Introduction
Kawasaki disease (KD) is an acute, systemic
vasculitis that is the leading cause of acquired heart disease in
children in the developed world (1). Vascular inflammation ultimately
leads to coronary artery abnormalities (CAA), including aneurysm,
myocardial fibrosis, and aortic root dilatation (2,3).
Within the first 10 days after onset of fever, CAA develops in 25%
of untreated patients and in 3–5% of patients treated with
intravenous immunoglobulin (IVIG) (4). Histological examination of tissues
from KD autopsies revealed significant vascular lesions. Regressive
changes, such as degeneration, desquamation or necrosis, were
observed in the endothelial cells of the damaged artery (5). Endothelial cell injuries appear to
cause edematous and inflammatory cell infiltration in the
subendothelial space, which eventually leads to coronary artery
anomalies (6,7). A follow-up study of KD demonstrated
that a subset of patients had persistent endothelial injury many
years following KD, both with and without CAA (8). Much effort has been spent on the
study of the biological mechanism of KD, and the clinical events of
arteritis are well-characterized (9). However, the molecular mechanisms
underlying the development of KD remain poorly understood.
MicroRNAs (miRNAs) are a class of endogenous, small,
noncoding RNAs that control gene expression either by inducing
transcript degradation or by blocking translation (10). Recently, miRNAs have been reported
to be present in human plasma in a remarkably stable form, and to
be involved in the regulation of diverse developmental and
pathological processes (11,12). Previous studies have demonstrated
that miRNAs act as new promising diagnostic and prognostic
biomarkers for various diseases, including KD. Yun et al
(13) and Shimizu et al
(14) demonstrated that miRNAs
were significantly upregulated in the sera or peripheral blood of
patients with KD compared with healthy controls. They concluded
that the possible pathways of these miRNAs predominantly involved
inflammatory responses and the transforming growth factor (TGF)-β
pathway. Rowley et al (15) used a custom quantitative nuclease
protection assay to reveal a set of miRNAs that appear to be
dysregulated in KD coronary arteries. Rong et al (16) reported that miR-92a had a
relatively high degree of sensitivity and specificity in predicting
the incidence of coronary artery disease. It is worth mentioning
that He et al (17)
demonstrated that the Kruppel-like factor 4/miR-483/connective
tissue growth factor pathway may be useful in the evaluation of the
pro-inflammatory and pro-endothelial-to-mesenchymal transition
status of KD patients. Recently, Chu et al (18) determined the role of blood miR-223
in KD and KD-induced injuries in vascular endothelial cells, as
well as the mechanisms involved.
In our previous study, differentially expressed
miRNAs were profiled in KD serum (18). The results of the previous study
demonstrated that miR-186 is among the highly expressed miRNAs in
the serum of a patient with KD. Recent studies on miR-186 have
focused on its role in carcinogenesis, by regulating cell
proliferation, apoptosis and metastasis (19,20). However, very little is known about
the role and underlying mechanism of miR-186 in KD. In the present
study, miR-186 was demonstrated to be enriched in acute KD serum
compared with healthy controls and febrile controls, and its
expression was decreased to normal levels in serum from
convalescent KD (3 weeks following fever onset). Overexpression of
miR-186 mimic promoted apoptosis in human umbilical vein
endothelial cells (HUVECs). Furthermore, miR-186 overex-pression
promoted mitogen-activated protein kinase (MAPK) activation in
HUVECs by targeting and inhibiting SMAD family member 6 (SMAD6). By
contrast, miR-186 inhibitor limited the effect of KD serum on HUVEC
apoptosis. The present results suggested that miR-186 present in KD
serum may act as a treatment target for the disease.
Materials and methods
Subjects
The demographic and clinical characteristics of the
subjects involved in the present study are presented in Table I. All patients diagnosed with KD
had fever for at least three days and met at least four of the five
clinical criteria for KD (rash, conjunctival injection, cervical
lymphadenopathy, oral mucosal changes, and changes in the
extremities), or three of the five criteria along with coronary
artery abnormalities documented by echocardiogram (21). Patients diagnosed with acute upper
respiratory infection and herpangina, which mimic many of the
clinical and laboratory findings in acute KD, were prospectively
enrolled as febrile control subjects. In the present study, the
sera of 21 acute KD children, 25 healthy controls, 17 febrile
children and 11 convalescent KD children were collected from the
second affiliated Hospital of WenZhou Medical University (WenZhou,
China) between May 2014 and May 2016. All serum samples were stored
at −80°C until usage. The study protocol was approved by The Second
Affiliated Hospital of WenZhou Medical University Ethical
Committee.
 | Table ICharacteristics of the clinical index
independent validation cohort in different groups. |
Table I
Characteristics of the clinical index
independent validation cohort in different groups.
| Groups | Healthy
control | Febrile
children | Convalescent
KD | Acute KD |
|---|
| Number | 25 | 17 | 11 | 21 |
| Male | 17 (68%) | 10 (59%) | 6 (55%) | 16 (76%) |
| Female | 8 (32%) | 7 (41%) | 5 (45%) | 5 (24%) |
| Age (months) | 50.4±28.35 | 49.27±32.78 | 30.68±27.18 | 32.95±16.18 |
| WBC
(×109/l) | 7.98±2.47 | 10.86±6.04 | 8.78±3.18 | 16.16±6.33 |
| PLT
(×109) | 331.24±76.75 | 262.4±68.1 | 529.18±212.96 | 378.47±102.88 |
| CRP (mg/l) | md | 20.85±18.04 | 3.95±2.94 | 78.61±57.41 |
| ALT (U/l) | 16.11±9.62 | 15.47±7.95 | 100.1±131.6 | 87.13±116.15 |
| AST (U/l) | 33.33±9.8 | 33.4±10.74 | 47.9±21.83 | 37.5±43.4 |
| NT-proBNP
(pg/ml) | md | md | 179.73±133.4 | 1,657.2±3,268 |
| D-dimmer
(μg/ml) | md | md | 0.9±0.46 | 1.41±0.86 |
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were
purchased from American Type Culture Collection (Manassas, VA, USA)
and cultured in Roswell Park Memorial Institute 1640 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% (vol/vol) fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Cells were maintained at 37°C
in a humidified atmosphere with 5% CO2. HUVECs were
transiently transfected with miR-186 mimics, miR-186 inhibitors or
negative control oligonucleotides using Lipofectamine 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. RNA oligos were chemically synthesized
and purified by Ribo Bio Co., Ltd. (Guangzhou, China). The sequence
of human miR-186 mimic was 5′-CAAAGAAUUCUCCUUUUGGGC U-3′. The
sequence of the human miR-186 inhibitor was
5′-GUUUCUUAAGAGGAAAACCCGA-3′. The negative control oligonucleotide
for the miRNA mimic was 5′-UUUGUACUACACAAAAGUACUG-3′. Cells were
treated for the indicated time with TGF-β1 (5 ng/ml) after 24 h of
transfection with miR-186 mimic (10 ng/ml). In other experiments,
cells were treated for 24 h with healthy control serum, KD serum
alone, or KD serum with miR-186 inhibitor (30 ng/ml).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from 200 μl serum
specimen or cells using TRIzol reagent (Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The extracted RNA
was reverse transcribed into cDNA using ReverTra Ace qPCR RT kit
(Toyobo Co., Ltd., Osaka, Japan). qPCR was performed using Power
SYBR-Green PCR Master Mix (Thermo Fisher Scientific, Inc.) on an
Bio-Rad CFX 96 Real-Time PCR System (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Briefly, samples were incubated at 95°C for 10
min for an initial denaturation, followed by 40 PCR cycles of at
95°C for 15 sec and then 60°C for 1 min. The specificity of SYBR
Green based PCR reaction was validated by post-amplification
melting curve analysis. For cell samples, U6 was used as an
internal control for miRNA template normalization and GADPH was
used for other template normalizations. For serum samples, the
internal control was a spiked-in control (cel-miR-39). Fluorescent
signals were normalized to the internal reference, and the
threshold cycle was set within the exponential phase of the PCR.
miRNA and mRNA expression levels were determined using the
2−ΔΔCT method (22).
