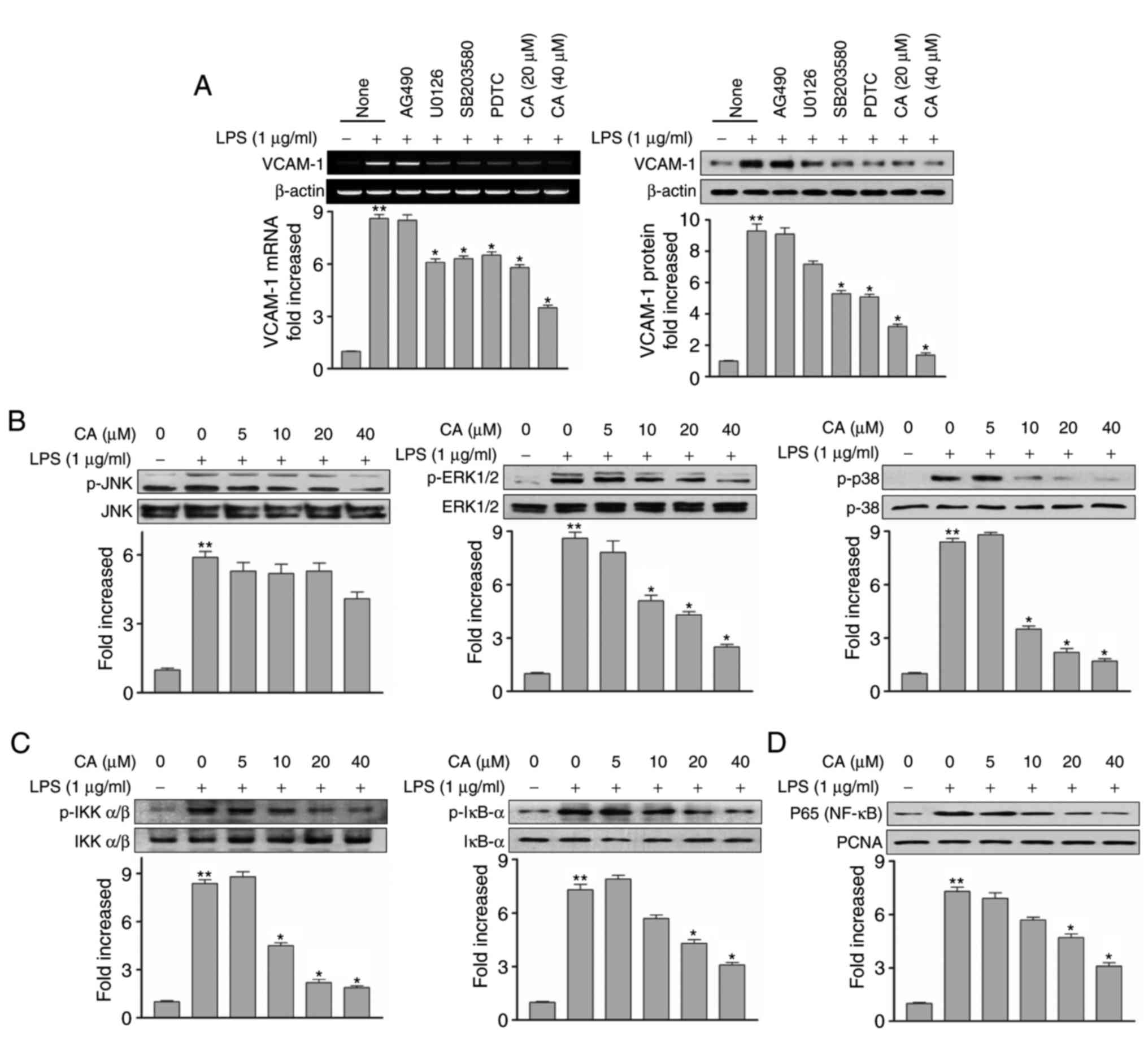 | Figure 3Effects of CA on the phosphorylation
of mitogen-activated protein kinase/IKK/IκB and analysis of the
translocation of p65 into the nucleus of LPS-activated HUVECs. (A)
HUVECs were pretreated with the indicated signal inhibitor, AG490
(10 µM), U0126 (10 µM), SB203580 (10 µM) or
PDTC (10 µM) for 1 h, and stimulated with LPS with or
without CA for 24 h. mRNA and protein expression levels of VCAM-1
were detected using reverse transcription-quantitative polymerase
chain reaction and western blot analyses, respectively. (B) HUVECs
were starved for 6 h and then pre-treated with CA for 1 h, followed
by activation by LPS for 30 min. Phosphorylation patterns were
analyzed using western blot analysis. (C) HUVECs were starved for 6
h and then pre-treated with CA for 1 h, followed by activation by
LPS for 30 min. The activation levels of p-IKK, p-IκB-α, IKK and
IκB-α were detected using western blot analysis. (D) HUVECs were
starved for 6 h and then pre-treated with CA for 1 h, followed by
activation with LPS for 2 h. Proteins in the nuclear extract
fraction were prepared and NF-κB p65 subunit translocation levels
were analyzed using western blot analysis. **P<0.01,
compared with the negative control; *P<0.05, compared
with treatment with LPS alone. HUVECs, human umbilical vascular
endothelial cells; CA, cynandione A; LPS, lipopolysaccharide;
VCAM-1, vascular adhesion molecule-1; NF-κB, nuclear factor-κB;
IκB, inhibitor of NF-κB; IKK, IκB kinase; JNK, c-Jun N-terminal
kinase; ERK, extracellular signal-regulated kinase; p-,
phosphorylated. |
Subsequently, the present study examined the effect
of CA on cellular signaling molecules activated by LPS-stimulation
in HUVECs. The inhibitory activity of CA on the phosphorylation of
the most well known MAPK pathways, including JNK, ERK1/2 and p38
proteins, were then confirmed in LPS-induced HUVECs. The results
indicated that CA decreased the phosphorylation of ERK1/2 and p38
proteins in the LPS-stimulated HUVECs in a concentration-dependent
manner, whereas CA had minimal effect on the phosphorylation of JNK
(Fig. 3B). These results
suggested that the inhibitory activity of CA on the expression of
VCAM-1 may be associated with inhibition of the phosphorylation of
ERK1/2 and p38 kinase in LPS-stimulated HUVECs.
To analyze the effect of CA on MAPK downstream
signaling pathways, the present study confirmed the activity of CA
on the phosphorylation of IKK/IκB-α signal transduction. The NF-κB
p65 subunit is an important transcription factor for the expression
of VCAM-1 in LPS-stimulated HUVECs (25). The nuclear translocation of the
p65 subunit is activated by the phosphorylation of IκB-α.
