Introduction
Heterogeneous disorders affecting kidney structure
and function, chronic kidney disease has been treated as a threat
to the health of the people all over the world (1). Due to the recurrent hospitalization,
accelerated death and morbidity, increasing research has been
devoted to investigate the underlying molecular mechanism. As the
final manifestation of chronic kidney disease, renal fibrosis is
the common pathway for chronic kidney disease towards kidney
failure (2,3). It is characterized by an excessive
accumulation and deposition of extracellular matrix components.
Fibrosis formation could be stimulated by chronic
glomerulonephritis, chronic pyelonephritis, obstructive
nephropathy, systemic lupus; hereditary kidney disease, diabetic
nephropathy, hypertensive kidney disease, drug-induced kidney
disease, hepatitis B or the HIV caused kidney disease and renal
transplantation (4–7). Among these inducements, with the
development of economy and the improvement of people's living
standard, eating habits caused kidney disease has been widely
received attention by scholars. In previous reports, diet-induced
increase in plasma oxidized LDL could promote early fibrosis in a
renal porcine auto-transplantation model (8,9).
Moreover, high fat diet-induced obesity would change kidney-related
gene expression and lead to chronic kidney disease formation
(10,11). These changes alter normal
physiological activity and promote type II diabetes, in turn;
diabetes enhancement will increase kidney injury development and
fibrosis formation (12).
However, though the medical substantial progress has expanded our
knowledge of kidney related diseases, there has been little in the
way of new therapies. Hence, the additional intervention, including
treatment with chemoprevention from natural products, for the
treatment of kidney injury.
To our knowledge, in the development of kidney
inflammation, TGF-β1/Smads pathway has been regarded as the most
significant in the promotion for fibrosis formation and kidney
injury (13). High fat
diet-induced kidney damage also has been thought to be associated
with inflammation development. As previously reports, kidney injury
as a typical inflammatory response, nuclear factor-κB (NF-κB)
signaling pathway was directly involved in the developmental
process of lung injury (14,15). Thus, how to inhibit the activation
of NF-κB pathway may be the key point for the treatment of renal
inflammation. CX3CL1-CX3CR1 interaction has been indicated as
important in many inflammation-related disorder, including
hepatitis and fibrosis, nerve damage and tumors (16,17). CX3CL1-CX3CR1 axis has the ability
to enhance the levels of phosphorylated NF-κB, which in turn, could
continually upregulate CX3CL1-CX3CR1 interaction. Crosstalk of
CX3CL1-CX3CR1 axis and NF-κB pathways play a critical role in the
inflammatory formation and development (15,17). Hence, we investigated whether
CX3CR1 knock out inhibits high fructose-stimulated acute kidney
injury by interfering with CX3CL1-CX3CR1 axis expression and NF-κB
pathways activation. Furthermore, podocyte cells were used as a
model to evaluate the effects of CX3CR1 on inflammatory
responses.
Materials and methods
Reagents and animals
Fructose (CAS: 53188-23-1, HPLC analysis ≥99%) was
purchased from the National Institute for the Control of
Pharmaceutical and Biological Products (Beijing, China).
Interleukin-1β (IL-1β) (freeze-dried powder) was obtained from
Nanjing Biohelper Co., Ltd. (Nanjing, China) and prepared in
phosphate-buffered saline (PBS) at the concentration of 100
µg/ml. The antibodies used in this study were obtained from
CST Technology Co. (Danvers, MA, USA). The mouse enzyme-linked
immunosorbent assay (ELISA) kits were purchased from BioLegend (San
Diego, CA, USA). Male C57BL/6 mice with CX3CR1 gene-knock out were
purchased from Beijing Biocytogen Co., Ltd. (Beijing, China). The
mice weighting 20–25 g were housed in a temperature and
humidity-controlled environment (25±2°C, 50±10% humidity) with a
standard 12 h light/dark cycle with food and water. This study was
approved by the Ethics Committee on Animal Research at Department
of Nephrology, Navy General Hospital of Chinese PLA. The mice were
randomly divided into 3 groups: i) wild-type (WT); ii) Fructose-fed
wild-type (Fru-WT); iii) Fructose-fed CX3CR1-knock out mice
(Fru-CX3CR1-KO). Fru-WT and Fru-CX3CR1-KO group mice were orally
administered with 30% fructose water/day; and the WT mice were
orally fed with equal amount of PBS. The entire experimental period
was 56 days. All animal experiments were performed in accordance
with the guide for the Care and Use of Laboratory Animals
established by the US National Institutes of Health.
Cell culture
The conditionally immortalized mouse podocyte cell
line was obtained from Institute of Biochemistry and Cell Biology,
SIBS, CAS (Shanghai, China). Cells were grown and maintained at
33°C in RPMI-1640 medium containing 10% fetal bovine serum (FBS),
recombinant interferon-γ (R&D Systems, Minneapolis, MN, USA)
and 1% penicillin/streptomycin. Recombinant lent viral particles
encoding CX3CR1 and short hairpin RNA targeting CX3CR1 were
produced, concentrated, and titrated. The short hairpin RNA
oligonucleotide sequences of CX3CR1 were described earlier
(18). Cells were seeded at
3×104 cells/ml and were infected with recombinant lent
virus twice with an interval of 12 h and incubated for 24 h. After
24 h, the medium was refreshed and the cells were cultured for
another 24 h. Cells were serum starved for 36 h and stimulated with
IL-1β (75 ng/ml) for the indicated time periods.
