Introduction
Acute myocardial infarction (AMI) is still one of
the leading causes of morbidity and mortality although medical and
device therapy has been markedly improved (1). AMI causes the dysfunction of
cardiomyocytes and induces damage to mitochondria (2). Dysregulated mitochondria can lead to
the gross production of reactive oxygen species (ROS) as an
inevitable by-product, which could cause damage to cellular DNA and
protein, even programmed cell death (3). Therefore, clearing away the damaged
mitochondria is essential for mitochondrial quality control and
normal cellular functions (4,5).
Pigment epithelial-derived factor (PEDF), a
multifunctional glycoprotein (6),
protects against hypoxia-induced apoptosis and necroptosis in
primary cardiomyocytes (7). In
the study of PEDF protecting hypoxic cardiomyocytes, we first
observed that the mitochondrial density of cardiomyocytes treated
with PEDF was decreased significantly comparing with that under
hypoxia only. Previous studies have shown that enhancement of
mitophagy or mitochondrial fusion is impacted by the decrease of
mitochondrial density (8,9). Studies have linked damaged
mitochondria to progression of heart failure and age associated
cardiac pathologies (10,11). Recent studies have suggested that
mitophagy plays a specific role in eliciting cardioprotective
benefits by removing damaged mitochondria (12,13). However, whether PEDF could
decrease the mitochondrial density of hypoxic cardiomyocytes via
mitophagy and/or mitochondrial fusion and whether this effect is
associated with cardioprotection remain unclear.
Recent study revealed adipose triglyceride lipase
(ATGL) as a receptor for PEDF in retinal epithelial cells and
cardiomyocytes (14,15). Based on the study of Subramanian
et al, we used PEDF-R for the 80-kDa receptor protein which
interacts with PEDF on the cell surface (16). Notari et al used a
cell-free system to show that this interaction induced lipase
activity (14). PEDF-R is the key
enzyme of lipid catabolism and catalyzes the lipid lipolysis
cascade, generating free fatty acids (FFAs) and diacylglycerol
(DAG) (17). In an earlier study
we demonstrated that PEDF could increase the level of FFAs in
cardiomyocytes after AMI via PEDF-R (15). Although Tan et al have
reported induction of autophagy by palmitic acid (PA) via DAG-PKC-α
pathway (18), the signaling
pathway linking PA stimulation to mitophagy in response to PEDF
remains to be determined.
The protein kinase C (PKC) family plays a role
downstream of many signal transduction pathways (19,20) and PKC-α is the predominant
conventional PKC isoform expressed in the mouse, rat, and human
heart (21,22). Mitophagy plays a major role in
mitochondrial quality control through three pathways, involving
Parkin, Nix (also known as BNIP3L) and FUNDC1 (23–25). Particularly, recent studies
suggest that ULK1 and SRC have a more specific effect on mitophagy
through interacting with its substrate FUNDC1 (24,26). However, it is not clear whether
PKC-α could modulate mitophagy or not.
Here, we first found PEDF decreased the
mitochondrial density of cardiomyocytes via promoting mitophagy
under hypoxic condition. In addition, PEDF-mediated mitophagy was
found to serve as a survival mechanism of cardiomyocytes under
hypoxic environment. Furthermore, we identified a novel signaling
pathway, PEDF/PEDF-R/PA/DAG/PKC-α/ULK1/FUNDC1, associated with
PEDF-mediated mitophagy.
Materials and methods
Antibodies and reagents
Anti-LC3B antibody was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The Go6976 and bafilomycin A1
(BAF1) were obtained from Selleck, Inc. (Houston, TX, USA).
Anti-phospho-PKC-α antibody was purchased from Millipore
(Billerica, MA, USA). Diacylglycerol (DAG) enzyme-linked
immunosorbent assay (ELISA) kit was purchased from USCN, Inc.
(Wuhan, China). MitoTracker® Red CMXRos was purchased
from Invitrogen, Inc. (Paisley, UK). Bromodeoxyuridine (BRDU) was
purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Anti-PKC-α
antibody was purchased from Bioworld Technology, Inc. (St. Louis
Park, MN, USA). Anti-Mfn1, Mfn2, Opa1, Nix, Parkin and ULK1
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Alexa Fluor 488 was purchased from Jackson
ImmunoResearch Inc. (West Grove, PA, USA). Anti-FUNDC1 polyclonal
antibody was purchased from Aviva PLC, Inc. (San Diego, CA, USA).
Cell counting kit-8 (CCK-8) was purchased from Dojindo Molecular
Technologies, Inc. (Kumamoto, Japan). LDH cytotoxicity assay kit
was purchased from Keygen Biotech, Co. (Nanjing, China).
Recombinant lentivirus constructs and
viral production
Recombinant lentivirus (LV) was prepared as
described previously (27). PEDF
overexpression plasmids and the RNAi vector PEDF-R-RNAi_LV of the
PEDF-R gene producing PEDF-R shRNA were successfully constructed
and then successfully packaged in 293T cells. The concentrated
titer of virus suspension was 2×1012 TU/l.
Induction of AMI
All the procedures were performed following the
guidelines of the Directive 2010/63/EU of the European Parliament,
and have been approved by the Ethics Committee for Animal
Experimentation of the Institutions where experiments were carried
out. Adult male Sprague-Dawley (SD) rats (200–250 g) were purchased
from the Experimental Animal Center of Xuzhou Medical University.
Myocardial ischemia was induced by ligation of the left-anterior
descending coronary artery (LAD) in anesthetized rats, as described
previously (28). The animal
models were randomly divided into four groups: normal; normal+PEDF,
PEDF-lentivirus was transferred before surgery; AMI (1, 2, 3 and 4
h); AMI+PEDF (1, 2, 3 and 4 h). The rats were anaesthetized with
ketamine at 100 mg/kg and xylazine at 10 mg/kg and maintained under
anesthesia using isoflurane (1.5–2.0%) mixed with air. During the
surgical procedure, the absence of the pedal reflex was used as an
indication that a surgical plane of anesthesia was maintained. With
the animal lying flat, left thoracotomy was performed through the
fourth intercostal space, PEDF-lentivirus (2×107 TU) in
20 µl enhanced infection solution (ENIS, pH 7.4) was
delivered with a 20-µl syringe and 25-gauge needle into the
myocardium along LAD. Control animals received an equivalent volume
of lentivirus vector in ENIS. The chest cavity was then closed.
After reinstallation of spontaneous respiration, animals were
extubated and allowed to recover from anesthesia. Buprenorphine was
administered at 0.5 mg/kg for postoperative analgesia. In the same
way, LAD was ligated with 6-0 silk suture (Ethicon; Johnson and
Johnson, Somerville, NJ, USA) after 7 days. Animals were sacrificed
with an overdose of sodium pentobarbitone (100 mg/kg, i.v.), and
their hearts were harvested at 1, 2, 3 and 4 h after induction of
MI for further analysis. Sham-operated rats underwent the same
procedure, excluding coronary artery ligation.
Primary cardiomyocyte isolation, culture
and infection
Cardiomyocytes were obtained from 1–3-day-old
neonatal SD rats as previously described (7,29,30). Briefly, neonatal rats were
sacrificed by rapid decapitation and hearts were rapidly removed
and placed into dishes on ice, then hearts were dissected and
minced into 1 mm3 pieces with sharp scissors, then
transferred to a sterile tube. The minced tissue was digested in a
phosphate-buffered saline (PBS) solution supplemented with 0.5%
trypsin, 0.1% collagenase and 0.02% glucose for 5 min at 37°C. Then
cells were incubated for 1 h in the presence of 0.1 mmol/l
bromodeoxyuridine to selectively enrich cardiomyocytes. The
inclusion of BRDU resulted in inhibition of the growth of cardiac
fibroblasts. The resultant cell suspension (10,000–12,000
cells/cm2) was plated onto 48 well culture plate in
DMEM/low glucose (HyClone) supplemented with 10% fetal bovine serum
and 100 mg/ml penicillin/streptomycin at 37°C in a humidified
atmosphere containing 5% CO2. Hypoxia was achieved by
culturing the cells in D-Hank's liquid with glucose deprivation in
a tri-gas incubator (Heal Force, Shanghai, China) saturated with 5%
CO2/1% O2 at 37°C for the indicated
times.