Therefore, the effect of CA on the phosphorylation of IKK was
examined. The results demonstrated that CA significantly inhibited
the phosphorylation of IKK in LPS-stimulated HUVECs. In addition,
it was found that CA inhibited the phosphorylation of IκB-α protein
and suppressed the degradation of IκB-α protein in the cell
cytosolic fraction (Fig. 3C).
These results suggested that CA significantly inhibited IKK/IκB-α
signal transduction. The protein levels of p65 in the nucleus of
cells treated with CA in the LPS-stimulated HUVECs were examined.
In the nuclei of resting state cells, a basic level of p65 protein
was identified, and in the cells treated with LPS alone, the
nuclear translocation of p65 subunit was significantly increased.
In cells stimulated with LPS in the presence of CA, it was
confirmed that the protein expression of p65 was decreased in a
concentration-dependent manner. The reduced protein expression of
p65 was similar to that in the non-stimulated cells (Fig. 3D). The inhibitory activity of CA
on the expression of VCAM-1 and pro-inflammatory cytokines may be
due to the decrease in nuclear p65 translocation through inhibition
of the phosphorylation of MAPK/IKK/IκB-α and degradation of
IκB-α.
CA inhibits LPS-induced transcription of
the VCAM-1 promoter and activation of NF-κB
The present study investigated whether CA regulates
the transcriptional activity of the VCAM-1 gene using VCAM-1
luciferase reporter gene constructs. The transfected HUVECs were
treated with or without CA and then activated with LPS. Stimulation
of the transfected cells with LPS promoted luciferase activity by
~5–6-fold over that in the mock transfected cells. CA significantly
reduced the LPS-activated induction of VCAM-1 in the transfected
HUVECs at the transcriptional level (Fig. 4A). The regulatory activity of CA
on transcriptional activation was examined using the established
pNF-κB/Luc reporter gene construct by LPS stimulation. As shown
Fig. 4B, the results demonstrated
that CA reduced transcriptional activity by LPS in a dose-dependent
manner in the HUVECs transfected with the pNF-κB/Luc reporter gene
construct. To analyze the activity of CA on the translocation of
the p65 subunit into the nucleus, fluorescence microscopy was used
to analyze the HUVECs treated with or without CA. It was confirmed
that the fluorescence intensity in the nucleus was increased in
cells treated with LPS alone, and a large quantity of p65-FITC
antibody was present in the nucleus. By contrast, CA decreased
fluorescence intensity in the nucleus by LPS stimulation, and a
similar result was observed in the cells treated with PDTC, which
is an NF-κB specific signaling inhibitor (Fig. 4C).
