Introduction
Glucocorticoids (GCs) are naturally produced steroid
hormones or synthetic compounds that are frequently used as
anti-inflammatory and immune-suppressive drugs to treat a variety
of diseases. However, there is evidence that prolonged or excessive
use of GCs is one of the leading causes of osteoporosis and
osteonecrosis (1–3). It has been demonstrated that GCs
induce fractures in 30–50%, and osteonecrosis in 9–40% of patients
receiving long-term therapy (4).
GCs not only directly suppress the osteogenic differentiation of
osteoblasts, but they also induce osteoblast and osteocyte
apoptosis (5,6). Furthermore, apoptosis of osteocytes
is the dominant mechanism of GC-induced osteoporosis (7).
Reactive oxygen species (ROS) are the derivatives of
biological aerobic metabolism, comprising hydrogen peroxide,
hydroxyl radicals and superoxide. ROS destroy and oxidize proteins,
lipids and DNA, leading to altered cell function. It is thought
that ROS are generated under various physiological conditions, but
also contribute to the pathogenesis of bone loss, including that in
osteoporosis (8–11). Autophagy is a major intracellular
degradation process, by which cytoplasmic proteins and organelles
are sequestered in double-membrane vesicles and degraded upon
fusion with lysosomes (12).
Autophagy principally acts as a form of cytoprotection via
maintaining the homeostasis of intracellular nutrients and energy.
Autophagy is associated with a large number of physiological
processes, as well as pathophysiological conditions and diseases,
including neurodegeneration and microbial infection (13).
Dexamethasone (DEX), a synthetic GC hormone, has
been identified to inhibit the synthesis of fibronectin and
collagen, and to activate collagenase synthesis. Evidence has
indicated that DEX promotes osteoblast apoptosis by activating the
expression of caspase family proteins (14). However, the involvement of ROS and
autophagy in the apoptosis of osteoblasts caused by GCs and the
exact underlying mechanisms have remained to be fully elucidated.
Therefore, the purpose of the present study was to measure the
pharmacological effect of DEX on ROS and autophagy in the
osteoblast-like MC3T3-E1 cell line. Furthermore, it was
investigated whether DEX induced apoptosis of MC3T3-E1 cells via
triggering ROS production and autophagy.
Materials and methods
Cell culture
MC3T3-E1 cells (American Type Culture Collection,
Manassas, VA, USA) were grown in Dulbecco's modified Eagle's medium
(high glucose; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and penicillin-streptomycin solution (100 U/ml
penicillin; 100 μg/ml streptomycin; Gibco; Thermo Fisher
Scientific, Inc.), in a humidified atmosphere with 5%
CO2 at 37°C.
Cell viability assay
Cell viability assessment was performed using a Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). MC3T3-E1 cells (1×104/well) were
seeded in 96-well plates, incubated overnight and then treated with
DEX at various concentrations 0.1, 1 and 10 μM for 24 h. The
absorbance values at 450 nm were measured using a microplate reader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Furthermore, in
order to assess the connection of DEX-induced autophagy with
apoptosis, 3-methyladenine (3-MA; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used as a common anti-autophagy factor.
Cells were divided into four groups, including normal control, 1
μM DEX, 5 mM 3-MA (cells cultured with 5 mM 3-MA for 24 h)
and 5 mM 3-MA + 1 μM DEX group (cells cultured with 5 mM
3-MA + 1 μM DEX for 24 h). In addition, in order to
investigate the association between ROS induced by DEX and cell
apoptosis, 10 mM N-acetylcysteine (NAC; Beyotime Institute of
Biotechnology, Haimen, China) was added to MC3T3-E1 cells treated
with DEX. NAC is a commonly used antioxidant. Cells were divided
into normal group, 1 μM DEX group, 10 mM NAC group (cells
cultured with 10 mM NAC for 24 h) and 10 mM NAC + 1 μM DEX
group (cells cultured with 10 mM NAC + 1 μM DEX for 24
h).
Analysis of cellular apoptosis
An Annexin V-fluorescein isothiocyanate (FITC)
apoptosis Detection kit (KeyGen Biotech Co., Ltd., Nanjing,
Jiangsu, China) was used to determine the level of cellular
apoptosis. MC3T3-E1 cells (4×106/well) were cultured in
6-well plates overnight and then treated as described above. In
brief, cells were suspended in 500 μl 1X binding buffer and
incubated in the dark with 5 μl Annexin V-FITC and 5
μl propidium iodide (PI) for 10 min at room temperature. A
flow cytometer (FACSCalibur; BD Biosciences, Franklin Lakes, NJ,
USA) was used to analyze cellular apoptosis.
Analysis of the cell cycle
A Cell Cycle Detection kit (KeyGen Biotech Co.,
Ltd.) was used to assess the cell cycle. Following treatment with
DEX, the MC3T3-E1 cells were trypsinized, collected and washed with
PBS. Subsequently, 70% cold ethanol was added to fix the cells for
2 h at room temperature or overnight at 4°C, and PBS was used to
wash away the fixing solution. Cells were incubated with 0.4 ml PI
containing 0.1 ml RNase A at 37°C for 30 min. Finally, cell cycle
distribution was analyzed by measuring the DNA content using a flow
cytometer.
Monodansylcadaverine (MDC) staining
Autophagy is characterized by the formation of
acidic vesicles, as described previously, and MDC is often used as
a selective fluorescent marker for autophagic vacuoles in
vivo (15). MC3T3-E1 cells
(4×106/well) were seeded onto sterile coverslips placed
in a 6-well plate and allowed to attach for 24 h. As
aforementioned, 0.05 mmol/l MDC in PBS was added to label the
autophagic vacuoles for 10 min at 37°C, and cells were washed three
times with PBS. MC3T3-E1 cells were observed under a fluorescence
microscope (Olympus BX53; Olympus Corp., Tokyo, Japan). Excitation
wavelength was 335 nm, and emission wavelength was 512 nm. Average
fluorescence intensity analysis was performed using Image-Pro Plus
v6.0 software (Media Cybernetics Inc., Rockville, MD, USA).
ROS detection
A ROS Assay kit (KeyGen Biotech Co., Ltd.) was used
for active ROS detection using the fluorescent probe
2′,7′-dichlorofluorescin diacetate (DCFH-DA), which is a type of
ROS-sensitive dye (16). The
DCFH-DA reagent must be diluted to 10 μmol/l in serum-free
medium prior to use. Following the experimental treatments, 10
μmol/l DCFH-DA was added to cells with subsequent incubation
for 30 min at 37°C in a humidified incubator, and cells were washed
three times with serum-free medium and collected. A flow cytometer
was used to quantify the relative fluorescence intensities.