Western blotting
For western blot analysis the cells were solubilized
in lysis buffer [100 mmol/l Tris-HCl, 4% sodium dodecyl sulfate
(SDS), 20% glycerine, 200 mmol/l DTT and protease inhibitors, pH
6.8]. Protein extraction of both the cytosolic and mitochondrial
fractions was performed using a multiple centrifugation method as
described previously (31,32).
Medium from treated cells was harvested, spun at 800 × g for 5 min
and supernatant filtered (0.45 mm). Membrane fraction lysates were
prepared as described previously. Proteins were precipitated with
trichloroacetic acid. Protein samples were denatured by boiling for
5 min with an equal volume of 2X Tris-glycine SDS buffer. Protein
was separated by 7–12% SDS-PAGE and transferred to nitrocellulose
membrane (Millipore). After blocking with 5% non-fat milk/PBS-T for
3 h at room temperature, the membranes were incubated with the
primary antibodies overnight at 4°C. Then, fluorescently labeled
secondary antibody (Rockland, Limerick, PA, USA) was added for 1 h
and subsequently scanned by the Odyssey Infrared Imaging system
(LI-COR Biosciences, Waltham, MA, USA). In these experiments,
β-actin and COX IV were used as loading controls for the whole
cellular and mitochondrial proteins, respectively.
Immunofluorescence
Cardiomyocytes were grown in 48-well plates. After
respective treatments for 4 h, cardiomyocytes were washed twice
with PBS, and fixed with freshly prepared 4% paraformaldehyde at
room temperature for 15 min. Antigen accessibility was increased by
treatment with 2% Triton X-100 for 10 min. Then cardiomyocytes were
blocked with 3% BSA for 30 min. Following incubation with primary
and secondary antibodies, cardiomyocytes were incubated with
primary antibodies overnight at 4°C. After washing, cardiomyocytes
were stained with a secondary antibody for 1 h at room temperature.
Each time after the operation, cardiomyocytes were washed thrice
with PBS, and each time for 5 min. Cardiomyocytes were captured and
analyzed using TCS SP8 STED 3X (Leica, Wetzlar, Germany).
Analysis of PA using gas
chromatography-mass spectrometry (GC-MS)
GC-MS analysis was performed on an Agilent 7890A gas
chromatograph coupled with an Agilent 5975C Series MSD (Agilent
Technologies, Palo Alto, CA, USA). An Agilent DB-23 column (60 m ×
0.25 mm × 0.15 µm) was used for separation. The initial oven
temperature was 50°C for 1 min and then raised to 178°C at 8°C/min
for 4 min, followed by further increases at 4°C/min to 186°C,
1°C/min to 190°C for 1 min and 15°C/min to 220°C for 10 min. The
injection column was 1 µl in splitless mode. The helium
carrier gas flow rate was set at 1 ml/min. Detector voltage of EI
was 70eV and the mass range was set at m/z 50-550.
Transmission electron microscopy
(TEM)
Samples of heart tissue were fixed with 2.5%
glutaraldehyde overnight. Subsequently, samples were incubated
while protected from light 1% osmium tetroxide for 2 h. After
washing in distilled water, the samples were incubated in 2% uranyl
acetate for 2 h at room temperature and then dehydrated in grades
ethanol concentrations. Finally, the samples were embedded in molds
with fresh resin. Ultrathin sections were obtained with an EM UC7
(Leica), stained with lead citrate and examined with a Tecnai G2
T12 (FEI, USA).
ELISA analysis
After respective treatments, cardiomyocytes were
collected and, after addition of phenylmethylsulfonyl fluoride
(PMSF), then the medium from treated cardiomyocytes was harvested,
spun at 800 × g for 5 min and supernatant filtered (0.45
µm). Samples were transferred to antibody-coated plates. The
concentration of DAG was determined by competitive inhibition
ELISA. Plate preparation and assay procedure were performed
according to the manufacturer's recommendations. The absorbance was
read with a reference wavelength of 450 nm. DAG concentration for
each sample was calculated after generating a standard curve by a
microplate reader (BioTek Synergy2; BioTek, Winooski, VT, USA).
Cardiomyocyte viability tests
Cardiomyocytes were seeded in 96-well plates at a
concentration of 1×104 cells/ml. After treatment, cell
viability was tested by using the CCK-8 kit. Absorbance at 450 nm
was measured with a microplate reader (BioTek Synergy2;
BioTek).
LDH release assay
The cardiomyocytes were seeded in 96-well plates
(1×104 cells/ml) and treated. The activity of LDH in
cardiomyocytes released into the medium following treatment was
assessed as previously described by a microplate reader (BioTek
Synergy2; BioTek) analysis at 440 nm using an LDH cytotoxicity
assay kit, according to the manufacturer's instructions.
Three-dimensional surface reconstruction
and mitochondrial volume calculations
Three-dimensional surface-reconstruction image raw
data sets were collected by spinning-disc confocal microscopy. The
Z-Stack was deconvolved and three-dimensional surface
reconstruction was carried out with IMARIS 7.0.0 software
(Bitplane, South Windsor, CT, USA). All confocal microscopy was
carried out on a confocal laser scanning microscope (Olympus FV10i;
Olympus, Tokyo, Japan). Confocal slice thickness was typically kept
at 0.6 µm consistently for each fluorescence channel, with
ten slices typically being taken to encompass the three-dimensional
entirety of the cells in the field of view. Maximum-intensity
projections of each region were calculated for subsequent
quantification and analysis. To quantify mitochondrial volume,
images were deconvolved with ImageJ and Z-Stack analysis of the
threshold images volume-reconstituted using the VolumeJ plug-in,
and volumes of mitochondria were quantified using the ImageJ-3D
object counter plug-in. Care was taken to ensure consistency of
thresholding over multiple fields of view and samples. Once this
process was complete, Object Counter 3D under the particle analysis
algorithm within ImageJ was employed to measure the volume of
mitochondria and area of cells within a specified region of
interest. Calculation for the adjusted total mitochondrial volume
per cell was as follows: (percentage of total volume of
mitochondria)/(percentage of total area of cell).
Statistical analysis
The results are expressed as the mean ± standard
deviation (SD). Statistical analysis of the results was carried out
using the repeated-measures analysis of the variance (ANOVA) or
two-way ANOVA followed by the Tukey's Honestly Significant
Difference (HSD) test for multiple comparisons. The significance
level was set at p<0.05.
Results
PEDF decreases the mitochondrial density
of primary cardiomyocytes under hypoxic condition
We have previously documented that PEDF has a
protective effect against hypoxia-induced apoptosis and necroptosis
in primary cardiomyocytes and H9c2 cells (7,33).
To further confirm the observation that PEDF exerts a protective
effect in hypoxic primary cardiomyocytes, CCK-8 assays were
performed. As shown in Fig. 1A,
onset of hypoxia up to 12 h led to an immediate decrease in the
viability of cardiomyocytes, while treatment with PEDF resulted in
a marked increase in the viability of cardiomyocytes compared with
hypoxia group. Compared with hypoxia group at the indicated times,
cardiomyocytes treated with PEDF resulted in a marked decrease in
the level of COX IV (Fig. 1B),
which is used as an index of mitochondrial density. Besides,
mitochondrial density analyzed by immunostaining with MitoTracker
Red in PEDF-treated cardiomyocytes was much less than that in
control groups (Fig. 1C). We also
quantitatively measured the total mitochondrial volume within the
cardiomyocytes by a commonly used three-dimensional imaging
technique followed by analysis with ImageJ software. As expected,
the total mitochondrial volume was significantly reduced after
addition of PEDF compared with hypoxia group (Fig. 1D). Taken together, the effect of
PEDF on decreasing mitochondrial density of hypoxic cardiomyocytes
were first observed and the results here suggest that compared with
hypoxia group, PEDF could further reduce the mitochondrial density
of primary cardiomyocytes.