The RT primer and forward and reverse primer pairs for miR-186 were
designed by Generay Biotech Co., Ltd. (Shanghai, China). Primer
sequences are listed in Table
II.
| Table IIPrimers used in the present
study. |
Table II
Primers used in the present
study.
| Name | Primer sequence
(5′-3′) |
|---|
| U6 RT |
AACGCTTCACGAATTTGCGT |
| U6 F |
CTCGCTTCGGCAGCACA |
| U6 R |
GTGCAGGGTCCGAGGT |
| GAPDH F |
AACTCTGGTAAAGTGGATATTG |
| GAPDH R |
GGTGGAATCATATTGGAACA |
| Cel-miR-39 RT |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAAGCT |
| Cel-miR-39 F |
GCCGCTCACCGGGTGTAAATC |
| Cel-miR-39 R |
GTGCAGGGTCCGAGGT |
| SMAD6 F |
CTGGAGTTGTTGAGCAGCC |
| SMAD6 R |
GTGCGTCTTTCTTGTTTTGTCC |
| SMAD6(Luci)WT
F |
AAACTAGTGCACTTTGGCTTATAATTCTTTCAATACAG |
| SMAD6(Luci)WT
R |
GGAAGCTTCAAACCCAGGCTTTTCCACC |
| SMAD6(Luci)Mut
F |
AAACTAGTGCACTTTGGCTTATATAAGAATCAATACAG |
| SMAD6(Luci)Mut
R |
GGAAGCTTCAAACCCAGGCTTT TCCACC |
| miR-186 RT |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCCCA |
| miR-186 F |
GCCGCCAAAGAATTCTCCTTT |
| miR-186 R |
GTGCAGGGTCCGAGGT |
Western blotting
The cells were lysed with RIPA buffer containing
protease and phosphatase inhibitors (complete ULTRA tablets; Roche
Molecular Diagnostics, Branchburg, NJ, USA). Following
centrifugation (12,000 × g, 4°C, 30 min), the supernatant was
collected and quantified. The proteins were quantified using a BCA
Kit (Beyotime Institute of Biotechnology, Jiangsu, China). A total
of 30 μg of protein was loaded per lane in the 10% gel and
separated by SDS-PAGE, then transferred to nitrocellulose
membranes. After blocking with 5% non-fat milk at room temperature
for 2 h, the membranes were probed with antibodies against SMAD6
(cat. no. sc-7004; Santa Cruz Biotechnology, Dallas, TX, USA), TNF
receptor-associated factor 6 (TRAF6; cat. no. ab40675; Abcam,
Cambridge, MA, USA), phosphorylated (p-) TGF-β-activated kinase 1
(TAK1)/total TAK1 (cat. no. 5206, cat. no. 9339; Cell Signaling
Technology, Danvers, MA, USA), p-P38/total P38 MAPK (cat. no. 4511,
cat. no. 8690; Cell Signaling Technology), p-c-Jun N-terminal
kinase (JNK)/total JNK (cat. no. 4668, cat. no. 9252; Cell
Signaling Technology), BCL2 associated X (cat. no. 2772, Bax; Cell
Signaling Technology), BCL2 apoptosis regulator (cat. no. 3498,
Bcl-2; Cell Signaling Technology), or cleaved Caspase-3 (cat. no.
9664, Cell Signaling Technology) overnight at 4°C. Membranes were
then incubated with a fluorescence-labeled secondary antibody at
room temperature for 2 h. GAPDH antibody (cat. no. 5174, Cell
Signaling Technology) was used as a loading control. Secondary
antibodies included HRP-conjugated against rabbit (cat. no. 7074,
Cell Signaling Technology, Inc.) and Rabbit anti-Goat peroxidase
conjugated H+L (cat. no. BL004A, Zhihong Biotechnology Co., Ltd.,
Hefei, China). Immunoblots were visualized using Biorad ChemiDoc
XRS+ with Image Lab Software (version 5.2.1, Hangzhou Baocheng
Biotechnology Co., Ltd., Hangzhou, China). Densitometry of Western
blots was quantified with Image Lab software (version 5.2.1,
Hangzhou Baocheng Biotechnology Co., Ltd.).
Luciferase reporter assay
A luciferase reporter construct was generated by
cloning the human SMAD6 mRNA sequence into the pMIR-Report vector
(Ambion; Thermo Fisher Scientific, Inc.). Wild-type or mutant SMAD6
mRNA fragments were amplified and cloned into the luciferase
reporter via SpeI and HindIII sites. All the primers
are listed in Table II. HUVECs
plated in a 48-well plate were co-transfected with 10 nM miRNA
mimics or negative control oligonucleotides, 50 ng of firefly
luciferase reporter and 10 ng of pRL-TK (Promega Corporation,
Madison, WI, USA) using the JetPRIME reagent
(Polyplus-transfection). Cells were collected 36 h after last
transfection and analyzed using Dual-Luciferase Reporter Assay
System (Promega Corporation).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
TUNEL staining was used to detect DNA fragmentation
of individual cells using a TUNEL fluorescence fluorescein
isothiocyanate (FITC) kit (Roche Diagnostics, Indianapolis, IN,
USA). HUVECs grown on coverslips were fixed with 4%
paraformaldehyde at 37°C for 1 h followed by permeabilization with
0.1% Triton X-100. Then, cells were incubated with TUNEL reaction
mixture at 37°C for 1 h. The stained cells were examined under a
fluorescence microscope.
