Introduction
Diabetes is a chronic macrovascular and
microvascular disease induced by hyperglycemia, affecting millions
of individuals globally (1).
Approximately 80% of diabetes-related mortality is caused by
cardiovascular diseases, including atherosclerosis, hypertension
and cardiomyopathy (2). Moreover,
endothelial dysfunction, characterized by inflammation and
oxidative stress, is one of the most common causes of morbidity and
mortality in diabetes-related cardiovascular diseases (1,3).
It is known that inflammation serves a critical role
in the development and progression of diabetes (3,4).
An excessive or inappropriate production of pro-inflammatory
cytokines, such as interleukin-1β (IL-1β), could induce the
expression of inflammatory mediators [e.g., intercellular adhesion
molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)]
as a positive feedback mechanism and subsequently result in
endothelial cell activation, which eventually leads to endothelial
dysfunction (1,5). In addition, high glucose-induced
oxidative stress, primarily associated with the production of
reactive oxygen species (ROS), also serves a pivotal role in
endothelial dysfunction during the development of diabetes
(6). ROS function as signaling
molecules to activate a variety of stress pathways and inflammatory
pathways, which in turn further enhance inflammatory responses
(7,8). Furthermore, inhibition of ROS with
antioxidant agents can attenuate inflammation and rescue
endothelial cells (9). However,
the mechanisms by which high glucose induces inflammation and
oxidative stress are not fully understood.
Mounting evidence has suggested that nuclear
factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK)
signaling are the classical pathways to regulate ROS production and
inflammation (10–12). Activation of these pathways can
increase ROS production and accelerate the inflammation progress,
and thus cause endothelial dysfunction (13). It is worthy to note that high
glucose can induce NF-κB nuclear translocation, MAPK activation and
inflammatory biomediator production in the endothelial cells of
diabetic patients (14).
Therefore, these signaling pathways and inflammation may be pivotal
pathogenic mechanisms underlying endothelial dysfunction caused by
hyperglycemia.
Cystic fibrosis (CF) is a common autosomal recessive
disorder due to mutations of the CF transmembrane conductance
regulator (CFTR) gene, which is located on the long arm of
chromosome 7 (15). Previous
studies have shown massive infiltration of neutrophils and
excessive production of pro-inflammatory cytokines in the airways
of CF patients (16,17), suggesting that CF may be a
hallmark of pulmonary inflammation. CFTR loss or aberration in the
lungs leads to bacterial infection in association with inflammation
as a consequence of abnormal reabsorption of sodium and water, and
impairment of mucociliary clearance (18). Even if multiple studies have
suggested that defective or dysfunctional CFTR can also result in
bacterial colonization, inflammation usually occurs in the earliest
stage of lung damage prior to bacterial infection in CF patients
(19,20), indicating the direct role of CFTR
in the inflammatory process. Moreover, NF-κB and MAPK signaling
pathways have been suggested to be implicated in the regulation of
the inflammatory response of CF airway epithelia (16,19,21). Although these findings have
provided evidence that CFTR defects are likely to contribute to
inflammation, little attention has been devoted to the
investigation of the role of CFTR in hyperglycemia-induced vascular
endothelial cell inflammation. The present study aims to establish
whether high glucose regulates CFTR expression in endothelial cells
and whether such changes are causally associated with high
glucose-induced endothelial cell oxidative stress and
inflammation.
Materials and methods
Materials and reagents
Endothelial basal medium (EBM), fetal bovine serum
(FBS), penicillin, streptomycin, L-glutamine, heparin and vascular
endothelial growth factor were obtained from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Mannitol, glucose, PD98059,
SP600125, BAY11, apocynin and N-acetyl-L-cysteine (NAC) were
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
CFTRinh-172 was purchased from Selleck Chemicals
(Houston, TX, USA).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
purchased from the American Type Culture Collection (Rockville, MD,
USA) and maintained in EBM supplemented with heat-inactivated 10%
FBS, 100 U/ml penicillin, 100 U/ml streptomycin, 2 mM L-glutamine,
25 U/ml heparin and 5 ng/ml vascular endothelial growth factor, in
a humidified atmosphere of 5% CO2 at 37°C.
Western blot analysis
Western blot analysis was performed as previously
described (19). HUVECs were
washed with phosphate-buffered saline (PBS) and harvested in
mammalian protein extraction reagent (Thermo Fisher Scientific,
Inc.) containing 1 mM protease inhibitor (Roche Diagnostics, Laval,
QC, Canada). To detect the expression of the NF-κB p65 subunit in
the nuclei and cytoplasm, nuclear and cytosolic proteins were
isolated with a Nuclear/Cytosol Fractionation kit (BioVision, Inc.,
Milpitas, CA, USA) according to the manufacturer's instructions.
The protein content was measured using the Bio-Rad protein assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). A total of 50
µg protein was separated on 8 or 10% sodium dodecyl
sulfate-polyacrylamide gels according to protein molecular weights
and was then electrotransferred onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
blocked in 5% non-fat milk at room temperature for 1 h and
incubated with the following primary antibodies overnight at 4°C:
CFTR (#78335), p65 (#8242) and lamin B (#12586) (diluted 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), phosphorylated
Janus kinase (p-JNK; sc-293136), JNK (sc-572), p-extracellular
signal-regulated kinase (ERK), ERK (sc-514302) and β-actin
(sc-130300) (diluted 1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA). Following incubation with horseradish
peroxidase-conjugated anti-rabbit (ab6721) or anti-goat (ab6885)
secondary antibodies (diluted 1:5,000; Abcam, Cambridge, MA, USA)
for 1 h at room temperature, bands were detected with ECL™ western
blot detection reagents (GE Healthcare, Chicago, IL, USA) and
quantified by Quantity One software (version 4.6.9; Bio-Rad
Laboratories, Inc.).
Immunofluorescence staining
To detect NF-κB nuclear translocation in endothelial
cells, cells were fixed with 4% paraformaldehyde for 10 min at room
temperature and labeled with rabbit-anti-p65 antibodies (diluted
1:100; #8242; Cell Signaling Technology, Inc.) overnight at 4°C.