Western blot analysis
Total protein was extracted from treated MC3T3-E1
cells with radioimmunoprecipitation assay lysis buffer (Tris 50 mM,
NaCl 150 mM, Nonidet P-40 1%, sodium deoxycholate 0.5% and SDS
0.1%; Beyotime Institute of Biotechnology, Haimen, China)
containing 1% phenylmethylsulfonyl fluoride (100 mM; Beyotime
Institute of Biotechnology). The lysates were centrifuged at 12,000
× g for 15 min at 4°C. Supernatants were collected and the
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology) was used to detect the protein concentration.
Equivalent amounts (25 μg) of protein were separated by 12%
SDS-PAGE and subsequently transferred onto a polyvinylidene
difluoride membrane (Merck KGaA). Membranes were blocked with 5%
non-fat milk in Tris-buffered saline with Tween-20 (0.05%) at room
temperature for 1 h and incubated overnight with primary
antibodies. Following incubation with the corresponding secondary
antibody, horseradish peroxidase-conjugated anti-rabbit
immunoglobulin G (1:10,000 dilution; cat. no. 7074P2; Cell
Signaling Technology, Inc., Danvers, MA, USA), at room temperature
for 2 h, membranes were washed three times with PBS. Finally,
according to the manufacture's protocols, the blotting proteins
were visualized with ECL plus reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Antibodies used for the western blot analysis in
the present study were as follows: Rabbit anti-activating
transcription factor 4 (ATF4; 1:1,000 dilution; cat. no. 11815),
rabbit anti-CCAAT/enhancer-binding protein homologous protein
(CHOP; 1:1,000 dilution; cat. no. 2895), rabbit anti-poly[ADP
ribose] polymerase (total PARP; 1:2,000 dilution; cat. no. 9542),
rabbit anti-caspase-3 (total caspase-3; 600 dilution; cat. no.
96621) were obtained from Cell Signaling Technology, Inc. In
addition, rabbit anti-Beclin1 (1:1,000 dilution, cat. no. ab62557),
rabbit anti-LC3B (1:1,000 dilution, cat. no. ab51520), rabbit
anti-P62 (1:1,000 dilution; cat. no. ab91526) and rabbit
anti-cyclin-dependent kinase 2 (CDK2; 1:1,000 dilution; cat. no.
ab32147) were obtained from Abcam (Cambridge, UK). Additionally,
rabbit anti-GAPDH (1:1,000 dilution; cat. no. AB-P-R 001; Hangzhou
Goodhere Biotechnology Co., Ltd., Hangzhou, China) was used as
loading control. ImageJ v1.47 software (National Institutes of
Health, Bethesda, MD, USA) was used to perform relative intensity
of each protein band analysis.
Statistical analysis
All experiments were performed three times.
Statistical analysis was performed with SPSS 19.0 statistical
software (IBM Corp., Armonk, NY, USA). One-way analysis of variance
followed by Tukey's post hoc test was used for multiple
comparisons, and Student's t-test for comparisons between two
groups. Values are expressed as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
DEX inhibits viability and enhances
apoptosis via endoplasmic reticulum (ER) stress in MC3T3-E1
cells
The effect of DEX on cell viability was evaluated
with a CCK-8 assay. As illustrated in Fig. 1A, the amount of viable MC3T3-E1
cells was significantly inhibited by DEX in a dose-dependent manner
(all P<0.01), and at 10 μM DEX, the cell viability
reached 70% of that of the control group. In addition, the cellular
apoptosis assay demonstrated that compared with that in the control
group (2.73%), the apoptotic rates in the DEX-treated groups were
7.9, 17.2 and 21.07% at DEX concentrations of 0.1, 1 and 10
μM, respectively (P<0.05, P<0.01 and P<0.01,
respectively; Fig. 1B and C).
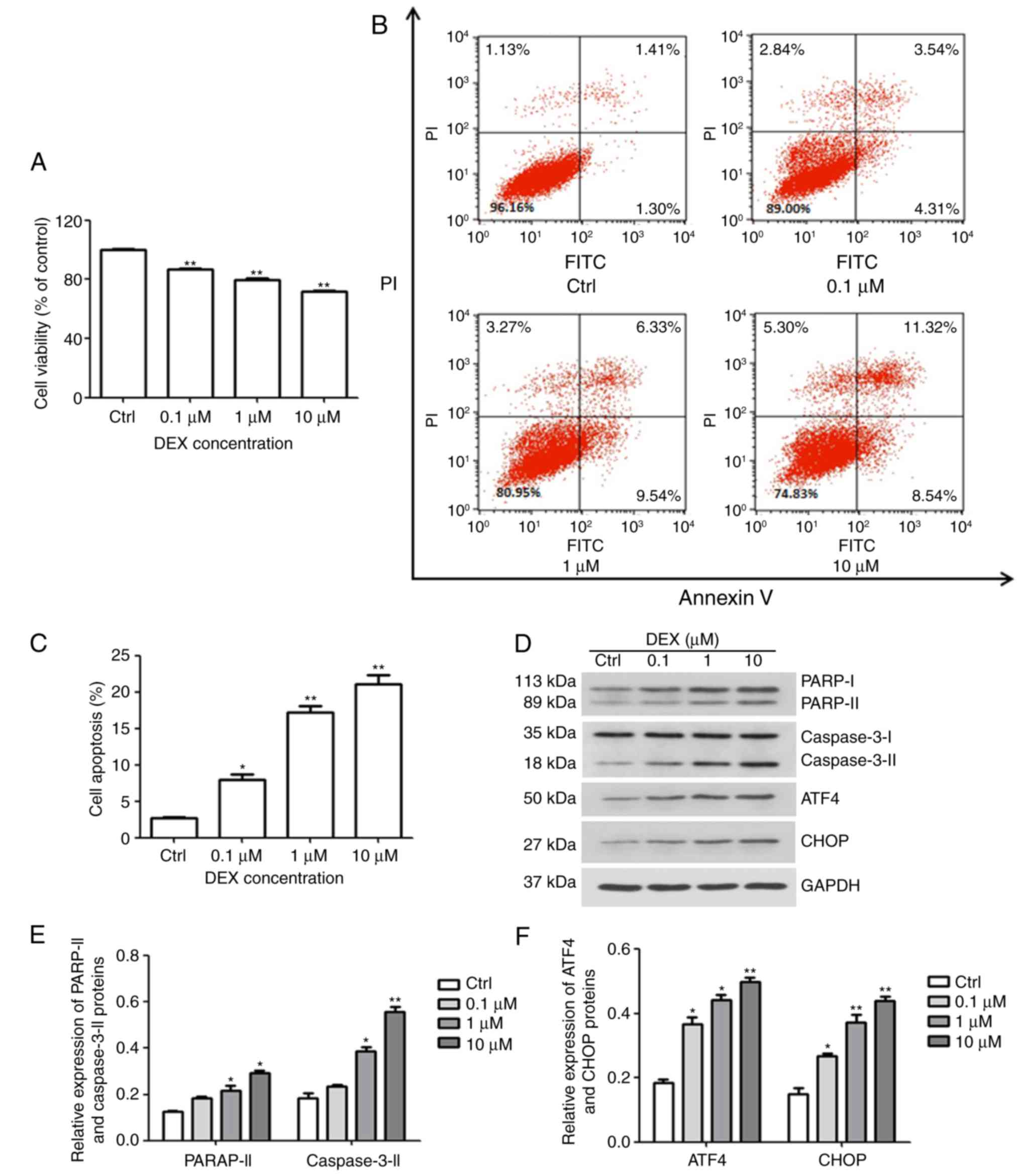 | Figure 1DEX induces ER stress and apoptosis
in MC3T3-E1 cells. (A) The viability and (B and C) apoptosis of
MC3T3-E1 cells were affected by DEX in a dose-dependent manner, as
determined by a Cell Counting Kit-8 assay and Annexin V-FITC/PI
staining, respectively. (D) Western blot analysis of ER
stress-associated proteins (ATF4 and CHOP) and apoptosis-associated
proteins (PARP and caspase-3). Values are expressed as the mean ±
standard deviation (n=3). (E and F) Data from intensities analyses
are presented as the relative expression of each protein to GAPDH.