Statistical analysis
Results are presented as the mean ± standard error
of the mean. Differences among groups were analyzed using one-away
analysis of variance accompanied with Turkey multiple-comparisons
test. Two-tailed Student's t-test was used for comparison between
two groups. Statistical analyses were performed with GraphPad Prism
version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
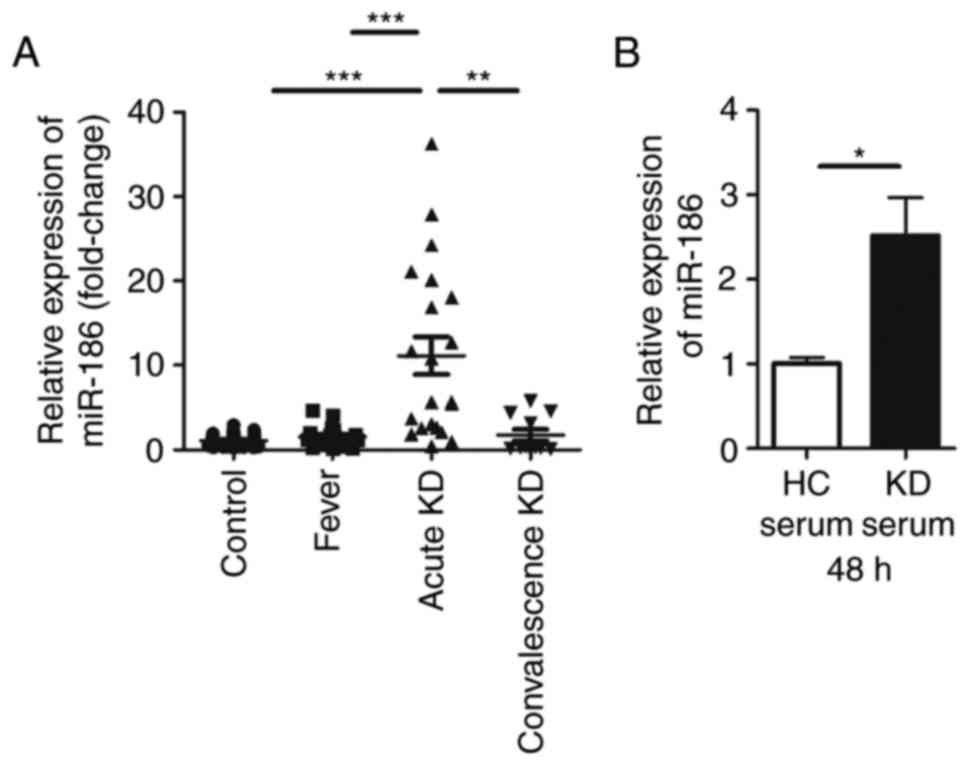
miR-186 is upregulated in KD serum and in
HUVECs stimulated with KD serum
Although our previous study revealed that miR-186
was differentially expressed in KD serum compared with healthy
children (18), the function of
miR-186 in KD remained unclear. In the present study, RT-qPCR was
used to confirm that miR-186 was significantly upregulated in KD
serum compared with serum from healthy children as well as serum
from febrile children (Fig. 1A).
The results demonstrated that the expression of miR-186 in the KD
serum was ~10-fold higher compared with healthy controls and
febrile children. In serum sampled from children with convalescence
KD, miR-186 exhibited a tendency of decreasing back to normal
levels (Fig. 1A).
KD is an acute rash autoimmune disease, primarily
affecting blood vessels and inducing systemic vasculitis (1). As previously reported, endothelial
damage is the first step in the development of coronary artery
aneurysm during the course of KD (6,21).
Since previous studies of KD have reported that HUVECs can be used
to study the vascular endothelium (23,24), these cells were used as the in
vitro model in the present study. In order to examine whether
KD serum could increase miR-186 in endothelial cells, HUVECs were
cultured with 20% KD serum or 20% healthy control (HC) serum. To
prevent the immune response of these sera, they were inactivated in
a 56°C water bath for 30 min prior to experiments. miR-186 levels
were then determined in HUVECs 48 h following HC or KD serum
incubation. A significant increase of miR-186 levels was identified
in cells cultured with KD serum, but not in cells cultured with HC
serum (Fig. 1B). As a result, it
was speculated that miR-186 could affect endothelial cells in the
pathological process of KD.
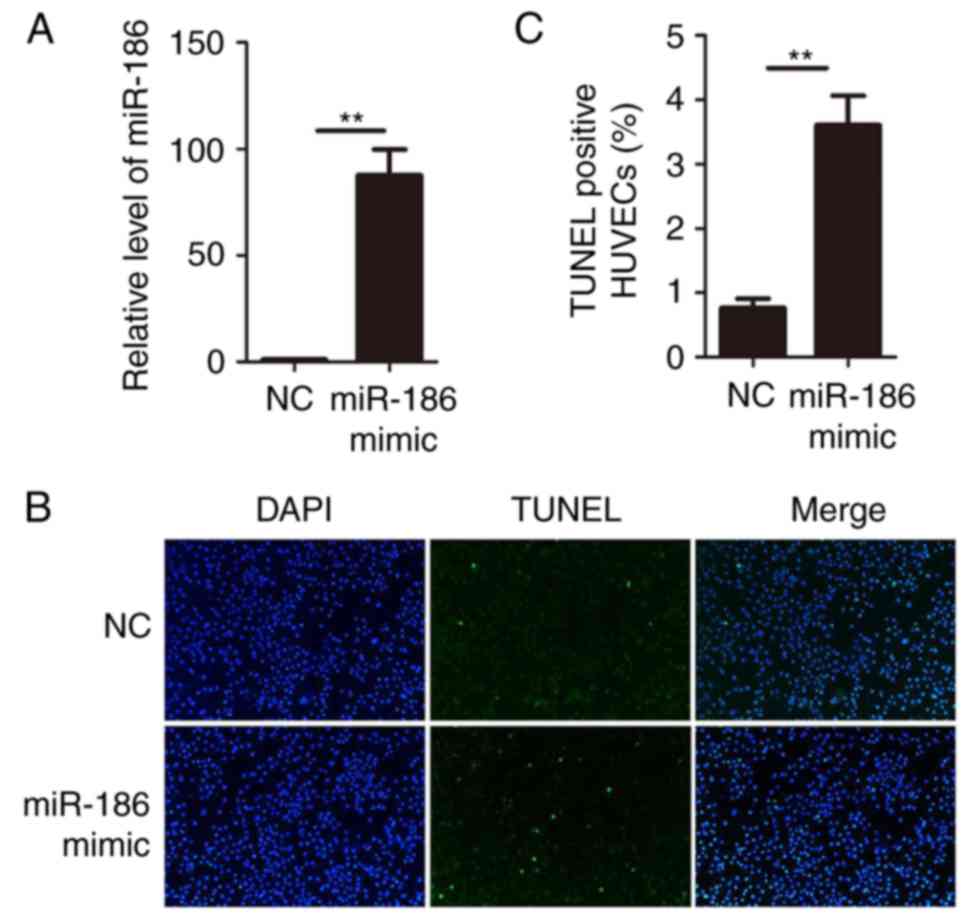
Overexpression of miR-186 induces HUVEC
apoptosis
Since miR-186 was upregulated in KD serum and KD
serum could upregulate miR-186 in HUVECs, gain-of-function studies
were performed in HUVECs. miR-186 mimics or negative control
oligonucleotides were transiently transfected into HUVECs.
Expression of miR-186 was determined by RT-qPCR. The results
confirmed that transfection of miR-186 mimic significantly
increased its expression in HUVECs (Fig. 2A). The functional role of miR-186
was then examined on HUVECs by performing TUNEL assays.
Overexpression of miR-186 in HUVECs resulted in a significant
increase of cell apoptosis compared with the negative control
oligonucleotide transfection (Fig. 2B
and C). These data suggested that miR-186 modulated HUVEC
viability and that overexpression of miR-186 promoted HUVEC
apoptosis.
miR-186 directly targets SMAD6
To understand the mechanisms by which miR-186
induces endothelial cell apoptosis, several miRNA target prediction
algorithms, including Microcosm (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/)
(25), starBase (http://starbase.sysu.edu.cn/) (26), Pictar (http://pictar.mdcberlin.de/) (27) and TargetScan (http://www.targetscan.org/) (12) were used to identify the potential
target genes of miR-186. A total of 14 target genes were predicted
by all four prediction algorithms used in the present study
(Fig. 3A; Table III). Among these genes, SMAD6
was also demonstrated to be targeted by miR-186 from experimental
data in the DIANA-TarBase v7.0 database (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/)
(28), and therefore we focused
on SMAD6. To determine whether SMAD6 was a genuine target of
miR-186, a set of functional experiments was performed. First,
miR-186 mimic was transfected into HUVECs, and SMAD6 expression was
determined at both the mRNA and protein levels. The expression of
SMAD6 was downregulated following miR-186 mimic overexpression in
HUVECs (Fig. 3B and C). To
further confirm that miR-186 could directly bind to SMAD6 and
inhibit its expression, a luciferase reporter plasmid was generated
with wild-type or mutant sequence of the SMAD6 predicted mRNA
target fragment (Fig. 3D). The
reporter constructs were co-transfected with miR-186 mimics or
negative control oligonucleotides into HUVECs for 48 h, and then
luciferase activity was measured in the transfected cells. The
results confirmed that the reporter construct with wild-type
targeting sequence of SMAD6 mRNA caused a significant decrease in
luciferase activity in cells transfected with miR-186, whereas the
reporter construct with mutant sequence of SMAD6 produced no change
in luciferase activity (Fig. 3E).