Subsequent to incubation with the primary antibody overnight at
4°C, the cells were then washed three times with PBS for 3 min and
incubated with anti-goat FITC antibody for labeling CFTR or with
anti-rabbit Cy3 antibody for labeling p65 (diluted 1:200; Beyotime,
Jiangsu, China) for 1 h at room temperature. Fluorescence images
were acquired using the Zeiss Axioplan 2 fluorescence microscope
(Carl Zeiss AG, Oberkochen, Germany).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from HUVECs using TRIzol
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. cDNA was synthesized using the ReverTra
ACE qPCR RT kit (Toyobo Life Science, Osaka, Japan) in a
20-µl reaction mixture. qPCR was performed with a CFX 96
Connect real-time PCR detection system (Bio-Rad Laboratories, Inc.)
using SYBR-Green PCR master mix reagents (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Samples were denatured at 94°C for
4 min, followed by 32 cycles of 95°C for 10 sec and 60°C for 30
sec. The specific primers (Invitrogen; Thermo Fisher Scientific,
Inc.) used were as follows: CFTR forward, 5′-AGAAGGCTGGGGCTCATT-3′
and reverse, 5′-GGGCCATCCACAGTCTTCT-3′; ICAM-1 forward,
5′-GCAGACAGTGACCATCTACAGCTT-3′ and reverse,
5′-CTTCTGAGACCTCTGGCTTCGT-3′; VCAM-1 forward,
5′-GGCAGGCTGTAAAAGAATTGCA-3′ and reverse,
5′-GTCATGGTCACAGAGCCACCTT-3′; E-selectin forward,
5′-AAGCCACATGTGAAGCTGT-3′ and reverse, 5′-CTCCAATAGGGGAATGAGCA-3′;
IL-1β forward, 5′-CTGAGCTCGCCAGTGAAATG-3′ and reverse,
5′-TGTCCATGGCCACAACAACT-3′; and 18S rRNA (reference gene) forward,
5′-CGGCTACCACATCCAAGGAA-3′ and reverse, 5′-CTGGAATTACCGCGGCT-3′.
The data were calculated using the 2−ΔΔCT method
(20).
ROS measurement
ROS level in the HUVECs was measured using
2′,7′-dichlorofluorescin diacetate (H2DCF-DA;
Invitrogen; Thermo Fisher Scientific, Inc.), as previously
described (7). Briefly, HUVECs
(2×104 cells/well) were seeded in 96-well clear-bottom
black plates and then incubated with different concentrations (10,
15, 20 and 25 mM) of glucose for 24 h at 37°C. Following treatment,
the cells were stained with H2DCF-DA (10 µM) in
serum-free EBM for 30 min at 37°C and washed with PBS three times.
ROS production was determined by a fluorescence microplate reader
(Tecan Group, Ltd., Männedorf, Switzerland) at 488 nm excitation
and 525 nm emission. The fluorescence intensities were normalized
to protein concentrations.
Adenovirus infection
The recombinant adenoviral vector (Ad) encoding
human CFTR was constructed by SunBio Biotechnology (Shanghai,
China). The recombinant Ad expressing LacZ (Clontech Laboratories,
Inc., Mountainview, CA, USA) was used as a control. When cells
reached 60–70% confluence, the medium was removed and replaced with
serum-free medium containing 50 multiplicity of infection LacZ or
Ad-CFTR. HUVECs were infected with LacZ or Ad-CFTR for 24 h prior
to high-glucose treatment.
Pharmacological treatment
HUVECs were pretreated with Ad-CFTR or
CFTRinh-172 (10 µM) for 24 h, followed by BAY11
(20 µM), PD98059 (10 µM), SP600125 (10 µM),
apocynin (1 µM) or NAC (5 µM) treatment for another
24 h in the presence of high glucose (25 mM).
Enzyme-linked immunosorbent assay
(ELISA)
The concentration of ICAM-1 (EK0370), VCAM-1
(EK0537), E-selectin (EK0501) and IL-1β (EK0392) was measured in
the supernatants of HUVECs using an ELISA kit (Wuhan Boster
Biological Technology, Ltd., Wuhan, China). All measurements were
performed as recommended by the manufacturer's protocols.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. N represents the number of independent experiments on
different batches of cells. The statistical significance between
samples was evaluated by the unpaired two-tailed Student's t-test
or by one-way analysis of variance using SPSS 16.0 system (SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
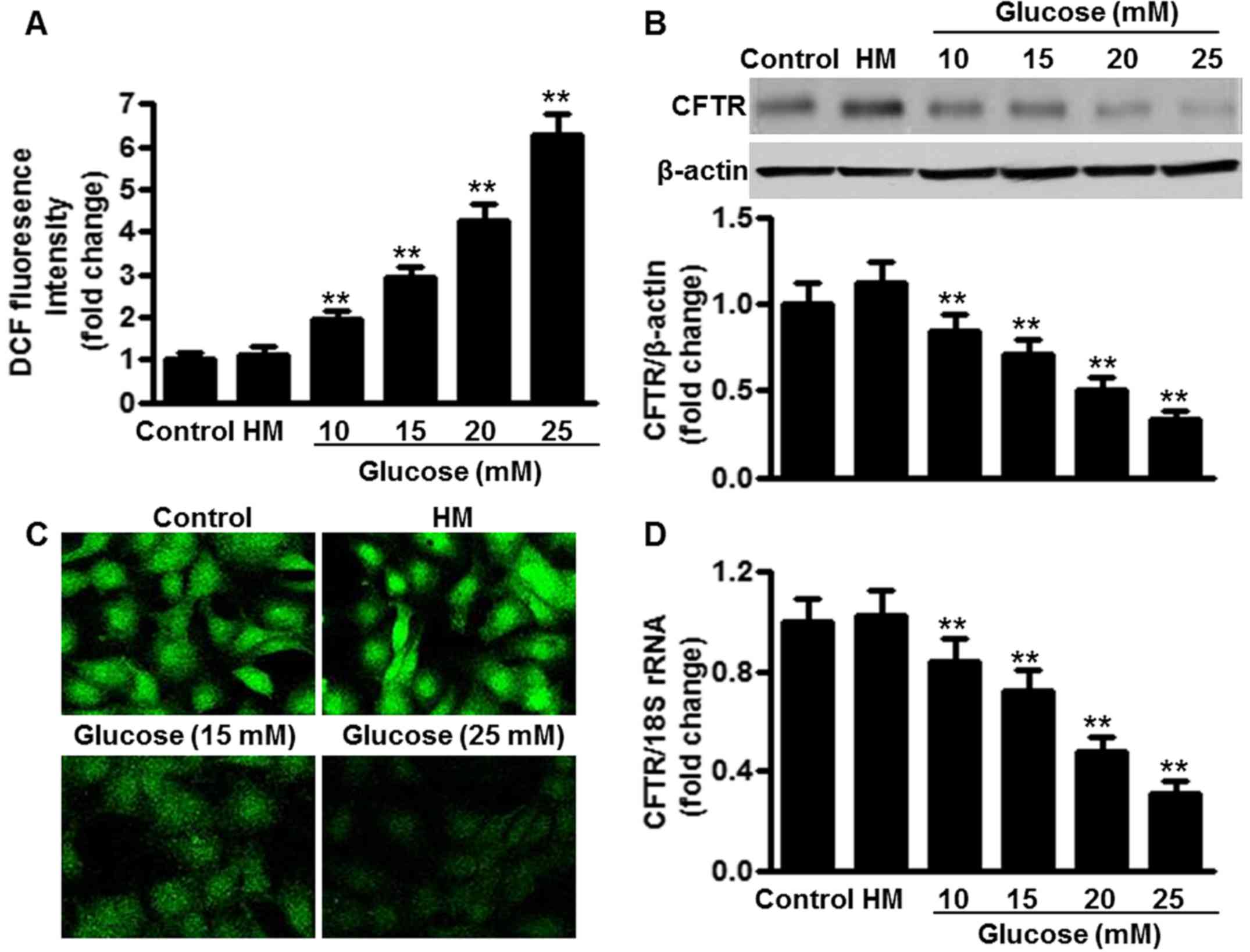
High glucose-induced endothelial cell ROS
production is associated with decreased CFTR expression
High glucose has been documented to increase
endothelial cell oxidative stress (22,23). To confirm these results of
previous studies, in the present study, HUVECs were incubated with
different concentrations of glucose (10, 15, 20 and 25 mM) for 24
h. As depicted in Fig. 1A,
increasing concentrations of glucose enhanced ROS production in the
HUVECs. Glucose concentrations of 15 and 20 mM induced ROS
production by 3.1- and 4.4-fold, respectively, compared with the
control group (5.5 mM glucose). A glucose concentration of 25 mM
further stimulated ROS production by 6.3-fold. High mannitol (25
mM), which served as an osmotic control, produced no significant
effects on ROS production. Notably, it was found that the
glucose-induced ROS production was associated with the inhibition
of the expression of CFTR. Glucose at 11 mM decreased CFTR protein
expression in the HUVECs to 70.2±8.3% relative to the control,
while at 20 mM glucose, CFTR protein expression was 51.6±6.8% of
the control, and at 25 mM, glucose decreased the CFTR protein
expression to 34.0±4.3% of the control (Fig. 1B). Similar results were obtained
in CFTR immunofluorescence staining of HUVECs treated with or
without high glucose (Fig. 1C).
