Introduction
Although the incidence of gastric cancer (GC) is
declining, it remains the second most common cause of
cancer-associated mortality worldwide (1–3).
Dysregulation of numerous oncogenic signaling pathways, including
E2F, K-RAS, p53 and Wnt/β-catenin, occurs frequently in GC
(4–7), thus suggesting that GC is a
molecularly heterogeneous disease. Non-resectable or metastatic GC
is associated with poor prognosis, and systemic chemotherapeutic
approaches provide minimal benefit (8,9).
Therefore, there is a significant need to develop novel therapeutic
approaches for the systemic therapy of this disease.
Cancer is a disease associated with dysregulated
proliferation; therefore, compounds that induce cell cycle are
promising candidates for the treatment and prevention of
malignancy. Progression through the cell cycle is driven by
cyclin-dependent kinases (CDKs) in cooperation with cyclins, which
can be specifically inhibited by CDK inhibitors (10). Previous studies have detected
aberrant coactivation of CDK4-cyclin D1 or CDK4/CDK6-cyclin D2 in
GC (11–13).
PD0332991 (also known as palbociclib and
Ibrance®) is an orally available small molecule that
potently and specifically inhibits CDK4/6 in a reversible manner
(4,10,14). It has emerged as a promising agent
for cell cycle-based therapy, due to its ability to rapidly and
specifically inhibit CDK4 and CDK6 activity. In breast cancer,
PD0332991 exhibited marked effects and was recently approved by the
US Food and Drug Administration (FDA) to treat advanced breast
cancer (15–17). Its effects have also been detected
in other tumor types, including primary human multiple myeloma
cells (5,10), solid tumor cell lines (4), mantle cell lymphoma and acute
myeloid leukemia cells (7,10,18).
However, its effects on GC remain to be determined.
The present study investigated whether PD0332991
exerts anticancer activity in GC cells and the underlying molecular
mechanisms. The results indicated that PD0332991 could effectively
inhibit proliferation in all tested GC lines. Furthermore,
according to the results of a protein pathway array (PPA), several
unknown downstream targets of PD0332991 were identified in GC.
Materials and methods
Chemicals and drugs
PD0332991 was obtained from Selleck Chemicals
(Houston, TX, USA). A 10 µM solution was prepared in 100%
dimethyl sulfoxide (DMSO), stored in small aliquots at −20°C and
was diluted as required in cell culture medium. Fluorouracil (5-FU)
was obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany),
dissolved in 100% DMSO to 50 mM, and stored in small aliquots at
−20°C and was then diluted as required in cell culture medium.
Cell lines and cell culture
Human GC cell lines AGS, KATO-III, NCI-N87, and
HS746T were obtained from Professor Goutham at Genetics and Genomic
Sciences of Icahn School of Medicine at Mount Sinai (New York, NY,
USA). AGS cells (gastric adenocarcinoma) were cultured in F-12K
medium (Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal
bovine serum (FBS; Gibco, Grand Island, NY, USA). KATO-III cells
(signet ring cell carcinoma) were cultured in Iscove's modified
Dulbecco's medium (Lonza BioWhittaker, Verviers, Belgium)
supplemented with 20% FBS. NCI-N87 cells (gastric carcinoma) were
cultured in RPMI-1640 medium (Corning Cellgro, Manassas, VA, USA)
supplemented with 10% FBS (Gibco). HS746T (gastric carcinoma) were
cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented
with 10% FBS (Gibco). All media were also supplemented with 100
U/ml penicillin and 100 µg/ml streptomycin. All cell lines
were cultured at 37°C in a humidified incubator containing 5%
CO2.
Cell viability assay
Cell viability of GC cells was monitored using the
MTT assay (Sigma-Aldrich). Approximately 5×103 cells
were seeded in each well of a 96-well plate and were incubated for
24 h at 37°C. Cells were left untreated, or were treated with
various concentrations of PD0332991 or 5-FU for 72 h at 37°C.
Subsequently, 10 µl MTT (5 mg/ml) was added to each well and
the cells were incubated for an additional 3 h, after which the
supernatant was discarded. Finally, 100 µl DMSO was added to
the wells to dissolve the precipitate. Optical density was measured
at a wavelength of 570 nm using an ELx800 (BioTek Instruments,
Inc., Winooski, VT, USA).
Flow cytometric analysis
Cells were treated with PD0332991 in a
dose-dependent manner for 72 h. The cells were then washed,
centrifuged at 1,000 rpm for 10 min, fixed with cold 70% ethanol
for ≥30 min, and incubated with 100 µg/ml RNase A and 50
µg/ml propidium iodide at room temperature for 30 min.
Samples were immediately analyzed by flow cytometry (BD
Biosciences, San Jose, CA, USA) and cell cycle phase distribution
was determined using CellQuest Pro software version 5.1 (BD
Biosciences, Franklin Lakes, NJ, USA).
Western blotting
Cells were lysed using 1X cell lysis buffer (Cell
Signaling Technology, Inc., Danvers, MA, USA) containing 20 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA-Na2, 1 mM
EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM Na3VO4 and 1
µg/ml leupeptin in the presence of 1X Protease Inhibitor
Cocktail and 1X Phosphatase Inhibitor Cocktail (both from Roche
Applied Science, Madison, WI, USA). Protein concentration was
determined using the Pierce Bicinchoninic Acid (BCA) Protein Assay
kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Equal amounts of cell lysate (20 µl, 1 µg/µl)
were then subjected to 10% SDS-PAGE under reducing conditions.
Prior to loading onto the gel, all lysates were boiled for 5 min
and immediately cooled on ice. The proteins were then transferred
onto Immobilon-P membranes (EMD Millipore, Billerica, MA, USA)
previously soaked in methanol and transfer buffer using the
Trans-Blot SD semi-dry transfer cell (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Following completion of the transfer process,
the membranes were allowed to dry, soaked in methanol, and
incubated with blocking buffer [5% dry non-fat milk in 1X TBS-0.1%
Tween-20 (TBST)] for 1 h at room temperature. Membranes were then
incubated with primary antibodies against phosphorylated
(p)-retinoblastoma (Rb) (Ser780) (1:1,000; #9307; Cell Signaling
Technology, Inc.), CDK6 (1:1,000 sc-177; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), Cyclin D1 (1:1,000; sc-718; Santa Cruz
Biotechnology, Inc.), P53 (1:1,000; sc-126; Santa Cruz
Biotechnology, Inc.), P27 (1:1,000; sc-528; Santa Cruz
Biotechnology, Inc.) and β-actin (1:10,000; #A5316; Sigma-Aldrich;
Merck KGaA), overnight at 4°C. The membranes were washed with 1X
TBS and 1X TBST, and were incubated with secondary anti-rabbit
(#1705046) or anti-mouse (#1705047) antibodies conjugated with
horseradish peroxidase (HRP) (1:10,000; Bio-Rad Laboratories, Inc.)
for 1 h at room temperature. The membrane was developed using a
chemiluminescence substrate (Immun-Star™ HRP Peroxide
Buffer/Immun-Star™ HRP Luminol Enhancer; Bio-Rad Laboratories,
Inc.), and chemiluminescent signals were captured using the
ChemiDoc XRS system (Bio-Rad Laboratories, Inc.). The signal
intensity of each protein was determined by densitometric scanning
(Quantity One software package version 4.6.2; Bio-Rad Laboratories,
Inc.).