Biochemical analysis
At the end of all experiments, rats were sacrificed
by decapitation at 9:30–10:30 a.m. after a 16-h fast in order to
avoid the fluctuation of hormone levels due to circadian rhythms.
Before this, the insulin tolerance test (ITT) and oral glucose
tolerance test (OGTT) were performed in accordance with a
perviously described method (20).
Inflammatory cell counts of kidney
tissue
The kidney samples were centrifuged (4°C, 3,000 rpm
for 10 min) to pellet the cells. The cell pellets were resuspended
in PBS for the total cell counts using a hemacytometer, and
cytospins were prepared for differential cell counts by staining
with the Wright-Giemsa staining method.
Histopathological examination of kidney
tissues and flow cytometry analysis
Histopathological evaluation was performed by
Nanjing Biohelper Co., Ltd. (Nanjing, China). Kidneys were fixed
with 10% buffered formalin, imbedded in paraffin, and sliced. After
hematoxylin and eosin (H&E) staining, Red Oil O, Masson and EM
pathological changes of tissues were observed under a light
microscope or electron microscope. Some samples were subjected to
immunohistochemical staining (CX3CL1, CX3CR1 and NF-κB) according
to CST technology Co. introduction and performed by Shanghai Zhenda
Biotechnology, Co., Ltd. (Shanghai, China). For flow cytometry
analysis, the cells were obtained through shearing kidney tissue,
separating by collagenase type II (Invitrogen, Carlsbad, CA, USA)
digestion and suspended in RPMI-1640 medium (Gibco-BRL, Grand
Island, NY, USA). Cell suspensions were centrifuged at 1,000 rpm
for 5 min to remove cellular debris and impurities. Then, the
entire cells were harvested and washed twice in Hank's buffer
(Gibco-BRL). According to the protocol of R&D kit systems
(R&D Systems) for flow cytometry, add anti-CD45 FITC and
anti-CD80 FITC antibody to the flow cytometry tube containing
single-cell suspension, and these tubes were analyzed by Cytomics™
FC 500 MCL of Beckman Coulter (Brea, CA, USA).
ELISA measurement and biochemical
analysis
At the end of the experiments, blood were extracted
from the eyeball, the different serum concentration of the
inflammatory cytokines such as tumor necrosis factor-α (TNF-α),
interferon-γ (IFN-γ), IL-2, IL-1β and IL-6 were measured using
ELISA kits according to the manufacturer's instructions (R&D
Systems). The other indicators shown in Table I were also performed by Shanghai
Zhenda Biotechnology, Co., Ltd.
 | Table IEffects of fructose on physiological
indexes and biochemical parameters in serum of CX3CR1-KO mice. |
Table I
Effects of fructose on physiological
indexes and biochemical parameters in serum of CX3CR1-KO mice.
| Items | WT | Fru-WT | Fru-CX3CR1-KO |
|---|
| Serum uric acid
(mg/dl) | 3.28±70.79 | 6.91±71.50a | 4.73±70.19d |
| Urinary uric acid
(mg/dl) | 41.77±71.52 |
32.15±73.79b | 40.70±72.34e |
| Serum creatinine
(mg/dl) | 0.96±70.24 | 1.94±70.26c | 0.84±71.18e |
| Urinary creatinine
(mg/dl) | 37.53±72.89 | 27.41±73.76a | 34.97±71.96d |
| Blood urea nitrogen
(mg/dl) | 32.95±71.61 | 42.47±72.74a | 32.93±74.38d |
| FEUA (%) | 19.17±70.34 |
12.37±70.25b | 16.85±71.30e |
| Serum triglyceride
(mg/dl) | 119.84±5.68 | 175.30±4.58a | 125.17±6.96d |
| Serum total
cholesterol (mg/dl) | 72.09±1.21 | 85.36±1.87a | 72.80±1.21d |
| Serum very-low
density lipoprotein (μg/ml) | 93.42±4.98 | 137.97±6.62c | 95.98±6.56d |
| Serum insulin
(µg/ml) | 2.33±0.19 | 3.90±0.46c | 2.02±0.23d |
| Serum leptin
(µg/l) | 0.48±0.02 | 0.64±0.03a | 0.46±0.05e |
Western blotting and quantitative RT-PCR
(qRT-PCR) analysis
Proteins were extracted from the kidney using T-PER
Tissue Protein Extraction Reagent kit (Thermo Fisher Scientific,
Waltham, MA, USA) according to the manufacturer's instructions.
Protein concentrations were determined by BCA protein assay kit,
and equal amounts of protein were loaded per well on a 10% sodium
dodecyl sulphate-polyacrylamide gel. Subsequently, proteins were
transferred onto polyvinylidene difluoride membrane. The resulting
membrane was blocked with Tris-buffered saline containing 0.05%
Tween-20 (TBS-T), supplemented with 5% skim milk (Sigma, St. Louis,
MO, USA) at room temperature for 2 h on a rotary shaker, and
followed by TBS-T washing. The specific primary antibody, diluted
in TBST, was incubated with the membrane at 4°C overnight.
Subsequently, the membrane was washed with TBS-T followed by
incubation with the peroxidase-conjugated secondary antibody at
room temperature for 1 h. The immunoactive proteins were detected
by using an enhanced chemiluminescence western blotting detection
kit. Western blot bands were observed using GE Healthcare ECL
western blotting analysis system and exposed to Kodak X-ray film.