*P<0.05 and **P<0.01 vs. Ctrl. Ctrl,
control; ATF, activating transcription factor; CHOP,
CCAAT/enhancer-binding protein homologous protein; DEX,
dexamethasone; ER, endoplasmic reticulum; PARP, poly[ADP ribose]
polymerase; PI, propidium iodide; FITC, fluorescein
isothiocyanate. |
Furthermore, western blotting was performed to
evaluate the expression of PARP and caspase-3, which are early
markers of apoptosis. As presented in Fig. 1D and E, DEX increased the
activation of caspase-3 and PARP, which further confirmed that DEX
promoted apoptosis of MC3T3-E1 cells. These results demonstrated
that DEX not only reduces cell viability, but also promotes
apoptosis. In addition, CHOP and ATF4 are key factors of ER stress.
The expression of CHOP and ATF4 was gradually enhanced with
increasing concentration of DEX (Fig.
1D and F). These results indicated that DEX may promote
apoptosis by activating ER stress.
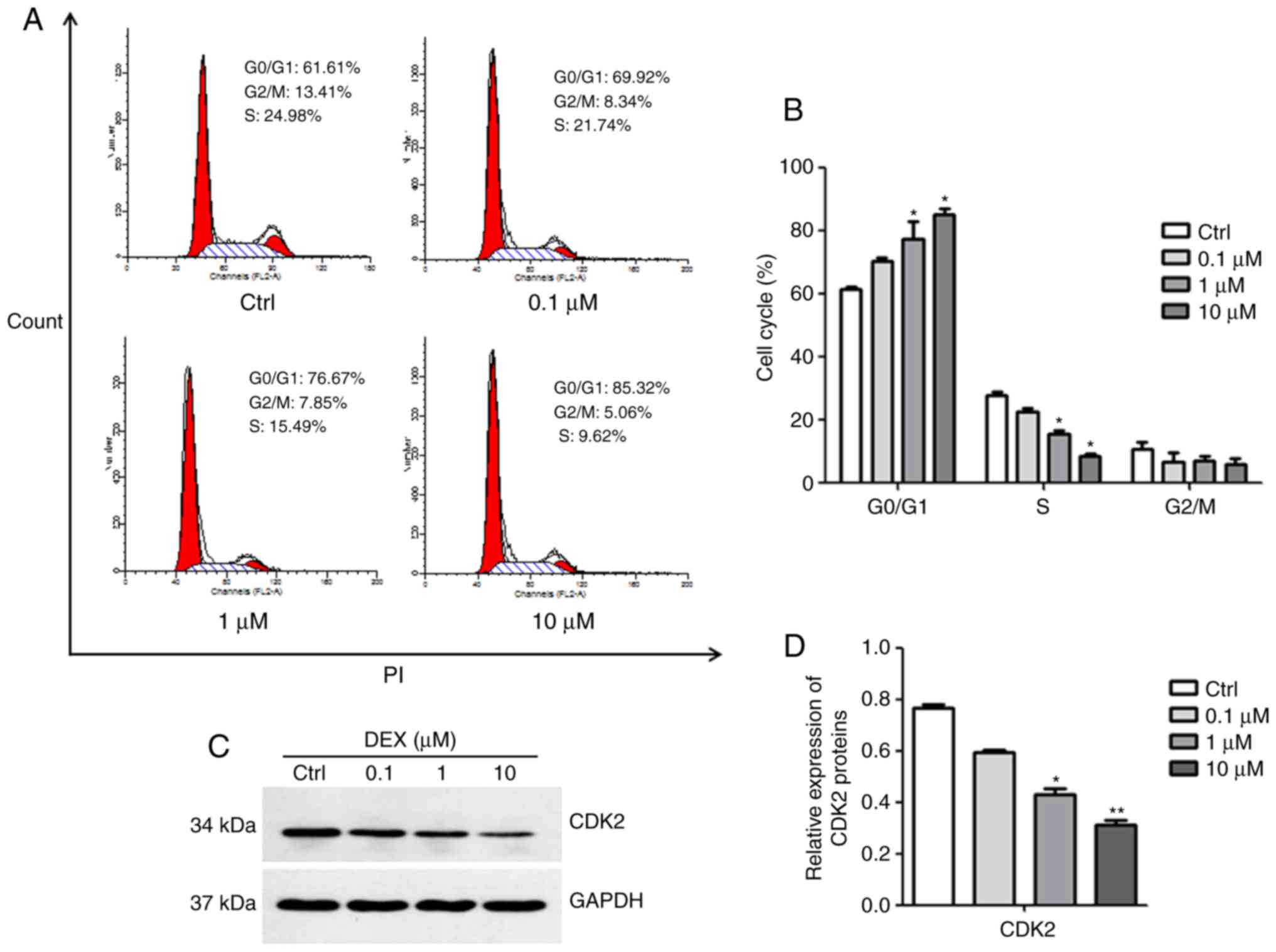
DEX causes G0/G1 arrest of MC3T3-E1
cells
The results of the cell cycle analysis are presented
in Fig. 2A and B. DEX at
concentrations of 1 and 10 μM induced a significant increase
in the number of cells in G0/G1 phase, but decreased the number of
cells in S phase, indicating that DEX inhibited cell cycle
progression (both P<0.05). Subsequently, the expression of CDK2,
which is essential for the G1/S transition, was detected. As
illustrated in Fig. 2C and D, the
expression of CDK2 was inhibited in MC3T3-E1 cells after treatment
with DEX at concentrations of 1 and 10 μM (P<0.05,
P<0.01). These results suggested that DEX may induce G0/G1
arrest in MC3T3-E1 cells due to decreased CDK2 protein
expression.