Furthermore, RT-qPCR results showed that CFTR mRNA expression was
also decreased following glucose challenge, in a similar
concentration-dependent manner (Fig.
1D).
CFTR attenuates ROS production in
endothelial cells under high glucose conditions
To determine the role of CFTR in ROS production in
endothelial cells, HUVECs were infected with adenoviral vector
encoding human CFTR (Ad-CFTR) for 24 h. Western blotting showed
that CFTR protein expression was significantly increased following
overexpression of CFTR compared with that in the control group.
LacZ exhibited no effect on CFTR expression. By contrast, CFTR
protein expression was markedly inhibited following treatment with
CFTR inhibitor, CFTRinh-172 (Fig. 2A). As shown in Fig. 2B, a 25 mM glucose concentration
(high glucose) was used to induce endothelial cell ROS production.
The H2DCF-DA fluorescence intensity of LacZ-infected
cells was ~6.5-fold higher than that of the control group. When
cells were infected with Ad-CFTR, the ability of high glucose to
induce ROS production was markedly inhibited. However, CFTR
inhibition markedly enhanced high glucose-induced ROS production in
the HUVECs. In addition, neither overexpression nor inhibition of
CFTR affected ROS production under the basal level (data not
shown).
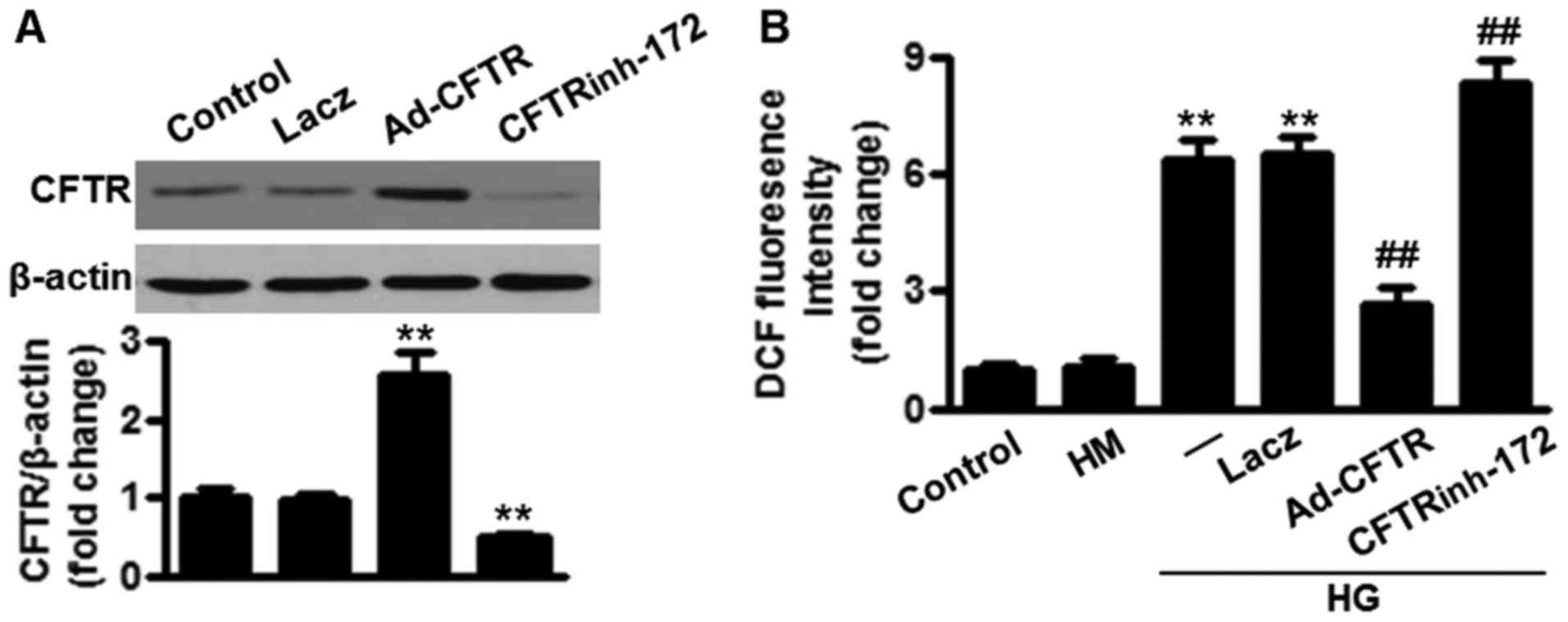 | Figure 2Overexpression of CFTR inhibits high
glucose-induced ROS production in HUVECs. (A) Cells were treated
with LacZ (50 MOI), Ad-CFTR (50 MOI) or CFTRinh-172 (10
µM) for 24 h. The infection efficiency and inhibition
efficiency were assessed by western blotting and data are shown as
the ratio of CFTR/β-actin. **P<0.01 vs. control
(n=4). (B) Following treatment, HUVECs were incubated with high
glucose (HG, 25 mM) for another 24 h and then ROS production was
measured. **P<0.01 vs. control;
##P<0.01 vs. high glucose alone (n=6). ROS, reactive
oxygen species; CFTR, cystic fibrosis transmembrane conductance
regulator; HUVECs, human umbilical vein endothelial cells;HM, high
mannitol; H2DCF-DA/DCF, 2′,7′-dichlorofluorescin
diacetate; MOI, multiplicity of infection; Ad, adenoviral vector;
HG, high glucose. |
Inhibition of CFTR promotes a high
glucose-induced inflammatory response in endothelial cells
In addition to oxidative stress, diabetes is also
associated with vascular inflammation (22,23). Therefore, the present study next
determined the role of CFTR in modulating endothelial cell
inflammation. The mRNA expression of the inflammatory mediators
ICAM-1 and VCAM-1, endothelial cell activation marker E-selectin,
and pro-inflammatory cytokine IL-1β were examined by RT-qPCR. In
HUVECs, the mRNA expression of ICAM-1, VCAM-1, E-selectin and IL-1β
was significantly increased upon high glucose treatment, and was
markedly attenuated following overexpression of CFTR. However,
CFTR-specific inhibitor further increased the mRNA expression of
ICAM-1, VCAM-1, E-selectin and IL-1β under high glucose condition
(Fig. 3A-D). Consistent with
these results, overexpression of CFTR inhibited, whereas inhibition
of CFTR enhanced, the concentration of these inflammatory
biomediators in HUVECs, as determined by ELISA assay (Fig. 3E-H). Similar to the effect of CFTR
on ROS production, overexpression or inhibition of CFTR also did
not alter the expression and concentration of ICAM-1, VCAM-1,
E-selectin and IL-1β mRNA under the basal level (data not shown).