For qRT-PCR experiments, quantitative RT-PCR (qRT-PCR) and western
blot analysis were performed as described previously (17).
Statistical analysis
Data are expressed as means ± SEM. Treated cells,
tissues and the corresponding controls were compared using GraphPad
Prism (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA) by
one-way ANOVA with Dunn's least significant difference tests.
Differences between groups were considered significant at
p<0.05.
Results
CX3CR1 deficiency ameliorates high
fructose-induced metabolic disorder responses and inflammatory cell
counts
We investigated that the effect of CX3CR1 deficiency
mice on fructose-stimulated kidney injury. As shown in Table I, major kidney injury-related
indicators in serum such as uric acid, blood urea nitrogen and
serum total cholesterol, were significantly increased with the
fructose administration, compared to WT group. In contrast, in
CX3CR1-KO with fructose treatment mice, these were downregulated,
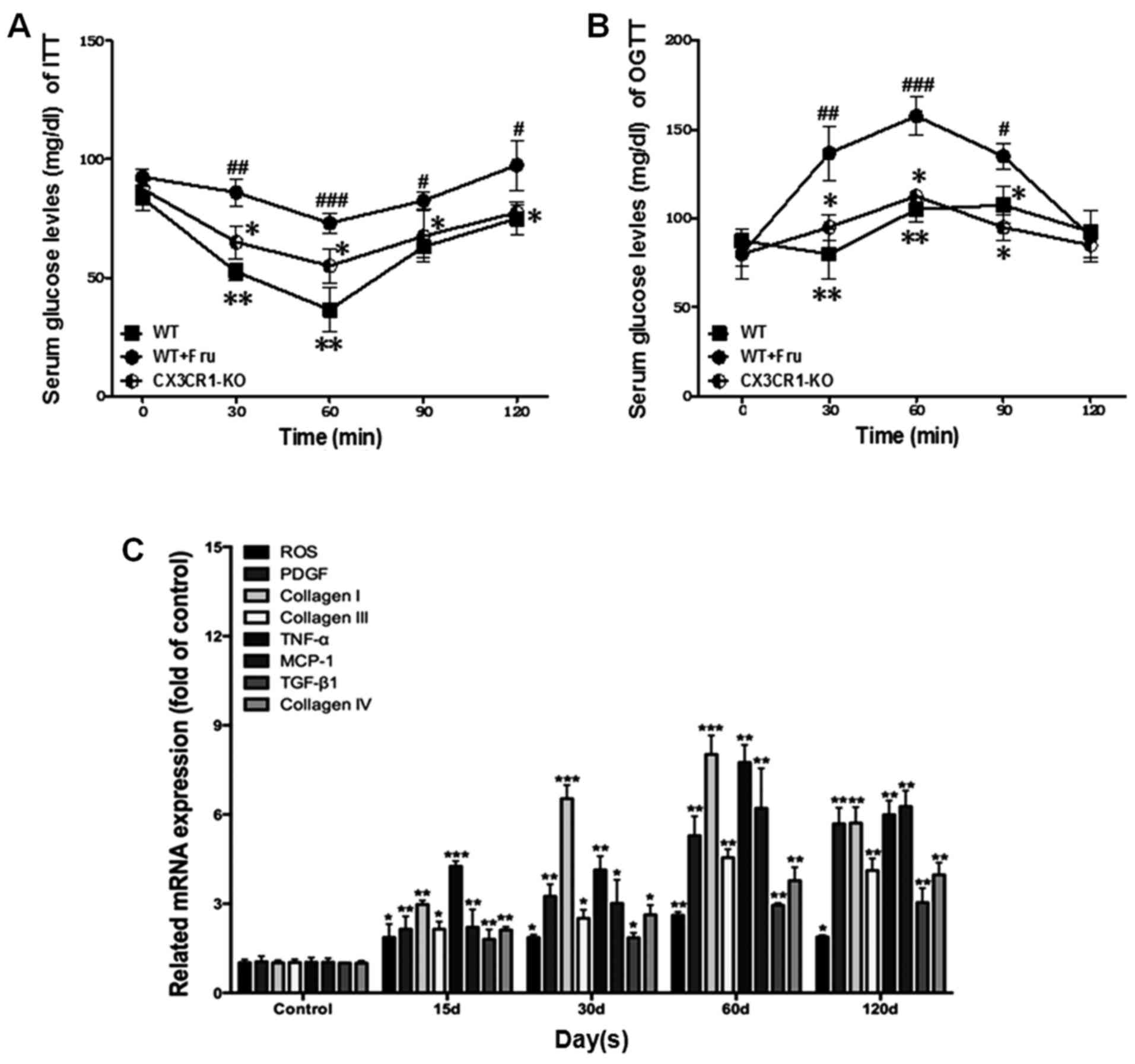
compared with the model. Also, as shown in Fig. 1A and B, the serum glucose levels
tested by ITT and OGTT were increased in WT+Fru group, compared
with CX3CR1-KO. Thus, high fructose is capable of changing
metabolic activities and weaken renal function. Fig. 1C shows that inflammation-related
cytokines in WT+Fru were significantly upregulated with the
increase of treatment in a time-independent manner. Of note, The
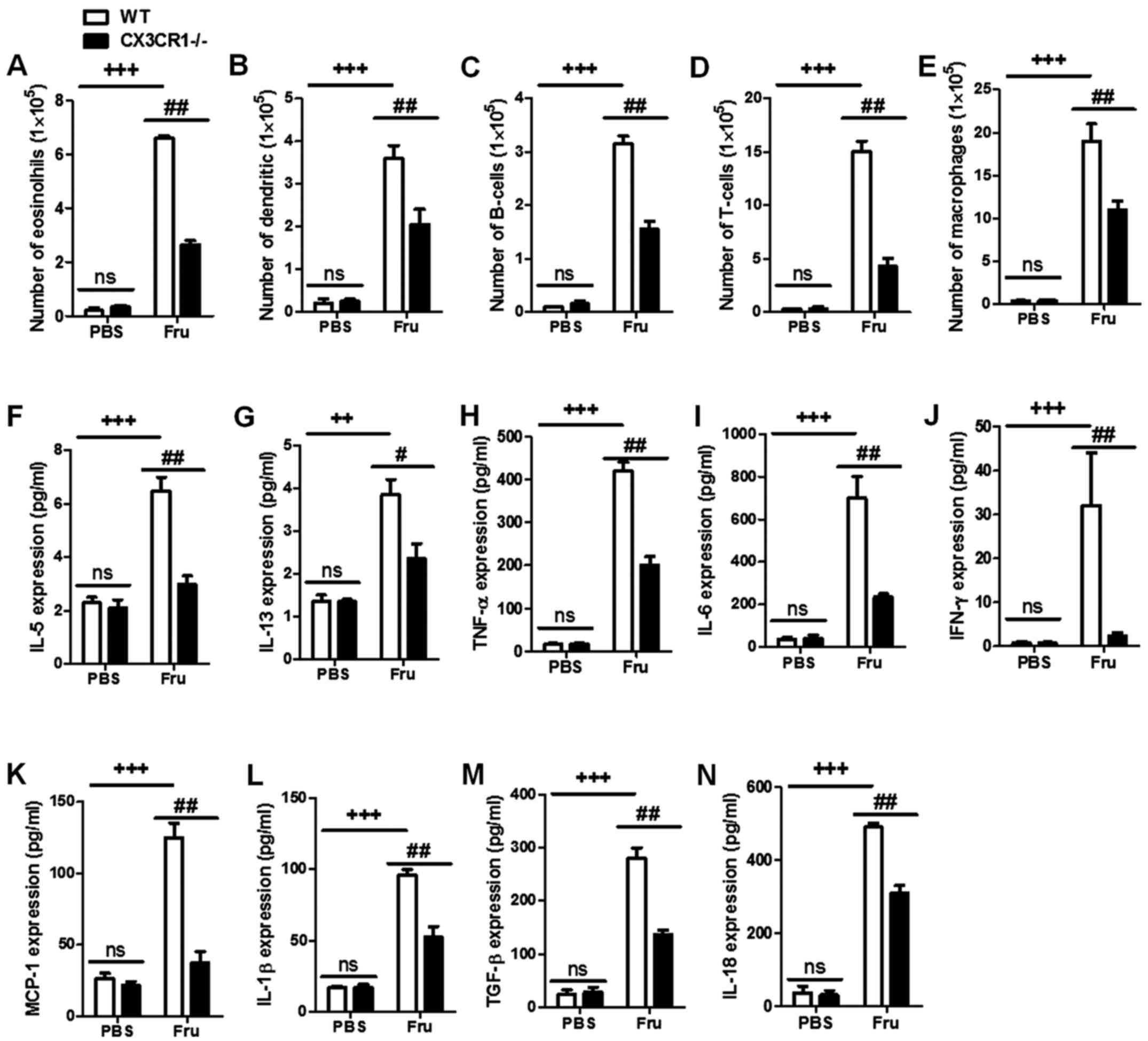
data of Fig. 2A-E indicated that
CX3CR1-KO may suppress the inflammation-related cells in kidneys,
including neutrophils and macrophages caused by fructose
administration-induced renal injury. These basic biomarkers
suggested that CX3CR1 deficiency is capable of suppressing kidney
damage with fructose administration.
Influence of CX3CR1 deficiency on
inflammation-related cytokines expression in fructose-induced
kidney injury
As indicated in Fig.
