Introduction
Pancreatic β cells are key regulators of the process
of glucose tolerance, and proper β cell function is essential for
the maintenance of blood glucose homeostasis. Notably, defects in
the mass and function of β cells are required for type 2 diabetes
to develop (1). In the early
phase of glucose metabolism abnormality, the levels of blood sugar
are kept relatively stable through the compensatory function of
islet β cells. When the limits of this compensatory function are
exceeded, apoptosis of the pancreatic β cells increases (2). The mechanism may be associated with
disorders of mitochondrial function, oxidative stress and elevated
levels of proinflammatory cytokines (3). In vivo, estrogen treatment
has been demonstrated to decrease β cell apoptosis and restore a
certain degree of insulin secretion in mice (4). Estrogen is a steroid hormone that
serves an important role in physiological processes by binding with
intracellular receptors. However, estrogen receptors on the
membranes of pancreatic β cells are not yet completely understood.
The existence of a non-classical membrane estrogen receptor has
been described by Nadal et al (5) and Ropero et al (6). Other researchers have suggested a
possible role for G-protein coupled receptor 30 (GPR30) as an
estrogen receptor involved in the effects of estrogen in the
endocrine pancreas (7,8). In the review conducted by Ropero
et al (9), a model for the
roles of estrogen receptor β and GPR30 in the physiology of the
endocrine pancreas was presented, and it was suggested that the
GPR30 is expressed in mice and rat pancreatic β cells. GPR30 is a
17β-estradiol (17β-E2)-binding receptor, which is also known as the
G protein-coupled estrogen receptor (GPER) (10). The 17β-E2-mediated activation of
the GPER stimulates intracellular calcium mobilization and
phosphoinositide 3-kinase (PI3K) activation (11). The functions and importance of the
GPER in pancreatic function and glucose metabolism have been
elucidated, which has revealed the therapeutic potential of GPER
activity (12).
The PI3K/Akt pathway is normally activated by
extracellular signals in physiological conditions, such as growth
factors, cytokines and hormones. It has been demonstrated that G
protein-coupled receptors directly stimulate the p110β and p110γ
isoforms of PI3K via the βγ subunits of heterotrimeric G proteins,
and activate the p110δ isoform of PI3K in β cells by an unknown
mechanism (13–15). For this reason, it would be
interesting to detect whether 17β-E2 activates the PI3K/Akt
signaling pathway via the GPER in an insulin-secreting β-cell line
(INS-1).
Autophagy is a biological process by which
cytoplasmic macromolecules and unnecessary organelles are degraded
in membranous vesicles, and is a widely present biological
phenomenaonin eukaryotes (16,17). Autophagy serves an important role
in various human diseases (18).
It is becoming clear that the regulation of autophagic activity is
associated with tumor formation and progression, and is also
important to cancer therapy (19). In cancer cells, chemical
inhibitors of autophagy increase the apoptosis induced by
active-site mechanistic target of rapamycin (mTOR) inhibitors or
dual PI3K/mTOR inhibitors, which suggests that the PI3K/Akt pathway
has an important role in the process of autophagy (20,21). Autophagy of the mitochondria, also
known as mitophagy, is important for mitochondrial quality control,
and thus is essential in cellular energy provision, calcium
homeostasis, redox signaling and apoptotic signaling (22,23). The health of the mitochondria is
important, because mitochondria are essential to major cell
metabolic pathways, and are a major cause of cell death (24). Mitochondrial dynamics, including
fission and fusion, serve a key role in mitophagic signaling
(25,26). 17β-E2 has been indicated to
increase mitochondrial fusion, decrease fission processes and
modify the normal development and function of mitochondria in MCF-7
breast cancer cells (27).
According to the review of Zhang (28), it is possible to visualize
mitophagy by the co-localization of mitochondrial proteins with
lysosomal markers.
The aim of the present study was to investigate
whether 17β-E2 regulates the PI3K/Akt pathway through the GPER and
whether 17β-E2 inhibits mitophagy by activating the GPER/PI3K/Akt
signaling pathway and thereby protects INS-1 cells.
Materials and methods
Reagents
17β-E2 was obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). RPMI-1640 medium and fetal bovine serum (FBS)
were obtained from Gibco (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The selective GPER antagonist (G15) was bought from
Tocris Bioscience (Minneapolis, MA, USA). The PI3K inhibitor
(LY294002) and 4,6-diamidino-2-phenylindole (DAPI) were purchased
from Beyotime Institute of Biotechnology (Jiangsu, China). GPER
antibody [sc-48524-R; polyclonal, rabbit anti-mouse, rat, human;
western blotting (WB) 1:200; immunofluorescence (IF) 1:50],
lysosomal-associated membrane protein 2 (LAMP2) antibody (sc-8100;
polyclonal, goat anti mouse, rat and human; IF 1:50), the
translocase of the mitochondrial outer membrane complex 20 (TOM20)
antibody (sc-11415; polyclonal, rabbit anti mouse, rat and human;
WB 1:200, IF 1:50), microtubule-associated protein-1 light chain 3
(LC3) antibody (sc-376404; monoclonal, mouse anti mouse and rat; WB
1:100), and the mitochondrial heat-shock protein 60 (Hsp60)
antibody (sc-1052; polyclonal, goat anti mouse, rat and human; WB
1:200) were all obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Total/phospho-Akt (t-Akt/p-Akt) polyclonal
antibody (1:1,000; Ab8805/Ab8932) was obtained from Abcam
(Cambridge, MA, USA). β-actin antibody (AF0003; monoclonal, mouse
anti-human/mouse/rat; WB 1:1,000) was acquired from Beijing
Biosynthesis Biotechnology Co., Ltd. (Beijing, China). An
immunofluorescence staining kit with Alexa Fluor 555-labeled donkey
anti-rabbit immunoglobulin G (lgG) (P0179) was purchased from
Beyotime Institute of Biotechnology. IFKine® Green
conjugated donkey anti-goat IgG (A24231) was obtained from Abbkine
Scientific Co., Ltd. (Redlands, CA, USA).
Cell culture
The rat insulin-secreting β-cell line (INS-1) was
obtained from China Infrastructure of Cell Line Resources (Beijing,
China) and maintained in RPMI-1640 medium supplemented with 10%
FBS, L-glutamine, 50 mg/ml penicillin and 100 mg/ml streptomycin
(Beyotime Institute of Biotechnology). The cells were grown in a
humidified atmosphere containing 5% CO2 at 37°C. Prior
to the experiment, the INS-1 cells were grown in Petri dishes in a
serum-free medium for 24 h. The following day, the INS-1 cells were
treated with different concentrations of 17β-E2 (0, 1, 10 and 100
nM), or with 100 nM 17β-E2 plus 15 µM G15 or 20 µM
LY294002 for 24 h, respectively. For the combination treatments,
the cells were pretreated with G15 or LY294002 for 30 min prior to
treatment with 17β-E2.