These results suggested that miR-186 could bind to SMAD6 directly
and inhibit its expression.
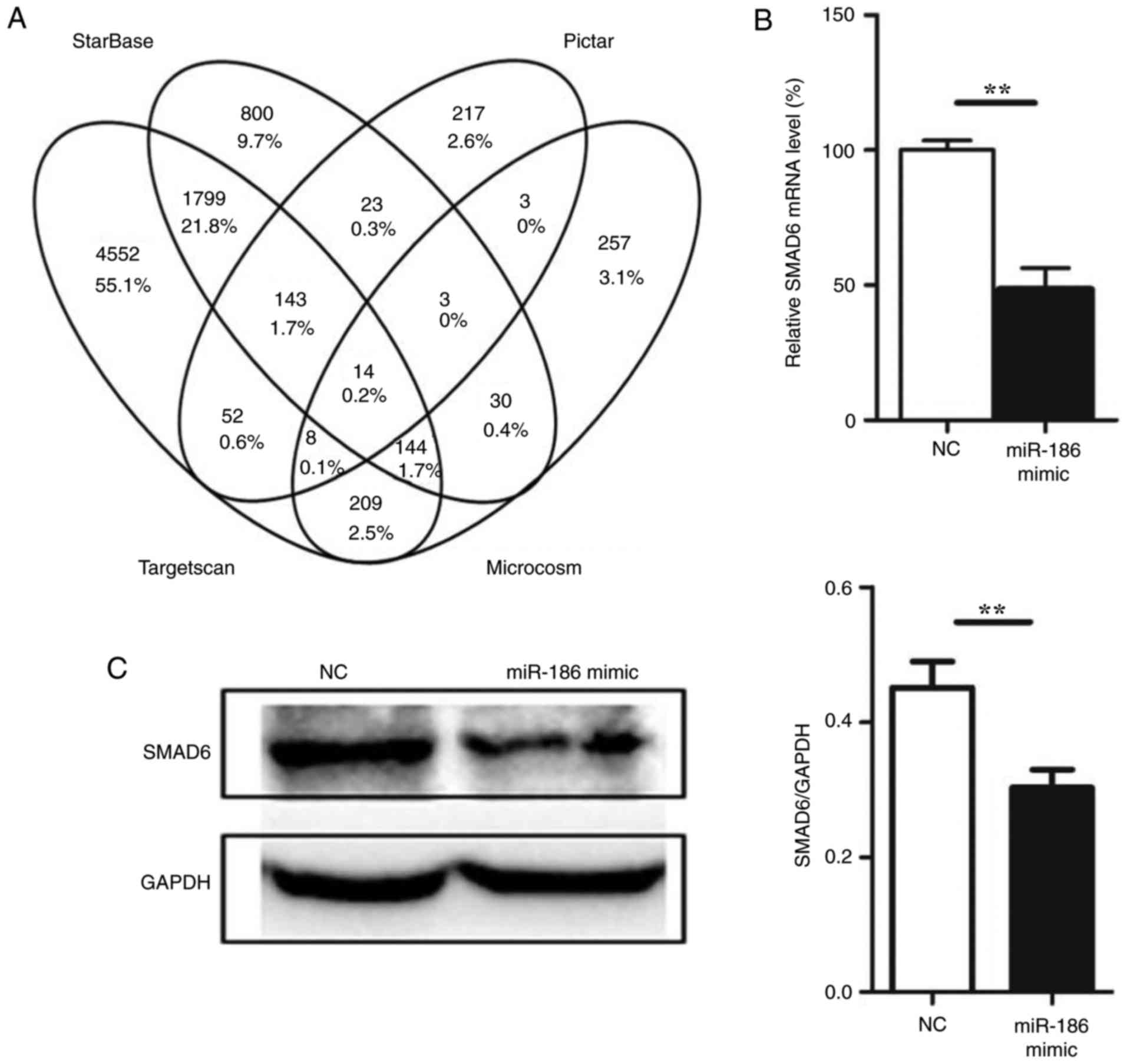 | Figure 3miR-186 directly targets SMAD6. (A)
Venn diagram of the results from the gene target prediction
algorithms. (B) Effect of miR-186 mimic overexpression on the
endogenous SMAD6 mRNA levels (n=3). (C) Effect of miR-186 mimic
overexpression on the endogenous SMAD6 protein levels (n=3). (D)
Schematic of the predicted miR-186 binding sequence on the SMAD6
mRNA 3′-UTR. Luciferase reporter construct was made by cloning the
human SMAD6 mRNA sequence into pMIR-Report construct. Wild-type or
mutant SMAD6 mRNA fragments (from 2,769 to 2,792) were amplified
and cloned into the luciferase reporter via SpeI and
HindIII sites. (E) HUVECs were co-transfected with the
reporter constructs bearing the wild-type and mutant SMAD6
sequences as indicated in Fig.
3D, and with miR-186 mimics or negative control
oligonucleotides. After 36 h, firefly luciferase activity was
measured and normalized to Renilla luciferase activity. Data are
presented as the mean ± standard error of the mean.
**P<0.01, and ***P<0.001, with
comparisons indicated by lines. SMAD6, SMAD family member 6; UTR,
untranslated region; HUVECs, human umbilical vein endothelial
cells; NC, negative control; WT, wild-type; MUT, mutant. |
| Table IIIList of the 14 predicted target genes
for miR-186. |
Table III
List of the 14 predicted target genes
for miR-186.
| Gene name | Gene Ensembl
ID |
|---|
| PSPH |
ENSG00000146733 |
| YY1 |
ENSG00000100811 |
| PABPC3 |
ENSG00000151846 |
| DNM3 |
ENSG00000197959 |
| CNTNAP1 |
ENSG00000108797 |
| PNN |
ENSG00000100941 |
| AHCYL1 |
ENSG00000168710 |
| EP300 |
ENSG00000100393 |
| NASP |
ENSG00000132780 |
| PUM2 |
ENSG00000055917 |
| SMAD6 |
ENSG00000137834 |
| RBM26 |
ENSG00000139746 |
| CETN2 |
ENSG00000147400 |
| GPBP1 |
ENSG00000062194 |
miR-186 enhances apoptosis in HUVECs by
targeting SMAD6
The results demonstrated that overexpression of
miR-186 induced apoptosis in HUVECs, and that SMAD6 was a direct
target of miR-186. Therefore, it was hypothesized that miR-186 may
affect the apoptosis of vascular endothelial cells through SMAD6.