2F-N, the fructose administration significantly enhance renal
injury levels compared to PBS group. Moreover, CX3CR1−/−
mice had a lower inflammatory cytokine expression than fructose-fed
WT mice. We further investigated the mRNA level expression of
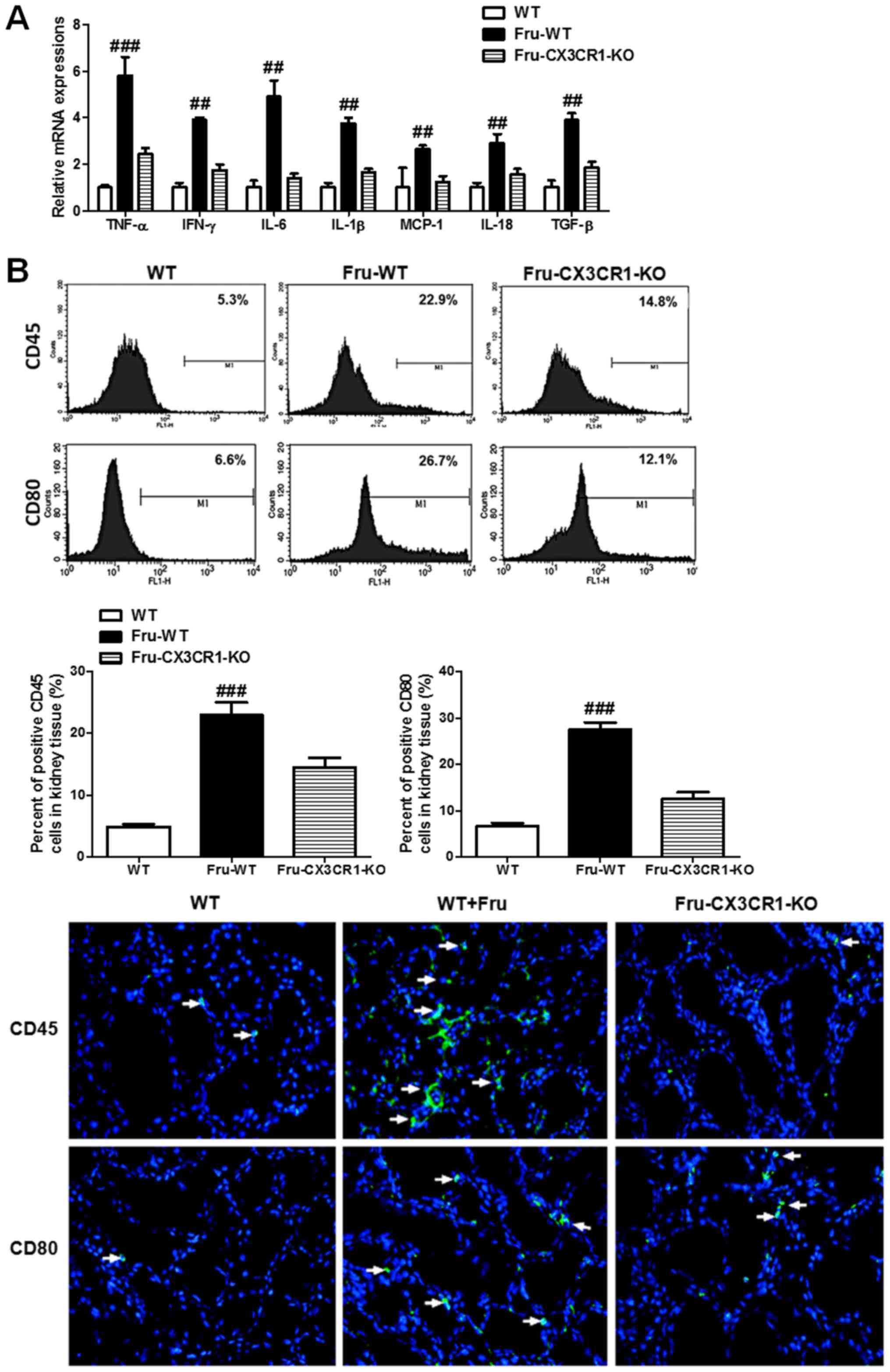
inflammatory cytokines. Data in Fig.
3A illustrate that the administration of fructose can increase
inflammatory cytokine expression in mRNA level, and downregulate in
CX3CR1 deficiency mice. Fig. 3B
shows that fructose significantly enhance the ratio of CD45 and
CD80 positive cells. In contrast, in CX3CR1-KO mice a lower ratio
of positive cells in CD45 and CD80 than fructose-fed WT was seen.
The histopathologic examination of kidney tissues in Fig. 3C show the effects of fructose on
renal injury. The results indicated that fructose caused lipid
accumulation, inflammatory cell infiltration, a certain degree of
fibrosis and damage of the tissues structure.
Effects of CX3CR1-KO on CX3CL1/CX3CR1
axis-stimulated NF-κB signaling pathway
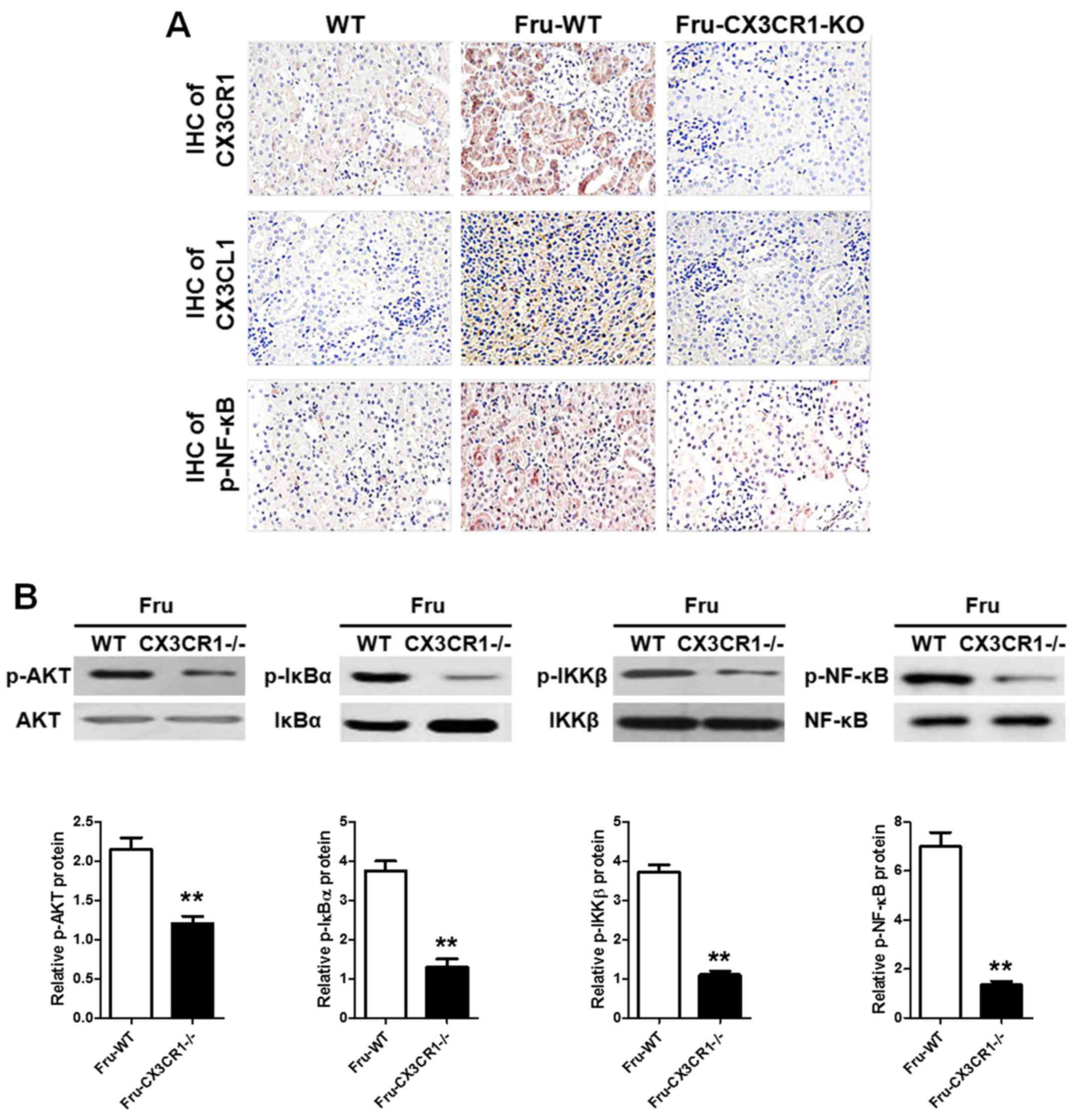
As showed in Fig.