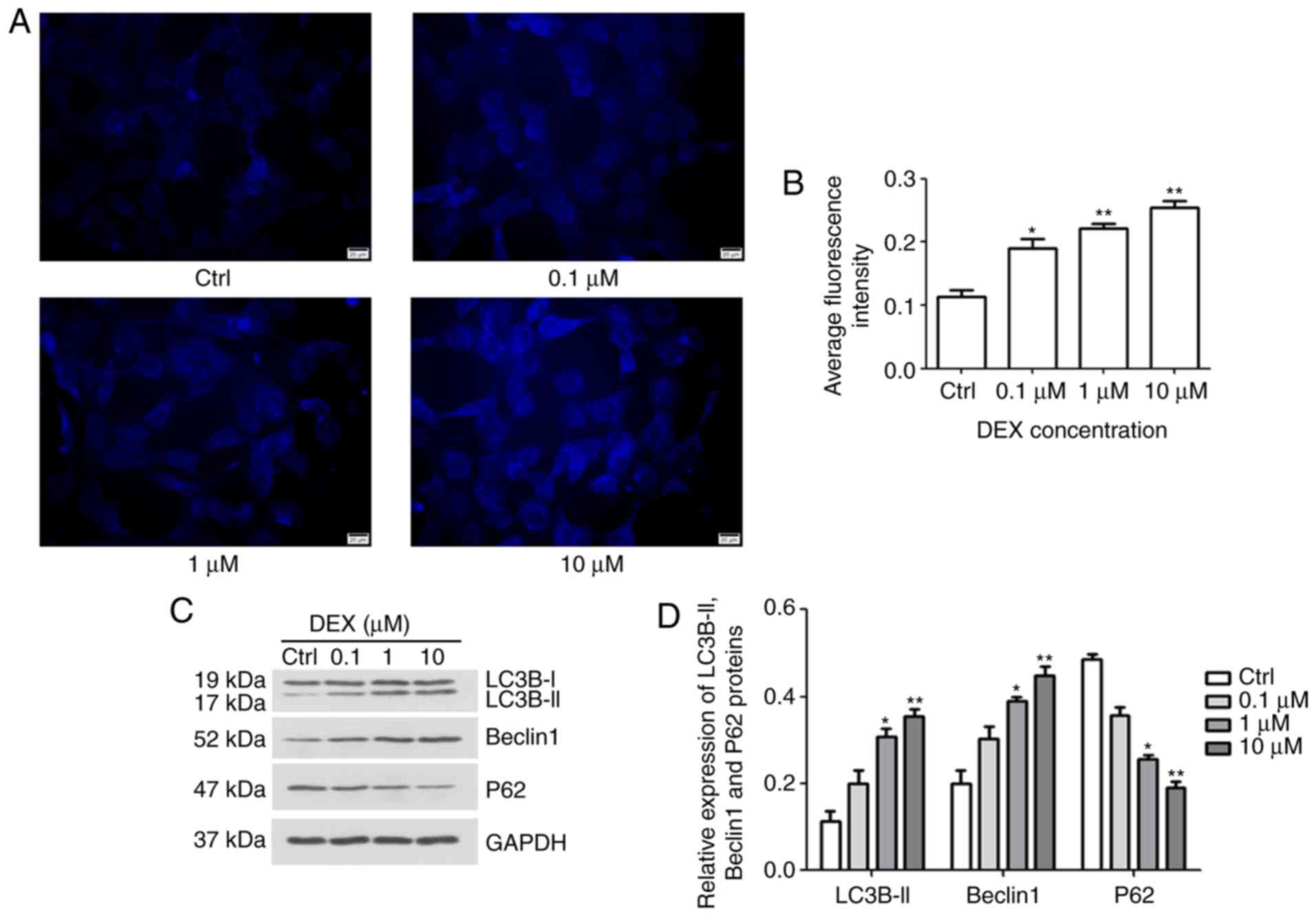
Effect of DEX on autophagy of MC3T3-E1
cells
To investigate the effect of DEX on autophagy in
MC3T3-E1 cells, MDC staining and western blotting were performed.
After treatment with DEX for 24 h, fluorescence intensity was
analyzed (Fig. 3A and B). The
average fluorescence intensities in the 0.1, 1 and 10 μM
DEX-treated groups were increased to 0.18, 0.22 and 0.25,
respectively, which were significantly different from the average
fluorescence intensity of the control group, 0.11 (P<0.05,
P<0.01 and P<0.01, respectively). Furthermore, in DEX (1
μM) and DEX (10 μM) groups, the expression levels of
autophagy markers, including LC3B-II, Beclin1 and P62, were
gradually increased, but the levels of P62 were reduced, indicating
the stimulatory role of DEX on autophagic flux (P<0.05,
P<0.01; Fig. 3C and D,
respectively). These results indicated that DEX may promote the
autophagy of MC3T3-E1 cells.
Association between autophagy and
apoptosis in MC3T3-E1 cells
3-MA inhibits autophagy by blocking autophagosome
formation via the inhibition of class III phosphoinositide-3 kinase
(17). To clarify the possible
association between autophagy and apoptosis after treatment with
DEX, the effect of 3-MA on viability and apoptosis was
investigated. MC3T3-E1 cells were treated with DEX (1 μM),
3-MA (5 mM) or 3-MA (5 mM) + DEX (1 μM). As demonstrated in
Fig. 4A, the viability of
MC3T3-E1 cells decreased to 76.5% in the presence of DEX (1
μM); however, after treatment with 3-MA (5 mM) + DEX (1
μM), the viability was significantly increased to 90.1%
(P<0.001). Furthermore, as presented in Fig. 4B and C, the apoptotic rate of
MC3T3-E1 cells treated with DEX (1 μM) was 24.3%, while it
significantly decreased to 18.56% in the 3-MA (5 mM) + DEX (1
μM) group (P<0.05). Inconsistencies in the apoptotic
proportion in the DEX (1 μM) group between Figs. 1C and Fig. 4C may be due to technical issues,
including different batches of kit. These results indicated that
3-MA reversed the DEX-induced inhibition of viability and promotion
of apoptosis in MC3T3-E1 cells.