These data indicate that overexpression of CFTR will be effective
in reducing high glucose-induced inflammation in endothelial
cells.
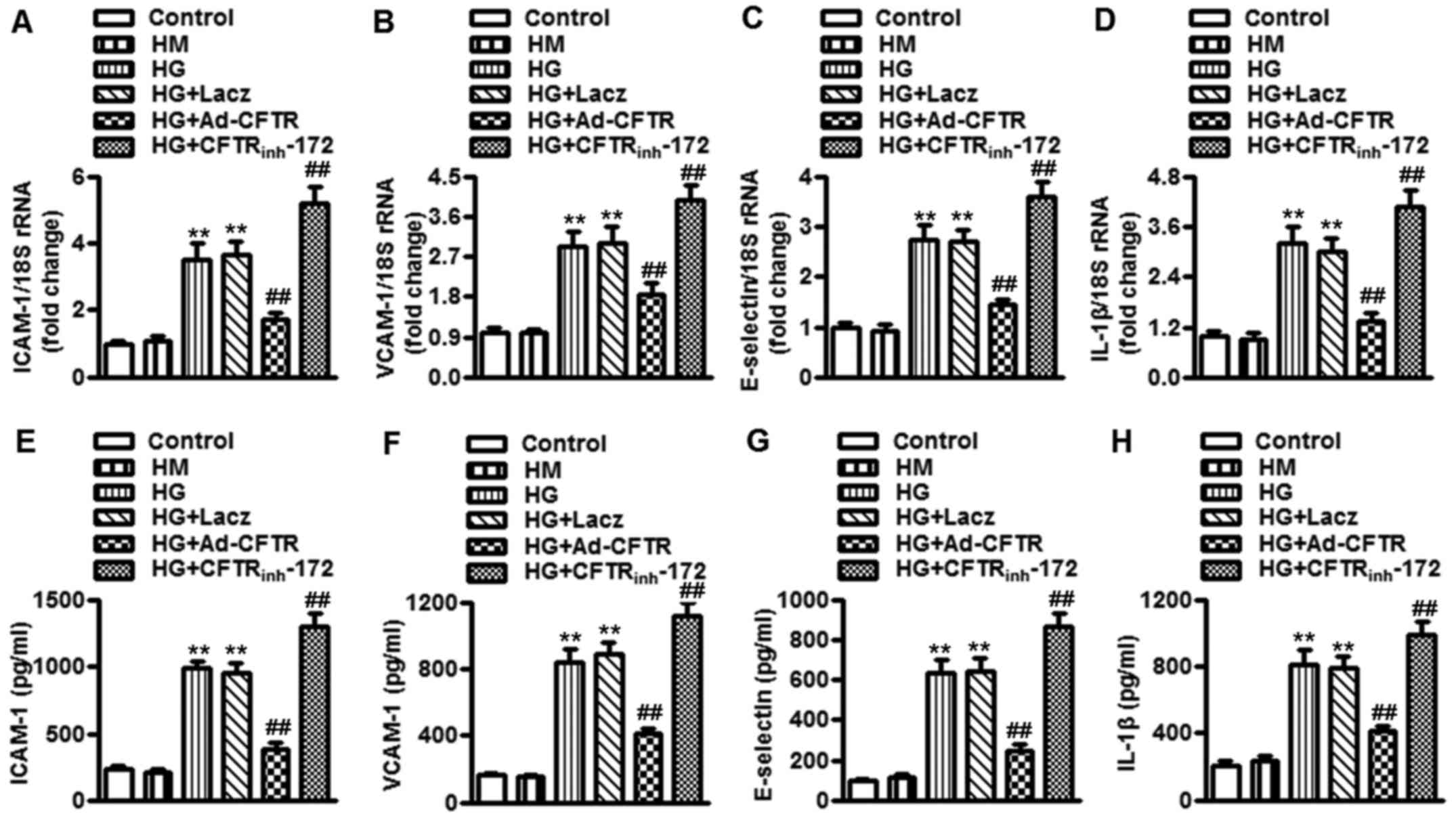 | Figure 3CFTR inhibition enhances high
glucose-induced secretion of inflammatory biomediators in
endothelial cells. (A-D) HUVECs were pretreated with LacZ (50 MOI),
Ad-CFTR (50 MOI) or CFTRinh-172 (10 µM) for 24 h
in prior to incubation of high glucose (25 mM) for another 24 h.
The mRNA expression of (A) ICAM-1, (B) VCAM-1, (C) E-selectin and
(D) IL-1β was determined by reverse transcription-quantitative
polymerase chain reaction analysis. (B) The concentration of (E)
ICAM-1, (F) VCAM-1, (G) E-selectin and (H) IL-1β in HUVECs were
measured by enzyme-linked immunosorbent assay. All data are
expressed as the mean ± standrad error of the mean.