4A, the IHC analysis given the AKT and NF-κB pathways were
significantly activated with the administration of fructose. Also,
in CX3CR1−/− mice, the phosphorylation of NF-κB is lower
than it in WT group. In addition, the western blot data (Fig. 4B) showed that AKT-stimulated NF-κB
signaling pathway in CX3CR1−/− mice with a significant
difference compared with WT. These findings demonstrated that AKT
pathway could be a link in the development of fructose-induced
inflammation of renal injury, which was directly activated by
CX3CL1-CX3CR1 axis. The AKT-stimulated NF-κB signaling pathway
activation was downregulated when the CX3CR1−/− mice
were challenged with fructose administration.
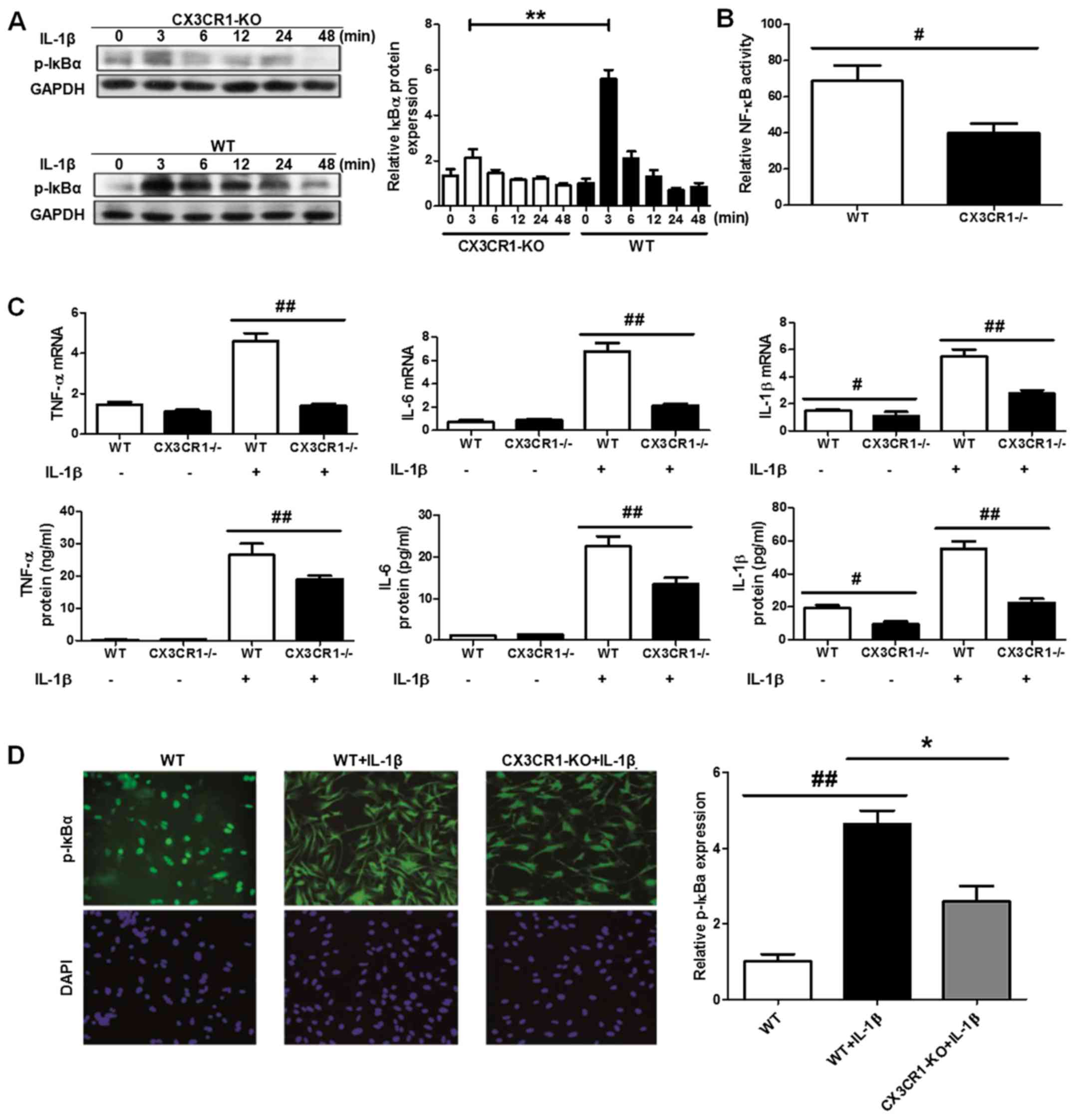
CX3CR1 deficiency inhibits inflammatory
response through NF-κB pathway in podocyte cells
Research has demonstrated that the activation of the
NF-κB pathway is involved in inflammation-related diseases both in
experimental models and in humans. Moreover, CX3CL1-CX3CR1 axis has
been shown to regulate the NF-κB pathway in multiple cell systems.