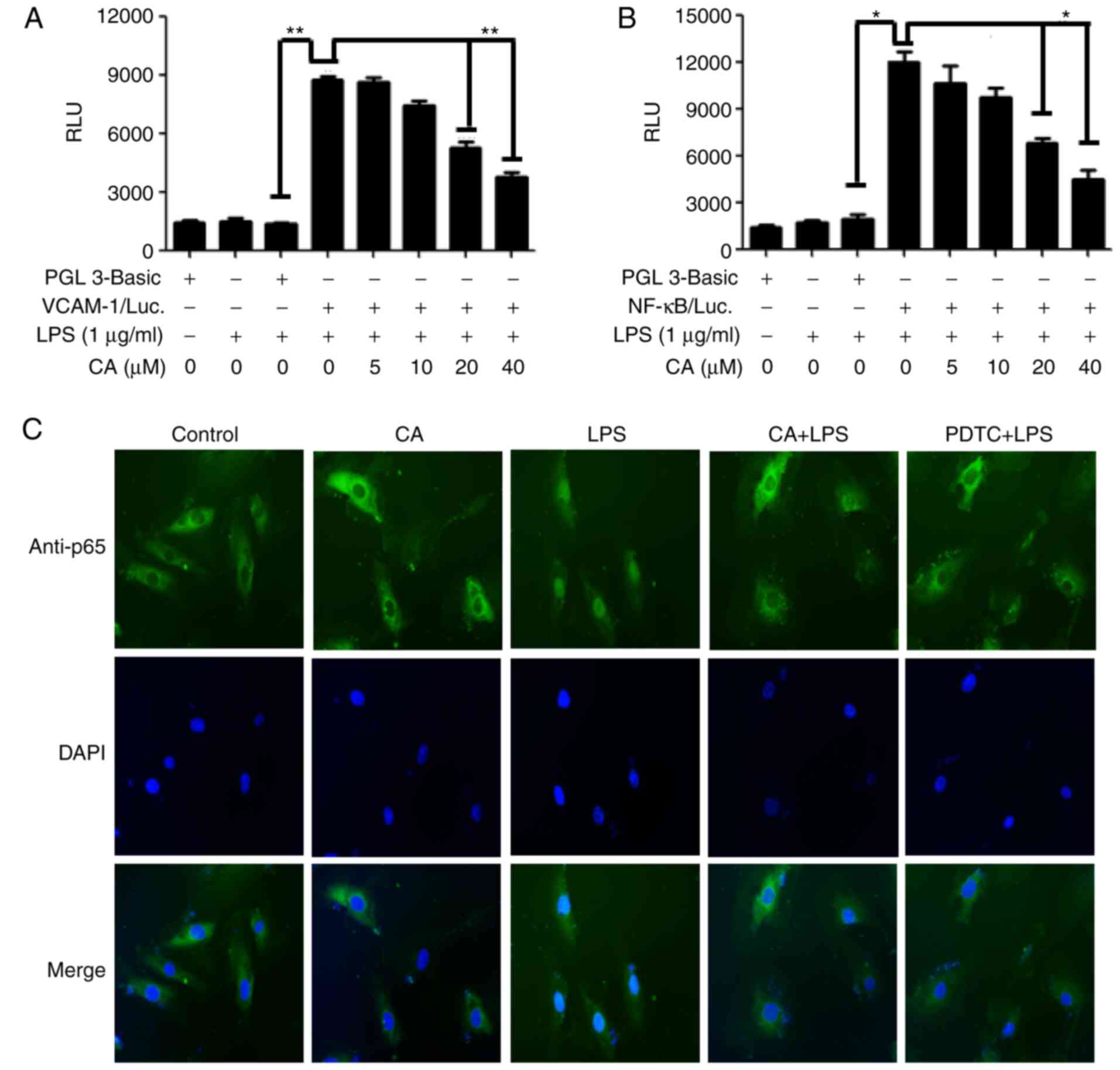 | Figure 4Effects of CA on the transcriptional
activity of NF-κB and luciferase reporter genes in transiently
transfected HUVECs. (A) pVCAM-1/Luc trans-fected HUVECs were
cultured for 24 h, and treated with indicated concentrations of CA
and 1 µg/ml LPS for 24 h. The values for relative luciferase
intensity are shown as the mean ± standard deviation of three
independent experiments (n=3). **P<0.01, compared
with the mock transfectant; **P<0.01, compared with
treatment with LPS alone. (B) pNF-κB/Luc transfected HUVECs were
cultured for 24 h, and treated with indicated concentrations of CA
and 1 µg/ml LPS for 24 h. The values for relative luciferase
intensity are shown as the mean ± standard deviation from three
independent experiments (n=3). *P<0.05, compared with
the mock transfectant; *P<0.05, compared with
treatment with LPS alone. (C) Nuclear translocation of NF-κB p65
subunit was analyzed following CA (40 µM) and PDTC (10
µM) treatment for 1 h. Cells were activated for 2 h with LPS
and then observed via fluorescence microscopy (magnification,
×100). HUVECs, human umbilical vascular endothelial cells; CA,