PPA
Cells were treated with PD0332991 at 75% inhibitory
concentration (IC75) for 48 h. Total cellular proteins
were extracted from cells using cell lysis buffer (Cell Signaling
Technology, Inc.) containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl,
1 mM EDTA-Na2, 1 mM EGTA, 1% Triton, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4 and 1 µg/ml leupeptin in the
presence of 1X Proteinase Inhibitor Cocktail and 1X Phosphatase
Inhibitor Cocktail (both from Roche Applied Science). The cell
lysates were sonicated twice for 15 sec, and centrifuged at 18,000
× g for 30 min at 4°C. The protein concentration was determined
using the BCA Protein Assay kit (Pierce; Thermo Fisher Scientific,
Inc.). Isolated proteins were separated by 10% SDS-PAGE. Cell
extract containing 300 µg protein was loaded into one well
across the entire width of the gel. After running the gel, the
proteins were electrophoretically transferred to a nitrocellulose
membrane. Subsequently, the membrane was blocked for 1 h with 5%
milk or 3% bovine serum albumin (BSA; Thermo Fisher Scientific,
Inc.)
at room temperature
The membrane was then clamped with a western
blotting manifold in order to isolate 20 channels across the
membrane (Bio-Rad Laboratories, Inc.). Two or three primary
antibodies were added to each channel and allowed to bind to
proteins overnight at 4°C. Different sets of antibodies were used
for each membrane (Table I). The
blot was washed and hybridized for 45 min with horseradish
peroxidase-conjugated secondary antibodies [1:10,000; ant-rabbit
(#1705046) and anti mouse (#1705047); Bio-Rad Laboratories, Inc.]
at room temperature. The membranes were washed and
chemiluminescence signals were captured using the ChemiDoc XRS
system (Bio-Rad Laboratories, Inc.). Differences in protein level
were estimated by densitometric scanning (Quantity One software
package version 4.6.2; Bio-Rad Laboratories, Inc.) and were
normalized using internal standards.
 | Table IAntibodies included in the protein
pathway array analysis. |
Table I
Antibodies included in the protein
pathway array analysis.
| Antibodies specific
for phosphorylation |
|
| p-PKCα (Ser657),
p-PDK1 (Ser241), p-PKC α/βII (Thr638/641), p-p53 (Ser392), p-AKT
(Ser473), p-PTEN (Ser380), p-Rb (Ser780),
p-β-catenin(Ser33/37/Thr41), p-c-Jun (Ser73), p-Stat3 (Ser727),
p-ERK (Thr202/Tyr204), p-GSK-3α/β (Ser21/9), p-p70 S6 kinase
(Thr389), p-eIF4B (Ser422), p-HGF R/c-MET (Y1234/Y1235), p-Smad
(Ser463/465), p-ERK5 (Thr218/Tyr220), p-p90RSK (Ser380), p-CREB
(Ser133), p-CDC2 (Tyr15), p-PKCδ (Thr505), p-FAK (Tyr397), p-Rb
(Ser807/811), p-p38 (Thr180/Tyr182) |
|
| Antibodies for
signal transduction proteins |
|
| FAS (C-20), FOXM1
(H-92), Erα (HC-20), Syk (LR), MetRS, Twist (H-81), Lyn, KLF6,
CaMKKα, SK3 (H-45), Stat1 (42H3), cyclin B1, cyclin D1, Cdk6,
CDC25B, cyclin E, CDK2, p27, TDP1 (H-300), Cdk4, HER2 (ErbB2),
14-3-3β, cPKCα, ERK/EGFR, SLUG (H-140), Cdc25C, Hsp90, CHK1, MDM2,
CDC2 p34, E2F-1, PCNA, p63, p38, Rap 1 (121), β-catenin, Akt, HCAM
(H-300), XIAP, Bcl-2, patched (H-267), HIF-1α, HIF-2α, TTF-1, p53,
Notch4 (L5C5), PTEN, SRC-1, Eg5 (H-300), HIF-3α, Bax, N-cadherin,
TNF-α, cdc42, eIF4B, Vimentin, OPN, Survivin, E-cadherin, TGF-β,
Erb (H-150), p27, WT1, Mesothelin, VEGF, ATF-1, Ep-CAM (KS1/4), Bad
(C-7), NF-κB p52, NF-κB p50, Calretinin, IL-1β, H-Ras, Bcl-6,
K-Ras, α-tubulin, NF-κB p65, CREB, BID (C-20), Maspin (C-20), DRG1
(C-20), Factor XIII B (I-20), IGFBP5 (T-17), HCAM (DF1485), ICAM-1,
Estrogen Receptor α (62A3), c-Flip, PSM (k1H7), Rab 7 (H-50),
VCAM-1 (HAE-2z), FGF-8 (H-181), NEP(CD 10), Bcl-xL (54H6), Endoglin
(H-300), Bak (G-23), TFIIH p89 (S-19), Nkx-3.1 (M-96), RIP
(D94C12), NM23, c-IAP2 (H-85), Epo (H-162), uPA, PDEF (H-250),
Stat3, ERCC1 (FL-297), uPAR, KAI1, L-selectin (H-149), PSCA,
E-selectin |
Statistical analysis
All experiments were biologically repeated three
times. PPA was performed in duplicate. Data are expressed as the
means ± SD. Statistical comparisons of results were made using
one-way analysis of variance by SPSS 17.0. P<0.05 was considered
to indicate a statistically significant difference. The human
pathway lists from PPA data determined by the 'Ingenuity System
Database' were selected (Qiagen, Inc., Valencia, CA, USA).
Ingenuity Pathway Analysis (IPA) is a system that transforms large
data sets into a group of relevant networks containing direct and
indirect relationships between genes based on known interactions in
the literature. The gene names of differentially expressed proteins
were input into the IPA system. According to IPA, a score of 3
indicates that there is a 1/1,000 chance that the focus genes are
in a network due to random chance; therefore, scores >3 have a
99.9% confidence of not being generated by random chance. This
score was used as the cut-off for identifying gene networks that
were significantly affected by the drug. Differentially expressed
proteins were mapped to canonical pathways, functions and tested by
the Fisher's exact test. Significance threshold was set at
P=0.001.