Hence, we investigated the effect of CX3CL1-CX3CR1 axis on NF-κB
pathway and the induced inflammatory responses with fructose
administration. As showed in Fig. 5A,
B and D, the IκBα degradation level was significantly
upregulated in CX3CR1 deficiency, compared with WT mice. The
increase of IκBα phosphorylation level results in the upregulation
of NF-κB pathway activation, compared to CX3CR1 deficiency mice.
The activity of NF-κB is also downregulated in the deficiency mice.
Of note, in podocyte cells with CX3CR1 deficiency, the inflammatory
cytokines have a lower release levels than WT group (Fig. 5C). Thus, these findings
demonstrated that CX3CR1 deficiency has a key role in the
inhibitory effect of fructose-induced inflammatory responses.
Discussion
Kidney injury syndrome consisting of chronic
inflammatory responses, has been emphasized as a serious health
threat world-wide (1,19). Moreover, as a serious syndrome,
the formation and development of kidney diseases are associated
with high morbidity and mortality. It is characterized by an
excessive accumulation and deposition of extracellular matrix
components. Kidney injury formation could be stimulated by chronic
glomerulonephritis, chronic pyelonephritis, obstructive
nephropathy, systemic lupus; hereditary kidney disease, diabetic
nephropathy, hypertensive kidney disease, drug-induced kidney
disease, hepatitis B or the HIV caused kidney disease and renal
transplantation. However, the molecular mechanism of high fructose
intake-induced chronic kidney disease, even kidney fibrosis is not
absolutely explained and reported. In previous studies, renal
damage as a typical inflammatory response, NF-κB signaling pathway
was directly involved in the developmental process of injury
(19,20). Thus, how to inhibit the activation
of NF-κB pathway may be the key point for the treatment of renal
damage. CX3CL1-CX3CR1 interaction has been treated as important in
inflammation-related disorders, including hepatitis and fibrosis,
nerve damage and tumor (21).
CX3CL1-CX3CR1 axis has the ability to enhance the levels of
phosphorylated NF-κB; in turn, NF-κB activation could continually
upregulate CX3CL1-CX3CR1 interaction (17,18). Crosstalk of CX3CL1-CX3CR1 axis and
NF-κB pathways play a critical role in the inflammation and
development (17). Hence, we
investigated whether CX3CR1-KO inhibits fructose-stimulated kidney
injury by interfering CX3CL1-CX3CR1 axis expression and NF-κB
pathways activation.
We used C57BL/6 mice to construct fructose-induced
renal injury model in order to investigate the effects of
CX3CL1-CX3CR1 axis. The results of our data indicated that fructose
administration could cause inflammatory cytokine releases including
TNF-α, IL-1β, IL-6, TGF-β and IL-18 in serum. Moreover, the
inflammatory cell counts of fructose-induced renal tissues were
significantly enhanced. Furthermore, we tested the crucial role of
CX3CL1/CX3CR1 signaling pathway in fructose-induced kidney injury.
The results showed that CX3CL1/CX3CR1 axis is involved in the
process of renal damage and could be activated via fructose
administration. The activation of CX3CL1/CX3CR1 axis will further
enhance the NF-κB pathway phosphorylation levels. As previous
reported the increase of NF-κB pathway phosphorylation leads to
inflammatory responses and cytokines production. Researchers have
indicated that the activation of CX3CL1/CX3CR1 axis was associated
with AKT pathway (ref. ?). Thus, we further assessed the
relationship of AKT pathway and CX3CL1/CX3CR1 axis. The data
demonstrated that fructose administration has the ability to
activate AKT pathway and increase the levels of phosphorylated AKT
expression. Phosphorylated AKT expression could significantly
enhance the activation of CX3CL1/CX3CR1 pathway and increase the
NF-κB pathway activation.