cynandione A; LPS, lipopolysaccharide; NF-κB, nuclear factor κB;
VCAM-1, vascular adhesion molecule-1; Luc, luciferase. |
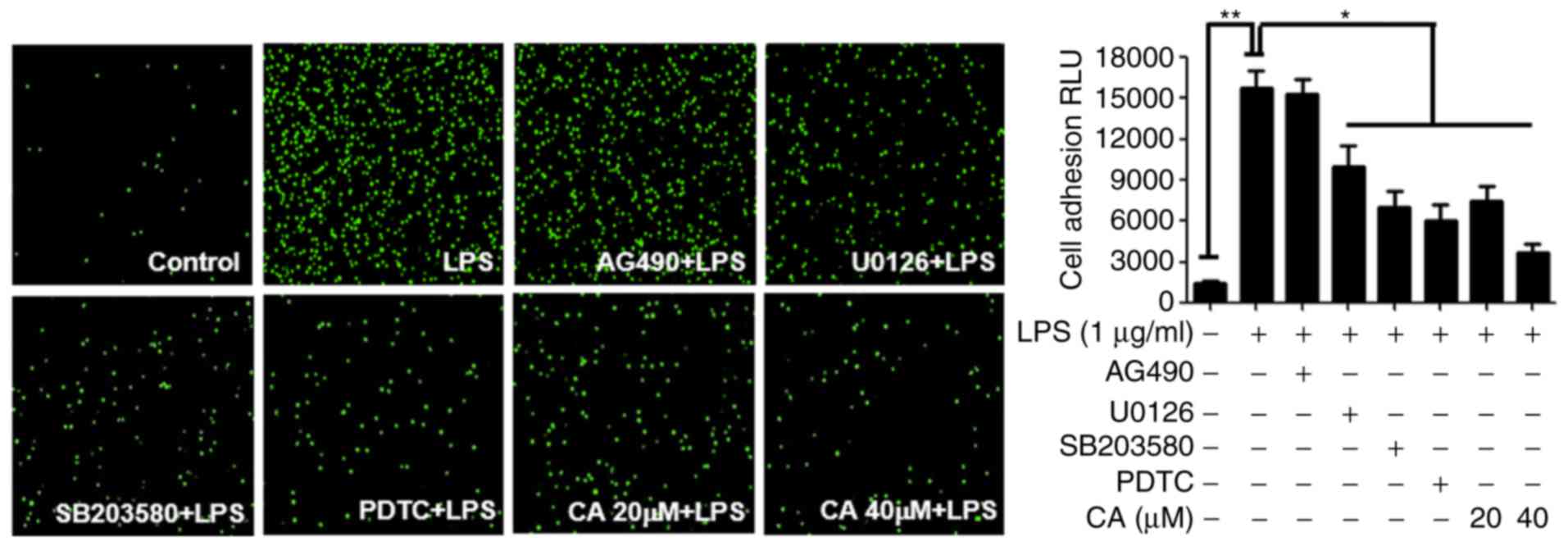
CA inhibits the LPS-induced adhesion of
U937 cells to HUVECs
The overproduction of cell adhesion molecules,
including VCAM-1, by LPS likely increases the adherence of
monocytes to endothelial cells in the early stages of vascular
inflammation. Therefore, in order to demonstrate the effect of CA
on U937 cell adhesion to endothelial cells, the present study
compared cell adhesion activity with various signal inhibitors in
the presence of CA in the LPS-induced HUVECs. In the case of cells
treated with LPS alone, the adhesion activity of the U937 cells was
markedly increased compared with that in the control cells. The
data showed that U0126, SB203580 and PDTC significantly inhibited
U937 adhesion in the LPS-induced HUVECs, whereas AG490 had no
effect. The LPS-activated HUVECs were treated with CA (20 or 40
µM), and it was shown that CA inhibited U937 adhesion to the
HUVECs (Fig. 5). The data showed
that CA inhibited the LPS-induced expression of VCAM-1 and
suppressed U937 adhesion to HUVECs.