IF analysis
The INS-1 cells were fixed in 4% paraformaldehyde
buffered with 0.1 M phosphate (pH 7.3) for 30 min at room
temperature and then washed with phosphate-buffered saline (PBS).
The cells were permeabilized with 0.1% Triton X-100 for 30 min and
washed with PBS. Following blocking with 5% bovine serum albumin
(Gibco) in Tris-buffered saline with Tween-20 (TBST) for 30 min at
room temperature, the INS-1 cells were incubated with specific
primary antibodies targeting LAMP2, TOM20 and GPER overnight at
4°C. The next day, the INS-1 cells were washed with PBS and then
incubated with donkey anti-goat IgG (1:1,000) or donkey anti-rabbit
IgG (1:1,000) secondary antibody for 30 min at 37°C. Subsequently,
the cells were stained with DAPI for 5 min at room temperature.
After washing with PBS for 15 min, the stained cells were viewed
using an Olympus FV1000 confocal laser-scanning microscope (Olympus
Corporation, Tokyo, Japan) with peak emission wavelengths of 518 nm
(green) and 565 nm (red).
Analysis using transmission electron
microscopy (TEM)
The cells were treated and collected by
trypsinization, fixed with 2.5% phosphate-buffered glutaraldehyde
at room temperature for 4 h, and post-fixed in 1%
phosphate-buffered osmium tetroxide at 4°C for 1.5 h. The cells
were embedded using Epon 812 epoxy resin at room temperature
overnight, sectioned, double stained with uranyl acetate and lead
citrate at room temperature for 23 min, and analyzed using a
JEM-1200EX transmission electron microscope (Jeol, Ltd., Tokyo,
Japan).
Protein preparation and western blot
analysis
The INS-1 cells were washed with cold PBS and
harvested in radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology) containing protease inhibitors,
including phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology) and phosphatase inhibitors (Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China). The cell lysates were incubated on ice
for 30 min, and then collected and centrifuged at 12,000 × g for 10
min at 4°C. The supernatants were collected, mixed with 5X loading
buffer and denatured by boiling for 10 min. The samples were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (10% gel used) and transferred onto polyvinylidene
fluoride membranes at 70 V for 1.5 h in a transfer buffer
containing Tris (20 mM; bioWORLD, Dublin, OH, USA), 150 mM glycine
(Beijing Solarbio Science and Technology Co., Ltd., Beijing, China)
and 20% methanol (Liaoning Xinxing Chemical Group Co., Ltd.,
Liaoning, China). The protein determination of the samples was made
using the Bicinchoninic Acid (BCA) method; 50 µg samples
were loaded per lane. The membranes were incubated in non-fat dry
milk for 120 min at room temperature, and were then washed thrice
with TBST for 30 min. The membranes were incubated with primary
antibodies in TBST overnight at 4°C. The membranes were then washed
and incubated with horseradish peroxidase-conjugated anti-species
secondary antibodies (1:1,000; A0208 and A0216; Beyotime Institute
of Biotechnology) for 2 h at room temperature, and were washed
thrice with TBST for 30 min. Proteins were visualized using the
BeyoECL plus kit (Beyotime Institute of Biotechnology) and
quantified using Quantity One software (Bio-Rad Laboratories Inc.,
Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean, and the differences between the means were analyzed by
one-way analysis of variance followed by a Dunnett post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. Statistical analysis was performed using the
Statistical Package for the Social Sciences (SPSS) 15.0 software
package (SPSS, Inc., Chicago, IL, USA).
Results
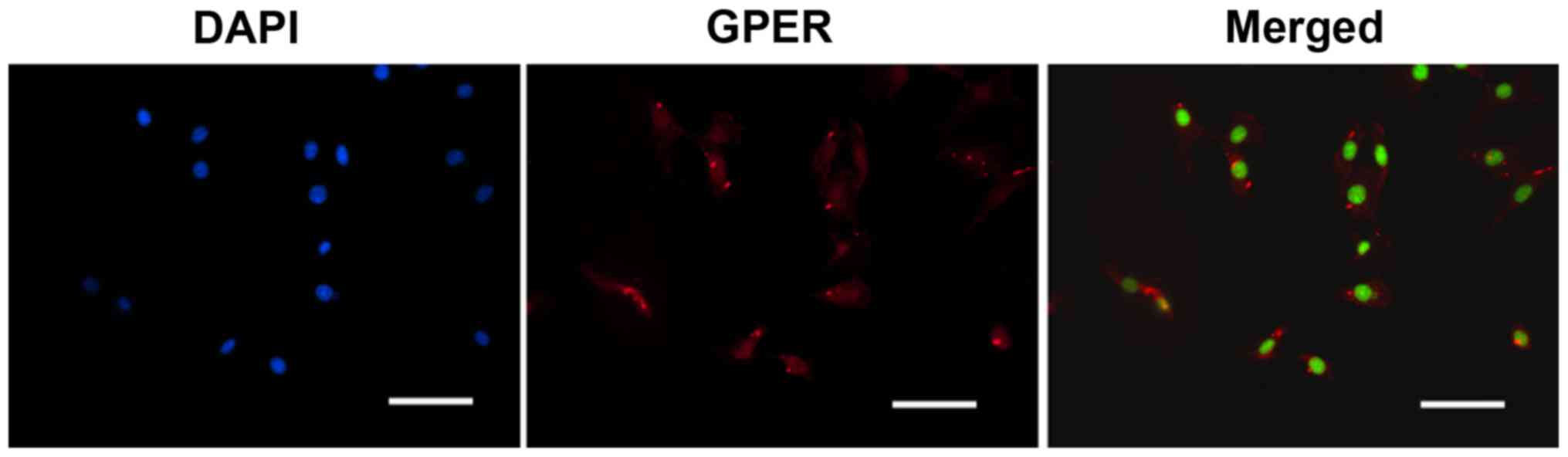
GPERs are expressed in INS-1 cells
Since it has been shown GPERs are expressed in MIN6
cells (9), a mouse β-cell line,
the present study aimed to detect the presence of GPERs in rat
INS-1 cells. The presence of GPERs in the INS-1 cells was
investigated using IF staining. As shown in Fig. 1, GPERs were present in the INS-1
cells. No GPER expression was detected in the negative controls
lacking primary antibody (data not shown).