**P<0.01 vs. control; ##P<0.01 vs. high
glucose alone (n=6). CFTR, cystic fibrosis transmembrane
conductance regulator; HUVECs, human umbilical vein endothelial
cells;HM, high mannitol; H2DCF-DA/DCF,
2′,7′-dichlorofluorescin diacetate; MOI, multiplicity of infection;
Ad, adenoviral vector; HG, high glucose; ICAM-1, intercellular
adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; IL,
interleukin. |
Role of NF-κB and MAPK signaling in
CFTR-attenuated inflammation induced by high glucose in endothelial
cells
Since NF-κB and MAPK signaling pathways serve
important roles in the regulation of inflammation (11,12), the present study proceeded to
investigate whether CFTR regulates high glucose-induced
inflammation via NF-κB and/or MAPK signaling. Western blotting
results showed that the translocation of p65 from the cytoplasm to
nucleus was increased following high glucose treatment for 24 h,
indicating the activation of NF-κB. Following CFTR overexpression,
the ability of high glucose to induce p65 nuclear translocation was
almost abolished. However, inhibition of CFTR further enhanced
NF-κB nuclear accumulation in the presence of high glucose
(Fig. 4A and B). These results
were further confirmed by immunofluorescence staining using
anti-p65 antibody (Fig. 4C). In
addition, high glucose also activated MAPK signaling, as evidenced
by significant phosphorylation of ERK and JNK. In Ad-CFTR-infected
cells, ERK and JNK signaling in HUVECs was no longer activated by
high glucose, whereas the phosphorylation of ERK and JNK was
further enhanced by the addition of CFTRinh-172
(Fig. 4D and E). To further
confirm whether NF-κB and/or MAPK serve a key role in CFTR-mediated
inflammation under high glucose conditions, pharmacological
inhibitors of NF-κB, ERK and JNK were employed, and then their
effects on the secretion of inflammatory biomediators were
measured. The enhanced effects of CFTR inhibition on inflammatory
biomediator secretion were completely inhibited by NF-κB inhibitor
(BAY11), ERK inhibitor (PD98059) or JNK inhibitor (SP600125) alone
(Fig. 4F), suggesting that CFTR
ameliorates the high glucose-induced inflammatory response through
inhibition of NF-κB and MAPK signaling. Moreover, western blotting
results revealed that PD98059 and SP600125 blocked CFTR
inhibition-induced NF-κB activation under high glucose conditions,
as shown by significantly inhibiting p65 nuclear translocation
(Fig. 4G and H). Nevertheless,
pharmacological inhibition of NF-κB with its selective inhibitor,
BAY11, did not alter the phosphorylation of ERK and JNK (Fig. 4I and J). These data indicate that
MAPK may be upstream of NF-κB in CFTR-mediated inflammation under
high glucose conditions.
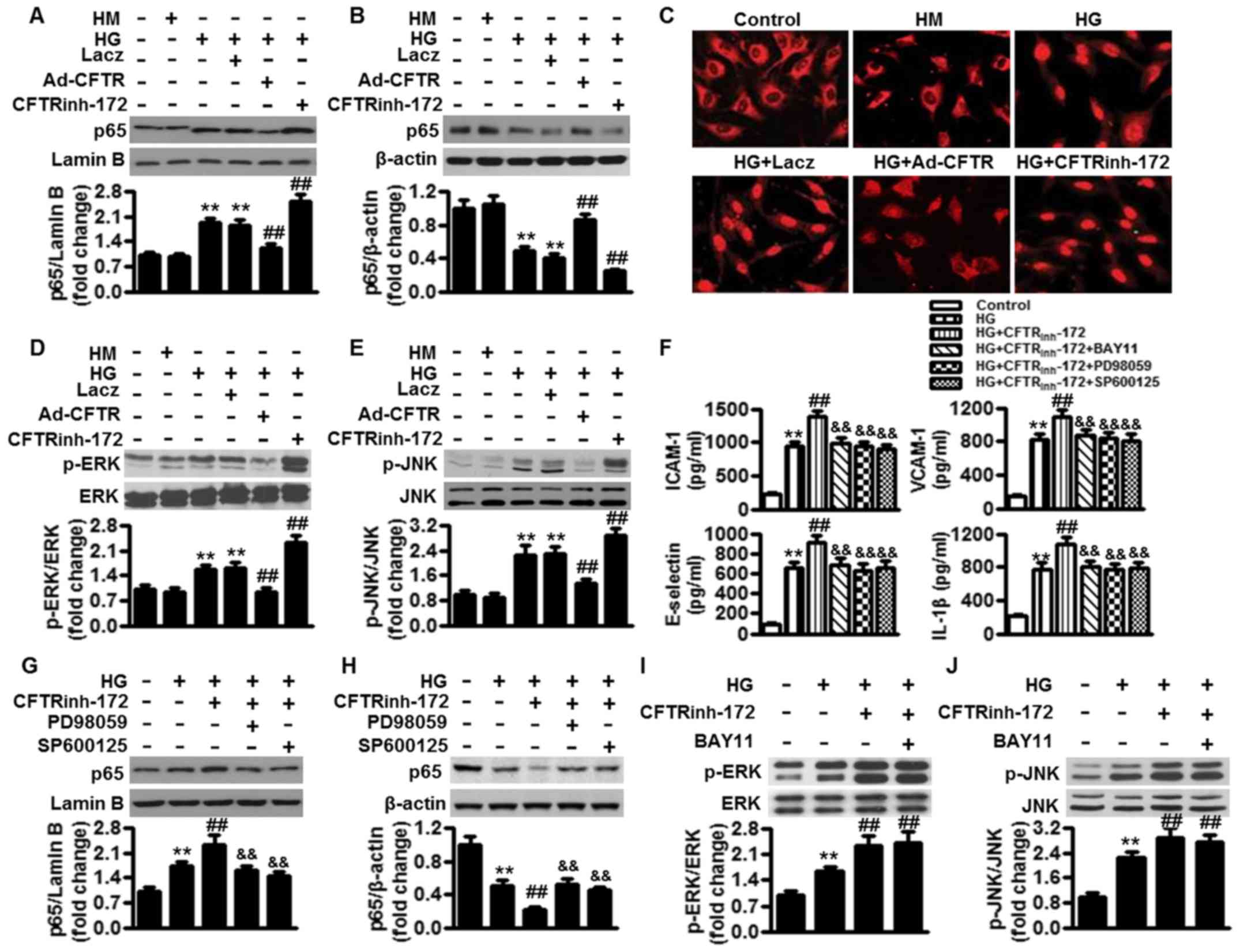 | Figure 4CFTR attenuates inflammatory response
under high glucose conditions through the inhibition of NF-κB and
MAPK signaling. (A and B) HUVECs were pretreated with LacZ (50
MOI), Ad-CFTR (50 MOI) or CFTRinh-172 (10 µM) for
24 h, followed by high glucose (25 mM) treatment for another 24 h.
(A) Nuclear and (B) cytosolic proteins were isolated and detected
by western blotting using p65 antibody. **P<0.01 vs.
control; ##P<0.01 vs. high glucose alone (n=6). (C)
Representative images of p65 distribution in HUVECs. (D) ERK and
(E) JNK phosphorylation was determined by western blotting.
**P<0.01 vs. control; ##P<0.01 vs. high
glucose alone (n=6). (F) Cells were pretreated with
CFTRinh-172 (10 µM) for 24 h, followed by
incubation with BAY11 (20 µM), PD98059 (10 µM) or
SP600125 (10 µM) for another 24 h under high glucose
conditions. The concentrations of ICAM-1, VCAM-1, E-selectin and
IL-1β were measured by enzyme-linked immunosorbent assay. (G-J) p65
expression in (G) nuclear and (H) cytosolic fractions, and (I) ERK
and (J) JNK phosphorylation were assessed by western blotting.