Discussion
In the present study, it was found that CA had
anti-adhesive activity with regard to the adhesion of U937
monocytes to HUVECs via suppressing the expression of VCAM-1 and
pro-inflammatory mediators in LPS-stimulated HUVECs. The inhibitory
activity of CA on the expression of cell adhesion molecules was
examined in LPS-induced HUVECs, and it was demonstrated that CA
treatment inhibited the mRNA and protein expression levels of
VCAM-1. The inhibition of VCAM-1 protein expression in LPS-induced
HUVECs was due to the fact that CA inhibited the phosphorylation
and degradation of IκB-α, the activation of NF-κB, and the
trans-location of NF-κB p65 subunit to the nucleus. Therefore, the
anti-adhesion effect of U937 monocytes to vascular endothelial
cells by CA was the result of the inhibition of vascular
inflammation, which prevented activation of the NF-κB signaling
pathway in HUVECs.
Chronic vascular inflammation is a main cause of
various vascular inflammatory diseases. In vascular inflammatory
sites, endothelial cells stimulated by inflammatory stimuli,
including LPS, are known to interact with other leukocytes through
a variety of adhesion molecules, including VCAM-1, inflammatory
cytokines and chemokines (2,26).
The firm adhesion of leukocytes to the endothelium is critical in
early atherosclerotic plaque development and in vascular
inflammation. LPS is a component of the Gram-negative bacterial
cell membrane and is known to induce inflammation in vivo.
According to previous reports, LPS is known to increase the
expression and secretion of cytokines, including IL-1β, IL-6, IL-8,
MCP-1 and TNF-α, in HUVECs (27–29). IL-8 and MCP-1 are chemoattractant
cytokines, which are involved in the accumulation of monocytes in
vascular inflammation plaque lesions (30). TNF-α and other inflammatory
stimuli have been reported to accrete the production of VCAM-1 in
endothelial cell membranes, which can promote
leukocyte-to-endothelium adhesion in vascular plaques (31). Therefore, inhibiting the secretion
of pro-inflammatory cytokines and the expression of cell adhesion
molecules may be important for the development of therapeutic
agents for atherosclerosis caused by vascular inflammation.
The expression of VCAM-1, pro-inflammatory cytokines
and chemoattractant cytokines, including IL-8 and MCP-1, in
LPS-induced cells is regulated by the MAPK/IKK/IκB signaling
cascade and activation of NF-κB (9,10,32). In addition, interactions with
transcription factor binding elements, including NF-κB, can affect
the activation of cell adhesion molecules and inflammatory
mediators by LPS stimulation (32). In inflammatory conditions, the
present study confirmed that CA inhibited the expression of VCAM-1
and pro-inflammatory cytokines. The results following signal
inhibitor treatment demonstrated that ERK1/2 and p38 kinase were
more important than JAK2 in the LPS-induced expression of VCAM-1.
To characterize the mechanism underlying the effect of CA in
LPS-induced HUVECs, the present study analyzed the phosphorylation
pattern of the MAPK/NF-κB signaling pathway. CA markedly inhibited
the phosphorylation of ERK1/2 and p38, and weakly inhibited the
phosphorylation of JNK. This result indicated that CA served as a
selective phosphorylation inhibitor of ERK1/2 and p38 kinase. It
was also shown that CA affected the inhibition of the
phosphorylation of IKK/IκB-α, which is a downstream signaling
pathway of MAPKs (14,15). These data demonstrated that CA
acted as a specific phosphorylation inhibitor of upstream
molecules, including IKK and MAPKs, in the NF-κB pathway.
In the resting state, the NF-κB protein has two
subunits, p65 and p50, which exist as a heterotrimer with IκB
protein in the cytoplasm. In the presence of external stimuli, the
IκB protein is activated via phosphorylation, and the p65 subunit
is released and translocated to the nucleus for target gene
activation (33). The present
study showed that CA inhibited nuclear translocation of the p65
subunit, and the results suggested that CA had an inhibitory effect
on the transcriptional activity of NF-κB using a NF-κB/Luc reporter
gene. The luciferase reporter assays demonstrated that CA
suppressed the expression of the VCAM-1/Luc reporter gene in the
LPS-stimulated HUVECs. The upregulation of VCAM-1 had functional
effects, specifically the adhesion of monocytes to the endothelium
in vascular inflammatory lesions. It was found that CA suppressed
U937 monocyte adhesion to activated HUVECs by the inhibition of
MAPK/IKK/IκB-α, which correlated with the decreased expression of
VCAM-1. These results provided additional evidence for the
involvement of the MAPK/IKK/IκB/NF-κB signaling pathway in
LPS-induced monocyte adhesion.