17β-E2 regulates the PI3K/Akt pathway via
the GPER in INS-1 cells
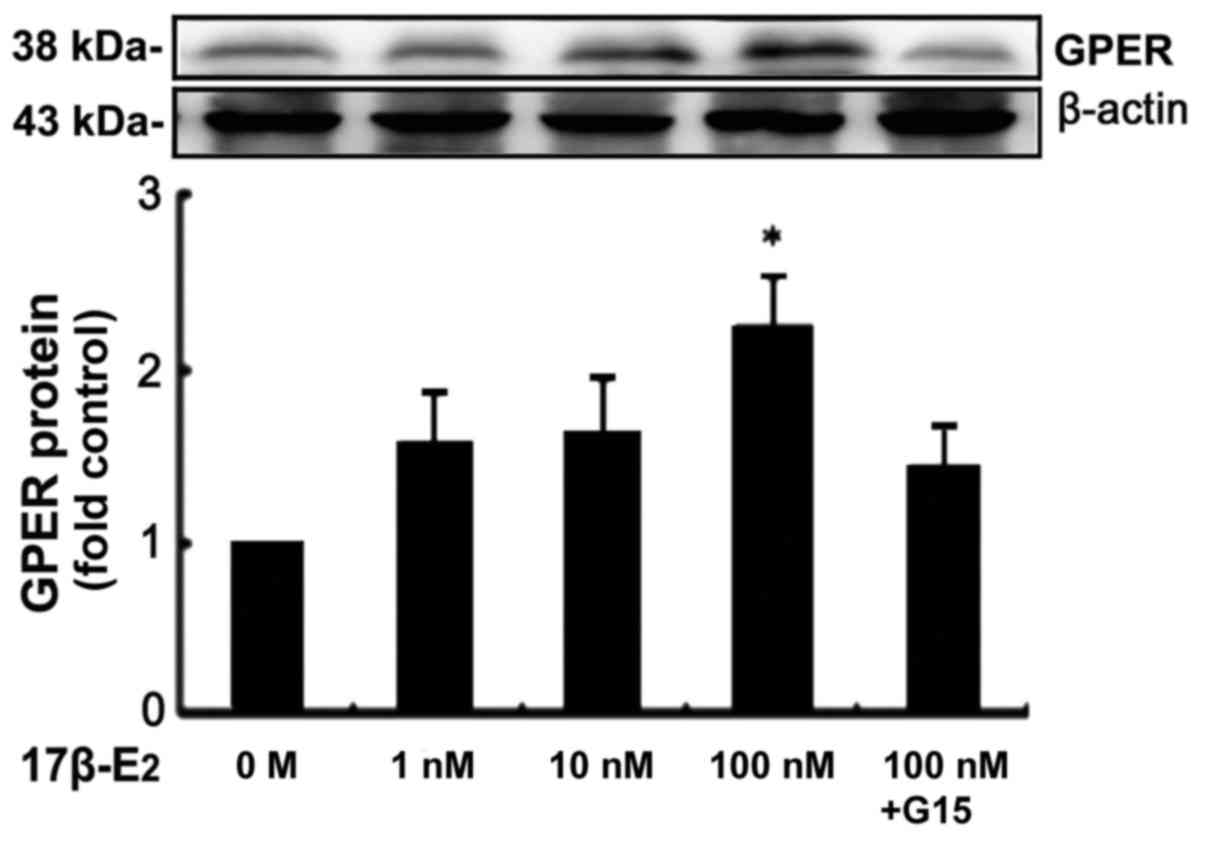
17β-E2 (1, 10 and 100 nM) caused the expression
levels of GPER protein to increase in an apparently dose-dependent
manner, with a significant increase detected at a concentration of
100 nM (P<0.05; Fig. 2). In
the 100 nM group, the expression level of GPER protein was
increased 2.2-fold in comparison with the control. However, the
stimulatory effect of 17β-E2 was eradicated by 15 µM G15, a
GPER-specific antagonist (Fig.
2).
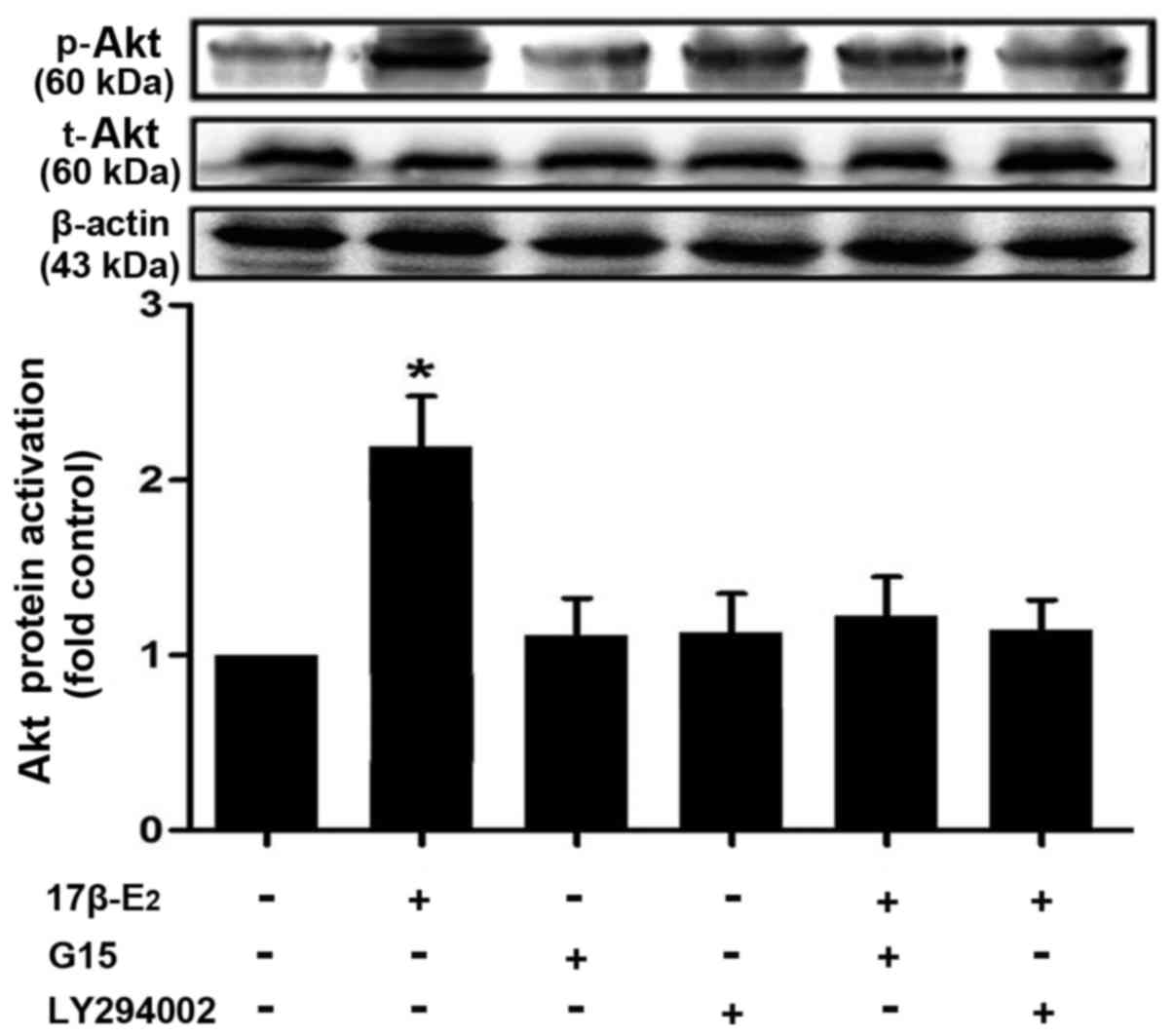
The ability of 17β-E2 to regulate the PI3K/Akt
pathway via the GPER in INS-1 cells was investigated via western
blot analysis. The results demonstrated that 100 nM 17β-E2
significantly increased the protein levels of p-Akt in INS-1 cells
(P<0.05; Fig. 3). However, the
protein expression level of t-Akt remained unchanged. The effect of