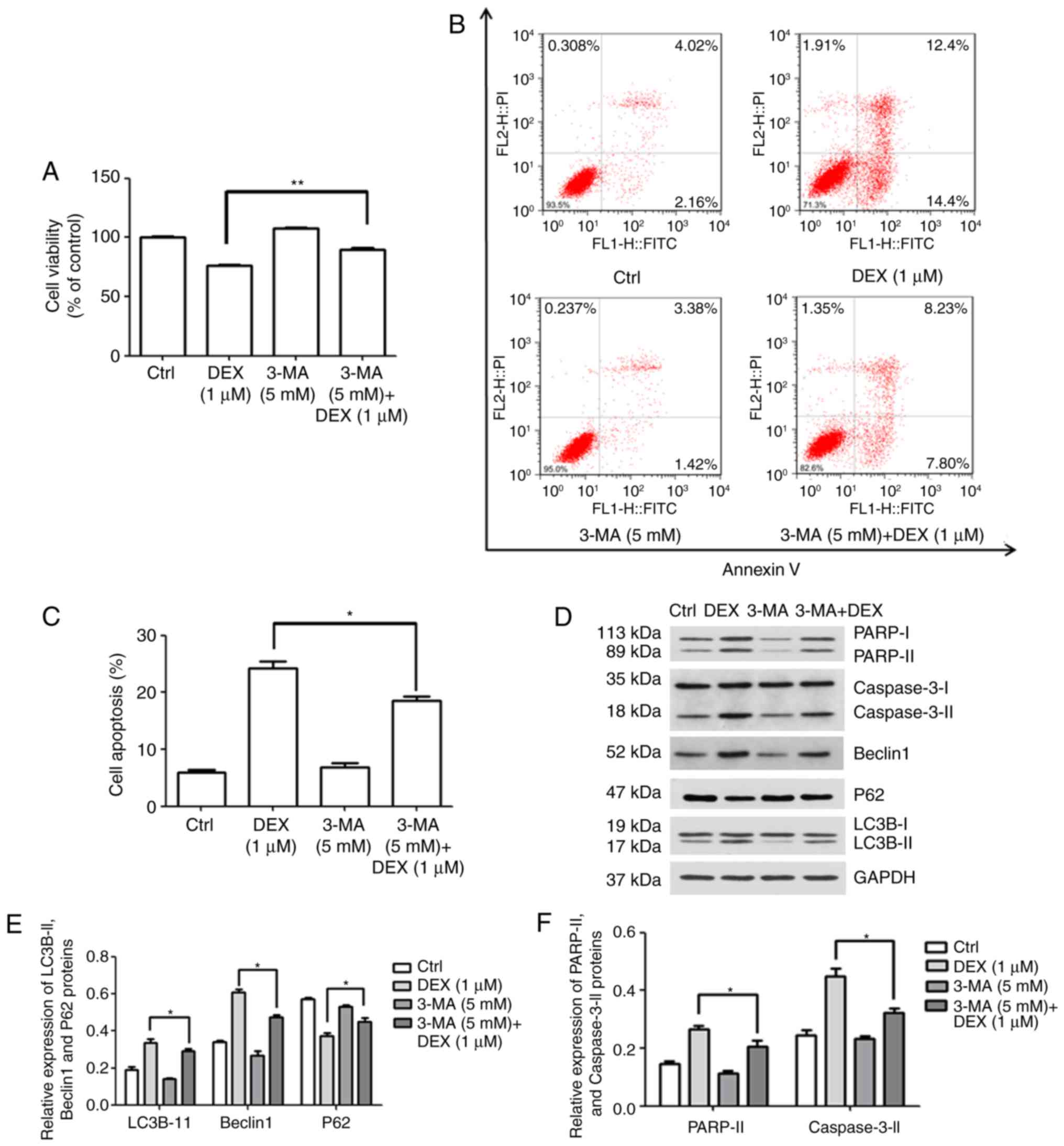 | Figure 4DEX promotes apoptosis by activating
autophagy in MC3T3-E1 cells. (A–C) MC3T3-E1 cells were treated with
DEX (1 μM), 3-MA (5 mM) or 3-MA (5 mM) + DEX (1 μM)
and (A) the viability and (B and C) the apoptotic rate were
measured using a Cell Counting Kit-8 assay. (D) Western blot
analysis of apoptosis-associated proteins (PARP and caspase-3) and
autophagy-associated proteins (P62, LC3B and Beclin1). (E and F)
The expression of each protein normalized to the GAPDH was
evaluated. Values are expressed as the mean ± standard deviation
(n=3). *P<0.05 and **P<0.01 vs. DEX (1
μM) group. Ctrl, control; ATF, activating transcription
factor; DEX, dexamethasone; MA, 3-methyladenine; LC3B,
microtubule-associated proteins 1A/1B light chain; PARP,
poly[ADP-ribose] polymerase; FITC, fluorescein isothiocyanate; PI,
propidium iodide. |
Furthermore, the expression levels of apoptosis- and
autophagy-associated proteins were detected (Fig. 4D). The expression levels of the
autophagy-associated proteins LC3B-II and Beclin1 were increased in
the DEX (1 μM) group, which was inhibited by simultaneous
treatment with 3-MA (5 mM; P<0.05; Fig. 4E). In addition, it was
demonstrated that the activation/expression of the
apoptosis-associated proteins PARP and caspase-3 was weakened after
treatment with 3-MA (5 mM) + DEX (1 μM), compared with that
in the DEX (1 μM) group (P<0.05; Fig. 4F). These results suggested that
DEX may promote MC3T3-E1 cell apoptosis by inducing autophagy.
DEX triggers ROS-dependent ER stress and
autophagy to induce apoptosis
To examine how DEX affects the production of ROS in
MC3T3-E1 cells, flow cytometry was performed to quantify the
fluorescence intensity. In this experiment, NAC was used as an
antioxidant. As illustrated in Fig.
5A and B, the mean ROS-associated fluorescence intensity in the
DEX (1 μM) group was increased compared with that in the
control group, which was significantly inhibited by simultaneous
treatment with NAC (10 mM; P<0.01). Furthermore, the cell
viability in the NAC (10 mM) + DEX (1 μM) group was 89.3% of
that in the control group, which was significantly higher than that
in the DEX (1 μM) group (77.8% of control; P<0.01;
Fig. 5C). These results
demonstrate that NAC inhibited the effect of DEX on MC3T3-E1 cell
proliferation following co-treatment. Furthermore, cellular
apoptosis was detected (Fig. 5D and
E), revealing that the apoptotic rate of MC3T3-E1 cells treated
with NAC (10 mM) + DEX (1 μM) was 17.7%, which was
significantly decreased compared with that in the DEX (1 μM)
group (25.97%; P<0.01). These results indicated that activation
of intracellular ROS was involved in the DEX-induced inhibition of
viability and the promotion of apoptosis in MC3T3-E1 cells.