**P<0.01 vs. control; ##P<0.01 vs. high
glucose alone; &&P<0.01 vs. high glucose +
CFTRinh-172 (n=6). CFTR, cystic fibrosis transmembrane
conductance regulator; HUVECs, human umbilical vein endothelial
cells;HM, high mannitol; MOI, multiplicity of infection; Ad,
adenoviral vector; HG, high glucose; NF-κB, nuclear factor-κB;
MAPK, mitogen-activated protein kinase; ERK, extracellular
signal-regulated kinase; JNK, Janus kinase; p-, phosphorylated. |
Inhibition of oxidative stress mitigates
high glucose-induced inflammation by reducing CFTR expression
Previous studies have demonstrated the role of high
glucose-induced oxidative stress in inflammation (3,6).
To investigate the role of oxidative stress on high
glucose-mediated CFTR expression, in the present study, HUVECs were
treated with the antioxidants apocynin (1 µM) and NAC (5
µM). Addition of the each antioxidant significantly
inhibited high glucose-induced downregulation of CFTR expression,
respectively (Fig. 5A). Next, the
effects of ROS inhibition on the secretion of inflammatory
biomediators were observed. HUVECs treated with apocynin or NAC
following high glucose treatment showed a marked reduction in the
concentration of inflammatory biomediators, including ICAM-1,
VCAM-1, E-selectin and IL-1β, in comparison to cells treated with
high glucose (Fig. 5B).
Concomitantly, western blotting results showed that following
treatment of high glucose-challenged HUVECs with apopcynin or NAC,
p65 translocation and ERK and JNK phosphorylation were
significantly inhibited as compared with that of the high
glucose-treated group without antioxidant treatment (Fig. 5C–F). These results suggested that
the anti-inflammatory effect of oxidative stress inhibition was
mediated, at least partially, by restoration of CFTR
expression.
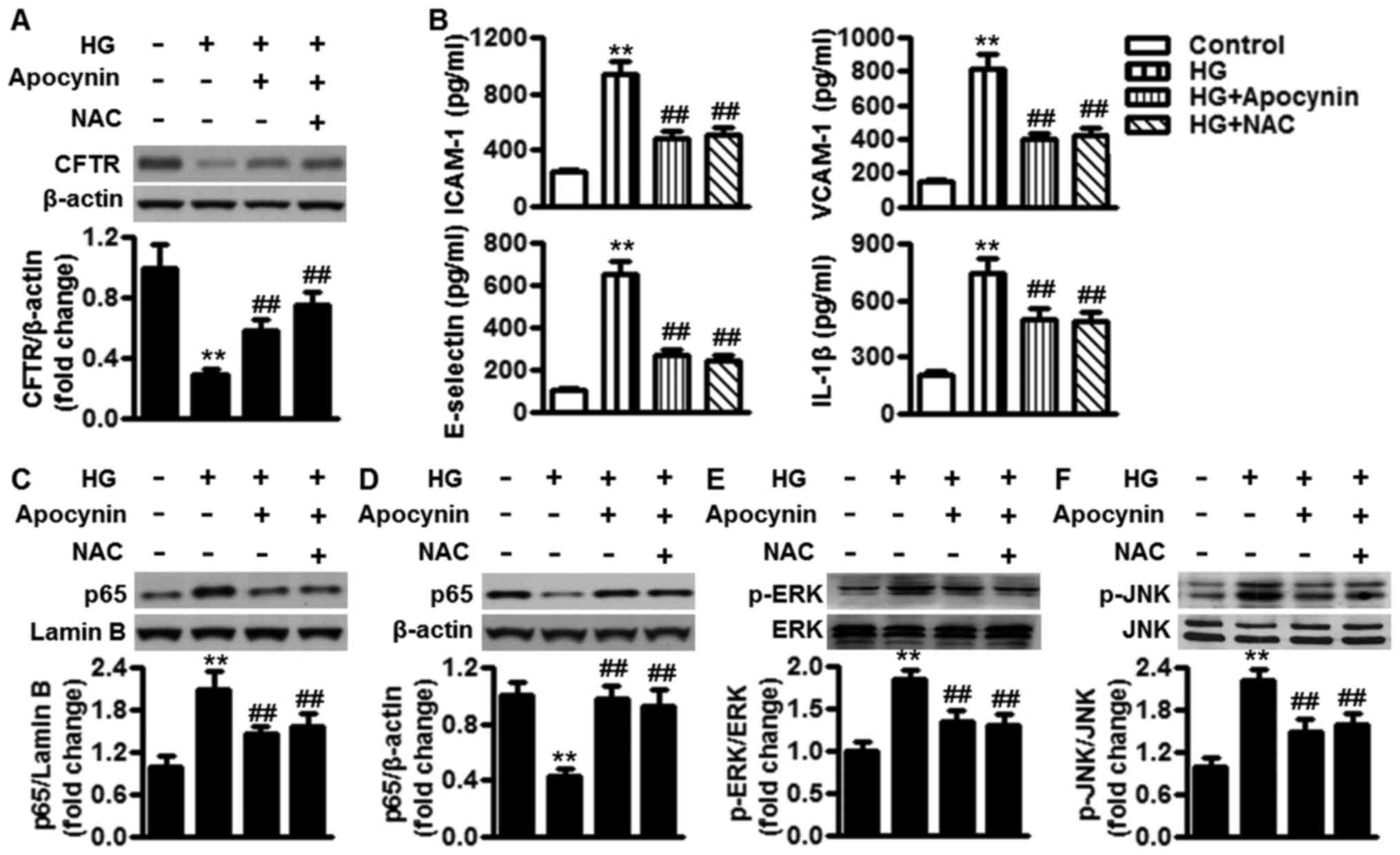 | Figure 5Inhibition of oxidative stress
ameliorates high glucose-induced inflammation by reducingCFTR
expression. (A) HUVECs were treated with apocynin (1 µM) or
NAC (5 µM) in the presence of high glucose (25 mM) for 24 h.
CFTR protein expression was examined by western blotting. (B) The
concentrations of ICAM-1, VCAM-1, E-selectin and IL-1β were
measured by enzyme-linked immunosorbent assay. (Cand D) p65 nuclear
translocation and (E) ERK and (F) JNK phosphorylation were
determined by western blotting. All data are expressed as the mean
± standard error of the mean. **P<0.01 vs. control;
##P<0.01 vs. high glucose alone (n=6). CFTR, cystic
fibrosis transmembrane conductance regulator; HUVECs, human
umbilical vein endothelial cells; HG, high glucose; NAC,
N-acetyl-L-cysteine; ICAM-1, intercellular adhesion molecule-1;
VCAM-1, vascular cell adhesion molecule-1; IL, interleukin; ERK,
extracellular signal-regulated kinase; JNK, Janus kinase; p-,
phosphorylated. |
Discussion
To the best of our knowledge, the current study is
the first to present the following novel findings: i) CFTR
expression is inhibited by glucose in a concentration-dependent
manner and is negatively associated with ROS production in
endothelial cells; ii) the downregulation of CFTR induced by high
glucose leads to activation of NF-κB and MAPK signaling pathways,
and excessive production of inflammatory biomediators, leading to
endothelial cell inflammation; and iii) inhibition of oxidative
stress ameliorates high glucose-induced endothelial cell
inflammation, at least partially by elevating CFTR expression.