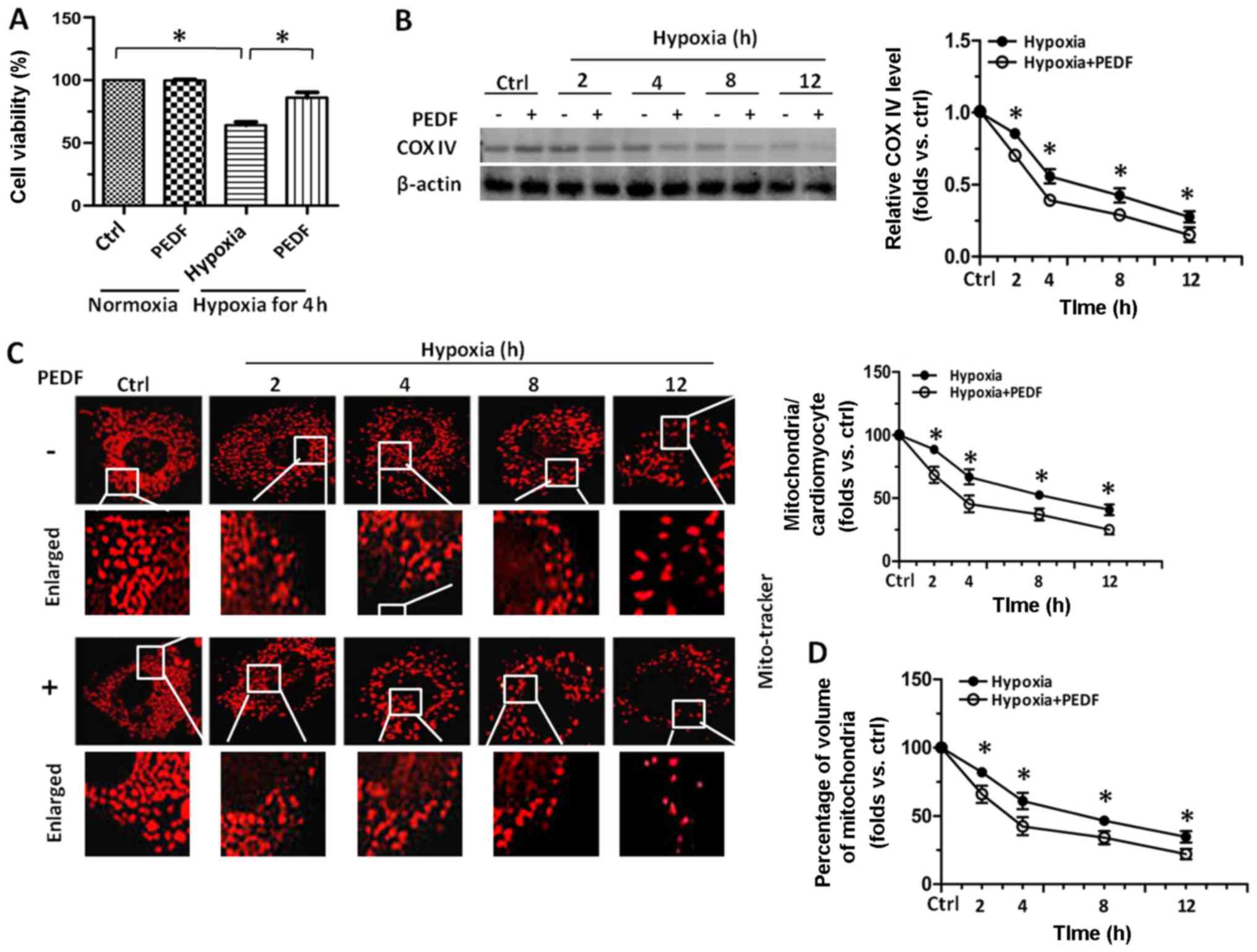 | Figure 1Pigment epithelial-derived factor
(PEDF) decreased the mitochondrial density of primary
cardiomyocytes under hypoxic condition. Primary cardiomyocytes were
maintained in normoxic or hypoxic conditions for 0, 2, 4, 8 and 12
h with or without PEDF (10 nM). (A) Cell viability was assayed by
the cell counting kit-8 (CCK-8). Approximately 1×104
cells were grown in each well of 96-well plates with 100 µl
medium and the absorbance at 450 nm was directly proportional to
the number of viable cells (n=4, *p<0.05). (B)
Samples were collected for western blotting to analyze the level of
mitochondrial protein COX IV by ImageJ software (n=4,
*p<0.05). (C) Samples were stained with MitoTracker
Red. For each indicated times, total mitochondrial number per
cultured cardiomyocytes was quantified from stacks of images
through the entire thickness of cardiomyocytes by ImageJ software
(*p<0.05, n=90 cardiomyocytes from three independent
experiments; scale bar, 20 µm). (D) Five randomly picked
regions of each sample were captured by confocal z-axis scanning
and the total volume of mitochondria was calculated and quantified
(*p<0.05, n=90 cardiomyocytes from three independent
experiments; scale bar, 20 µm). Data are expressed in fold
induction, relative to control. Values are means ± SD. |
PEDF plays a protective role in primary
cardiomyocytes through promoting mitophagy, and had no effect on
mitochondrial fusion
Mitophagy and mitochondrial fusion are closely
related with the change of mitochondrial number (8,9,34,35). Therefore, we investigated the role
of mitophagy and mitochondrial fusion in PEDF-decreased
mitochondrial density of hypoxic cardiomyocytes. As shown in
Fig. 2A, with the addition of
PEDF, we observed an increase of LC3-II level at the various time
points. PEDF groups reached a maximum after 4 h hypoxia, while
hypoxia groups reached a maximum after 8 h. However, the levels of
Opa1, Mfn1 and Mfn2 had no significant difference between the two
groups throughout the observation periods. Besides, we found that
PEDF could induce the increase of mitophagic flux (Fig. 2B). The results of CCK-8 assays
showed that treatment with PEDF up to 4 h resulted in a marked
increase in the viability of cardiomyocytes compared with hypoxia
group, while the addition of lysosome inhibitor BAF1 (24,26,36) significantly reduced the viability
of cardiomyocytes compared with PEDF-treated cardiomyocytes
(Fig. 2C). LDH released assays
obtained a similar conclusion (Fig.
2D). Fig. 2E shows the total
mitochondrial volume was markedly decreased after addition of PEDF
compared with hypoxia group, while the addition of BAF1
significantly increased the mitochondrial volume of cardiomyocytes.
The results demonstrated that PEDF can promote hypoxic
cardiomyocyte mitophagy, and has no significant effect on
mitochondrial fusion. More importantly, these results also suggest
the possibility that PEDF-induced mitophagy is important for cell
survival under hypoxic condition.
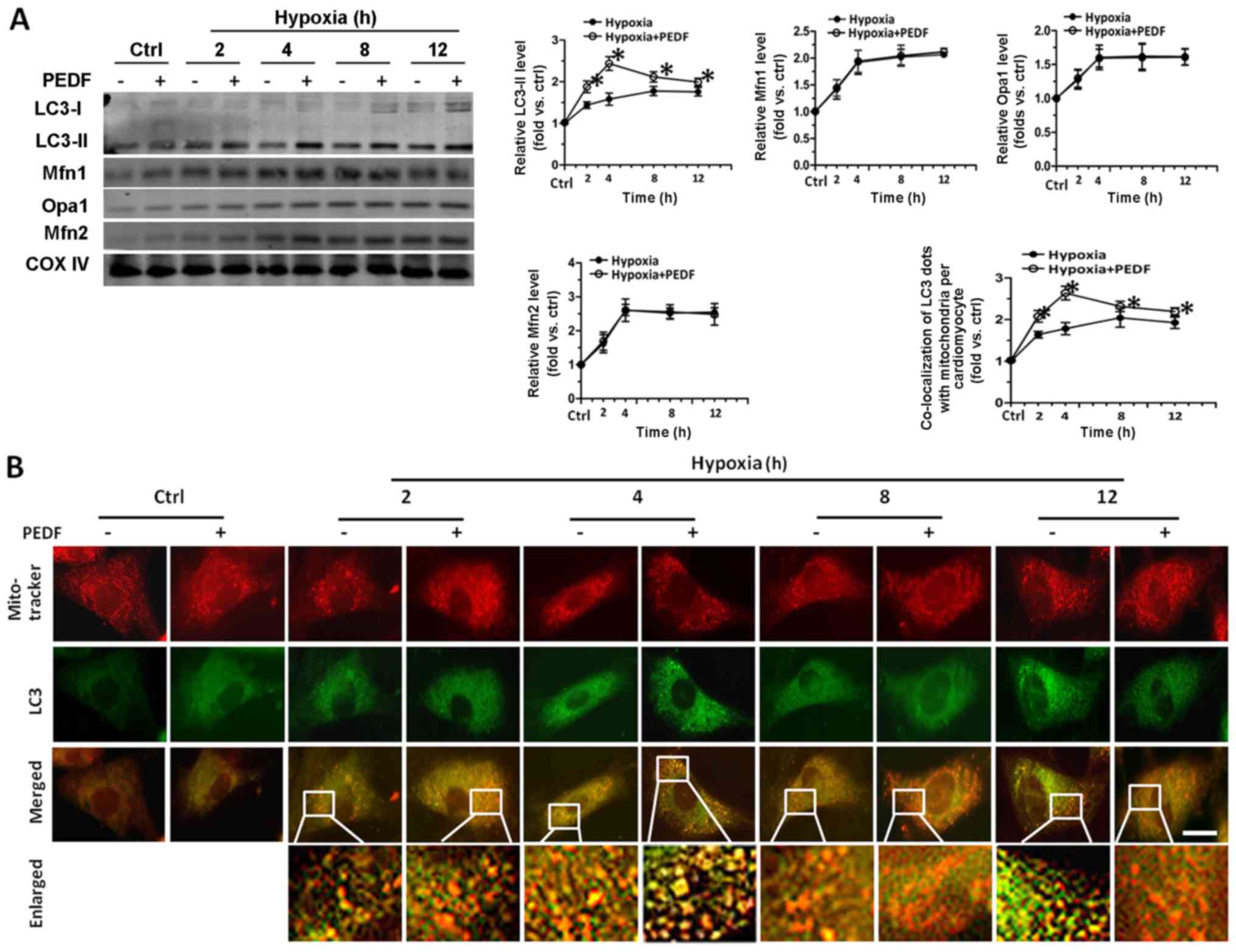 | Figure 2Pigment epithelial-derived factor
(PEDF) played a protective role in primary cardiomyocytes through
promoting mitophagy, and has no effect on mitochondrial fusion.