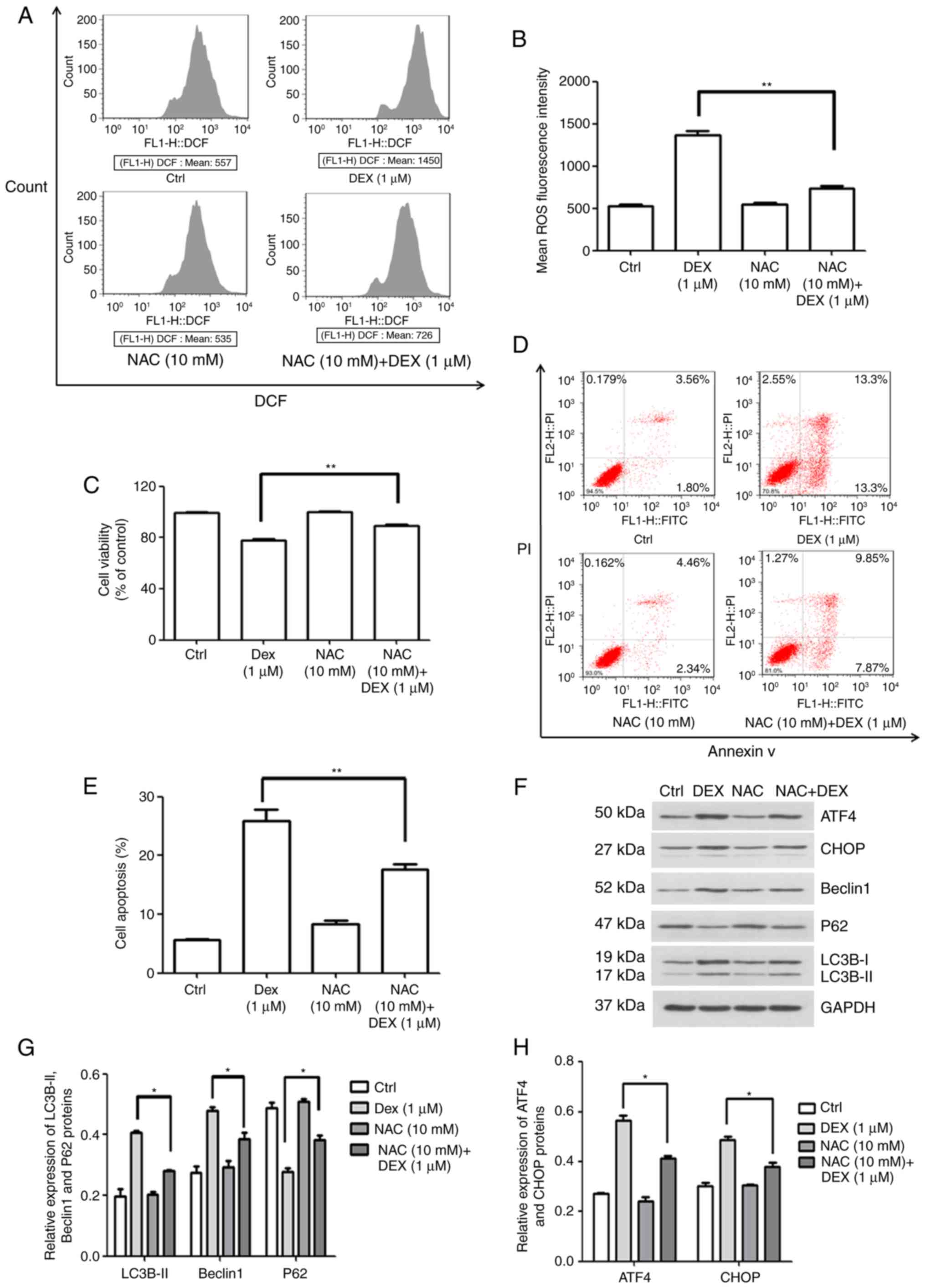 | Figure 5ROS production induced by DEX
promotes apoptosis in MC3T3-E1 cells through ER stress and
autophagy. (A and B) ROS production rate of MC3T3-E1 cells and
quantification of it, in the control, DEX (1 μM), NAC (10
mM) and NAC (10 mM) + DEX (1 μM) groups, as detected by flow
cytometry. (C) Viability and (D and E) apoptosis after treatment
with NAC. It was indicated that the activation of ROS was involved
in the inhibition of viability induced by DEX. (F) Western blot
analysis of autophagy-associated (LC3B, P62 and Beclin1) and ER
stress-associated proteins (ATF4 and CHOP). (G and H) A relative
quantification assay was performed to assess the expression of each
protein normalized to the GAPDH expression Values are expressed as
the mean ± standard deviation (n=3). *P<0.05 and
**P<0.01 vs. DEX (1 μM) group. Ctrl, control;
ATF, activating transcription factor; CHOP, CCAAT/enhancer-binding
protein homologous protein; DEX, dexamethasone; NAC,
N-acetylcysteine; ROS, reactive oxygen species; ER, endoplasmic
reticulum; DCF, dichlorofluorescin. |
Subsequently, the expression of autophagy- and ER
stress-associated proteins was quantified by western blot analysis
(Fig. 5F). As illustrated in
Fig. 5G, the protein levels of
LC3B-II and Beclin1 were decreased, but P62 expression was
increased in the NAC (10 mM) + DEX (1 μM) group when
compared with that in the DEX (1 μM) group (P<0.05). This
result indicated that DEX activated autophagy via inducing
ROS-associated stress. In addition, it was demonstrated that the
expression of ATF4 and CHOP was also inhibited by NAC, indicating
that ROS-associated stress also elevated ER stress (P<0.05;
Fig. 5H). Therefore, the effect
of DEX on cell viability, ROS stress, apoptosis and protein
expression was attenuated by NAC. These results demonstrated that
DEX promoted the production of ROS, which enhanced apoptosis
through the induction of autophagy and ER stress in MC3T3-E1
cells.
Discussion
GCs have excellent therapeutic effects and are among
the most important drugs used in the clinic (18). However, GC therapy is often
accompanied by severe side effects, including osteoporosis and
osteonecrosis (19–21). As a side effect of their clinical
applications, GCs cause bone mass loss, bone tissue degeneration
and apoptosis of osteoblasts. The precise molecular mechanisms
underlying DEX-induced apoptosis are yet to be fully elucidated. In
order to investigate whether ER stress, ROS and autophagy are
involved in this process, the production of ROS, ER stress and
autophagy were investigated, and the effects of 3-MA and NAC on
DEX-induced apoptosis in MC3T3-E1 osteoblast-like cells were
assessed. It was demonstrated that autophagy promoted apoptosis,
and that ROS induced ER stress and autophagy-mediated apoptosis.
The present study provides a theoretical foundation for the
mechanism of action of DEX, which comprises induction of apoptosis,
ER stress, ROS and autophagy in osteoblasts, and may provide an
alternative approach for molecular intervention against DEX-induced
apoptosis of MC3T3-E1 cells by inhibiting the production of ROS and
autophagy.