It has been reported that TGF stimulation induces cell apoptosis
through the MAPK signaling pathway, while SMAD6 negatively
regulates the TGF/MAPK signaling pathway (29,30). Therefore, in the present study,
first TGF-β1 was used to induce cell apoptosis, and then the
expression of SMAD6 was suppressed by miR-186. As expected,
compared with the TGF-β1-induced group, miR-186 significantly
enhanced TGF-β1-mediated cell apoptosis in HUVECs (Fig. 4A and B). In addition, miR-186
overexpression significantly enhanced the activation of the
TGF/MAPK signaling pathways in HUVECs (Fig. 4C). Taken together, these results
implied that miR-186 promoted the activation of MAPK signaling by
targeting the inhibition of SMAD6, thus inducing endothelial cell
apoptosis.
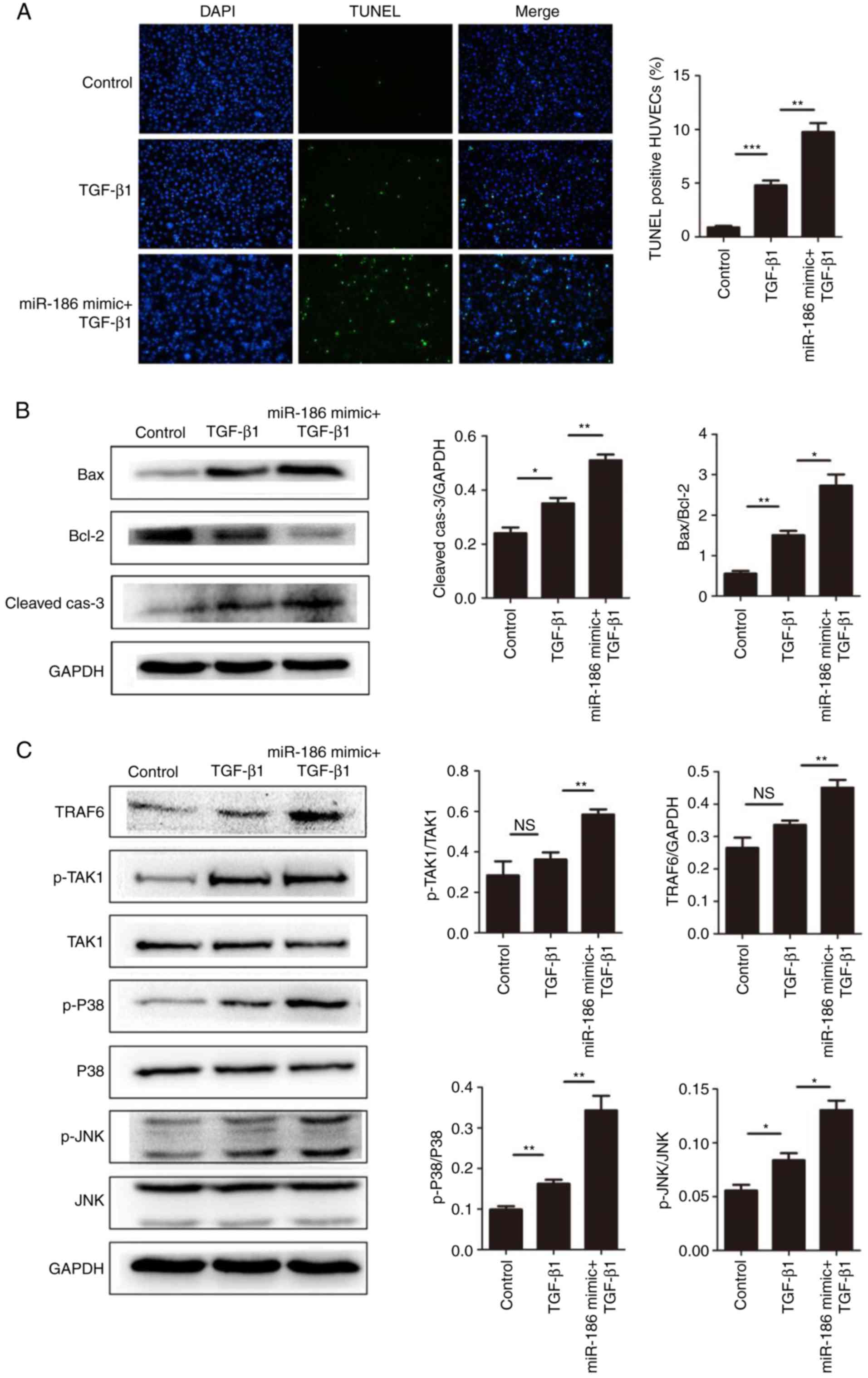 | Figure 4miR-186 overexpression enhances
TGF-β1-mediated apoptosis in HUVECs. (A) The effect of miR-186
overexpression on HUVEC apoptosis induced by TGF-β1 (5 ng/ml) was
measured by TUNEL assay. Representative images are shown at ×100
magnification and quantification of % of TUNEL + cells were
calculated in 10 random fields per slide (n=4). (B) miR-186
overexpression increased the TGF-β1-mediated expression of
apoptosis-related proteins. Representative blots and quantification
is shown. The expression levels of Bax/Bcl-2 in the TGF-β1 group
were higher compared with the control group (P=0.02), but in the
TGF-β1 + miR-186 mimic transfection group were higher compared with
the TGF-β1 alone group (P=0.006). The expression levels of
cleaved-caspase-3 (normalized to GAPDH) in the TGF-β1 group were
higher compared with the control group (P=0.023), but in the TGF-β1
+ miR-186 mimic transfection group were higher compared with the
TGF-β1 alone group (P=0.004). (C) Expression levels of MAPK
pathway-related proteins were detected by western blotting in
HUVECs induced by TGF-β1 with or without miR-186 mimic
transfection. Compared with the TGF-β1 alone group, TGF-β1 +
miR-186 mimic transfection significantly activated the MAPK pathway
(P<0.05). Data are presented as the mean ± standard error of the
mean. *P<0.05, **P<0.01, and
***P<0.001, with comparisons indicated by lines. TGF,
transforming growth factor; HUVECs, human umbilical vein
endothelial cells; TUNEL, terminal deoxynucleotidyl transferase
dUTP nick end labeling; Bcl-2, BCL2 apoptosis regulator; Bax, BCL2
associated X; MAPK, mitogen-activated protein kinase; p,
phosphorylated; TRAF6, TNF receptor-associated factor 6; TAK1,
TGF-β-activated kinase 1; JNK, c-Jun N-terminal kinase. |
miR-186 has a role in KD-induced
endothelial cell injury
The present study demonstrated that miR-186 was
increased in HUVECs following culture with KD serum. It was also
confirmed that miR-186 could promote HUVEC apoptosis. To
demonstrate whether KD serum mediates endothelial cell apoptosis
through miR-186, HUVECs were cultured with 20% serum either from
patients with KD or healthy controls (the sera were from the
patients listed in Table I). The
results demonstrated that KD serum induced HUVEC apoptosis
(Fig. 5A and B). However, KD
serum-induced apoptosis was significantly inhibited by addition of
the miR-186 inhibitor (30 nmol/l; Fig. 5A and B). In addition, in KD
serum-treated HUVECs, the SMAD6 protein levels were decreased, and
this effect was significantly reversed by addition of the miR-186
inhibitor (Fig. 5C). Similarly,
that the results demonstrated that KD serum could induce activation
of the TGF-β/MAPK pathway in HUVECs, whereas addition of the
miR-186 inhibitor significantly reversed the TGF-β/MAPK pathway
activation (Fig. 5D). The present
results suggested that miR-186 in the serum of patients with KD may
have an essential role in endothelial apoptosis by activating MAPK
signaling through targeting of the SMAD6 gene.