According to previous reports, natural compounds,
including flavonoids, phytosteroids and small phenolic molecules,
have various pharmacological activities, including
anti-inflammatory activity (34,35). CA is an acetophenone isolated from
the roots of Cynanchum extracts, and is known to be a
natural compound with effective anti-oxidant, anti-inflammatory and
neuroprotective effects (19,20).
In this present study, it was demonstrated that CA
markedly inhibited the expression of LPS-induced VCAM-1 in HUVECs.
The result showed that the LPS increased the expression levels of
ICAM-1 and VCAM-1 in the HUVECs. However, it was confirmed that CA
only inhibited the expression of VCAM-1 (Fig. 1). The activation of ICAM-1 by
endotoxins, including LPS, in HUVECs has been demonstrated in a
number of studies (36). In
particular, the inhibition of cell adhesion molecules using C.
wilfordii root ethanol extract and its active ingredients have
shown no effect on the expression of ICAM-1 (18). Taken together, the results of the
present and previous studies suggest that CA likely has no
significant effect on the expression of ICAM-1. The present study
further examined the inhibition of the expression of VCAM-1 at the
molecular levels and suggested a mode of action in the
anti-adhesion activity of CA. CA decreased the levels of IL-1β,
IL-6, TNF-α, IL-8 and MCP-1 induced by LPS. At the molecular level,
CA acted to inhibit the activation of NF-κB by inhibiting the
phosphorylation of IKK/IκB-α, and inhibiting p38 and ERK1/2 MAPKs.
In addition, CA substantially inhibited the adhesion of U937 to
HUVECs in a dose-dependent manner. These results suggested that CA
may be used as a novel drug candidate for the treatment of vascular
inflammation by the inhibition of monocyte infiltration.
Acknowledgments
The present study was supported by funding from the
Technology Commercialization Support Program (grant no.
814003031SB010), the Ministry of Agriculture Food and Rural Affairs
(grant no. NRF-2015R1D1A1A01058506) and the Region Industry Total
Information Service Program (grant no. R0004665), Republic of
Korea.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Binion DG, West GA, Ina K, Ziats NP,
Emancipator SN and Fiocchi C: Enhanced leukocyte binding by
intestinal micro-vascular endothelial cells in inflammatory bowel
disease. Gastroenterology. 112:1895–1907. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calixto JB, Campos MM, Otuki MF and Santos
AR: Anti-inflammatory compounds of plant origin. Part II modulation
of pro-inflammatory cytokines, chemokines and adhesion molecules.
Planta Med. 70:93–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu X, Pan L, Wang X, Gong Q and Zhu YZ:
Leonurine protects against tumor necrosis factor-α-mediated
inflammation in human umbilical vein endothelial cells.
Atherosclerosis. 222:34–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luscinskas FW, Ding H, Tan P, Cumming D,
Tedder TF and Gerritsen ME: L- and P-selectins, but not CD49d
(VLA-4) integrins, mediate monocyte initial attachment to
TNF-alpha-activated vascular endothelium under flow in vitro. J
Immunol. 157:326–335. 1996.PubMed/NCBI
|
|
5
|
Nizamutdinova IT, Kim YM, Jin H, Son KH,
Lee JH, Chang KC and Kim HJ: Tanshinone IIA inhibits TNF-α-mediated
induction of VCAM-1 but not ICAM-1 through the regulation of GATA-6
and IRF-1. Int Immunopharmacol. 14:650–657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alapati A, Deosarkar SP, Lanier OL, Qi C,
Carlson GE, Burdick MM, Schwartz FL, McCall KD, Bergmeier SC and
Goetz DJ: Simple modifications to methimazole that enhance its
inhibitory effect on tumor necrosis factor-α-induced vascular cell
adhesion molecule-1 expression by human endothelial cells. Eur J
Pharmacol. 751:59–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin P, Tang X, Elloso MM and Harnish DC:
Bile acids induce adhesion molecule expression in endothelial cells
through activation of reactive oxygen species, NF-kappaB, and p38.