To study the possible role of CX3CL1/CX3CR1 axis in
fructose-induced kidney injury of mice, we used CX3CR1-knock out
mice as model to investigate the effects of crosstalk of
CX3CL1-CX3CR1 axis and NF-κB pathways. IHC analysis of IκB of
fructose administration showed that IκB was involved in the
development of renal injury. Compared with WT group, the
CX3CR1−/− mice have a lower IκB activation when
challenged with fructose. As the inducer of CX3CR1, we tested the
CX3CL1 expression between WT and CX3CR1−/− mice by IHC.
The results indicated that CX3CR1 knockout directly impacts the
expression of CX3CL1. The AKT signaling pathway may directly or
indirectly associate with fructose-induced kidney injury. In
knockout mice, the p-AKT expression was determined to be involved
in the process of injury. The data indicated AKT as an important
upstream of CX3CL1-CX3CR1 axis may impact CX3CL1-mediated
inflammatory responses.
We investigated fructose-induced kidney injury model
from the point of CX3CL1-CX3CR1 axis and its-related indicators. We
found that fructose as functional food additive has the ability to
cause inflammation by enhancing CX3CL1-CX3CR1 axis and NF-κB
activation. Also, the phosphorylated AKT could be significantly
activated in fructose-induced renal injury via CX3CL1-CX3CR1 axis.
We further investigated the role of CX3CL1-CX3CR1 pathway in lung
injury. CX3CR1 expression between WT and CX3CR1−/− mice
were tested to establish their relationship to injury. Our results
indicated that CX3CR1 may be the central and major indicator in the
process of kidney injury, which mediate the CX3CL1 to activate AKT
pathway and further enhance the NF-κB activation.
Notes
[1] Competing
interests
Authors declare that they have no competing
interest.
References
|
1
|
Megyesi J, Andrade L, Vieira JM Jr,
Safirstein RL and Price PM: Coordination of the cell cycle is an
important determinant of the syndrome of acute renal failure. Am J
Physiol Renal Physiol. 283:F810–F816. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou H, Kato A, Yasuda H, Miyaji T,
Fujigaki Y, Yamamoto T, Yonemura K and Hishida A: The induction of
cell cycle regulatory and DNA repair proteins in cisplatin-induced
acute renal failure. Toxicol Appl Pharmacol. 200:111–120. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Price PM, Safirstein RL and Megyesi J:
Protection of renal cells from cisplatin toxicity by cell cycle
inhibitors. Am J Physiol Renal Physiol. 286:F378–F384. 2004.
View Article : Google Scholar
|
|
4
|
Megyesi J, Safirstein RL and Price PM:
Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the
course of cisplatin-induced acute renal failure. J Clin Invest.
101:777–782. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou H, Fujigaki Y, Kato A, Miyaji T,
Yasuda H, Tsuji T, Yamamoto T, Yonemura K and Hishida A: Inhibition
of p21 modifies the response of cortical proximal tubules to
cisplatin in rats. Am J Physiol Renal Physiol. 291:F225–F235. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nowak G, Price PM and Schnellmann RG: Lack
of a functional p21WAF1/CIP1 gene accelerates caspase-independent
apoptosis induced by cisplatin in renal cells. Am J Physiol Renal
Physiol. 285:F440–F450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nath KA: Provenance of the protective
property of p21. Am J Physiol Renal Physiol. 289:F512–F513. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hengst L and Reed SI: Inhibitors of the
Cip/Kip family. Curr Top Microbiol Immunol. 227:25–41.
1998.PubMed/NCBI
|
|
9
|
Sherr CJ and Roberts JM: Inhibitors of
mammalian G1 cyclin-dependent kinases. Genes Dev. 9:1149–1163.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Al-Mohanna MA, Manogaran PS, Al-Mukhalafi
Z, A Al-Hussein K and Aboussekhra A: The tumor suppressor
p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet
light and cisplatin. Oncogene. 23:201–212. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Le HV, Minn AJ and Massagué J:
Cyclin-dependent kinase inhibitors uncouple cell cycle progression
from mitochondrial apoptotic functions in DNA-damaged cancer cells.
J Biol Chem. 280:32018–32025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mori K: Tripartite management of unfolded
proteins in the endoplasmic reticulum. Cell. 101:451–454. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kleizen B and Braakman I: Protein folding
and quality control in the endoplasmic reticulum. Curr Opin Cell
Biol. 16:343–349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Price PM, Yu F, Kaldis P, Aleem E, Nowak
G, Safirstein RL and Megyesi J: Dependence of cisplatin-induced
cell death in vitro and in vivo on cyclin-dependent kinase 2. J Am
Soc Nephrol. 17:2434–2442. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu F, Megyesi J and Price PM: Cytoplasmic
initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol.
295:F44–F52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirai H, Roussel MF, Kato JY, Ashmun RA
and Sherr CJ: Novel INK4 proteins, p19 and p18 are specific
inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol
Cell Biol. 15:2672–2681. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cordon-Cardo C: Mutations of cell cycle
regulators. Biological and clinical implications for human
neoplasia. Am J Pathol. 147:545–560. 1995.PubMed/NCBI
|
|
19
|
Shankland SJ and Wolf G: Cell cycle
regulatory proteins in renal disease: Role in hypertrophy,
proliferation, and apoptosis. Am J Physiol Renal Physiol.
278:F515–F529. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Franklin DS, Godfrey VL, Lee H, Kovalev
GI, Schoonhoven R, Chen-Kiang S, Su L and Xiong Y: CDK inhibitors
p18(INK4c) and p27(Kip1) mediate two separate pathways to
collaboratively suppress pituitary tumorigenesis. Genes Dev.
12:2899–2911. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|