Primary cardiomyocytes were maintained in normoxic or hypoxic
conditions for 0, 2, 4, 8 and 12 h with or without PEDF (10 nM).
Lysosome inhibitor BAF1 (50 nM) was added. (A) LC3-II, Opa1, Mfn1
and Mfn2 proteins in the mitochondrial fractions were tested by
western blotting (n=4, *p<0.05). (B) Samples were
stained with MitoTracker Red and anti-LC3 (green) antibody.
Quantitative analysis of cardiomyocytes that contain
co-localization of LC3 dots with mitochondria per cardiomyocytes.
(*p<0.05, n=90 cardiomyocytes from three independent
experiments; scale bar, 20 µm). Lysosome inhibitor BAF1 (50
nM) was added. (C and D) Cell counting kit-8 (CCK-8) and LDH
released assays were employed to assessed cell viability and the
rate of cell death (n=4, *p<0.05). (E) Samples were
stained with MitoTracker Red. Five randomly picked regions of each
sample were captured by confocal z-axis scanning and the total
volume of mitochondria was calculated and quantified
(*p<0.05, n=90 cardiomyocytes from three independent
experiments; scale bar, 20 µm). Data were expressed in fold
induction, relative to control. Values are means ± SD. |
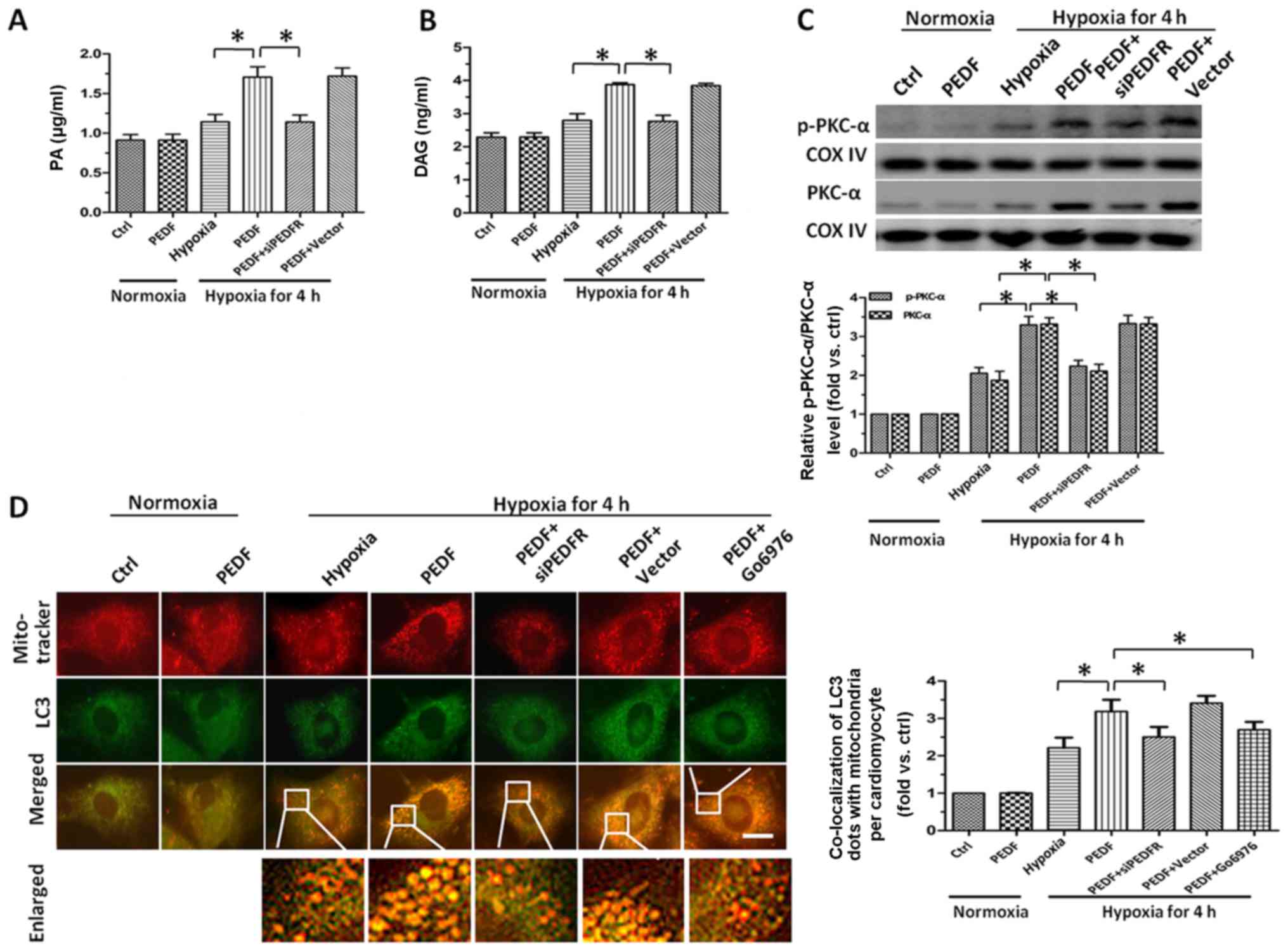
PEDF increases the level of PA and DAG
via PEDF-R, leading to PKC-α activation, which induced
mitophagy
Since we had confirmed PEDF could reduce the
mitochondrial density of cardiomyocytes through promoting
mitophagy, we attempted to identify the pathway underlying
PEDF-induced hypoxic cardiomyocytes mitophagy. An earlier study
showed that PEDF could stimulate cardiac lipid degradation via
PEDF-R (15). Therefore, we next
investigated the lipolysis of PEDF in hypoxic cardiomyocytes. After
4 h hypoxia, we found the level of PA increased significantly after
treated with PEDF compared with hypoxia group (Fig. 3A). Once taken by cells, excess
FFAs can be converted into their respective acyl-CoA derivatives
and then incorporated and stored in the cells as neutral lipids
like DAG (37–39). Therefore, we measured the level of
DAG in cardiomyocytes and found that treatment with PEDF led to a
marked increase of DAG (Fig. 3B).
As one of the well-established pathway activated by DAG is the PKC
family (40), we next evaluate
the expression and activation of PKC-α. As shown in Fig. 3C, we found PEDF not only
upregulated expression of PKC-α, but also increased its activation.