Results
PD0332991 inhibits growth of GC
cells
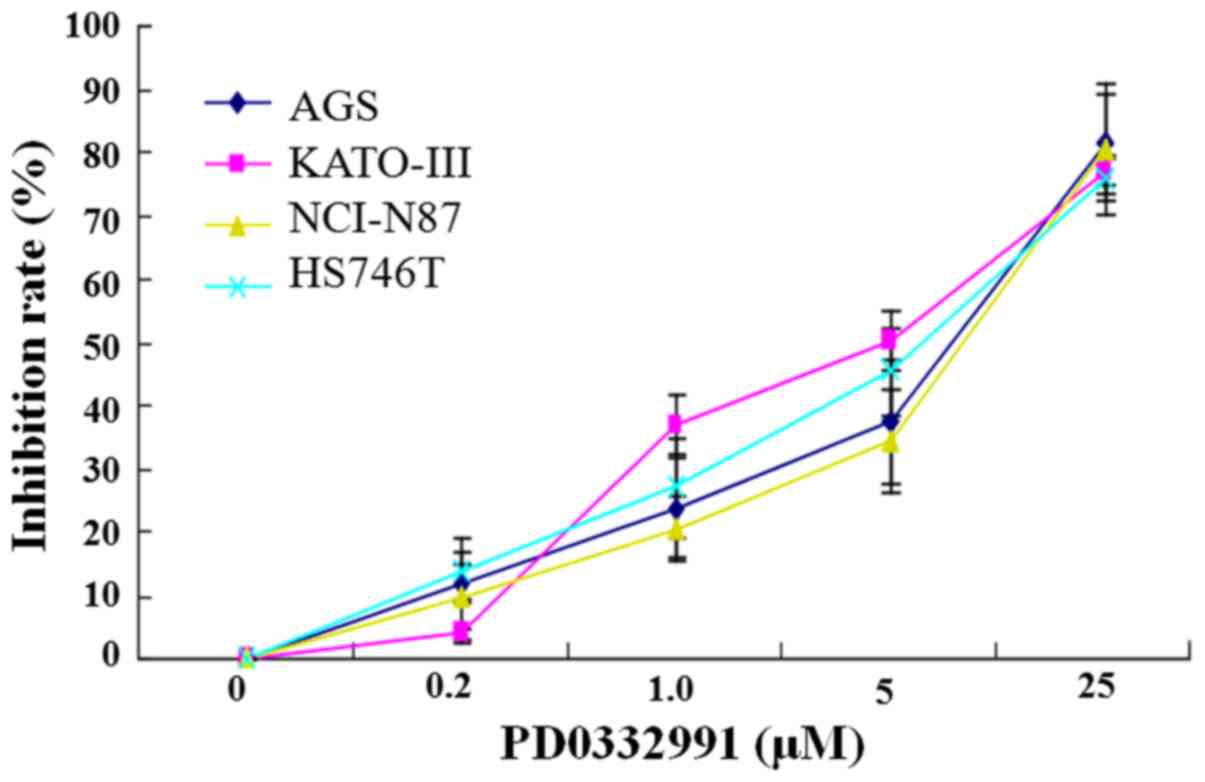
To determine whether PD0332991 inhibits
proliferation of GC cells, AGS, KATO-III, NCI-N87 and HS756T cells
were treated with varying concentrations of PD0332991 (0.2–25
µm) for 72 h and cell proliferation was measured by the MTT
assay. The inhibitory rate of PD0332991 was determined as a
percentage of viable cells in treated cultures compared with
control cells. As shown in Fig.
1, PD0332991 inhibited proliferation of GC cells in a
dose-dependent manner, with a 50% inhibitory concentration
(IC50) value of 12 µM in AGS cells, 5.0 µM
in KATO-III cells, 13 µM in NCI-N87 cells and 11 µM
in HS756T cells. The IC50 value was calculated from nine
independent experiments. These results indicated that PD0332991 may
inhibit the growth of GC cells.
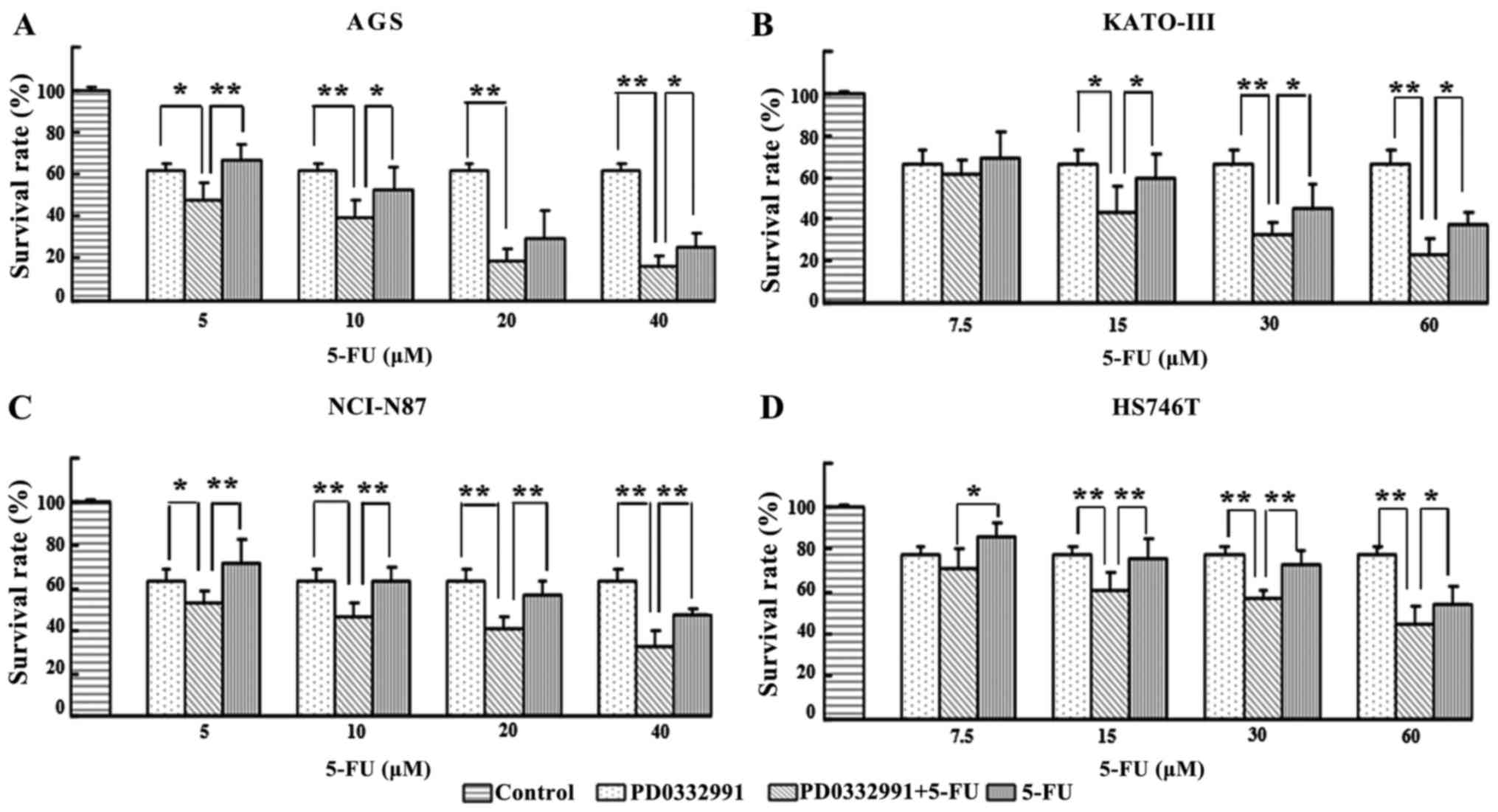
PD0332991 increases the efficiency of
5-FU in GC cells
The present study demonstrated that proliferation of
AGS, KATO-III, NCI-N87 and HS746T cells was significantly reduced
by PD0332991 (Fig. 1). Therefore,
the present study subsequently examined whether PD0332991 affects
the response of GC cells to 5-FU. Cells were treated with
increasing concentrations of 5-FU (5–60 µM), alone or in
combination with PD0332991 (at a concentration lower than
IC50). The results demonstrated that PD0332991 enhanced
the proliferation-inhibiting effects of 5-FU (Fig. 2). As presented in Fig. 3, the IC50 value of 5-FU
was significantly decreased by PD0332991 in all GC cell lines. The
inhibitory effects of the combination of PD0332991 and 5-FU were
stronger than the expected additive effects in all cell lines, thus
suggesting that PD0332991 and 5-FU synergize. Taken together, these
findings indicated that PD0332991 not only inhibits proliferation
of GC cells, but also enhances the cytotoxic effects of 5-FU, a
common chemotherapeutic drug used to treat GC.