In the present study, MC3T3-E1 cells were used to
investigate the effect of DEX on osteoblast viability, apoptosis
and cell cycle distribution. Previous studies suggested that DEX
inhibited the viability of MC3T3-E1 cells in a dose-dependent
manner, and that a concentration of 1 μM DEX significantly
decreased the viability of MC3T3-E1 cells (22–24). Additional studies revealed that 1
μM DEX not only caused apoptosis, but also G0/G1 phase
arrest (22,23). These results demonstrated that DEX
indeed inhibited the viability, and induced apoptosis and G0/G1
phase arrest in MC3T3-E1 cells. The present results are therefore
consistent with those of previous studies. In addition, the
expression of PARP and CDK2 was detected in the present study. PARP
is a downstream effector of the caspase family of proteins in the
apoptotic pathway (25), and the
cleavage of PARP after the activation of caspases has been
identified as a biochemical hallmark of cellular apoptosis. Studies
have demonstrated that DEX induces apoptosis and the expression of
PARP in MC3T3-E1 cells simultaneously (26). CDK2 serves an important role in
cell entry and transformation through S phase, as well as G2 phase
transition to mitosis (27). The
present results indicated that DEX may promote apoptosis by
activating the cleavage of PARP and caspase-3, and that increased
CDK2 expression may arrest the cell cycle of MC3T3-E1 cells at the
G0/G1 phase.
In the present study, the impact of DEX on the
production of ROS, ER stress and autophagy in MC3T3-E1 cells was
investigated. The ER is an important organelle with a crucial role
in a variety of cellular functions, including assembly, protein
folding and Ca2+ storage. ER stress is a state in which
the homeostasis of protein folding load and the capacity of the ER
is disrupted (28,29). Excess ER stress has been
demonstrated in MC3T3-E1 cells after treatment with DEX, and has
also been proven to induce apoptosis (30,31). Autophagy is known to be a
self-degradative process that is important for balancing sources of
energy at critical times during development, and in response to
nutrient stress (32). Studies
have demonstrated that autophagy is involved in a number of general
processes, including proliferation, development, apoptosis and cell
death (33,34). LC3BII, Beclin1 and P62 are closely
associated with the formation of autophagosomes, serving as markers
of autophagy (35). Under normal
physiological conditions, the cellular antioxidant system and the
production of ROS are in a dynamic equilibrium, which has been
demonstrated to serve a crucial role in the differentiation of
osteoblasts (36). Disrupting
this balance has certain harmful effects; for instance, an
increased production of ROS may inhibit the differentiation of
osteoblasts (24,37). The results of the present study
indicated that ER stress was activated by DEX via increasing the
expression of the associated proteins ATF4 and CHOP, which is in
accordance with the study by Yang et al (30). In addition, the results of the
autophagy analysis indicated that DEX caused autophagy by
increasing the levels of Beclin1 and LC3BII, and by decreasing the
expression of P62. Furthermore, it was demonstrated that DEX
increased the production of ROS in MC3T3-E1 cells, and this result
is similar to the result described by Zhang et al (38).
In the present study, inhibition of autophagy by
co-treatment with 3-MA significantly reduced the DEX-induced
decreases in MC3T3-E1 cell viability. In addition, the protein
activities of PARP and Caspase-3 were suppressed, indicating that
DEX promoted apoptosis by activating autophagy. Of note, previous
studies have revealed that autophagy is closely correlated with
apoptosis, and that they interact in three ways, comprising
cooperation, confrontation and promotion (39–41). Previous studies have indicated
that relatively excessive accumulation of ROS serves a crucial role
in the development and progression of apoptosis (42,43), and under these conditions,
cellular autophagy may be induced by transcriptional and
post-transcriptional regulation (44). However, the association between
autophagy and apoptosis induced by DEX in MC3T3-E1 cells has rarely
been reported. The present study demonstrated that, in the
experiment with the antioxidant NAC, the effects of DEX on
viability and apoptosis were significantly suppressed,
demonstrating that DEX promoted apoptosis by activating ROS
signaling pathways in MC3T3-E1 cells. Furthermore, the expression
of autophagy-associated proteins (Beclin1, LC3B-II and P62) and ER
stress-associated proteins (ATF4 and CHOP) was detected,
demonstrating that the effects of DEX on ER stress and autophagy in
MC3T3-E1 cells were attenuated by NAC. These results indicated that
DEX increased the production of ROS, which promoted apoptosis by
activating autophagy and ER stress. Further study is required to
confirm these results.
In conclusion, the present study demonstrated that
DEX mediated ER stress, G0/G1 phase arrest, the production of ROS,
autophagy and apoptosis in MC3T3-E1 osteoblast-like cells.
Furthermore, production of ROS induced by DEX increased the level
of autophagy, leading to MC3T3-E1 cellular apoptosis. Chloroquine,
a lysosomal protease inhibitor, will be used in future studies to
confirm the results of the present study. In addition, it is
required to further elucidate the detailed mechanisms underlying
the association between autophagy and apoptosis induced by DEX in
MC3T3-E1 cells.
Abbreviations:
|
DEX
|
dexamethasone
|
|
ROS
|
reactive oxygen species
|
|
ER
|
endoplasmic reticulum
|
|
NAC
|
N-acetylcysteine
|
Acknowledgments
This study was supported by the Natural Science
Foundation of China (grant nos. 81360273, 81460331 and
81560349).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Kenanidis E, Potoupnis ME, Kakoulidis P,
Leonidou A, Sakellariou GT, Sayegh FE and Tsiridis E: Management of
glucocorticoid-induced osteoporosis: Clinical data in relation to
disease demographics, bone mineral density and fracture risk.
Expert Opin Drug Saf. 14:1035–1053. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bultink IE, Baden M and Lems WF:
Glucocorticoid-induced osteoporosis: An update on current
pharmacotherapy and future directions. Exp Opinion Pharmacotherapy.
14:185–197. 2013. View Article : Google Scholar
|
|
3
|
Weinstein RS: Clinical practice.
Glucocorticoid-induced bone disease. N Engl J Med. 365:62–70. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weinstein RS: Glucocorticoid-induced
osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am.
41:595–611. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim J, Lee H, Kang KS, Chun KH and Hwang
GS: Protective effect of Korean Red Ginseng against
glucocorticoid-induced osteoporosis in vitro and in vivo. J Ginseng
Res. 39:46–53. 2015. View Article : Google Scholar
|
|
6
|
Yun SI, Yoon HY, Jeong SY and Chung YS:
Glucocorticoid induces apoptosis of osteoblast cells through the
activation of glycogen synthase kinase 3beta. J Bone Miner Metab.
27:140–148. 2009. View Article : Google Scholar
|
|
7
|
Gu G, Hentunen TA, Nars M, Härkönen PL and
Väänänen HK: Estrogen protects primary osteocytes against
glucocorticoid-induced apoptosis. Apoptosis. 10:583–595. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arai M, Shibata Y, Pugdee K, Abiko Y and
Ogata Y: Effects of reactive oxygen species (ROS) on antioxidant
system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB
Life. 59:27–33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schröder K: NADPH oxidases in bone
homeostasis and osteoporosis. Cell Mol Life Sci. 72:25–38. 2015.
View Article : Google Scholar
|
|
10
|
Filaire E and Toumi H: Reactive oxygen
species and exercise on bone metabolism: Friend or enemy. Joint
Bone Spine. 79:341–346. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wauquier F, Leotoing L, Coxam V, Guicheux
J and Wittrant Y: Oxidative stress in bone remodelling and disease.