After 4 h hypoxia, Fig. 3D showed
the number of LC3 and MitoTracker puncta in PEDF-treated
cardiomyocytes was significantly more than that in control group.
With the addition of PKC-α inhibitor Go6976, the puncta were
decreased. Taken together, such findings demonstrated that the
upregulation and activation of PKC-α resulted from the increase of
PA and accumulation of DAG is implicated in PEDF-induced mitophagy
and the activation of PKC-α is involved as upstream signal for the
promotion of mitophagy in primary cardiomyocytes stimulated with
PEDF under hypoxic condition.
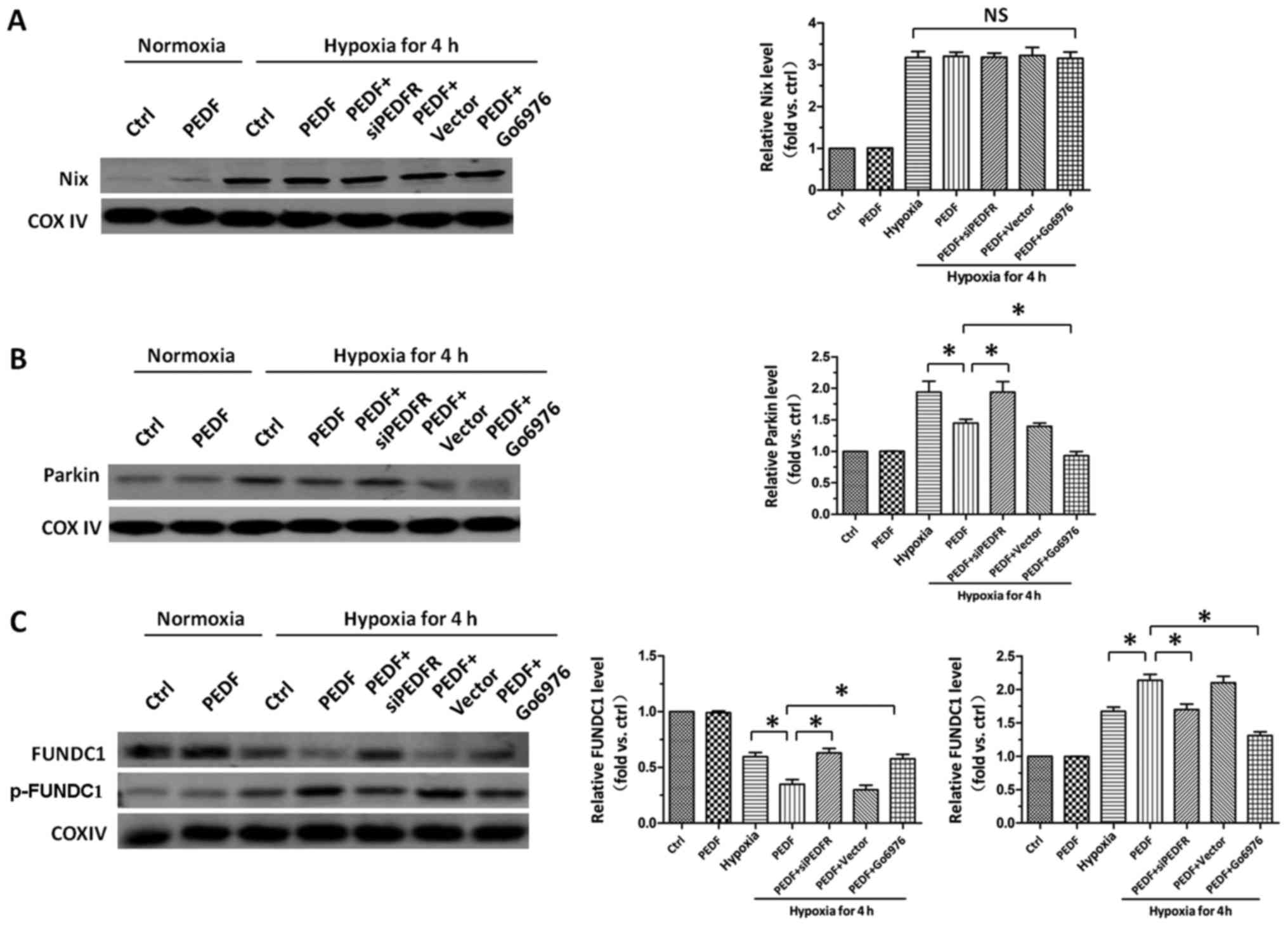
PEDF-activated PKC-α promotes
FUNDC1-mediated primary cardiomyocyte mitophagy under hypoxic
condition
After establishing the role of PKC-α in PEDF-induced
hypoxic cardiomyocytes mitophagy, we then tried to investigate the
downstream molecular mechanism underlying PKC-α-regulated
mitophagy. As shown in Fig. 4A,
we found hypoxia increased the level of Nix, but compared with
hypoxia group, PEDF failed to increase the level of Nix. It has
been established that decreased recruitment of Parkin to
mitochondria suppress mitophagy. The result showed PEDF
downregulated the level of Parkin, suggesting PEDF could inhibit
hypoxia-induced Parkin mitochondrial translocation, which indicated
PEDF suppress Parkin-medicated mitophagy (Fig. 4B). After precluding the effects of
Parkin and Nix, we then examined the effect of PEDF on FUNDC1 and
p-FUNDC1. We found that PEDF decreased the level of FUNDC1 and
increased the level of p-FUNDC1, while Go6976 (18) was capable of effectively
increasing the level of FUNDC1 and decreasing the level of p-FUNDC1
(Fig. 4C). Collectively, these
data indicate that PEDF is able to increase the level of p-FUNDC1
via activation of PKC-α and then induce mitophagy, rather than
through Parkin and Nix pathways.
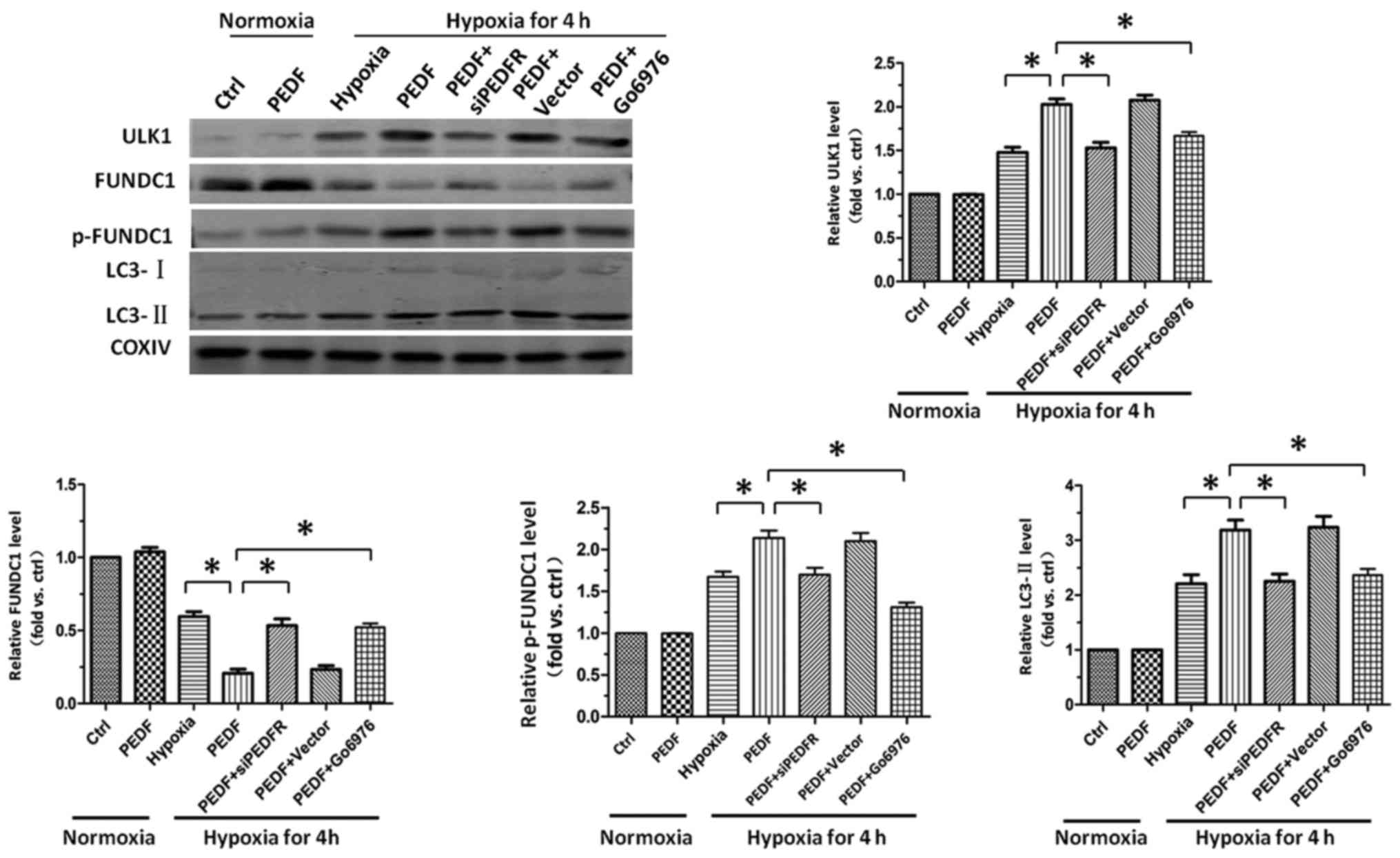
PEDF-induced hypoxic primary
cardiomyocyte mitophagy through FUNDC1 is dependent on ULK1
pathway
The ULK1 signaling pathway has been well established
as the crucial regulator of FUNDC1-induced mitophagy (26). Therefore, we next examined the
role of ULK1 signaling pathway in PEDF-induced mitophagy. Our data
demonstrated that PEDF could markedly increase the level of ULK1,
and significantly increase the level of p-FUNDC1 in hypoxic
cardiomyocytes. Besides, we also observed inhibition of PKC-α by