17β-E2 on Akt activation in the INS-1 cells was blocked by the PI3K
inhibitor LY294002 (20 µM) and the GPER inhibitor G15 (15
µM) (Fig. 3).
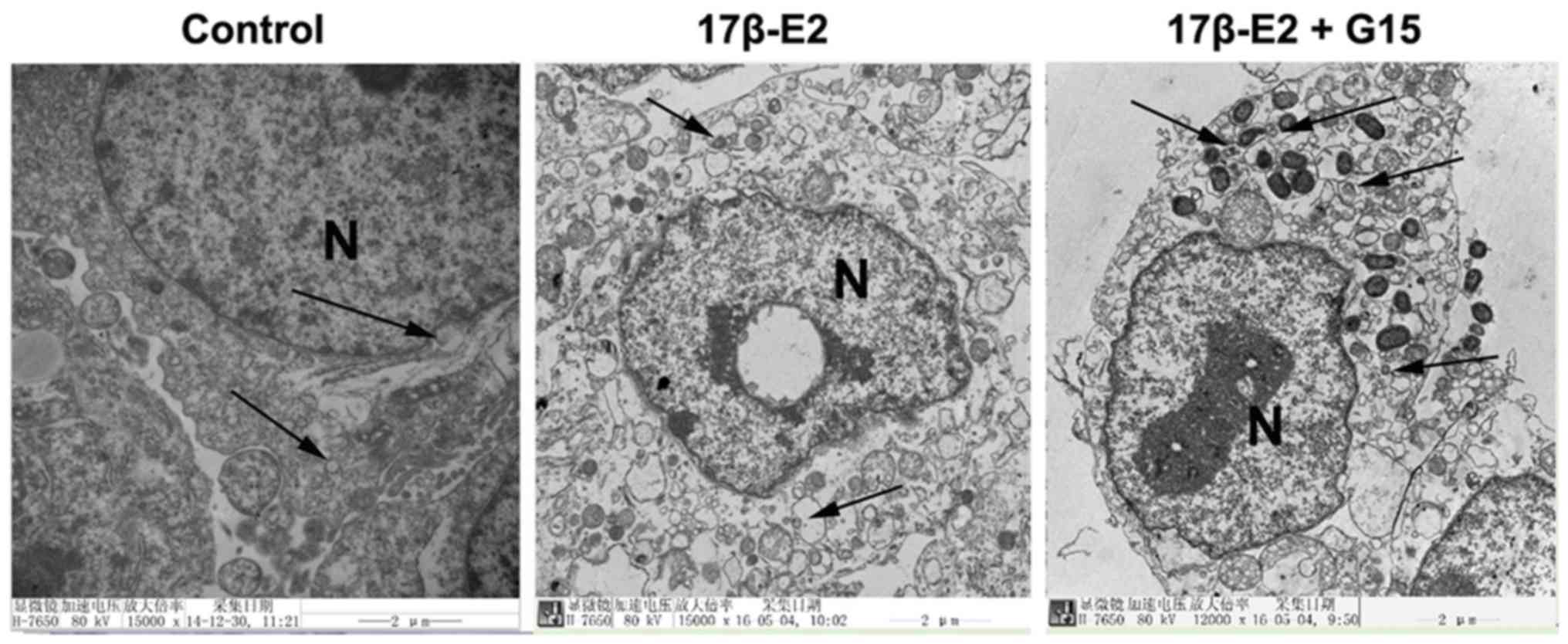
17β-E2 protects INS-1 cells from
mitophagy via the GPER
Mitophagy can be visualized by the detection of
mitophagosomes or autophagosomes using TEM or the co-localization
of lysosomes and mitochondria with mitophagic proteins by IF
staining. Whether 17β-E2 is involved in mitophagy in INS-1 cells
was investigated using these methods in the present study.
Mitophagosomes were detected in the INS-1 cells by
TEM, indicating that mitophagy occurs in INS-1 cells. Compared with
the control group, fewer mitophagosomes were observed in the INS-1
cells treated with 17β-E2 (Fig.
4). Greater numbers of double-membrane vacuoles were observed
in the INS-1 cells treated with 17β-E2 and G15 than that in the
cells with 17β-E2 only.