Am J Physiol Heart Circ Physiol. 291:H741–H747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ueno H, Pradhan S, Schlessel D, Hirasawa H
and Sumpio BE: Nicotine enhances human vascular endothelial cell
expression of ICAM-1 and VCAM-1 via protein kinase C, p38
mitogen-activated protein kinase, NF-kappaB, and AP-1. Cardiovasc
Toxicol. 6:39–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chai H, Wang Q, Huang L, Xie T and Fu Y:
Ginsenoside Rb1 inhibits tumor necrosis factor-alpha-induced
vascular cell adhesion molecule-1 expression in human endothelial
cells. Biol Pharm Bull. 31:2050–2056. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zapolska-Downar D and Naruszewicz M:
Propionate reduces the cytokine-induced VCAM-1 and ICAM-1
expression by inhibiting nuclear factor-kappa B (NF-kappaB)
activation. J Physiol Pharmacol. 60:123–131. 2009.PubMed/NCBI
|
|
11
|
Wang W, Deng M, Liu X, Ai W, Tang Q and Hu
J: TLR4 activation induces nontolerant inflammatory response in
endothelial cells. Inflammation. 34:509–518. 2011. View Article : Google Scholar
|
|
12
|
Li B, Zhang R, Li J, Zhang L, Ding G, Luo
P, He S, Dong Y, Jiang W, Lu Y, et al: Antimalarial artesunate
protects sepsis model mice against heat-killed Escherichia coli
challenge by decreasing TLR4, TLR9 mRNA expressions and
transcription factor NF-kappa B activation. Int Immunopharmacol.
8:379–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mo SJ, Son EW, Lee SR, Lee SM, Shin DH and
Pyo S: CML-1 inhibits TNF-alpha-induced NF-kappaB activation and
adhesion molecule expression in endothelial cells through
inhibition of IκBalpha kinase. J Ethnopharmacol. 109:78–86. 2007.
View Article : Google Scholar
|
|
14
|
Madonna R, Massaro M, Pandolfi A, Consoli
A and De Caterina R: The prominent role of p38 mitogen-activated
protein kinase in insulin-mediated enhancement of VCAM-1 expression
in endothelial cells. Int J Immunopathol Pharmacol. 20:539–555.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Suna S, Sakata Y, Shimizu M, Nakatani D,
Usami M, Matsumoto S, Mizuno H, Ozaki K, Takashima S, Takeda H, et
al: Lymphotoxin-alpha3 mediates monocyte-endothelial interaction by
TNFR I/NF-kappaB signaling. Biochem Biophys Res Commun.
379:374–378. 2009. View Article : Google Scholar
|
|
16
|
Kim MS, Baek JH, Park JA, Hwang BY, Kim
SE, Lee JJ and Kim KW: Wilfoside K1N isolated from Cynanchum
wilfordii inhibits angiogenesis and tumor cell invasion. Int J
Oncol. 26:1533–1539. 2005.PubMed/NCBI
|
|
17
|
Choi DH, Lee YJ, Kim JS, Kang DG and Lee
HS: Cynanchum wilfordii ameliorates hypertension and endothelial
dysfunction in rats fed with high fat/cholesterol diets.
Immunopharmacol Immunotoxicol. 34:4–11. 2012. View Article : Google Scholar
|
|
18
|
Koo HJ, Sohn EH, Pyo S, Woo HG, Park DW,
Ham YM, Jang SA, Park SY and Kang SC: An ethanol root extract of
Cynanchum wilfordii containing acetophenones suppresses the
expression of VCAM-1 and ICAM-1 in TNF-α-stimulated human aortic
smooth muscle cells through the NF-κB pathway. Int J Mol Med.
35:915–924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hwang BY, Kim YH, Ro JS, Lee KS and Lee
JJ: Acetophenones from the roots of Cynanchum wilfordii H(EMSLEY).
Arch Pharm Res. 22:72–74. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang Y, Choi HG, Li Y, Park YM, Lee JH,
Kim DH, Lee JH, Son JK, Na M and Lee SH: Chemical constituents of
Cynanchum wilfordii and the chemotaxonomy of two species of the
family Asclepiadacease, C. wilfordii and C. auriculatum. Arch Pharm
Res. 34:2021–2027. 2011. View Article : Google Scholar
|
|
21
|
Lee MK, Yeo H, Kim J and Kim YC:
Protection of rat hepatocytes exposed to CCl4 in-vitro by
cynandione A, a biacetophenone from Cynanchum wilfordii. J Pharm
Pharmacol. 52:341–345. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang SB, Lee SM, Park JH, Lee TH, Baek NI,
Park HJ, Lee H and Kim J: Cynandione A from Cynanchum wilfordii
attenuates the production of inflammatory mediators in LPS-induced
BV-2 microglial cells via NF-κB inactivation. Biol Pharm Bull.