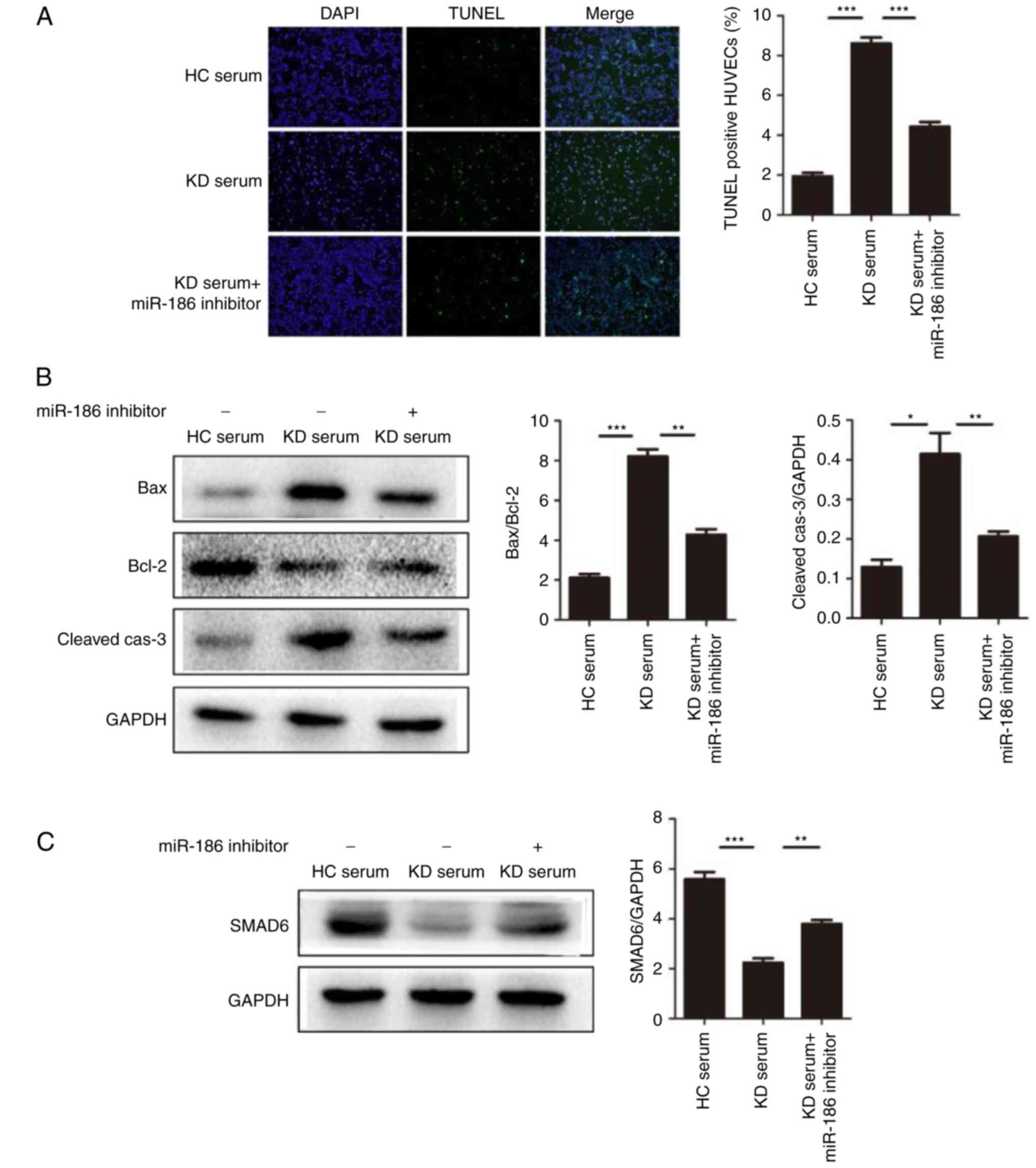 | Figure 5miR-186 promotes cell apoptosis
induced by KD serum. (A) HUVEC apoptosis was induced by culture
with KD serum and the effect of miR-186 was examined by the
addition of the miR-186 inhibitor (30 nmol/l; n=3). Representative
TUNEL images (magnification, ×100) are shown. (B) Representative
images and quantification from western blot analysis of the
expression levels of apoptosis-related proteins. KD serum promoted
the expression of apoptosis proteins, such as Bax/Bcl-2
(P=0.000013) and cleaved-caspase-3 (P=0.00221), while these effects
were significantly blocked by the miR-186 inhibitor (P<0.05).
(C) Representative images and quantification from western blot
analysis of the SMAD6 protein expression levels in HUVECs induced
by KD serum with (P=0.005023) or without (P=0.000079) miR-186
inhibitor. (D) KD serum alone activated the MAPK pathway, while KD
serum with miR-186 inhibitor transfection significantly prevented
this effect (P<0.05). Data are presented as the mean ± standard
error of the mean. *P<0.05, **P<0.01,
and ***P<0.001, with comparisons indicated by lines.
KD, Kawasaki disease; HUVECs, human umbilical vein endothelial
cells; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end
labeling; Bcl-2, BCL2 apoptosis regulator; Bax, BCL2 associated X;
SMAD6, SMAD family member 6; MAPK, mitogen-activated protein
kinase; HC, healthy control; p, phosphorylated; TRAF6, TNF receptor
associated factor 6; TAK1, TGF-β-activated kinase 1; JNK, c-Jun
N-terminal kinase. |
Discussion
The present study demonstrated that overexpression
of miR-186 may be a promoting factor for endothelial injury in KD.
The results revealed that miR-186 was upregulated in KD serum and
KD serum could increase miR-186 levels in HUVECs. Overexpression of
miR-186 in HUVECs induced apoptosis, and the underlying mechanism
likely involved the repression of SMAD6 and the subsequent
activation of the TGF/MAPK pathway. In addition, the results
demonstrated that KD serum induced HUVEC apoptosis through miR-186.
The present study provides evidence that miR-186 may be a
therapeutic target for KD.
miR-186 has an important role in a variety of
diseases, especially in cancer. Hua et al (31) reported that miR-186 inhibited cell
proliferation through targeting the oncogene GOLPH3 in prostate
cancer. Liu et al (32)
demonstrated that ectopic expression of miR-186 significantly
inhibited cell growth in multiple myeloma. Notably, Wang et
al (33) reported that
circulating miR-186 could be considered a promising novel
diagnostic biomarkers for the early phase of acute myocardial
infarction. In the present study, miR-186 was demonstrated to
inhibit expression of SMAD6 and activate MAPK signaling in HUVECs.
In summary, the present results determined a new mechanism for
miR-186 participating in the pathological process of KD.
KD is an important febrile illness causing
multi-system vasculitis in childhood. The mechanisms involved in
the pathogenesis of KD are not clearly understood and confirmatory
diagnosis has not been established. Endothelial injury, an early
risk marker of cardiovascular diseases, refers to loss of normal
homeostatic function in blood vessels, and is charac-terized by
inflammatory activity (34,35). In KD, the long-term cardiovascular
outcome for pediatric survivors is of important concern.