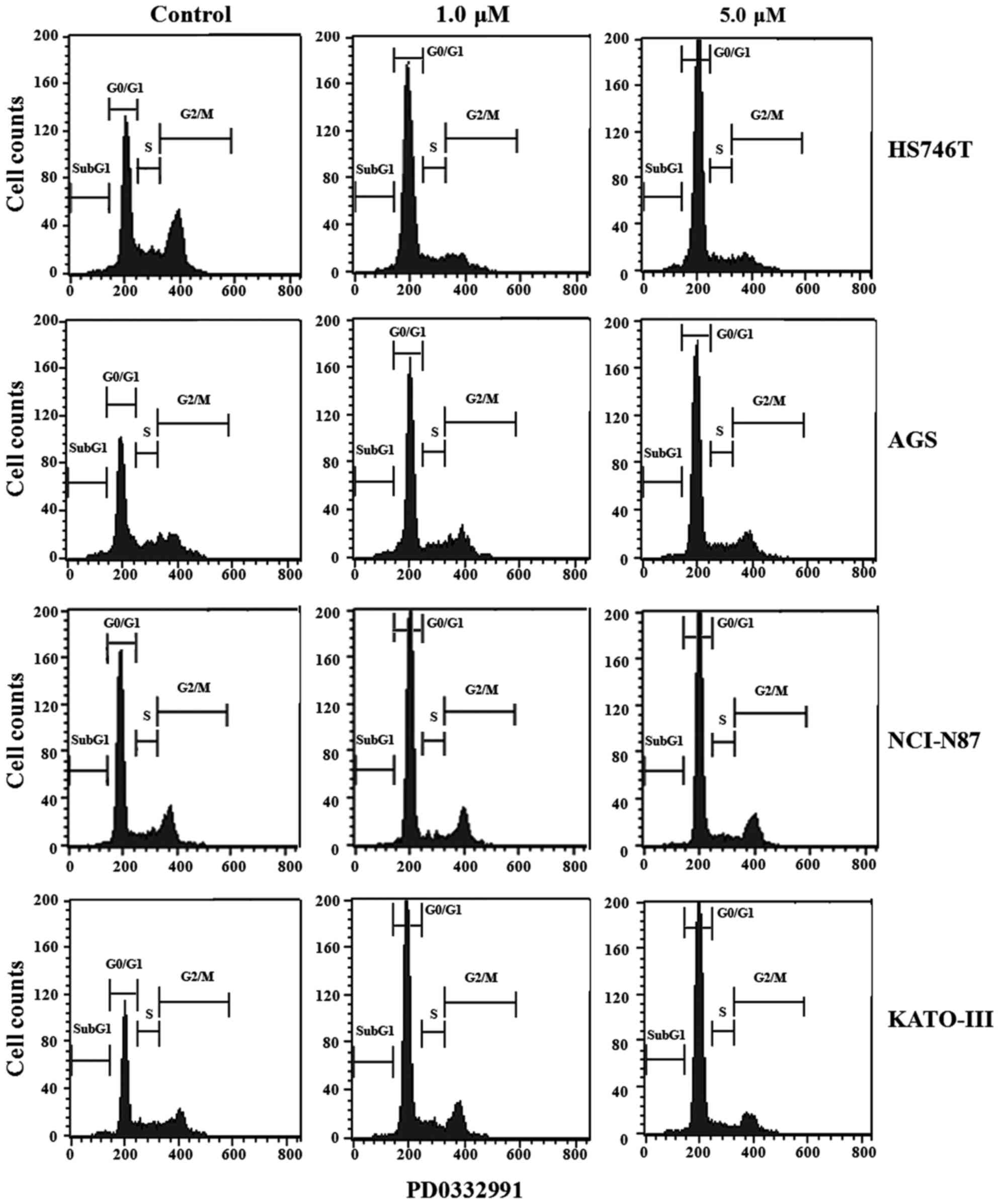
PD0332991 induces G1 cell
cycle arrest in GC cells
To understand the molecular mechanism underlying the
effects of PD0332991, the present study analyzed the effects of
PD0332991 on cell cycle progression in four GC cell lines. AGS,
KATO-III, NCI-N87 and HS746T cells were treated with 1.0 or 5.0
µM PD0332991 for 72 h. The results demonstrated that
PD0332991 triggered arrest of cells in G1 phase, with a
concurrent decrease of cells in G2/M phase (Fig. 4, Table II). Upon treatment with PD0332991
the percentage of cells in G1 phase increased from 53.03
to 79.2% (1 µM) and to 82.25% (5 µM) in HS746T cells.
PD0332991 increased G1 cells from 63.35 to 68.48% (1
µM) and to 75.15% (5 µM) in AGS cells. In KATO-III
cells, PD0332991 increased the percentage of cells in G1
phase from 60.32 to 72.72% (1 µM) and to 82.74% (5.0
µM). In NCI-N87 cells, the percentage of cells in
G1 phase was increased from 65.60 to 75.60% in cells
treated with 1.0 µM PD0332991, and to 74.65% in cells
treated with 5.0 µM. The percentage of cells in S phase was
decreased by PD0332991 in all cell lines (Fig. 4, Table II). These results suggested that
the antiproliferative effects of PD0332991 may be due to cell cycle
arrest.
| Table IIResults of cell cycle analysis in
cell lines following treatment with PD0332991, as determined using
flow cytometry. |
Table II
Results of cell cycle analysis in
cell lines following treatment with PD0332991, as determined using
flow cytometry.