Trends Mol Med. 15:468–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chu CC, Chua BH, Chen Z, Landy C and Hamdy
RC: Dexamethasone induces caspase activation in murine osteoblastic
MC3T3-E1 cells. Biochim Biophys Acta. 1642:79–85. 2003. View Article : Google Scholar
|
|
15
|
Biederbick A, Kern HF and Elsässer HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
16
|
Eruslanov E and Kusmartsev S:
Identification of ROS using oxidized DCFDA and flow-cytometry.
Methods Mol Biol. 594:57–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heckmann BL, Yang X, Zhang X and Liu J:
The autophagic inhibitor 3-methyladenine potently stimulates
PKA-dependent lipolysis in adipocytes. Br J Pharmacol. 168:163–171.
2013. View Article : Google Scholar :
|
|
18
|
de Quervain D, Schwabe L and Roozendaal B:
Stress, gluco-corticoids and memory: Implications for treating
fear-related disorders. Nat Rev Neurosci. 18:7–19. 2017. View Article : Google Scholar
|
|
19
|
Fowler TW, Acevedo C, Mazur CM, Hall-Glenn
F, Fields AJ, Bale HA, Ritchie RO, Lotz JC, Vail TP and Alliston T:
Glucocorticoid suppression of osteocyte perilacunar remodeling is
associated with subchondral bone degeneration in osteonecrosis. Sci
Rep. 7:446182017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woolf AD: An update on
glucocorticoid-induced osteoporosis. Curr Opin Rheumatol.
19:370–375. 2007.PubMed/NCBI
|
|
21
|
Henneicke H, Gasparini SJ,
Brennan-Speranza TC, Zhou H and Seibel MJ: Glucocorticoids and
bone: Local effects and systemic implications. Trends Endocrinol
Metab. 25:197–211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qian Li H, Weng WX, Wu Z, Li H, Zhuang Q,
Feng B and Bian Y: Glucocorticoid receptor and sequential P53
activation by dexamethasone mediates apoptosis and cell cycle
arrest of osteoblastic MC3T3-E1 cells. PLoS One. 7:e370302012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin H, Wei B, Li G, Zheng J, Sun J, Chu J,
Zeng R and Niu Y: Sulforaphane reverses glucocorticoid-induced
apoptosis in osteoblastic cells through regulation of the Nrf2
pathway. Drug Des Devel Ther. 8:973–982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin H, Gao X, Chen G, Sun J, Chu J, Jing
K, Li P, Zeng R and Wei B: Indole-3-carbinol as inhibitors of
glucocorticoid-induced apoptosis in osteoblastic cells through
blocking ROS-mediated Nrf2 pathway. Biochem Biophys Res Commun.
460:422–427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mcilwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 7(pii): a0086562015. View Article : Google Scholar
|
|
26
|
Li J, He C, Tong W, Zou Y, Li D, Zhang C
and Xu W: Tanshinone IIA blocks dexamethasone-induced apoptosis in
osteoblasts through inhibiting Nox4-derived ROS production. Int J
Clin Exp Pathol. 8:13695–13706. 2015.
|
|
27
|
Oakes V, Wang W, Harrington B, Lee WJ,
Beamish H, Chia KM, Pinder A, Goto H, Inagaki M, Pavey S and
Gabrielli B: Cyclin A/Cdk2 regulates Cdh1 and claspin during late
S/G2 phase of the cell cycle. Cell Cycle. 13:3302–3311. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar
|
|
29
|
Bánhegyi G, Baumeister P, Benedetti A,
Dong D, Fu Y, Lee AS, Li J, Mao C, Margittai E, Ni M, et al:
Endoplasmic reticulum stress. Ann NY Acad Sci. 1113:58–71. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang J, Wu Q, Lv J and Nie H: 4-Phenyl
butyric acid prevents glucocorticoid-induced osteoblast apoptosis
by attenuating endoplasmic reticulum stress. J Bone Miner Metab.
35:366–374. 2017. View Article : Google Scholar
|
|
31
|
Sato AY, Tu X, McAndrews KA, Plotkin LI
and Bellido T: Prevention of glucocorticoid induced-apoptosis of
osteoblasts and osteocytes by protecting against endoplasmic
reticulum (ER) stress in vitro and in vivo in Female Mice. Bone.
73:60–68. 2015. View Article : Google Scholar
|
|
32
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sui Y, Yao H, Li S, Jin L, Shi P, Li Z,
Wang G, Lin S, Wu Y, Li Y, et al: Delicaflavone induces autophagic
cell death in lung cancer via Akt/mTOR/70S6K signaling pathway. J
Mol Med. 95:311–322. 2017. View Article : Google Scholar
|
|
34
|
Yang M, Wang B, Miao L, Xu X and He X:
Autophagy is involved in aldosterone-induced mesangial cell
proliferation. Mol Med Rep. 14:4638–4642. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008. View Article : Google Scholar
|
|
36
|
Pawlowska E, Wysokiński D, Tokarz P,
Piastowska-Ciesielska A, Szczepanska J and Blasiak J: Dexamethasone
and 1,25-dihy-droxyvitamin D3 reduce oxidative stress-related DNA
damage in differentiating osteoblasts. Int J Mol Sci.
15:16649–16664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang Y, Su Y, Wang D, Chen Y, Wu T, Li G,
Sun X and Cui L: Tanshinol attenuates the deleterious effects of
oxidative stress on osteoblastic differentiation via Wnt/FoxO3a
signaling. Oxid Med Cell Longev. 2013:3518952013. View Article : Google Scholar
|
|
38
|
Zhang S, Li D, Yang JY and Yan TB:
Plumbagin protects against glucocorticoid-induced osteoporosis
through Nrf-2 pathway. Cell Stress Chaperones. 20:621–629. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ryter SW, Mizumura K and Choi AM: The
impact of autophagy on cell death modalities. Int J Cell Biol.
2014:5026762014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li M, Gao P and Zhang J: Crosstalk between
autophagy and apoptosis: Potential and emerging therapeutic targets
for cardiac diseases. Int J Mol Sci. 17:3322016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Z, Zheng L, Zhao Z, Shi J, Wang X
and Huang J: Grape seed proanthocyanidins inhibit
H2O2-induced osteoblastic MC3T3-E1 cell
apoptosis via ameliorating H2O2-induced mitochondrial dysfunction.
J Toxicol Sci. 39:803–813. 2014. View Article : Google Scholar
|
|
43
|
Suh KS, Choi EM, Lee YS and Kim YS:
Protective effect of albiflorin against oxidative-stress-mediated
toxicity in osteo-blast-like MC3T3-E1 cells. Fitoterapia. 89:33–41.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and Autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015. View Article : Google Scholar : PubMed/NCBI
|