Accelerated coronary atherosclerosis in KD-related lesions
occurring in young adults has been reported and results in acute
coronary syndrome or sudden death (36). Multiple studies exist on
KD-associated endothelial injury; for example, the levels of blood
cell-derived miR-223 in endothelial cells may act as a novel
endocrine genetic signal and may participate in vascular injury of
KD (18), while miR-125a-5p has
been demonstrated to induce apoptosis in HUVECs through direct
inhibition of MAP kinase kinase 7 levels resulting in Caspase-3
activation (37). However, the
specific mechanisms of KD-induced endothelial injury remain
unclear. The present study revealed that aberrantly expressed
miR-186 in KD serum could induce endothelial cell apoptosis, which
suggested that miR-186 could be the effector for impaired
endothelium in KD and may be a new target for KD treatment.
TGF-β is a multifunctional peptide that regulates
proliferation, differentiation, apoptosis, and migration in many
cell types (38).
TGF-β-associated signal transduction involves SMAD-dependent and
SMAD-independent pathways. The MAPK family of proteins consists of
extracellular signal-regulated kinase (ERK), JNK/stress-activated
protein kinase (SAPK) and p38 MAPK. The TGF-β/MAPK pathway has been
demonstrated to be involved in many processes in various diseases,
including epithelial-myofibroblast trans-differentiation (39,40), inflammatory (41) and apoptotic processes (42,43). In endothelial cells, TGF-β1
inhibits endothelial cell proliferation and migration (44), and induces endothelial cell
apoptosis by inhibiting expression of the anti-apoptotic protein
Bcl-2 and activating p38 MAPK (45). In KD, a genetic association study
has established that polymorphisms in the TGF-β pathway influence
disease susceptibility and coronary artery aneurysm formation
(46). In addition, the TGF-β
related genes SMAD3, TGFB2, and TGFBR2 were observed to have a
significant effect on KD susceptibility, severity, or treatment
response. A later study provided more evidence on the role of the
TGF-β/SMAD3 signaling pathway in the arteritis of KD (47). In the present study, miR-186 was
determined to directly target SMAD6 which then negatively regulated
the TGF-β/MAPK signaling pathway.
In conclusion, the present study confirmed that
miR-186 was upregulated in serum from patients with acute KD and
miR-186 upregulation appeared to be highly specific to the acute
phase of KD. Furthermore, miR-186 was demonstrated to regulate the
expression of SMAD6, which then negatively regulated TGF-β
signaling, resulting in feedback activation of the MAPK pathway to
induce endothelial cell injury. These results suggest that miR-186
might be used as a target of KD treatment.
Acknowledgments
This study was supported by grants from the Zhejiang
Provincial Medical and Health Science and Technology plan (grant
no. WKJ-ZJ-1725), the Zhejiang Provincial Medical and Health
Science and Technology plan (grant no. 2016KYB197), the Scientific
Research Foundation of Wenzhou (grant no. Y20150015), and the
Zhejiang Provincial Natural Science Foundation of China (grant no.
Q15H020015).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Newburger JW, Takahashi M and Burns JC:
Kawasaki Disease. J Am Coll Cardiol. 67:1738–1749. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gordon JB, Kahn AM and Burns JC: When
children with Kawasaki disease grow up: Myocardial and vascular
complications in adulthood. J Am Coll Cardiol. 54:1911–1920. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kato H, Sugimura T, Akagi T, Sato N,
Hashino K, Maeno Y, Kazue T, Eto G and Yamakawa R: Long-term
consequences of Kawasaki disease. A 10- to 21-year follow-up study
of 594 patients. Circulation. 94:1379–1385. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Newburger JW, Takahashi M, Beiser AS,
Burns JC, Bastian J, Chung KJ, Colan SD, Duffy CE, Fulton DR, Glode
MP, et al: A single intravenous infusion of gamma globulin as
compared with four infusions in the treatment of acute Kawasaki
syndrome. N Engl J Med. 324:1633–1639. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leung DY, Cotran RS, Kurt-Jones E, Burns
JC, Newburger JW and Pober JS: Endothelial cell activation and high
interleukin-1 secretion in the pathogenesis of acute Kawasaki
disease. Lancet. 2:1298–1302. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amano S, Hazama F and Hamashima Y:
Pathology of Kawasaki disease: I. Pathology and morphogenesis of
the vascular changes. Jpn Circ J. 43:633–643. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arkin A: A Clinical and pathological study
of periarteritis nodosa: A report of five cases, one histologically
healed. Am J Pathol. 6:401–426.5. 1930.PubMed/NCBI
|
|
8
|
Shah V, Christov G, Mukasa T, Brogan KS,
Wade A, Eleftheriou D, Levin M, Tulloh RM, Almeida B, Dillon MJ, et
al: Cardiovascular status after Kawasaki disease in the UK. Heart.
101:1646–1655. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsuda E, Hamaoka K, Suzuki H, Sakazaki H,
Murakami Y, Nakagawa M, Takasugi H and Yoshibayashi M: A survey of
the 3-decade outcome for patients with giant aneurysms caused by
Kawasaki disease. Am Heart J. 167:249–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang M: MicroRNA: A new entrance to the
broad paradigm of systems molecular medicine. Physiol Genomics.
38:113–115. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lagos-Quintana M, Rauhut R, Lendeckel W
and Tuschl T: Identification of novel genes coding for small
expressed RNAs. Science. 294:853–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yun KW, Lee JY, Yun SW, Lim IS and Choi
ES: Elevated serum level of microRNA (miRNA)-200c and miRNA-371-5p
in children with Kawasaki disease. Pediatr Cardiol. 35:745–752.
2014. View Article : Google Scholar
|
|
14
|
Shimizu C, Kim J, Stepanowsky P, Trinh C,
Lau HD, Akers JC, Chen C, Kanegaye JT, Tremoulet A, Ohno-Machado L
and Burns JC: Differential expression of miR-145 in children with
Kawasaki disease. PLoS One. 8:e581592013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rowley AH, Pink AJ, Reindel R, Innocentini
N, Baker SC, Shulman ST and Kim KY: A study of cardiovascular miRNA
biomarkers for Kawasaki disease. Pediatr Infect Dis J.
33:1296–1299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rong X, Jia L, Hong L, Pan L, Xue X, Zhang
C, Lu J, Jin Z, Qiu H, Wu R and Chu M: Serum miR-92a-3p as a new
potential biomarker for diagnosis of Kawasaki disease with coronary
artery lesions. J Cardiovasc Transl Res. 10:1–8. 2017. View Article : Google Scholar
|
|
17
|
He M, Chen Z, Martin M, Zhang J, Sangwung
P, Woo B, Tremoulet AH, Shimizu C, Jain MK, Burns JC and Shyy JY:
miR-483 targeting of CTGF suppresses endothelial-to-mesenchymal
transition: Therapeutic implications in Kawasaki disease. Circ Res.