Endothelial dysfunction in diabetes is characterized
by complex changes in the biochemical, mechanical and structural
properties of the endothelium, which may be responsible for the
development of various cardiovascular diseases (24,25). However, the precise mechanism of
endothelial dysfunction induced by hyperglycemia has not been fully
investigated. In the present experiment, in vitro, it was
found that CFTR expression in endothelial cells was inhibited
following high glucose challenge, indicating that the changes in
CFTR expression may serve as an important factor or mediator of
endothelial dysfunction in diabetes. The mechanisms by which high
glucose regulates CFTR expression in endothelial cells requires
further study.
Multiple hypotheses have been proposed to explain
the mechanisms of CF, among which oxidative stress and inflammation
are considered to be closely associated with CFTR mutations, as
chronic inflammation with excessive production of inflammatory
biomediators can be observed in the airways of CF patients
(16,17). Moreover, mutation of CFTR or loss
of function of CFTR has also been shown to directly affect the
intracellular redox status in CF lungs (26,27). Additionally, in CF mouse
intestines or CFTR-knockdown intestinal epithelial cells, there is
an upregulation of genes involved in oxidative stress and
inflammation (19,28). These findings collectively suggest
that oxidative stress and inflammation are implicated in the
pathophysiology of several disorders in CF subjects. Notably,
oxidative stress and inflammation are central in diabetes
complications, and vascular endothelial cells are critical in
orchestrating these effects (22,23). However, the contributions of CFTR
have been poorly investigated in endothelial cells under high
glucose conditions. The present study found that high glucose could
increase ROS production and induce the secretion of inflammatory
biomediators in endothelial cells. Pharmacological inhibition of
CFTR markedly augmented the effects of high glucose treatment,
while overexpression of CFTR markedly attenuated the high
glucose-induced responses. These results clearly suggest that high
glucose-induced CFTR downregulation is responsible for high
glucose-induced oxidative stress and inflammation.
To further investigate the mechanism of CFTR with
regard to protecting endothelial function, the involvement of NF-κB
signaling in damage responses triggered by high glucose in
endothelial cells was first investigated, as various studies have
indicated the intrinsic activation of this signaling in CF
(19,29). Moreover, NF-κB has been found to
be an essential regulator of various genes, including inflammatory
biomediators ICAM-1, VCAM-1, E-selectin and IL-1β (19,30). Once activated, the p65 subunit of
NF-κB is released and translocates into the nucleus to regulate the
target genes (31). In the
present study, it was found that forced CFTR expression blocked
high glucose-induced p65 nuclear translocation in endothelial
cells, whereas CFTR inhibition further enhanced the translocation.
In addition, the increase in pro-inflammatory cytokines and
oxidative stress under diabetic conditions can also activate MAPK
signaling (32). Importantly,
mutations of CFTR have been suggested to abnormally activate MAPK
(16,33). The present data showed that
overexpression of CFTR was decreased, whereas inhibition of CFTR
augmented the phosphorylation of ERK and JNK under high glucose
conditions. It is noteworthy that the pro-inflammatory effect of
CFTR inhibition was almost abolished by NF-κB, ERK and JNK
inhibitors, further supporting the fact that CFTR alleviates high
glucose-induced inflammation via inhibition of the NF-κB and MAPK
signaling pathways. It has also been reported that inhibition of
the MAPK signaling pathway can block NF-κB nuclear translocation,
and thus attenuate inflammatory response (29). In line with this, pharmacological
inhibition of ERK or JNK in the present study markedly blocked CFTR
inhibition-induced NF-κB nuclear translocation under high glucose
conditions. However, NF-κB inhibition failed to inhibit MAPK
activation, suggesting NF-κB is downstream of MAPK in CFTR-mediated
inflammatory responses.
Furthermore, to delineate the mechanism behind high
glucose-induced inflammation, given the reported involvement of
oxidative stress in regulating inflammation (7,8),
the role of ROS was examined. The present study showed that high
glucose decreased CFTR expression, which exhibited a reciprocal
pattern of concentration-dependent ROS production. More
importantly, apocynin and NAC, known antioxidants, not only
restored the high glucose-induced decrease in CFTR expression, but
also attenuated the pro-inflammatory effect of high glucose via
inactivation of the NF-κB and MAPK signaling pathways. These data
indicate that the anti-inflammatory effect of antioxidants may be
due to their effect on the upregulation of CFTR expression.
In conclusion, the present findings revealed a novel
function of CFTR as a regulator of endothelial cell oxidative
stress and inflammation, suggesting that CFTR overexpression may be
a feasible strategy for alleviating vascular endothelial
inflammation and diabetic cardiovascular diseases.
Acknowledgments
Not applicable.
Notes
[1]
Funding
No funding was received.
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
YF supervised the study. LS and CY participated in
study design and scientific discussion of the data. YF, MJ and QL
contributed to the scientific discussion of the data. YF and YX
contributed to the biochemical analysis of the experiments. All
authors read and approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Hamilton SJ and Watts GF: Endothelial
dysfunction in diabetes: Pathogenesis, significance, and treatment.
Rev Diabet Stud. 10:133–156. 2013. View Article : Google Scholar
|
|
2
|
Winer N and Sowers JR: Epidemiology of
diabetes. J Clin Pharmacol. 44:397–405. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Domingueti CP, Dusse LM, Carvalho M, de
Sousa LP, Gomes KB and Fernandes AP: Diabetes mellitus: The linkage
between oxidative stress, inflammation, hypercoagulability and
vascular complications. J Diabetes Complications. 30:738–745. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agrawal NK and Kant S: Targeting
inflammation in diabetes: Newer therapeutic options. World J
Diabetes. 5:697–710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Westermann D, Van Linthout S, Dhayat S,
Dhayat N, Escher F, Bücker-Gärtner C, Spillmann F, Noutsias M, Riad
A, Schultheiss HP, et al: Cardioprotective and anti-inflammatory
effects of interleukin converting enzyme inhibition in experimental
diabetic cardiomyopathy. Diabetes. 56:1834–1841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giugliano D, Ceriello A and Paolisso G:
Diabetes mellitus, hypertension, and cardiovascular disease: Which
role for oxidative stress? Metabolism. 44:363–368. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Safi SZ, Batumalaie K, Qvist R, Mohd Yusof
K and Ismail IS: Gelam honey attenuates the oxidative
stress-induced inflammatory pathways in pancreatic hamster cells.
Evid Based Complement Alternat Med. 2016:58436152016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Creager MA, Lüscher TF, Cosentino F and
Beckman JA: Diabetes and vascular disease: Pathophysiology,
clinical consequences, and medical therapy: Part I. Circulation.