| Cell line | PD0332991
(µM) | Cell cycle phase
|
|---|
| SubG1
(%) |
G0/G1 (%) | S (%) | G2/M
(%) |
|---|
| HS746T | 0 | 1.47±0.36 | 53.03±1.72 | 15.33±1.09 | 30.61±3.05 |
| 1.0 | 1.69±0.37 | 79.2±0.01a | 8.76±0.10 | 10.70±0.27 |
| 5.0 | 1.66±0.44 | 82.25±0.15b | 7.15±0.15 | 9.27±0.75 |
| AGS | 0 | 1.98±0.48 | 63.35±2.23 | 13.30±0.79 | 31.83±1.15 |
| 1.0 | 2.59±0.15 | 68.48±2.45a | 10.35±0.42 | 18.86±0.28 |
| 5.0 | 1.05±0.04 | 75.15±0.99b | 7.27±0.40 | 16.72±1.37 |
| KATO-III | 0 | 1.27±0.09 | 60.32±0.92 | 12.79±0.22 | 26.09±1.17 |
| 1.0 | 1.12±0.30 | 72.72±0.83b | 9.80±0.56 | 16.66±0.04 |
| 5.0 | 0.88±0.01 | 82.74±1.64b | 5.18±0.31 | 11.34±1.26 |
| NCI-N87 | 0 | 0.51±0.05 | 65.60±3.00 | 10.88±1.55 | 22.33±0.50 |
| 1.0 | 0.21±0.04 | 75.60±1.04b | 5.06±0.07 | 19.28±1.09 |
| 5.0 | 0.41±0.06 | 74.65±1.57a | 6.27±0.36 | 18.90±1.22 |
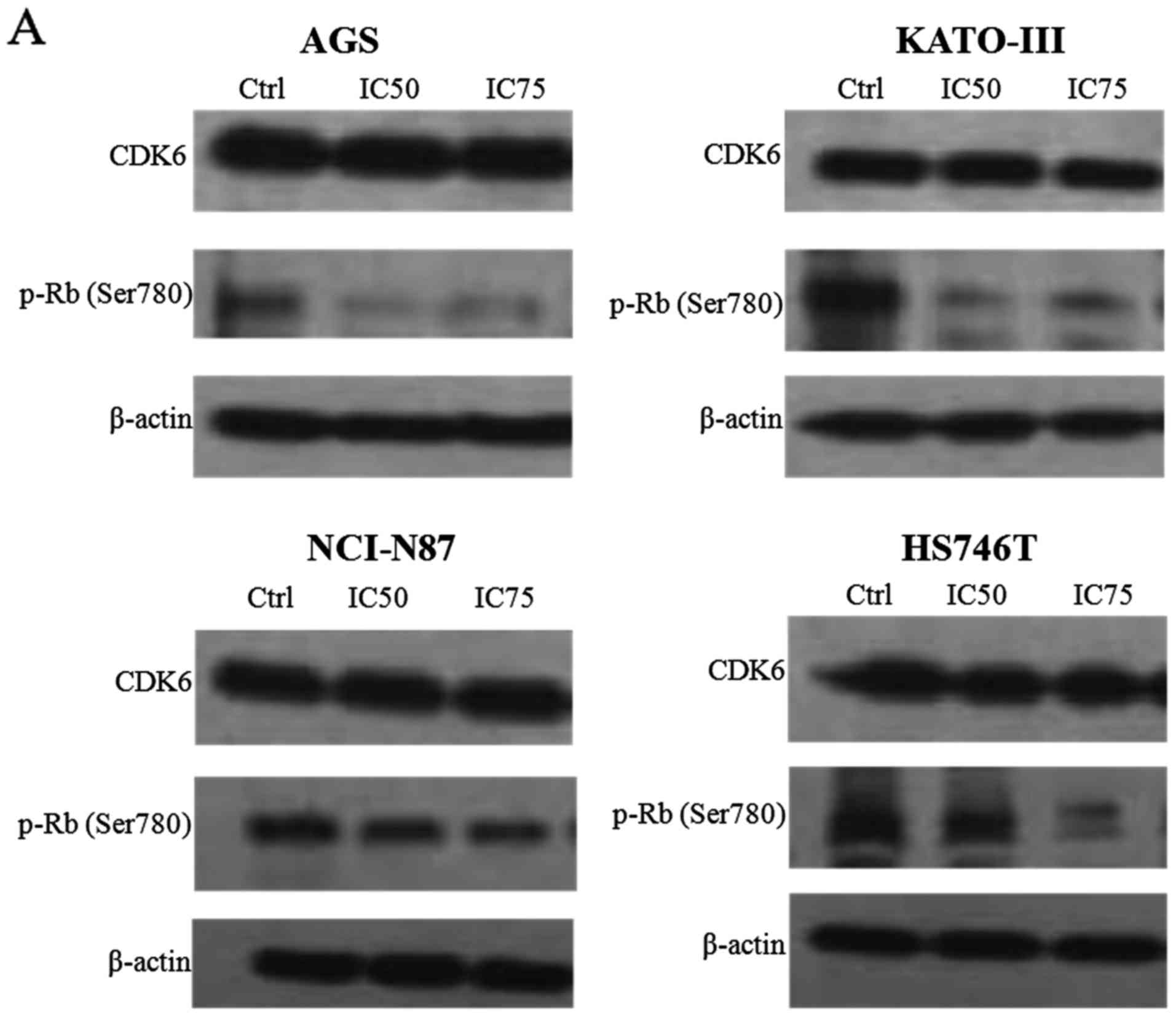
PD0332991 inhibits Rb phosphorylation in
GC cells on CDK6-specific sites
The present study demonstrated that PD0332991
induces G1 arrest in GC cell lines (Fig. 4). PD0332991 is a potent and highly
selective inhibitor of CDK6/4 kinase activity; the only known
substrates for CDK4/6 are the Rb family proteins. Of the 16 known
phosphorylation sites on Rb, two are specifically phosphorylated by
CDK4/6: Ser780 and Ser795. Therefore, phosphorylation status of Rb
at these specific sites represents an appropriate biomarker for the
activity of CDK4/6 in tumor cells and tissues. To determine the
inhibitory effects of PD0332991 on CDK6 in GC cells, the four GC
cell lines were treated with PD0332991 for 48 h. The results
demonstrated that PD0332991 did not alter the expression levels of
CDK6; however, it did significantly inhibit phosphorylation of Rb
on Ser780 (Fig. 5A). These
findings confirmed that PD0332991 may potently inhibit CDK4/6
activity in GC cells.
The present study also investigated the effects of
PD0332991 on other proteins involved in G0/G1
cell cycle arrest. KATO-III cells were treated with various doses
of PD0332991 (0, 0.2, 1, 5 and 25 µM) for 72 h. The ratio of
signal intensity for each protein was compared with β-actin. As
shown in Fig. 5B and C, treatment
of KATO-III cells with PD0332991 resulted in a marked decrease in
the expression of cyclin D1. In addition, PD0332991 induced the
expression of p53 and p27 in GC cells in a dose-dependent manner
(Fig. 5B, D and E). These results
suggested that p53 and p27 may be associated with the ability of
PD0332991 to induce G0/G1 arrest in GC
cells.