inhibitor Go6976 capable of effectively suppressing the increase of
ULK1, and inhibiting the increase of p-FUNDC1 (Fig. 5). These results showed that
FUNDC1-induced mitophagy which mediated by PEDF depends on the ULK1
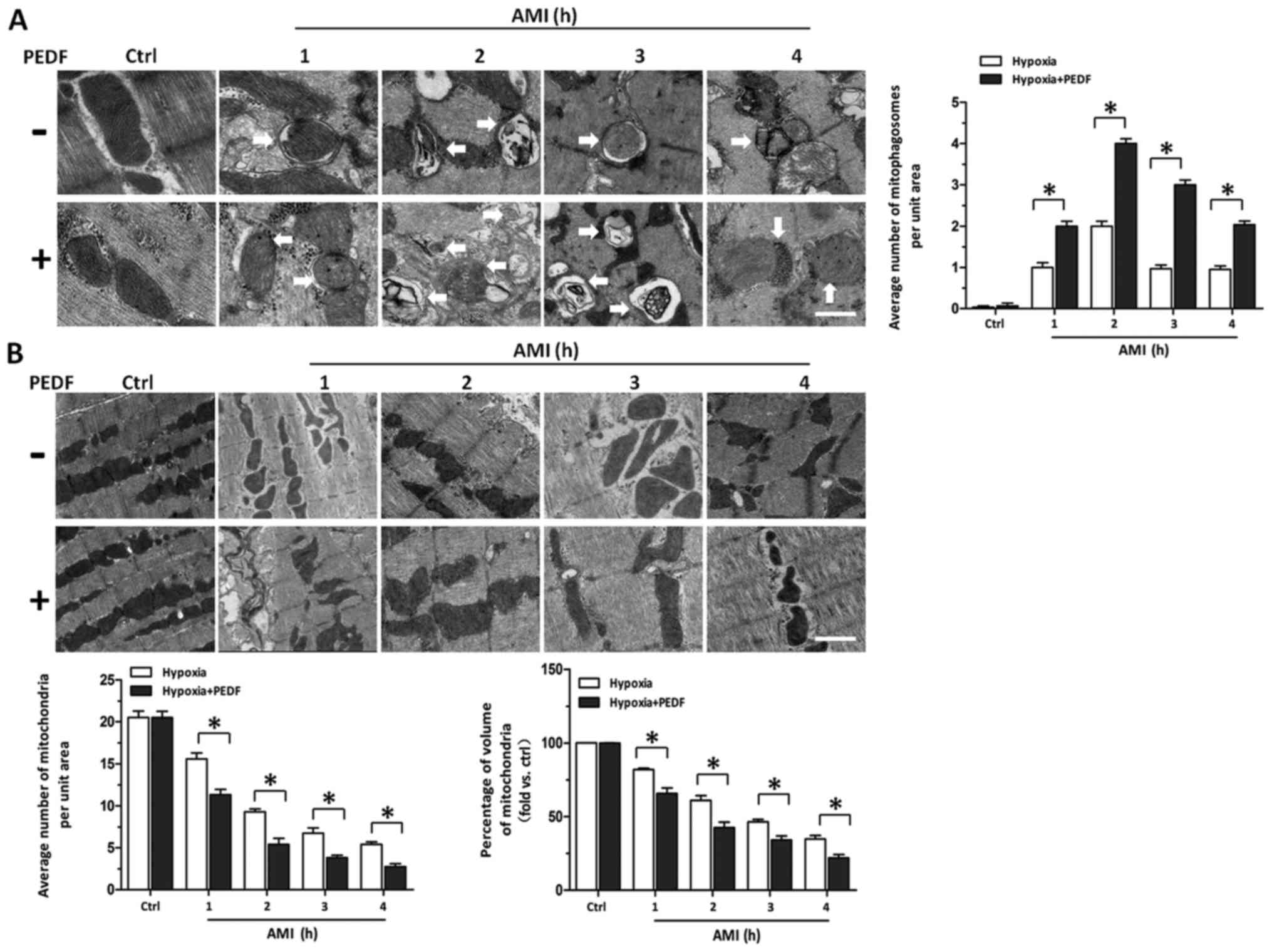
signaling pathway. PEDF decreased the mitochondrial density by
promoting mitophagy in vivo. After concluding PEDF could
decrease the mitochondrial density of cardiomyocytes via promoting
mitophagy in vitro, we tested whether PEDF could promote
cardiomyocytes mitophagy in vivo. Transmission Electron
Microscopic observation in rats treated with PEDF showed a marked
increase in the number of mitophagy compared with control group
(Fig. 6A). We also found that in
border zones the number of mitochondria and volume of mitochondria
in PEDF-treated rats was less than that in the control group
(Fig. 6B). These findings
indicate that under ischemic condition, PEDF could promote
mitophagy and decrease the mitochondrial density in in vivo
model of AMI.
Discussion
Both in vitro and in vivo studies, we
first found PEDF could promote mitophagy and decrease the
mitochondrial density of hypoxic primary cardiomyocytes.
Importantly, PEDF protects cardiomyocytes through promoting hypoxic
cardiomyocyte mitophagy. We identified a novel signaling pathway,
PEDF/PEDF-R/PA/DAG/PKC-α/ULK1/FUNDC1, in regulating PEDF-promoted
mitophagy. Based on the findings from this study, in conjunction
with those from previous studies (7,33),
it is conceivable that PEDF may represent a promising therapeutic
approach for ischemic heart disease.
Accumulating evidence has shown that hypoxia can
induce a loss of mitochondrial density (41–43). Mitophagy and mitochondrial fusion
are closely related with the change of mitochondrial number
(8,9,34,35). The number of mitochondria within a
cell are controlled by precisely regulated rates of organelle
fusion (35) and mitophagy is a
process through which damaged mitochondria are selectively
eliminated by autophagy (44). As
this study shows, hypoxia did reduce the total mitochondrial volume
and increase the level of fusion protein. However, there is no
significant difference between PEDF group and hypoxia group. So we
speculate that PEDF has no effect on mitochondrial fusion, and PEDF
decrease the mitochondrial density of hypoxic cardiomyocytes
through promoting mitophagy, rather than mitochondrial fusion.