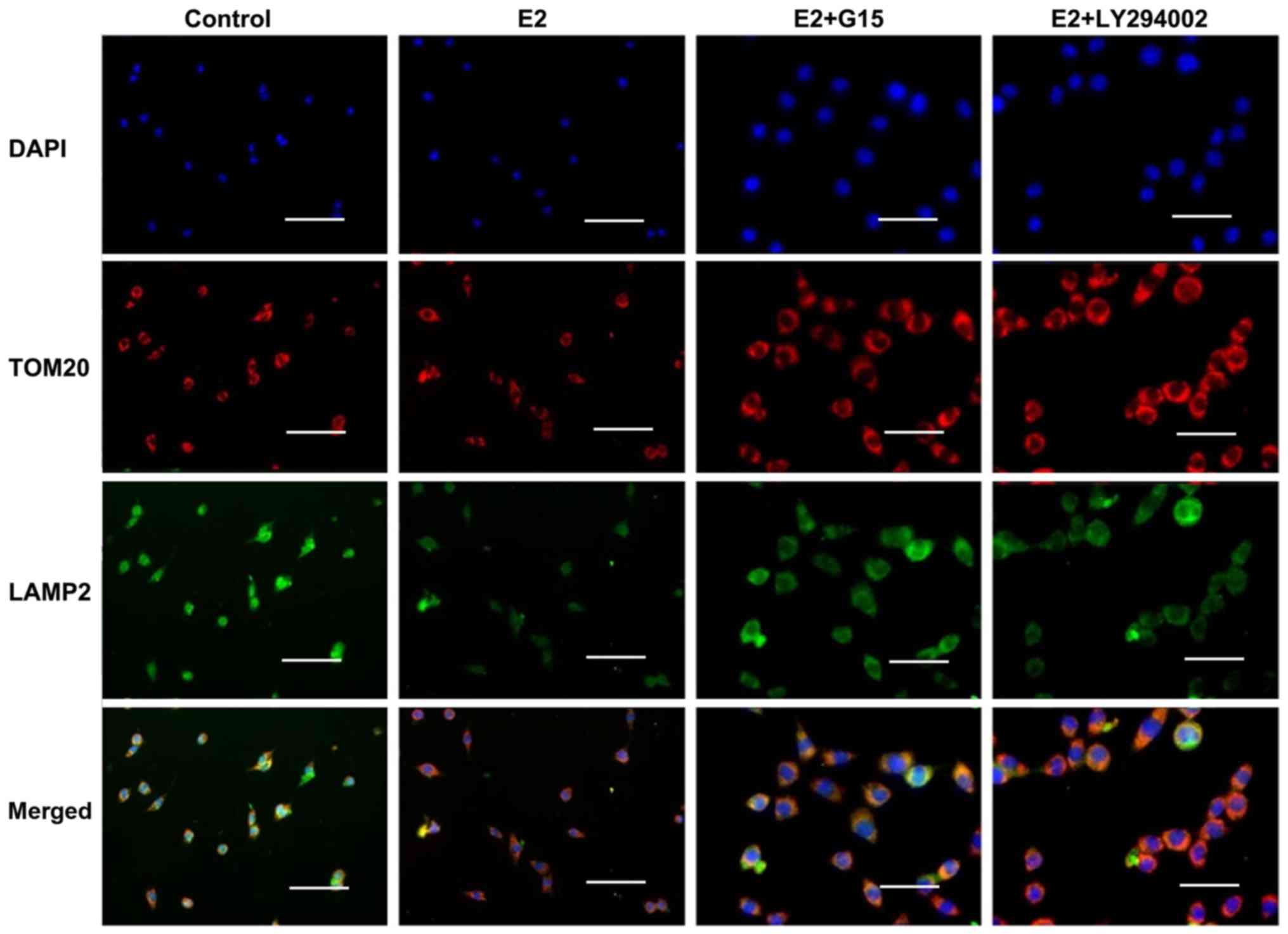
In addition, the co-localization of TOM20 with LAMP2
was detected by IF staining. The results demonstrated a reduction
in TOM20-positive granules and a co-localized reduction in LAMP2
expression in the INS-1 cells exposed to 17β-E2, indicating that
the numbers of mitophagosomes or autophagosomes were decreased
(Fig. 5). However, these effects
of 17β-E2 were eliminated by the presence of G15 (Fig. 5).
17β-E2 is involved in mitophagy through
the GPER/PI3K/Akt pathway
Whether 17β-E2 participates in mitophagy through the
PI3K/Akt pathway was then investigated. The results of IF staining
revealed that there were increased numbers of LAMP2-positive
granules with increased TOM20 expression in the INS-1 cells treated
with 17β-E2 and LY294002 compared with the cells treated with
17β-E2 alone (Fig. 5).
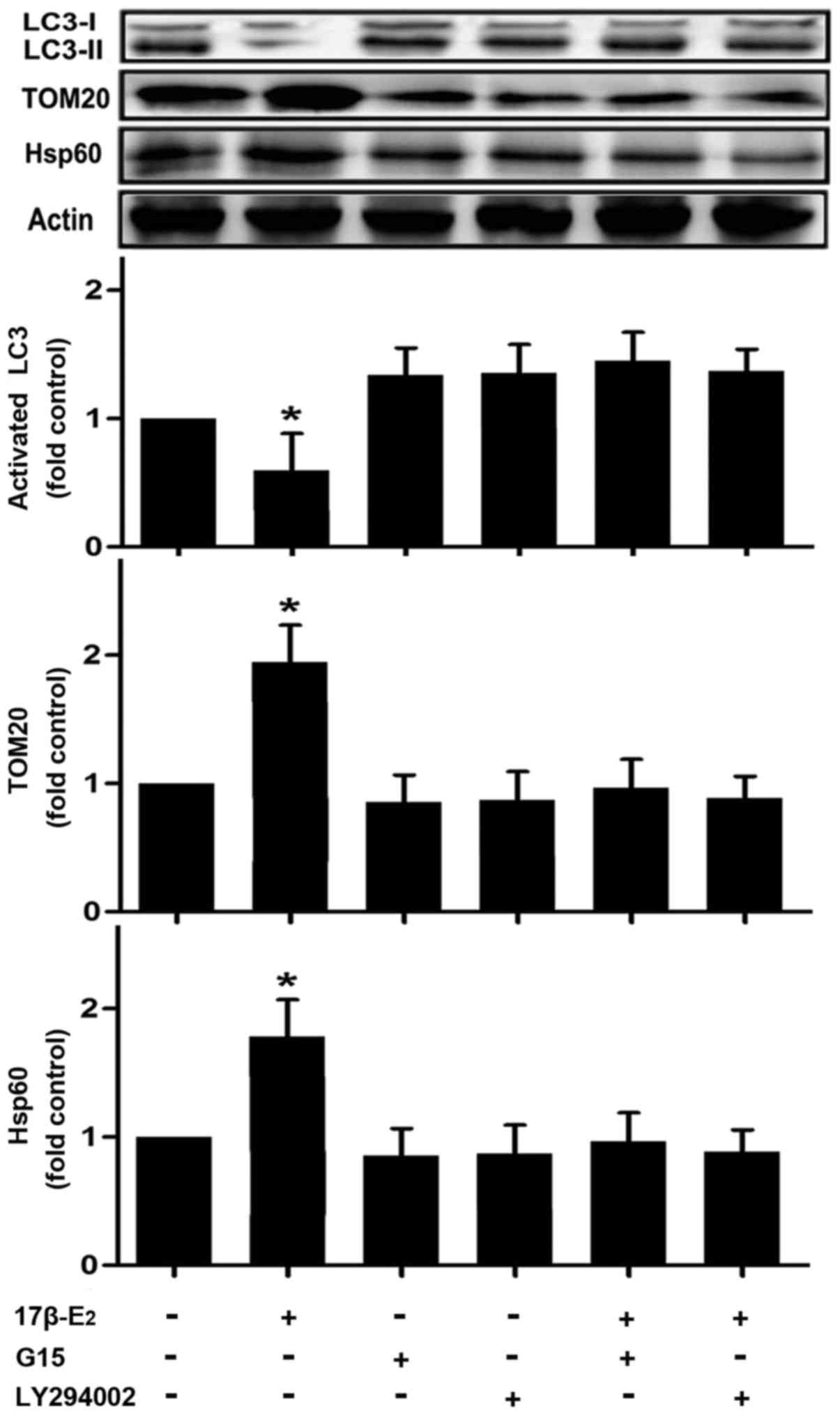
17β-E2 regulates the expression of LC3,
TOM20 and Hsp60 in INS-1 cells
The present study suggested that 17β-E2 may be
involved in mitophagy in INS-1 cells, on the basis of the detection
of mitophagosomes and autophagosomes by TEM and mitophagy-related
protein co-location by IF. To further investigate this, the
expression of LC3, TOM20 and Hsp60 was detected by western blot
analysis. LC3-II is converted from LC3-I, and serves as a typical
marker of completed autophagy (29). The exposure of INS-1 cells to
17β-E2 for 24 h resulted in the LC3-II protein levels being
significantly reduced compared with those of the control group
(Fig. 6). However, pretreatment
with LY294002 or G15 attenuated the 17β-E2-induced reduction in
LC3-II expression (Fig. 6). The
17β-E2 group also had significantly higher expression levels of
TOM20 and Hsp60 compared with the control group (Fig. 6). However, the effect of 17β-E2 on
these proteins was eradicated by pretreatment with LY294002 or G15
(Fig. 6).
Discussion
In the present study, the involvement of 17β-E2 in
the process of mitophagy in INS-1 cells was investigated. The
results indicated that 17β-E2 protects INS-1 cells from mitophagy,
with this regulatory effect likely occurring through the
GPER/PI3K/Akt pathway.
The PI3K/Akt signaling pathway is involved in almost
every aspect of the physiological and pathological functions of
cells, including growth, tumorigenesis, apoptosis and autophagy
(30–32). The study conducted by Ropero et
al (9) demonstrated that the
INS-1 rat β-cell line expresses GPERs. The present study confirmed
the expression of GPERs in INS-1 cells. It has been reported that
E2 stimulates insulin secretion and induces glucagon secretion via
the GPER in pancreatic islets (7). Therefore, the present study aimed to
detect whether 17β-E2 regulates the PI3K/Akt signaling pathway via