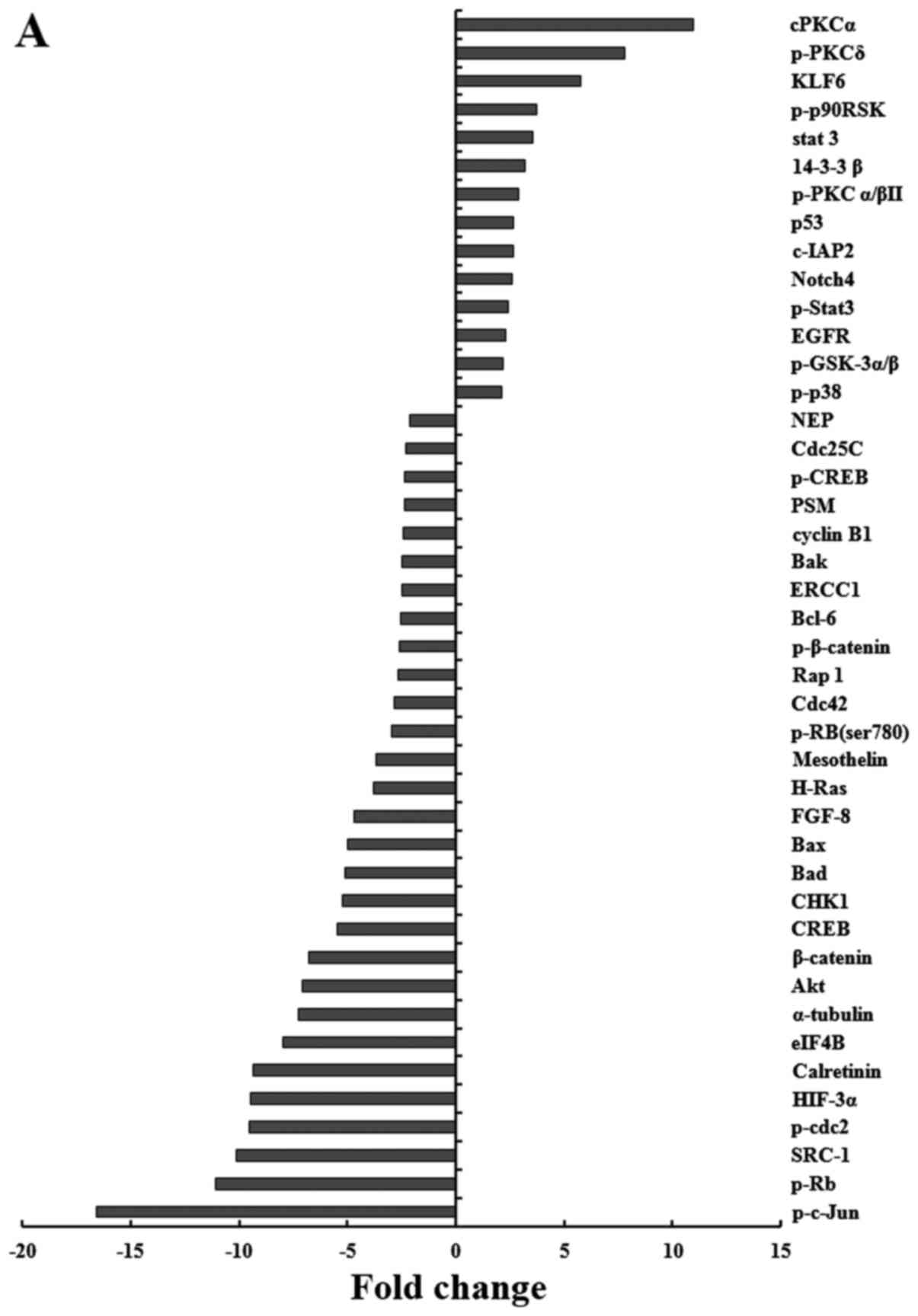
Effects of PD0332991 on the expression of
signaling proteins in GC cells
To further identify the molecular mechanisms by
which PD0332991 initiates G0/G1 arrest in GC
cells, the four GC cell lines were treated with PD0332991
(IC75) for 48 h. Subsequently, protein expression
patterns were analyzed using PPA analysis. Proteins that exhibited
a >2-fold alteration in expression in response to PD0332991 were
considered differentially expressed. A total of 43 proteins were
revealed to be differentially expressed in AGS cells (Fig. 6A), 32 in KATO-III cells (Fig. 6B), 29 in NCI-N87 cells (Fig. 6C) and 39 in HS746T cells (Fig. 6D). A total of 31 proteins were
similarly regulated in at least three cell lines. Pathway analysis
identified categories associated with cellular development (23
proteins, P=1.00×10−17−3.49×10−6), cell death
(23 proteins, P=5.47×10−17−3.25×10−6), cancer
(26 proteins, P=7.21×10−17−3.25×10−6), cell
cycle (21 proteins, P=7.63×10−17−3.49×10−6),
and cell growth and proliferation (24 proteins,
P=7.07×10−17−3.25×10−3). Notably, a large
number of genes regulating cell proliferation and cell cycle
progression were downregulated by PD0332991.
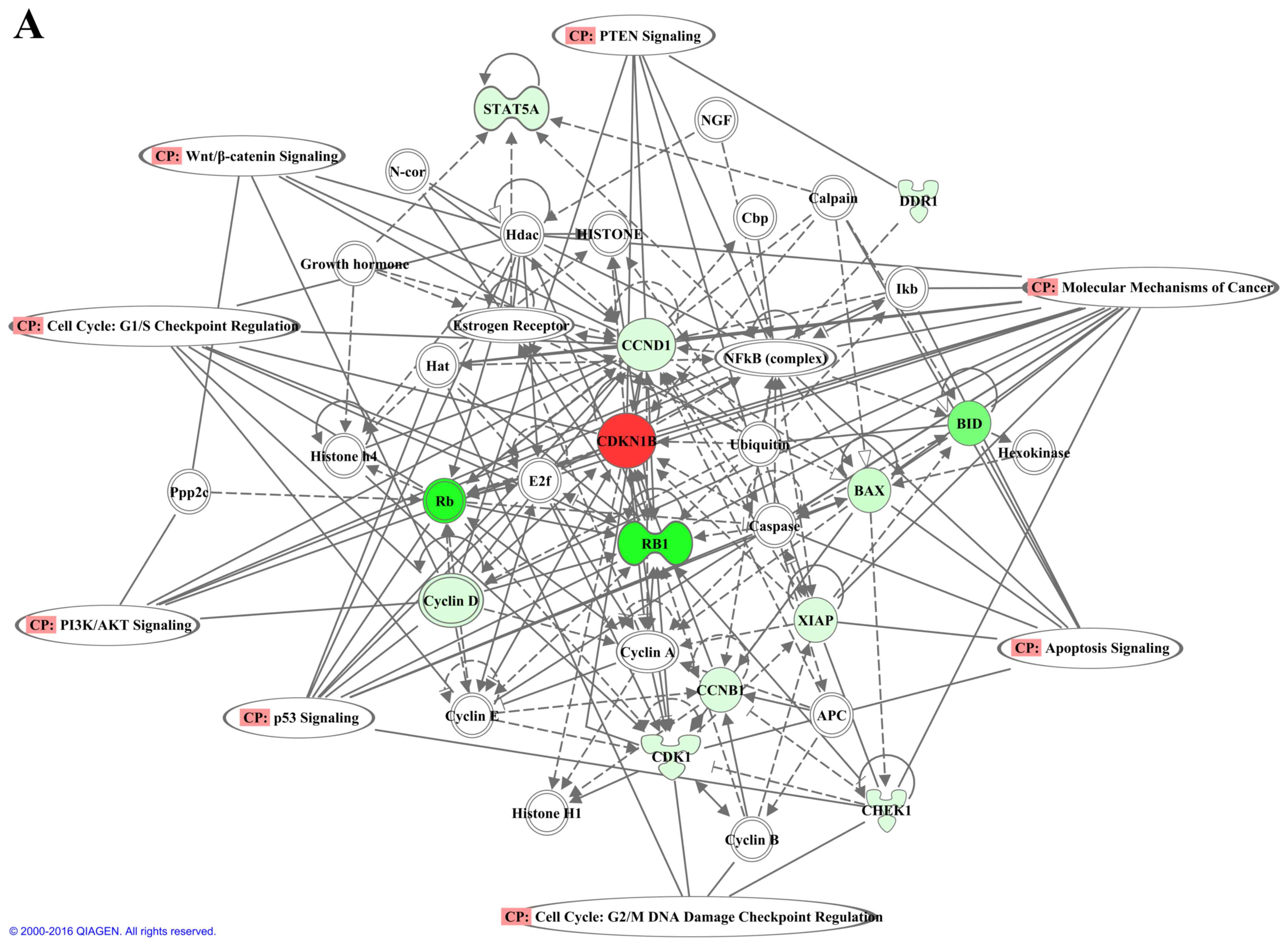
Identification of biological pathways
modulated by PD0332991 in GC cells
The present study further analyzed the PD0332991
target proteins, in order to determine key biological pathways
modulated by PD0332991 treatment. IPA was performed on 31 proteins
that were targeted by PD0332991 in GC cell lines. Five networks
were revealed to be altered by PD0332991. The top three networks
were mainly associated with cell death, cell cycle and molecular
mechanism of cancer (Fig. 7). The
results of IPA also indicated that PD0332991 target proteins are
involved in numerous canonical signaling pathways, including
molecular mechanisms of cancer (P=6.06×10−17), p53
signaling (P=1.46×10−11), phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (PKB) (PI3K/AKT) signaling
(P=1.07×10−10), apoptosis signaling (P=
4.58×10−10), phosphatase and tensin homolog signaling
(P=3.55×10−8), 14-3-3-mediated signaling
(p=1.14×10−7), Janus kinase/signal transducer and
activator of transcription signaling (P=2.04×10−7), cell
cycle: G2/M DNA damage checkpoint regulation
(P=1.52×10−6), epidermal growth factor signaling
(P=2.26×10−6), hypoxia-inducible factor 1α signaling
(P=2.44×10−6), cell cycle: G1/S checkpoint
regulation (P=5.7×10−6), fibroblast growth factor
signaling (P=3.02×10−5), Rac signaling
(P=7.11×10−5), p70S6 kinase signaling
(P=1.30×10−4), mammalian target of rapamycin signaling
(P=2.05×10−4), extracellular signal-regulated protein
kinase (ERK)5 signaling (P=4.25×10−4), retinoic acid
receptor activation (P=4.32X10-4), Wnt/β-catenin signaling
(P=4.42×10−4), ERK/mitogen-activated protein kinase
(MAPK) signaling (P=5.65×10−4), and stress-activated
protein kinase/c-Jun N-terminal kinase (JNK) signaling
(P=1.01×10−3) (Fig.
7).
Discussion
PD0332991, a pyrido[2,3-d]pyrimidin-7-one
inhibitor, is a selective inhibitor of CDK4 and CDK6 (6) that has been reported to inhibit the
growth of a panel of Rb-positive solid tumor cell lines (5,7).
Recently, PD0332991 was approved by the FDA to treat advanced
breast cancer. The present study demonstrated that PD0332991 exerts
potent antiproliferative activity in human GC cells. Consistent
with the established role of CDK4/6 in cell cycle progression,
PD0332991 induced G0/G1 arrest, and reduced
phosphorylation of Rb at the CDK4/6-specific Ser780 site.