In the endogenous protective effects of hypoxic
cells, mitophagy could attenuate the damage of ROS and cytochrome
c by eliminating the injured mitochondria (45). Moreover, mitophagy also could
reduce ATP consumption and maintain metabolic stability (46). In this study PEDF promotes and
enhances hypoxic cardiomyocyte mitophagy, which means PEDF could
eliminate the damaged mitochondria earlier and greatly via
promoting hypoxic cardiomyocyte mitophagy. Besides, considering the
role of mitochondria in cell ATP generation, we propose that
PEDF-promoted mitophagy may be a potentially protective mechanism
and play an earlier protective role in cardiomyocytes. Results from
this study showed that PEDF was able to decrease the mitochondrial
density of hypoxic cardiomyocytes earlier and greatly, compared
with hypoxia only, which means PEDF could alleviate the damage
caused by injured mitochondria. Therefore, it is significant that
PEDF reduces the mitochondrial density of hypoxic cardiomyocytes,
and plays a crucial role in cardiomyocyte protection.
In this study we found PEDF could promote the level
of PA. As is known, PA is a saturated fatty acid known to cause
lipotoxicity in cells (18), and
several factors such as ROS production (47) have been implicated in
lipotoxicity. However, in this study PA promotes hypoxic
cardiomyocyte mitophagy without lipotoxicity. We previously
reported that PEDF could increase the level of FFAs in
cardiomyocytes after AMI via binding to PEDF-R (15), a lipase-linked cell membrane
receptor for PEDF (14). When
taken by cells, excess FFAs can be converted into their respective
acyl-CoA derivatives (38) which
can be incorporated and stored in the cells similarly to DAG
(37,39). Consistent with this, we found the
increase of PA caused the accumulation of DAG.
Previous study noted that DAG could serve as a
natural agonist to recruit PKC proteins to membrane for activation
(40). The PKC family and/or
lipid-activated serine-threonine protein kinase function downstream
of many signal transduction pathway (19,20). PKC-α is the predominant
conventional PKC isoform expressed in the mouse, rat, and human
heart (21,22). Our findings showed DAG not only
upregulated PKC-α, but also induced its activation, which is
inconsistent with earlier study that DAG can only induce the
activation of PKC-α in mouse embryonic fibroblasts and HepG2 cells
(18). We postulate that the
conflicting results may be attributed to factors such as the cell
types used, the concentration and time of PA induced by PEDF. Taken
together, it is believed that PEDF can promote mitophagy via
activation of PKC-α dependent increase of PA and accumulation of
DAG.
In this study we also found that the activation of
PKC-α was associated with mitophagy. Mitophagy is mainly mediated
by three signaling pathways, namely, Parkin, Nix and FUNDC1
(23–25). Early studies have suggested that
endogenously or ectopically Parkin could be recruited towards the
depolarized mitochondria through interaction with PINK1 (23,48). As mitochondrial membrane potential
(Δψ) is lost, PINK1 is stabilized and accumulates on the outer
membrane of damaged mitochondria where it selectively recruits
Parkin (48). FUNDC1, an outer
mitochondrial membrane protein, promotes mitophagy through
interaction with LC3 in response to hypoxia (24). Nix is also a hypoxia-responsive
protein and acts as a mitophagy receptor which is necessary for
erythroid maturation through autophagic removal of unwanted
mitochondria (49). Data from
this study showed PEDF has no effect on Nix, and PEDF cannot
enhance mitophagy through Parkin, because PEDF could inhibit the
decline of mitochondrial membrane potential (33). The results revealed PEDF could
increase the level of p-FUNDC1 compared with hypoxia group, and
then promote mitophagy. So it is believed that PEDF could promote
mitophagy through FUNDC1 via activating PKC-α. It is beneficial to
further investigate mitophagy. Recent studies have suggested that
damaged and unwanted mitochondria can be removed by autophagy
(23). Therefore, based on the
findings from this study, in conjunction with those from previous
studies, it is conceivable that PEDF-promoted mitophagy through
FUNDC1 is essential for mitochondrial quality control in
cardiomyocytes under hypoxic condition. Besides, although recent
studies suggest SRC and ULK1 are critical for the FUNDC1-mediated
mitophagy, we found that PEDF induced mitophagy is dependent on
ULK1 pathway, rather than SRC pathway.
Taken together and to the best of our knowledge,
data presented in this study clearly demonstrate for the first time
that PEDF-promoted mitophagy constitutes an underlying protective
response against hypoxia in primary cardiomyocytes. However, future
studies are needed to fully understand whether PEDF-promoted
mitophagy represents a means to reduce ischemic injury in
vivo. Besides, this study did not pursue the search for
molecular mechanism in which PKC-α modulates the expression levels
of ULK1. Therefore, future investigation is needed to clarify these
detailed mechanisms. Because mitophagy has been linked to
cardioprotective benefit, data from this study also provide
insights into a promising future therapeutic strategy in cardiac
pathologies.
Acknowledgments
We would like to express our sincere gratitude to Dr
Xiaofang Yang (Laboratory of Clinical and Experimental Pathology,
Xuzhou Medical University) for providing the anti-p-FUNDC1 (Ser-17)
polyclonal antibody which was used for detecting phosphorylated
FUNDC1.
Notes
[1]
Funding
This study was supported by the National Nature
Science Foundation of China (nos. 81570242 and 81270173) and the
Technology Bureau of Xuzhou of China (grant no. KC14SH106).
[2] Availability
of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
All the authors conceived and designed the study
protocol. YL, ZL and YZ performed experiments. YL, ZL, YZ and XW
analyzed the data. YL, ZL, YZ, XW and PL interpreted the results of
the experiments. YL prepared the figures. YL, ZL, YZ, XW, PL, QZ,
HZ, ZW, HD and ZZ approved the final version of manuscript. HD and
ZZ conceived and designed the research. HD and ZZ edited and
revised manuscript. ZZ drafted the manuscript. All the authors gave
final read and approved the final manuscript.
[4] Ethics
approval and consent to participate
All the procedures were performed following the
guidelines of the Directive 2010/63/EU of the European Parliament,
and have been approved by the Ethics Committee for Animal
Experimentation of the Institutions where experiments were carried
out.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Lloyd-Jones D, Adams R, Carnethon M, De
Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund
K, et al American Heart Association Statistics Committee and Stroke
Statistics Subcommittee: Heart disease and stroke statistics - 2009
update: A report from the American Heart Association Statistics
Committee and Stroke Statistics Subcommittee. Circulation.
119:480–486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Begieneman MP, van de Goot FR, Fritz J,
Rozendaal R, Krijnen PA and Niessen HW: Validation of
ultrastructural analysis of mitochondrial deposits in
cardiomyocytes as a method of detecting early acute myocardial
infarction in humans. J Forensic Sci. 55:988–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawaguchi T, Yamagishi SI and Sata M:
Structure-function relationships of PEDF. Curr Mol Med. 10:302–311.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao X, Zhang H, Zhuang W, Yuan G, Sun T,
Jiang X, Zhou Z, Yuan H, Zhang Z and Dong H: PEDF and PEDF-derived
peptide 44mer protect cardiomyocytes against hypoxia-induced
apoptosis and necroptosis via anti-oxidative effect. Sci Rep.
4:56372014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodriguez-Enriquez S, Kai Y, Maldonado E,
Currin RT and Lemasters JJ: Roles of mitophagy and the
mitochondrial permeability transition in remodeling of cultured rat
hepatocytes. Autophagy. 5:1099–1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jain K, Prasad D, Singh SB and Kohli E:
Hypobaric hypoxia imbalances mitochondrial dynamics in rat brain
hippocampus. Neurol Res Int. 2015:7420592015.PubMed/NCBI
|
|
10
|
Ikeda Y, Sciarretta S, Nagarajan N,
Rubattu S, Volpe M, Frati G and Sadoshima J: New insights into the
role of mitochondrial dynamics and autophagy during oxidative
stress and aging in the heart. Oxid Med Cell Longev.
2014:2109342014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marzetti E, Csiszar A, Dutta D, Balagopal
G, Calvani R and Leeuwenburgh C: Role of mitochondrial dysfunction
and altered autophagy in cardiovascular aging and disease: From
mechanisms to therapeutics. Am J Physiol Heart Circ Physiol.