the GPER in INS-1 cells. Whether Akt phosphorylation levels are
modulated by 17β-E2 via a GPER-dependent pathway was tested in the
present study. The phosphorylation level of Akt was significantly
increased following stimulation with 17β-E2 for 24 h, and the
effect was blocked by pretreatment with the GPER and PI3K
antagonists G15 and LY294002, respectively, for 30 min, indicating
that the activation of the PI3K/Akt signaling pathway in INS-1
cells is regulated by 17β-E2 through the GPER. Additionally, 17β-E2
has been demonstrated to increase p-Akt levels via the GPER in
cardiomyocytes, which may rescue the heart from pathological
hypertrophy (33). The rapid
phosphorylation of Akt by E2 has been shown to upregulate miR144 in
SkBr3 breast cancer and HepG2 hepatocarcinoma cells, and this
process has been indicated to occur in cancer-associated
fibroblasts and cancer progression (34). Similar GPER-mediated modulatory
effects of E2 on Akt phosphorylation have been demonstrated in
endometrial cancer cells (35).
However, E2 did not activate the PI3K/Akt pathway via the GPER in
MCF-7 and MCF-10A cells (36).
Therefore, the mechanism by which 17β-E2 activates the PI3K/Akt
pathway remains unclear. To fully ascertain how 17β-E2 acts through
the GPER, the use of a specific agonist of this receptor (such as
G1) is planned in future research.
The association between 17β-E2 and mitophagy has
become a topic of particular research interest. Mitochondria in
pancreatic β cells are continuously recruited in fusion and fission
processes (37). In an animal
model of spinal cord injury, treatment with 17β-E2 significantly
attenuated cell death (38).
Furthermore, in a study conducted by Sastre-Serra et al
(27), 17β-E2 increased
mitochondrial fusion, decreased fission processes, and modified the
normal development and functions of mitochondria in MCF-7 breast
cancer cells through estrogen receptors. In the present study,
17β-E2 was demonstrated to protect INS-1 cells from mitophagy via
the GPER. The presence of mitochondria in autophagosomes
(mitophagosomes) was identified using TEM, which indicates that
mitophagy occurs in INS-1 cells. TEM analysis demonstrated that
there were few mitophagosomes and scarcely any autophagomes in the
INS-1 cells treated with 17β-E2. However, the number of
mitophagosomes and autophagomes was increased in the INS-1 cells
cultured with 17β-E2 following pretreatment with G15. Therefore, it
is suggested that the protective effect of 17β-E2 against mitophagy
may occur through the activation of the GPER. This observation may
be associated with inhibitory effect of 17β-E2 on the mitochondrial
fission process, and may also indicate that the GPER serves an
important role in this protective process.