Accordingly, the levels of cyclin D1 were decreased, whereas the
expression levels of p27 were increased following treatment with
PD0332991.
The combination of targeted therapeutic agents with
cytotoxic chemotherapy has become a standard therapeutic strategy
for the treatment of numerous types of cancer. 5-FU is a major
chemotherapy drug used to treat GC. In the present study, cells
were cotreated with PD0332991 and 5-FU; the results demonstrated
that PD0332991 increased the cytotoxic effects of 5-FU on all four
GC cell lines.
Using large scale proteomic analysis, the present
study identified numerous proteins that were altered in GC cells
upon treatment with PD0332991. Data analysis demonstrated that
PD0332991 altered the expression of proteins involved in the
regulation of cellular development, cell death, cell cycle, cell
growth and proliferation, and cell migration. Proteins regulated by
PD0332991 were involved in several canonical pathways, including
molecular mechanism of cancer, p53 signaling and PI3K/AKT
signaling, Ras-ERK pathway, JNK/MAPK pathway, Wnt/β-catenin pathway
and Smad signaling (Fig. 7).
c-Jun is a transcription factor, which serves a role
in the development of skin and liver tumors (18,19). c-Jun is a positive regulator of
cell proliferation, and c-Jun-deficient fibroblasts exhibited
marked defects in proliferation in vitro (20–22). In addition, the proliferation of
c-Jun-deficient hepatocytes is severely impaired during liver
regeneration in vivo (20). The c-Jun protein is activated by
JNKs (23). Subsequently, the
activated c-Jun-containing activator protein-1 complex induces
transcription of positive regulators of cell cycle progression,
including cyclin D1, or suppresses negative regulators, including
the tumor suppressor p53 and the CDK inhibitor INK4A. c-Jun can
also cooperate with activated Ras (24). The present study demonstrated that
following treatment with PD0332991, H-Ras, p-c-Jun and cyclin D1
were downregulated, whereas p53 was upregulated in GC cells. These
alterations suggested that the Ras/Jun pathway may participate in
PD0332991-induced growth inhibition and cell cycle arrest.
Hyperactivation of the Wnt/β-catenin pathway may
lead to aberrant cell growth (25) in various types of cancer. The
present study demonstrated that the expression levels of p-catenin
and unphosphorylated-catenin were decreased in GC cell lines
following treatment with PD0332991. Consistent with this finding,
the expression levels of cyclin D1, a target of Wnt signaling, were
also inhibited by PD0332991, thus suggesting that PD0332991 may
inhibit growth of GC cells by inhibiting Wnt/β-catenin signaling.
Furthermore, the expression levels of p-Smad were decreased in GC
cells following treatment with PD0332991, which may also contribute
to the growth inhibition of GC cells.
The transcription factor p53 is a critical component
in the normal cell response to cellular stress, including DNA
damage, oncogenic stimulation, nutrient deprivation or hypoxia
(26). Its role as a tumor
suppressor is exemplified by the fact that numerous types of cancer
are associated with selective inactivation of p53 and/or p53
pathways. p53 serves a critical role during the DNA damage-induced
G1/S cell cycle checkpoint; p53-deficient cells fail to
undergo G1/S arrest in response to genotoxic stress
(27–29). The present study demonstrated that
PD0332991 induced p53 expression, which may underlie the ability of
PD0332991 to induce G1/S arrest in GC cells.