120:354–365. 2017. View Article : Google Scholar :
|
|
18
|
Chu M, Wu R, Qin S, Hua W, Shan Z, Rong X,
Zeng J, Hong L, Sun Y, Liu Y, et al: Bone marrow-derived
microRNA-223 works as an endocrine genetic signal in vascular
endothelial cells and participates in vascular injury from Kawasaki
disease. J Am Heart Assoc. 6:pii: e0048782017. View Article : Google Scholar
|
|
19
|
Cao C, Sun D, Zhang L and Song L: miR-186
affects the proliferation, invasion and migration of human gastric
cancer by inhibition of Twist1. Oncotarget. 7:79956–79963. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong Y, Jin X, Sun Z, Zhao Y and Song X:
MiR-186 inhibited migration of NSCLC via targeting cdc42 and
effecting EMT process. Mol Cells. 40:195–201. 2017.PubMed/NCBI
|
|
21
|
McCrindle BW, Rowley AH, Newburger JW,
Burns JC, Bolger AF, Gewitz M, Baker AL, Jackson MA, Takahashi M,
Shah PB, et al: Diagnosis, treatment, and long-term management of
Kawasaki disease: A scientific statement for health professionals
from the American heart association. Circulation. 135:e927–e999.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
23
|
Higashi K, Terai M, Hamada H, Honda T,
Kanazawa M and Kohno Y: Impairment of angiogenic activity in the
serum from patients with coronary aneurysms due to Kawasaki
disease. Circ J. 71:1052–1059. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian J, Lv HT, An XJ, Ling N and Xu F:
Endothelial microparticles induce vascular endothelial cell injury
in children with Kawasaki disease. Eur Rev Med Pharmacol Sci.
20:1814–1818. 2016.PubMed/NCBI
|
|
25
|
He JH, Han ZP, Zou MX, Wang L, Lv YB, Zhou
JB, Cao MR and Li YG: Analyzing the LncRNA, miRNA, and mRNA
regulatory network in prostate cancer with bioinformatics software.
J Comput Biol. 2017.Epub ahead of print. PubMed/NCBI
|
|
26
|
Yang JH, Li JH, Shao P, Zhou H, Chen YQ
and Qu LH: starBase: A database for exploring microRNA-mRNA
interaction maps from Argonaute CLIP-Seq and Degradome-Seq data.
Nucleic Acids Res. 39:D202–D209. 2011. View Article : Google Scholar
|
|
27
|
Krek A, Grün D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky N: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
Indexing more than half a million experimentally supported miRNA:
mRNA interactions. Nucleic Acids Res. 43:D153–D159. 2015.
View Article : Google Scholar
|
|
29
|
Jung SM, Lee JH, Park J, Oh YS, Lee SK,
Park JS, Lee YS, Kim JH, Lee JY, Bae YS, et al: Smad6 inhibits
non-canonical TGF-β1 signalling by recruiting the deubiquitinase
A20 to TRAF6. Nat Commun. 4:25622013. View Article : Google Scholar
|
|
30
|
Lee MK, Pardoux C, Hall MC, Lee PS,
Warburton D, Qing J, Smith SM and Derynck R: TGF-beta activates Erk
MAP kinase signalling through direct phosphorylation of ShcA. EMBO
J. 26:3957–3967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hua X, Xiao Y, Pan W, Li M, Huang X, Liao
Z, Xian Q and Yu L: miR-186 inhibits cell proliferation of prostate
cancer by targeting GOLPH3. Am J Cancer Res. 6:1650–1660.
2016.PubMed/NCBI
|
|
32
|
Liu Z, Zhang G, Yu W, Gao N and Peng J:
miR-186 inhibits cell proliferation in multiple myeloma by
repressing Jagged1. Biochem Biophys Res Commun. 469:692–697. 2016.
View Article : Google Scholar
|
|
33
|
Wang KJ, Zhao X, Liu YZ, Zeng QT, Mao XB,
Li SN, Zhang M, Jiang C, Zhou Y, Qian C, et al: Circulating
miR-19b-3p, miR-134-5p and miR-186-5p are promising novel
biomarkers for early diagnosis of acute myocardial infarction. Cell
Physiol Biochem. 38:1015–1029. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(Suppl 1):
III27–III32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Horio E, Kadomatsu T, Miyata K, Arai Y,
Hosokawa K, Doi Y, Ninomiya T, Horiguchi H, Endo M, Tabata M, et
al: Role of endothelial cell-derived angptl2 in vascular
inflammation leading to endothelial dysfunction and atherosclerosis
progression. Arterioscler Thromb Vasc Biol. 34:790–800. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hartopo AB and Setianto BY: Coronary
artery sequel of Kawasaki disease in adulthood, a concern for
internists and cardiologists. Acta Med Indones. 45:69–75.
2013.PubMed/NCBI
|
|
37
|
Li Z, Jiang J, Tian L, Li X, Chen J, Li S,
Li C and Yang Z: A plasma mir-125a-5p as a novel biomarker for
Kawasaki disease and induces apoptosis in HUVECs. PLoS One.
12:e01754072017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ruiz-Ortega M, Rodriguez-Vita J,
Sanchez-Lopez E, Carvajal G and Egido J: TGF-beta signaling in
vascular fibrosis. Cardiovasc Res. 74:196–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu C, Chen F, Han X, Xu H and Wang Y:
Role of TGF-β1/p38 MAPK pathway in hepatitis B virus-induced
tubular epithelial-myofibroblast transdifferentiation. Int J Clin
Exp Pathol. 7:7923–7930. 2014.
|
|
40
|
Wei J, Li Z, Chen W, Ma C, Zhan F, Wu W
and Peng Y: AEG-1 participates in TGF-beta1-induced EMT through p38
MAPK activation. Cell Biol Int. 37:1016–1021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Malik S, Suchal K, Khan SI, Bhatia J,
Kishore K, Dinda AK and Arya DS: Apigenin ameliorates
streptozotocin-induced diabetic nephropathy in rats via
MAPK/NF-κB/TNF-α and TGF-β1/MAPK/fibronectin pathways. Am J Physiol
Renal Physiol. 2:F414–F422. 2017. View Article : Google Scholar
|
|
42
|
Hyman KM, Seghezzi G, Pintucci G, Stellari
G, Kim JH, Grossi EA, Galloway AC and Mignatti P: Transforming
growth factor-beta1 induces apoptosis in vascular endothelial cells
by activation of mitogen-activated protein kinase. Surgery.
132:173–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ferrari G, Terushkin V, Wolff MJ, Zhang X,
Valacca C, Poggio P, Pintucci G and Mignatti P: TGF-β1 induces
endothelial cell apoptosis by shifting VEGF activation of p38
(MAPK) from the prosurvival p38β to proapoptotic p38α. Mol Cancer
Res. 10:605–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pollman MJ, Naumovski L and Gibbons GH:
Vascular cell apoptosis: Cell type-specific modulation by
transforming growth factor-beta1 in endothelial cells versus smooth
muscle cells. Circulation. 99:2019–2026. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsukada T, Eguchi K, Migita K, Kawabe Y,
Kawakami A, Matsuoka N, Takashima H, Mizokami A and Nagataki S:
Transforming growth factor beta 1 induces apoptotic cell death in
cultured human umbilical vein endothelial cells with down-regulated
expression of bcl-2. Biochem Biophys Res Commun. 210:1076–1082.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shimizu C, Jain S, Davila S, Hibberd ML,
Lin KO, Molkara D, Frazer JR, Sun S, Baker AL, Newburger JW, et al:
Transforming growth factor-beta signaling pathway in patients with
Kawasaki disease. Circ Cardiovasc Genet. 4:16–25. 2011. View Article : Google Scholar
|
|
47
|
Shimizu C, Oharaseki T, Takahashi K,
Kottek A, Franco A and Burns JC: The role of TGF-β and
myofibroblasts in the arteritis of Kawasaki disease. Hum Pathol.
44:189–198. 2013. View Article : Google Scholar
|