It has been reported that PI3K/Akt signal
transduction prevents cells from undergoing apoptosis (30,39). The PI3K/Akt pathway, the p38
mitogen-activated protein kinase signaling pathway, and reactive
oxygen species participate in wogonoside-induced autophagy and
apoptosis (40). Inactivation of
the PI3K/Akt pathway prevents the translocation of damage-regulated
autophagy modulator to the mitochondria and induces apoptosis in
hepatocellular carcinoma cells by the mediation of mitophagy
(41). Furthermore, inhibition of
the PI3K/Akt/mTOR signaling pathway is accompanied by autophagy and
mitophagy in human glioblastoma cells, glioblastoma stem cells and
human prostate cancer (42,43). In the present study, the results
indicated that 17β-E2 regulated mitophagy in INS-1 cells through
the GPER/PI3K/Akt pathway. In addition, IF analysis of the INS-1
cells revealed that 17β-E2 reduced the number of LAMP2-labeled
lysosomes that were co-localized with TOM20-labeled mitochondria in
comparison with the cells treated with 17β-E2 following G15 or
LY294002 pretreatment, which exhibited an increased presence of
mitophagosomes, indicating that 17β-E2 alone reduced the formation
of mitophagosomes. These results are consistent with the TEM
results of the present study. Therefore, it is suggested that the
PI3K/Akt pathway is also involved in the mechanistic pathway by
which 17β-E2 exerts a protective effect on INS-1 cells via the
GPER. Furthermore, the results of western blot analysis also
supported this suggestion. As shown in Fig. 6, the expression of LC3-II was
decreased and the expression of TOM20 and Hsp60 was increased in
INS-1 cells exposed to 17β-E2, and this was partially consistent
with the observations concerning the 17β-E2-induced suppression of
mitophagy in INS-1 cells. mTOR is a downstream factor of the
PI3K/Akt signaling pathway, which has an important role in the
modulation of mitophagy (44–46). The inhibition of mTOR increases
mitophagic activity through a ubiquitin-like-conjugating enzyme
ATG3-dependent mechanism in natural killer cells (47). Therefore, it is hypothesized that
17β-E2 activates the PI3K/Akt signaling pathway by means of the
GPER, which may subsequently inhibit mitophagy via the regulation
of mTOR.
In conclusion, the results of the present study
demonstrate that 17β-E2 protects INS-1 rat insulinoma cells from
mitophagy via the GPER and acts through the PI3K/Akt signaling
pathway. These results provide novel insights for understanding the
pathophysiological functions of the GPER in pancreatic β cells.
Glossary
Abbreviations
Abbreviations:
|
17β-E2
|
17β-estradiol
|
|
GPER
|
G protein-coupled estrogen
receptor
|
|
INS-1 cells
|
insulin-secreting β-cell line
|
|
TOM20
|
the translocase of the mitochondrial
outer membrane complex 20
|
|
LAMP2
|
lysosomal-associated membrane protein
2
|
|
Hsp60
|
mitochondrial heat-shock protein
60
|
|
LC3
|
microtubule-associated protein-1 light
chain 3
|
Acknowledgments
The authors would like to thank the China Medical
University Affiliated Hospital Laboratory Center for kindly
providing the equipment. The present study was supported by the
National Natural Science Foundation of China (grant nos. 81470998,
81071460 and 81271996).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Kahn SE, Zraika S, Utzschneider KM and
Hull RL: The beta cell lesion in type 2 diabetes: There has to be a
primary functional abnormality. Diabetologia. 52:1003–1012. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marchetti P, Dotta F, Lauro D and Purrello
F: An overview of pancreatic beta-cell defects in human type 2
diabetes: Implications for treatment. Regul Pept. 146:4–11. 2008.
View Article : Google Scholar
|
|
3
|
Prentki M and Nolan CJ: Islet beta cell
failure in type 2 diabetes. J Clin Invest. 116:1802–1812. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geisler JG, Zawalich W, Zawalich K, Lakey
JR, Stukenbrok H, Milici AJ and Soeller WC: Estrogen can prevent or
reverse obesity and diabetes in mice expressing human islet amyloid
polypeptide. Diabetes. 51:2158–2169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nadal A, Ropero AB, Laribi O, Maillet M,
Fuentes E and Soria B: Nongenomic actions of estrogens and
xenoestrogens by binding at a plasma membrane receptor unrelated to
estrogen receptor alpha and estrogen receptor beta. Proc Natl Acad
Sci USA. 97:11603–11608. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ropero AB, Soria B and Nadal A: A
nonclassical estrogen membrane receptor triggers rapid differential
actions in the endocrine pancreas. Mol Endocrinol. 16:497–505.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mårtensson UE, Salehi SA, Windahl S, Gomez
MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N,
Hellstrand P, Grände PO, et al: Deletion of the G protein-coupled
receptor 30 impairs glucose tolerance, reduces bone growth,
increases blood pressure, and eliminates estradiol-stimulated
insulin release in female mice. Endocrinology. 150:687–698. 2009.
View Article : Google Scholar
|
|
8
|
Sharma G and Prossnitz ER: Mechanisms of
estradiol-induced insulin secretion by the G protein-coupled
estrogen receptor GPR30/GPER in pancreatic beta-cells.
Endocrinology. 152:3030–3039. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ropero AB, Pang Y, Alonso-Magdalena P,
Thomas P and Nadal A: Role of ERβ and GPR30 in the endocrine
pancreas: A matter of estrogen dose. Steroids. 77:951–958. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alexander SP, Davenport AP, Kelly E,
Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL,
Southan C, et al: CGTP Collaborators: The Concise Guide to
PHARMACOLOGY 2015/16: G protein-coupled receptors. Br J Pharmacol.
172:5744–5869. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Revankar CM, Cimino DF, Sklar LA,
Arterburn JB and Prossnitz ER: A transmembrane intracellular
estrogen receptor mediates rapid cell signaling. Science.
307:1625–1630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prossnitz ER and Barton M: The
G-protein-coupled estrogen receptor GPER in health and disease. Nat
Rev Endocrinol. 7:715–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suire S, Condliffe AM, Ferguson GJ, Ellson
CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers
ER, et al: Gbetagammas and the Ras binding domain of p110gamma are
both important regulators of PI(3)Kgamma signalling in neutrophils.