AKT protects cells from apoptosis by phosphorylating
downstream target proteins involved in the regulation of cell
growth and survival, including glycogen synthase kinase-3β, p21,
p27, X-linked inhibitor of apoptosis protein, B-cell lymphoma
2-associated death promoter and forkhead box O3α (30). Suppression of AKT activity has
been reported to lead to p53 activation, which in turn may lead to
growth arrest and activation of proapoptotic signaling pathways
(31). The present study
indicated that following treatment with PD0332991, AKT was
downregulated, and p53 and p27 were upregulated, thus suggesting
that the PI3K/AKT pathway may have an important role in the effects
of PD0332991 on GC cells.
In conclusion, the present study demonstrated that
PD0332991 inhibits cell proliferation via modulation of cell cycle
progression, and that numerous signaling pathways are regulated by
PD0332991. These results suggested that PD0332991 may be considered
a promising preventive and therapeutic agent for the treatment of
GC.
Acknowledgments
The present study was supported by National Natural
Science Foundation of China grants (grant nos. 81372295 and
81402374).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Carcas LP: Gastric cancer review. J
Carcinog. 13:142014. View Article : Google Scholar
|
|
2
|
Thiel A and Ristimäki A: Targeted therapy
in gastric cancer. APMIS. 123:365–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hohenberger P and Gretschel S: Gastric
cancer. Lancet. 362:305–315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
May P and May E: Twenty years of p53
research: Structural and functional aspects of the p53 protein.
Oncogene. 18:7621–7636. 1999. View Article : Google Scholar
|
|
5
|
Fry DW, Harvey PJ, Keller PR, Elliott WL,
Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et
al: Specific inhibition of cyclin-dependent kinase 4/6 by PD0332991
and associated antitumor activity in human tumor xenografts. Mol
Cancer Ther. 3:1427–1438. 2004.PubMed/NCBI
|
|
6
|
Toogood PL, Harvey PJ, Repine JT, Sheehan
DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T,
et al: Discovery of a potent and selective inhibitor of
cyclin-dependent kinase 4/6. J Med Chem. 48:2388–2406. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
VanderWel SN, Harvey PJ, McNamara DJ,
Repine JT, Keller PR, Quin J III, Booth RJ, Elliott WL, Dobrusin
EM, Fry DW, et al: Pyrido[2,3-d]pyrimidin-7-ones as specific
inhibitors of cyclin-dependent kinase 4. J Med Chem. 48:2371–2387.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Foo M and Leong T: Adjuvant therapy for
gastric cancer: Current and future directions. World J
Gastroenterol. 20:13718–13727. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanat O and O'Neil BH: Metastatic gastric
cancer treatment: A little slow but worthy progress. Med Oncol.
30:4642013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SR, Shin JW, Kim HO, Son BH, Yoo CH
and Shin JH: Determining the effect of transforming growth
factor-β1 on cdk4 and p27 in gastric cancer and cholangiocarcinoma.
Oncol Lett. 5:694–698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Motohashi M, Wakui S, Muto T, Suzuki Y,
Shirai M, Takahashi H and Hano H: Cyclin D1/cdk4, estrogen
receptors α and β, in N-methyl-N′-nitro-N-nitrosoguanidine-induced
rat gastric carcinogenesis: Immunohistochemical study. J Toxicol
Sci. 36:373–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takano Y, Kato Y, van Diest PJ, Masuda M,
Mitomi H and Okayasu I: Cyclin D2 overexpression and lack of p27
correlate positively and cyclin E inversely with a poor prognosis
in gastric cancer cases. Am J Pathol. 156:585–594. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dhillon S: Palbociclib: First global
approval. Drugs. 75:543–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carey LA and Perou CM: Palbociclib -
taking breast-cancer cells out of gear. N Engl J Med. 373:273–274.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dukelow T, Kishan D, Khasraw M and Murphy
CG: CDK4/6 inhibitors in breast cancer. Anticancer Drugs.
26:797–806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morikawa A and Henry L: Palbociclib for
the treatment of estrogen receptor-positive, HER2-negative
metastatic breast cancer. Clin Cancer Res. 21:3591–3596. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Young MR, Li JJ, Rincón M, Flavell RA,
Sathyanarayana BK, Hunziker R and Colburn N: Transgenic mice
demonstrate AP-1 (activator protein-1) transactivation is required
for tumor promotion. Proc Natl Acad Sci USA. 96:9827–9832. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eferl R, Ricci R, Kenner L, Zenz R, David
JP, Rath M and Wagner EF: Liver tumor development. c-Jun
antagonizes the proapoptotic activity of p53. Cell. 112:181–192.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Behrens A, Sibilia M, David JP,
Möhle-Steinlein U, Tronche F, Schütz G and Wagner EF: Impaired
postnatal hepatocyte proliferation and liver regeneration in mice
lacking c-jun in the liver. EMBO J. 21:1782–1790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schreiber M, Kolbus A, Piu F, Szabowski A,
Möhle-Steinlein U, Tian J, Karin M, Angel P and Wagner EF: Control
of cell cycle progression by c-Jun is p53 dependent. Genes Dev.
13:607–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wisdom R, Johnson RS and Moore C: c-Jun
regulates cell cycle progression and apoptosis by distinct
mechanisms. EMBO J. 18:188–197. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mechta F, Lallemand D, Pfarr CM and Yaniv
M: Transformation by ras modifies AP1 composition and activity.
Oncogene. 14:837–847. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amelio I and Melino G: The p53 family and
the hypoxia-inducible factors (HIFs): Determinants of cancer
progression. Trends Biochem Sci. 40:425–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baker SJ, Markowitz S, Fearon ER, Willson
JK and Vogelstein B: Suppression of human colorectal carcinoma cell
growth by wild-type p53. Science. 249:912–915. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diller L, Kassel J, Nelson CE, Gryka MA,
Litwak G, Gebhardt M, Bressac B, Ozturk M, Baker SJ and Vogelstein
B: p53 functions as a cell cycle control protein in osteosarcomas.
Mol Cell Biol. 10:5772–5781. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martinez J, Georgoff I, Martinez J and
Levine AJ: Cellular localization and cell cycle regulation by a
temperature-sensitive p53 protein. Genes Dev. 5:151–159. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steelman LS, Stadelman KM, Chappell WH,
Horn S, Bäsecke J, Cervello M, Nicoletti F, Libra M, Stivala F,
Martelli AM, et al: Akt as a therapeutic target in cancer. Expert
Opin Ther Targets. 12:1139–1165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fraser M, Leung BM, Yan X, Dan HC, Cheng
JQ and Tsang BK: p53 is a determinant of X-linked inhibitor of
apoptosis protein/Akt-mediated chemoresistance in human ovarian
cancer cells. Cancer Res. 63:7081–7088. 2003.PubMed/NCBI
|