305:H459–H476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dorn GW II: Mitochondrial pruning by Nix
and BNip3: An essential function for cardiac-expressed death
factors. J Cardiovasc Transl Res. 3:374–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoshino A, Mita Y, Okawa Y, Ariyoshi M,
Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T and Matoba S: Cytosolic
p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial
dysfunction in the mouse heart. Nat Commun. 4:23082013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Notari L, Baladron V, Aroca-Aguilar JD,
Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML,
Sanchez-Sanchez F, et al: Identification of a lipase-linked cell
membrane receptor for pigment epithelium-derived factor. J Biol
Chem. 281:38022–38037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Sun T, Jiang X, Yu H, Wang M, Wei
T, Cui H, Zhuang W, Liu Z, Zhang Z, et al: PEDF and PEDF-derived
peptide 44mer stimulate cardiac triglyceride degradation via ATGL.
J Transl Med. 13:682015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Subramanian P, Locatelli-Hoops S, Kenealey
J, DesJardin J, Notari L and Becerra SP: Pigment epithelium-derived
factor (PEDF) prevents retinal cell death via PEDF Receptor
(PEDF-R): Identification of a functional ligand binding site. J
Biol Chem. 288:23928–23942. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zimmermann R, Strauss JG, Haemmerle G,
Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger
G, Eisenhaber F, Hermetter A, et al: Fat mobilization in adipose
tissue is promoted by adipose triglyceride lipase. Science.
306:1383–1386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan SH, Shui G, Zhou J, Li JJ, Bay BH,
Wenk MR and Shen HM: Induction of autophagy by palmitic acid via
protein kinase C-mediated signaling pathway independent of mTOR
(mammalian target of rapamycin). J Biol Chem. 287:14364–14376.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steinberg SF: Structural basis of protein
kinase C isoform function. Physiol Rev. 88:1341–1378. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Churchill E, Budas G, Vallentin A,
Koyanagi T and Mochly-Rosen D: PKC isozymes in chronic cardiac
disease: Possible therapeutic targets? Annu Rev Pharmacol Toxicol.
48:569–599. 2008. View Article : Google Scholar
|
|
21
|
Ping P, Zhang J, Qiu Y, Tang XL,
Manchikalapudi S, Cao X and Bolli R: Ischemic preconditioning
induces selective translocation of protein kinase C isoforms
epsilon and eta in the heart of conscious rabbits without
subcellular redistribution of total protein kinase C activity. Circ
Res. 81:404–414. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hambleton M, Hahn H, Pleger ST, Kuhn MC,
Klevitsky R, Carr AN, Kimball TF, Hewett TE, Dorn GW II, Koch WJ,
et al: Pharmacological- and gene therapy-based inhibition of
protein kinase Calpha/beta enhances cardiac contractility and
attenuates heart failure. Circulation. 114:574–582. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Feng D, Chen G, Chen M, Zheng Q,
Song P, Ma Q, Zhu C, Wang R, Qi W, et al: Mitochondrial
outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in
mammalian cells. Nat Cell Biol. 14:177–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Novak I, Kirkin V, McEwan DG, Zhang J,
Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et
al: Nix is a selective autophagy receptor for mitochondrial
clearance. EMBO Rep. 11:45–51. 2010. View Article : Google Scholar :
|
|
26
|
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W,
Zhang X, Xue P, Zhou C, Liu L, et al: ULK1 translocates to
mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO
Rep. 15:566–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Wang Z, Feng SJ, Xu L, Shi HX,
Chen LL, Yuan GD, Yan W, Zhuang W, Zhang YQ, et al: PEDF improves
cardiac function in rats with acute myocardial infarction via
inhibiting vascular permeability and cardiomyocyte apoptosis. Int J
Mol Sci. 16:5618–5634. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khan M, Meduru S, Gogna R, Madan E, Citro
L, Kuppusamy ML, Sayyid M, Mostafa M, Hamlin RL and Kuppusamy P:
Oxygen cycling in conjunction with stem cell transplantation
induces NOS3 expression leading to attenuation of fibrosis and
improved cardiac function. Cardiovasc Res. 93:89–99. 2012.
View Article : Google Scholar
|
|
29
|
Fukuhara S, Tomita S, Yamashiro S,
Morisaki T, Yutani C, Kitamura S and Nakatani T: Direct cell-cell
interaction of cardiomyocytes is key for bone marrow stromal cells
to go into cardiac lineage in vitro. J Thorac Cardiovasc Surg.
125:1470–1480. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luedde M, Lutz M, Carter N, Sosna J,
Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F,
et al: RIP3, a kinase promoting necroptotic cell death, mediates
adverse remodelling after myocardial infarction. Cardiovasc Res.
103:206–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujimura M, Morita-Fujimura Y, Kawase M,
Copin JC, Calagui B, Epstein CJ and Chan PH: Manganese superoxide
dismutase mediates the early release of mitochondrial cytochrome c
and subsequent DNA fragmentation after permanent focal cerebral
ischemia in mice. J Neurosci. 19:3414–3422. 1999.PubMed/NCBI
|
|
32
|
Akbari M, Otterlei M, Peña-Diaz J and
Krokan HE: Different organization of base excision repair of uracil
in DNA in nuclei and mitochondria and selective upregulation of
mitochondrial uracil-DNA glycosylase after oxidative stress.
Neuroscience. 145:1201–1212. 2007. View Article : Google Scholar
|
|
33
|
Wang X, Zhang Y, Lu P, Zhang H, Li Y, Dong
H and Zhang Z: PEDF attenuates hypoxia-induced apoptosis and
necrosis in H9c2 cells by inhibiting p53 mitochondrial
translocation via PEDF-R. Biochem Biophys Res Commun. 465:394–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murray AJ: Metabolic adaptation of
skeletal muscle to high altitude hypoxia: How new technologies
could resolve the controversies. Genome Med. 1:1172009. View Article : Google Scholar
|
|
35
|
Karbowski M and Youle RJ: Dynamics of
mitochondrial morphology in healthy cells and during apoptosis.
Cell Death Differ. 10:870–880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li W, Zhang X, Zhuang H, Chen HG, Chen Y,
Tian W, Wu W, Li Y, Wang S, Zhang L, et al: MicroRNA-137 is a novel
hypoxia-responsive microRNA that inhibits mitophagy via regulation
of two mitophagy receptors FUNDC1 and NIX. J Biol Chem.
289:10691–10701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Listenberger LL, Han X, Lewis SE, Cases S,
Farese RV Jr, Ory DS and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:3077–3082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Coleman RA and Mashek DG: Mammalian
triacylglycerol metabolism: Synthesis, lipolysis, and signaling.
Chem Rev. 111:6359–6386. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li LO, Klett EL and Coleman RA: Acyl-CoA
synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta.
1801:246–251. 2010. View Article : Google Scholar :
|
|
40
|
Newton AC: Protein kinase C: Structural
and spatial regulation by phosphorylation, cofactors, and
macromolecular interactions. Chem Rev. 101:2353–2364. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferretti G: Limiting factors to oxygen
transport on Mount Everest 30 years after: A critique of Paolo
Cerretelli's contribution to the study of altitude physiology. Eur
J Appl Physiol. 90:344–350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Levett DZ, Radford EJ, Menassa DA, Graber
EF, Morash AJ, Hoppeler H, Clarke K, Martin DS, Ferguson-Smith AC,
Montgomery HE, et al: Caudwell Xtreme Everest Research Group:
Acclimatization of skeletal muscle mitochondria to high-altitude
hypoxia during an ascent of Everest. FASEB J. 26:1431–1441. 2012.
View Article : Google Scholar
|
|
43
|
Howald H and Hoppeler H: Performing at
extreme altitude: Muscle cellular and subcellular adaptations. Eur
J Appl Physiol. 90:360–364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim I, Rodriguez-Enriquez S and Lemasters
JJ: Selective degradation of mitochondria by mitophagy. Arch
Biochem Biophys. 462:245–253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar
|
|
46
|
Mammucari C and Rizzuto R: Signaling
pathways in mitochondrial dysfunction and aging. Mech Ageing Dev.
131:536–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Piro S, Anello M, Di Pietro C, Lizzio MN,
Patanè G, Rabuazzo AM, Vigneri R, Purrello M and Purrello F:
Chronic exposure to free fatty acids or high glucose induces
apoptosis in rat pancreatic islets: Possible role of oxidative
stress. Metabolism. 51:1340–1347. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sandoval H, Thiagarajan P, Dasgupta SK,
Schumacher A, Prchal JT, Chen M and Wang J: Essential role for Nix
in autophagic maturation of erythroid cells. Nature. 454:232–235.
2008. View Article : Google Scholar : PubMed/NCBI
|