Nat Cell Biol. 8:1303–1309. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dbouk HA, Vadas O, Shymanets A, Burke JE,
Salamon RS, Khalil BD, Barrett MO, Waldo GL, Surve C, Hsueh C, et
al: G protein-coupled receptor-mediated activation of p110β by Gβγ
is required for cellular transformation and invasiveness. Sci
Signal. 5:ra892012. View Article : Google Scholar
|
|
15
|
Durand CA, Hartvigsen K, Fogelstrand L,
Kim S, Iritani S, Vanhaesebroeck B, Witztum JL, Puri KD and Gold
MR: Phosphoinositide 3-kinase p110 delta regulates natural antibody
production, marginal zone and B-1 B cell function, and autoantibody
responses. J Immunol. 183:5673–5684. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Duve C and Wattiaux R: Functions of
lysosomes. Annu Rev Physiol. 28:435–492. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Clark SL Jr: Cellular differentiation in
the kidneys of newborn mice studies with the electron microscope. J
Biophys Biochem Cytol. 3:349–362. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carayol N, Vakana E, Sassano A, Kaur S,
Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, et
al: Critical roles for mTORC2- and rapamycin-insensitive
mTORC1-complexes in growth and survival of BCR-ABL-expressing
leukemic cells. Proc Natl Acad Sci USA. 107:12469–12474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan QW, Cheng C, Hackett C, Feldman M,
Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA,
Debnath J, et al: Akt and autophagy cooperate to promote survival
of drug-resistant glioma. Sci Signal. 3:ra812010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mitchell T, Chacko B, Ballinger SW, Bailey
SM, Zhang J and Darley-Usmar V: Convergent mechanisms for
dysregulation of mitochondrial quality control in metabolic
disease: Implications for mitochondrial therapeutics. Biochem Soc
Trans. 41:127–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Andersson SG, Zomorodipour A, Andersson
JO, Sicheritz-Pontén T, Alsmark UC, Podowski RM, Näslund AK,
Eriksson AS, Winkler HH and Kurland CG: The genome sequence of
Rickettsia prowazekii and the origin of mitochondria. Nature.
396:133–140. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim I and Lemasters JJ: Mitochondrial
degradation by autophagy (mitophagy) in GFP-LC3 transgenic
hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol.
300:C308–C317. 2011. View Article : Google Scholar :
|
|
26
|
Twig G and Shirihai OS: The interplay
between mitochondrial dynamics and mitophagy. Antioxid Redox
Signal. 14:1939–1951. 2011. View Article : Google Scholar :
|
|
27
|
Sastre-Serra J, Nadal-Serrano M, Pons DG,
Roca P and Oliver J: Mitochondrial dynamics is affected by
17β-estradiol in the MCF-7 breast cancer cell line. Effects on
fusion and fission related genes. Int J Biochem Cell Biol.
44:1901–1905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J: Teaching the basics of autophagy
and mitophagy to redox biologists - mechanisms and experimental
approaches. Redox Biol. 4:242–259. 2015. View Article : Google Scholar
|
|
29
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: A target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brazil DP, Yang ZZ and Hemmings BA:
Advances in protein kinase B signalling: AKTion on multiple fronts.
Trends Biochem Sci. 29:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee TM, Lin SZ and Chang NC: Both GPER and
membrane oestrogen receptor-α activation protect ventricular
remodelling in 17β oestradiol-treated ovariectomized infarcted
rats. J Cell Mol Med. 18:2454–2465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vivacqua A, De Marco P, Santolla MF,
Cirillo F, Pellegrino M, Panno ML, Abonante S and Maggiolini M:
Estrogenic gper signaling regulates mir144 expression in cancer
cells and cancer-associated fibroblasts (cafs). Oncotarget.
6:16573–16587. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsai CL, Wu HM, Lin CY, Lin YJ, Chao A,
Wang TH, Hsueh S, Lai CH and Wang HS: Estradiol and tamoxifen
induce cell migration through GPR30 and activation of focal
adhesion kinase (FAK) in endometrial cancers with low or without
nuclear estrogen receptor α (ERα). PLoS One. 8:e729992013.
View Article : Google Scholar
|
|
36
|
Wróbel AM and Gregoraszczuk EL: Action of
methyl-, propyl-and butylparaben on GPR30 gene and protein
expression, cAMP levels and activation of ERK1/2 and PI3K/Akt
signaling pathways in MCF-7 breast cancer cells and MCF-10A
non-transformed breast epithelial cells. Toxicol Lett. 238:110–116.
2015. View Article : Google Scholar
|
|
37
|
Stiles L and Shirihai OS: Mitochondrial
dynamics and morphology in beta-cells. Best Pract Res Clin
Endocrinol Metab. 26:725–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sribnick EA, Matzelle DD, Ray SK and Banik
NL: Estrogen treatment of spinal cord injury attenuates calpain
activation and apoptosis. J Neurosci Res. 84:1064–1075. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang H, Zhang J, Zhu H, Li H and Zhang X:
Nerve growth factor prevents the apoptosis-associated increase in
acetylcholinesterase activity after hydrogen peroxide treatment by
activating Akt. Acta Biochim Biophys Sin (Shanghai). 39:46–56.
2007. View Article : Google Scholar
|
|
40
|
Zhang L, Wang H, Cong Z, Xu J, Zhu J, Ji X
and Ding K: Wogonoside induces autophagy-related apoptosis in human
glioblastoma cells. Oncol Rep. 32:1179–1187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu K, Shi Y, Guo XH, Ouyang YB, Wang SS,
Liu DJ, Wang AN, Li N and Chen DX: Phosphorylated AKT inhibits the
apoptosis induced by DRAM-mediated mitophagy in hepatocellular
carcinoma by preventing the translocation of DRAM to mitochondria.
Cell Death Dis. 5:e10782014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ma J, Meng F, Li S, Liu L, Zhao L, Liu Y,
Hu Y, Li Z, Yao Y, Xi Z, et al: Autophagy induction by
endothelial-monocyte activating polypeptide II contributes to the
inhibition of malignant biological behaviors by the combination of
EMAP II with rapamycin in human glioblastoma. Front Mol Neurosci.
8:742015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu YQ, Ji Y, Li XZ, Tian KL, Young CY,
Lou HX and Yuan HQ: Retigeric acid B-induced mitophagy by oxidative
stress attenuates cell death against prostate cancer cells in
vitro. Acta Pharmacol Sin. 34:1183–1191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y,
Zhang T, Frank JA, Bower KA, Shi X, et al: Autophagy is a
protective response to ethanol neurotoxicity. Autophagy.
8:1577–1589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhuang W, Qin Z and Liang Z: The role of
autophagy in sensitizing malignant glioma cells to radiation
therapy. Acta Biochim Biophys Sin (Shanghai). 41:341–351. 2009.
View Article : Google Scholar
|
|
46
|
Edinger AL and Thompson CB: An activated
mTOR mutant supports growth factor-independent, nutrient-dependent
cell survival. Oncogene. 23:5654–5663. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
O'Sullivan TE, Johnson LR, Kang HH and Sun
JC: BNIP3- and BNIP3L-mediated mitophagy promotes the generation of
natural killer cell memory. Immunity. 43:331–342. 2015. View Article : Google Scholar : PubMed/NCBI
|