108:1527–1532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang DD, Niu HX, Peng FF, Long HB, Liu ZR,
Zhao H and Chen YH: Hypochlorite-modified albumin upregulates
ICAM-1 expression via a MAPK-NF-κB signaling cascade: Protective
effects of apocynin. Oxid Med Cell Longev. 2016:18523402016.
View Article : Google Scholar
|
|
10
|
Kuo WW, Wang WJ, Tsai CY, Way CL, Hsu HH
and Chen LM: Diallyl trisufide (DATS) suppresses high
glucose-induced cardiomyocyte apoptosis by inhibiting JNK/NFκB
signaling via attenuating ROS generation. Int J Cardiol.
168:270–280. 2013. View Article : Google Scholar
|
|
11
|
Zhou J, Xu G, Ma S, Li F, Yuan M, Xu H and
Huang K: Catalpol ameliorates high-fat diet-induced insulin
resistance and adipose tissue inflammation by suppressing the JNK
and NF-κB pathways. Biochem Biophys Res Commun. 467:853–858. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang
Y, Zhang J, Zhou S, Liu Q, Li X, et al: Inhibition of JNK
phosphorylation by a novel curcumin analog prevents high
glucose-induced inflammation and apoptosis in cardiomyocytes and
the development of diabetic cardiomyopathy. Diabetes. 63:3497–3511.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu X and Sun J: Endothelial cells
dysfunction induced by silica nanoparticles through oxidative
stress via JNK/P53 and NF-kappaB pathways. Biomaterials.
31:8198–8209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Caballo C, Palomo M, Cases A, Galán AM,
Molina P, Vera M, Bosch X, Escolar G and Diaz-Ricart M: NFκB in the
development of endothelial activation and damage in uremia: An in
vitro approach. PLoS One. 7:e433742012. View Article : Google Scholar
|
|
15
|
Stoltz DA, Meyerholz DK and Welsh MJ:
Origins of cystic fibrosis lung disease. N Engl J Med. 372:351–362.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bérubé J, Roussel L, Nattagh L and
Rousseau S: Loss of cystic fibrosis transmembrane conductance
regulator function enhances activation of p38 and ERK MAPKs,
increasing interleukin-6 synthesis in airway epithelial cells
exposed to Pseudomonas aeruginosa. J Biol Chem. 285:22299–22307.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitola S, Sorbello V, Ponte E, Copreni E,
Mascia C, Bardessono M, Goia M, Biasi F, Conese M, Poli G, et al:
Tumor necrosis factor-alpha in airway secretions from cystic
fibrosis patients upregulate endothelial adhesion molecules and
induce airway epithelial cell apoptosis: Implications for cystic
fibrosis lung disease. Int J Immunopathol Pharmacol. 21:851–865.
2008. View Article : Google Scholar
|
|
18
|
Cantin AM, White TB, Cross CE, Forman HJ,
Sokol RJ and Borowitz D: Antioxidants in cystic fibrosis.
Conclusions from the CF antioxidant workshop, Bethesda, Maryland,
November 11–12, 2003. Free Radic Biol Med. 42:15–31. 2007.
View Article : Google Scholar
|
|
19
|
Kleme ML, Sané AT, Garofalo C and Levy E:
Targeted CFTR gene disruption with zinc-finger nucleases in human
intestinal epithelial cells induces oxidative stress and
inflammation. Int J Biochem Cell Biol. 74:84–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Verhaeghe C, Remouchamps C, Hennuy B,
Vanderplasschen A, Chariot A, Tabruyn SP, Oury C and Bours V: Role
of IKK and ERK pathways in intrinsic inflammation of cystic
fibrosis airways. Biochem Pharmacol. 73:1982–1994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tomaru M and Matsuoka M: The role of
mitogen-activated protein kinases in crystalline silica-induced
cyclooxygenase-2 expression in A549 human lung epithelial cells.
Toxicol Mech Methods. 21:513–519. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Funk SD, Yurdagul A Jr and Orr AW:
Hyperglycemia and endothelial dysfunction in atherosclerosis:
Lessons from type 1 diabetes. Int J Vasc Med.
2012:5696542012.PubMed/NCBI
|
|
23
|
Taye A, Saad AH, Kumar AH and Morawietz H:
Effect of apocynin on NADPH oxidase-mediated oxidative stress-LOX
1-eNOS pathway in human endothelial cells exposed to high glucose.
Eur J Pharmacol. 627:42–48. 2010. View Article : Google Scholar
|
|
24
|
Singh P, Khullar S, Singh M, Kaur G and
Mastana S: Diabetes to cardiovascular disease: Is depression the
potential missing link? Med Hypotheses. 84:370–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prieto D, Contreras C and Sánchez A:
Endothelial dysfunction, obesity and insulin resistance. Curr Vasc
Pharmacol. 12:412–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Kinter M, Shank S, Cotton C,
Kelley TJ and Ziady AG: Dysfunction of Nrf-2 in CF epithelia leads
to excess intracellular H2O2 and inflammatory cytokine production.
PLoS One. 3:e33672008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kelly M, Trudel S, Brouillard F, Bouillaud
F, Colas J, Nguyen-Khoa T, Ollero M, Edelman A and Fritsch J:
Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172
and GlyH-101 target mitochondrial functions, independently of
chloride channel inhibition. J Pharmacol Exp Ther. 333:60–69. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Norkina O, Burnett TG and De Lisle RC:
Bacterial overgrowth in the cystic fibrosis transmembrane
conductance regulator null mouse small intestine. Infect Immun.
72:6040–6049. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong ZW, Chen J, Ruan YC, Zhou T, Chen Y,
Chen Y, Tsang LL, Chan HC and Peng YZ: CFTR-regulated MAPK/NF-κB
signaling in pulmonary inflammation in thermal inhalation injury.
Sci Rep. 5:159462015. View Article : Google Scholar
|
|
30
|
Chen J, Jiang XH, Chen H, Guo JH, Tsang
LL, Yu MK, Xu WM and Chan HC: CFTR negatively regulates
cyclooxygenase-2-PGE(2) positive feedback loop in inflammation. J
Cell Physiol. 227:2759–2766. 2012. View Article : Google Scholar
|
|
31
|
Mitchell S, Vargas J and Hoffmann A:
Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med.
8:227–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kaneto H, Nakatani Y, Kawamori D,
Miyatsuka T and Matsuoka TA: Involvement of oxidative stress and
the JNK pathway in glucose toxicity. Rev Diabet Stud. 1:165–174.
2004. View Article : Google Scholar
|
|
33
|
Roque T, Boncoeur E, Saint-Criq V, Bonvin
E, Clement A, Tabary O and Jacquot J: Proinflammatory effect of
sodium 4-phenylbutyrate in deltaF508-cystic fibrosis transmembrane
conductance regulator lung epithelial cells: Involvement of
extracellular signal-regulated protein kinase 1/2 and
c-Jun-NH2-terminal kinase signaling. J Pharmacol Exp Ther.
326:949–956. 2008. View Article : Google Scholar : PubMed/NCBI
|