37:1390–1396. 2014. View Article : Google Scholar
|
|
23
|
Kim SH, Lee TH, Lee SM, Park JH, Park KH,
Jung M, Jung H, Mohamed MA, Baek NI, Chung IS and Kim J: Cynandione
A attenuates lipopolysaccharide-induced production of inflammatory
mediators via MAPK inhibition and NF-κB inactivation in RAW264.7
macrophages and protects mice against endotoxin shock. Exp Biol
Med. 240:946–954. 2015. View Article : Google Scholar
|
|
24
|
Park KH, Lee TH, Kim CW and Kim J:
Enhancement of CCL15 expression and monocyte adhesion to
endothelial cells (ECs) after hypoxia/reoxygenation and induction
of ICAM-1 expression by CCL15 via the JAK2/STAT3 pathway in ECs. J
Immunol. 190:6550–6558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen CC, Rosenbloom CL, Anderson DC and
Manning AM: Selective inhibition of E-selectin, vascular cell
adhesion molecule-1, and intercellular adhesion molecule-1
expression by inhibitors of I kappa B-alpha phosphorylation. J
Immunol. 155:3538–3545. 1995.PubMed/NCBI
|
|
26
|
Choi JH, Yoo JY, Kim SO, Yoo SE and Oh GT:
KR-31543 reduces the production of proinflammatory molecules in
human endothelial cells and monocytes and attenuates
atherosclerosis in mouse model. Exp Mol Med. 44:733–739. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weglarz L, Dzierzewicz Z, Skop B, Orchel
A, Parfiniewicz B, Wiśniowska B, Swiatkowska L and Wilczok T:
Desulfovibrio desulfuricans lipopolysaccharides induce endothelial
cell IL-6 and IL-8 secretion and E-selectin and VCAM-1 expression.
Cell Mol Biol Lett. 8:991–1003. 2003.PubMed/NCBI
|
|
28
|
Makó V, Czúcz J, Weiszhár Z, Herczenik E,
Matkó J, Prohászka Z and Cervenak L: Proinflammatory activation
pattern of human umbilical vein endothelial cells induced by IL-1β,
TNF-α, and LPS. Cytometry A. 77:962–970. 2010. View Article : Google Scholar
|
|
29
|
Fernández-Pisonero I, Dueñas AI, Barreiro
O, Montero O, Sánchez-Madrid F and García-Rodríguez C:
Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce
inflammatory molecules and leukocyte adhesion in endothelial cells.
J Immunol. 189:5402–5410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lukacs NW, Strieter RM, Elner V, Evanoff
HL, Burdick MD and Kunkel SL: Production of chemokines,
interleukin-8 and monocyte chemoattractant protein-1, during
monocyte: Endothelial cell interactions. Blood. 86:2767–2773.
1995.PubMed/NCBI
|
|
31
|
Madonna R, Massaro M and De Caterina R:
Insulin potentiates cytokine-induced VCAM-1 expression in human
endothelial cells. Biochim Biophys Acta. 1782:511–516. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ming X, Ding M, Zhai B, Xiao L, Piao T and
Liu M: Biochanin A inhibits lipopolysaccharide-induced inflammation
in human umbilical vein endothelial cells. Life Sci. 136:36–41.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lush CW, Cepinskas G and Kvietys PR: LPS
tolerance in human endothelial cells: Reduced PMN adhesion,
E-selectin expression, and NF-kappaB mobilization. Am J Physiol
Heart Circ Physiol. 278:H853–H861. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung MR, Lee TH, Bang MH, Kim H, Son Y,
Chung DK and Kim J: Suppression of thymus- and activation-regulated
chemo-kine (TARC/CCL17) production by 3-O-β-D-glucopyanosylsp
inasterol via blocking NF-κB and STAT1 signaling pathways in TNF-α
and IFN-γ-induced HaCaT keratinocytes. Biochem Biophys Res Commun.
427:236–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee TH, Jung M, Bang MH, Chung DK and Kim
J: Inhibitory effects of a spinasterol glycoside on
lipopolysaccharide-induced production of nitric oxide and
proinflammatory cytokines via down-regulating MAP kinase pathways
and NF-κB activation in RAW264.7 macrophage cells. Int
Immunopharmacol. 13:264–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee CH, Reid YA, Jong JS and Kang YH:
Lipopolysaccharide-induced differential cell surface expression of
intercellular adhesion molecule-1 in cultured human umbilical cord
vein endothelial cells. Shock. 3:96–101. 1995. View Article : Google Scholar : PubMed/NCBI
|