Introduction
Renal interstitial fibrosis is the compulsory route
for chronic kidney disease (CKD) progressing to end-stage renal
disease (ESRD), and is closely related to the loss of renal
function. Its main pathological features are the multiplication of
fibroblast and the accumulation of extracellular matrix, which
leads to the formation of interstitial fibrosis. More and more
studies have demonstrated that epithelial-mesenchymal transition
(EMT) of renal tubular epithelial cell is one of the important
mechanisms of multiplication of renal tubular fibroblasts (1). So exploring the molecular mechanism
of EMT is important to delay the process of renal interstitial
fibrosis, to search effective prevention and control measures and
to prolong the life of patients. However, the mechanism of the
intracellular signaling pathways involved in EMT were complex and
incompletely understood.
EMT, a process characterized by downregulating
epithelial characteristics and acquiring mesenchymal
characteristics, is essential in development, wound healing and
stem cell behaviour, and contributes pathologically to fibrosis and
cancer progression. In the past several years, EMT has emerged as
an important pathway leading to generation of matrix-producing
fibroblasts and myofibroblasts in diseased kidney (2), but the signaling pathway underlying
this process are poorly understood.
Phosphatidylinositol 3-kinase/protein kinase B
(PI3K/AKT) pathway plays a central role in cell growth, survival,
and metabolism. The function of PI3K is to catalyze the cellar
phosphatidylinositol(3,4,5)-trisphosphate [PtdIns(3,4,5)P3], which can phosphorylate AKT, one
of the key pathways downstream of PI3K (3). Phosphatase and tensin homolog on
chromosome 10 (PTEN), a phosphatidylinositol 3′-phosphatase that
converts PtdIns(3,4,5)P3
to phosphatidylinositol 4,5-bisphosphate, has been for a long time
attributed to its lipid phosphatase activity against PI(3,4,5)P3,
the phospholipid product of the class I PI3Ks (4). Activation of PI3K/AKT/mTOR signaling
contributes to the pathogenesis of many tumor types (5). Some recent studies indicated that it
also acts as an vital mediator in organ fibrosis, including
pulmonary fibrosis (6), liver
fibrosis (7) and myocardial
fibrosis. A recent study demonstrated that fluorofenidone inhibits
nicotinamide adenine dinucleotide phosphate oxidase via PI3K/AKT
pathway in the pathogenesis of renal interstitial fibrosis,
suggesting that PI3K/AKT is involved in the EMT in the process of
interstitial fibrosis (8).
Angiotensin II (Ang II), the main effector of the
renin-angiotensin system, contributes to the development of renal
fibrosis by inducing epithelial-mesenchymal transition (9). Binding to Ang II type 1 receptor
(AT1R), Ang II may modulate the ECM deposition in NRK-52E via
complex intracellular signalling pathways including stimulation of
PI3K/AKT (8). Moreover, the
multiple effects mediated by Ang II require a dynamic modulation of
gene expression/protein translation, which is in part under the
control of microRNAs (miRNAs).
MicroRNAs (miRNAs) are endogenous small non-coding
RNAs that regulate gene expression by degrading target mRNA or
suppressing their translation at the 3′-untranslated regions
(3′-UTR). During recent decades, the understandings of miRNAs on
molecular mechanisms in various disease processes are expanding
rapidly (10,11). miR-29b, a member of miRNAs family,
also exert great influence on the pathological process. Increasing
evidence showed that miR-29b may act as an important pathogenic
modulator in fibrosis in organs, such as heart (12), lung (13) and liver (7).
In spite of these preliminary studies, there are few
reports regarding the role that the PI3K/AKT pathway plays in the
Ang II induced-EMT. Thus, further investigation of miR-29b
regulation in renal tubular epithelial cell is essential to uncover
the molecular mechanism underpinning EMT. In the present study, we
demonstrated the ability of miR-29b to prevent EMT development in
renal tubular epithelial cell via repressing PI3K/AKT signaling
pathway.
Materials and methods
Cell culture
The normal rat kidney epithelial cell line NRK-52E
was purchased from American Type Culture Collection (CRL-1571;
Manassas, VA, USA). The NRK-52E cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) (low glucose) containing 5% fetal
bovine serum (FBS) (both from Gibco-BRL, Gaithersburg, MD, USA) and
1% streptomycin (100 µg/ml; Invitrogen, Carlsbad, CA, USA) and
penicillin (100 U/ml) and incubated in a humidified atmosphere of
95% O2, 5% CO2 at 37°C in a CO2
incubator. The cells were seeded on 6-well culture plates to 60–70%
confluence in the complete medium containing 10% FBS for 24 h. One
day before treatment, cells were incubated with serum-free media
for 24 h to synchronize the cell growth. When the cells grew to 90%
confluency, they were then harvested by a brief exposure to 0.05%
trypsin-EDTA (Gibco-BRL) and passaged every 2 days.
Treatments and groups
Medium was changed every two days to maintain
sufficient nutrient and then the cells were treated with Ang II at
different concentrations (10, 100 and 1,000 nM) for 24 h. SF1670
group was treated with SF1670 at the concentration of 100, 500 and
1,000 nM for 60 min. Untreated cells served as the control and were
cultured in the same condition. The following experiments were
carried out after the cells were cultured at 37°C in an incubator
containing 5% CO2 for 24 or 48 h.
Western blot analysis
Cells were washed with cold phosphate-bovine serum
(PBS) and lysed with RIPA lysis buffer (Beyotime, Shanghai, China)
containing protease and a phosphatase inhibitor (both from Roche,
Mannheim, Germany). Bradford Protein Assay (Beyotime) was performed
to measure the total amount of protein content. Total cell lysates
were separated by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred on to a polyvinylidene difluoride
(PVDF) membrane. Non-specific binding was blocked with 5%
fatty-free milk for 2 h at room temperature and the membranes were
incubated overnight at 4°C with primary antibodies against PI3K,
p-AKT, vimentin and cytokeratin 18 (Cell Signaling Technology,
Danvers, MA, USA) followed by incubation with secondary antibodies
for at least 1 h at room temperature. The membrane was then washed
three times in TBS containing 0.1% Tween-20 for 10 min. The
antibodies were detected using a horseradish peroxidase linked
secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) and the ECL Western Blotting Detection system (Amersham,
Buckinghamshire, UK). The membrane was re-blotted with an
anti-actin antibody to verify equal loading of the protein in each
lane.
Quantitative real-time polymerase chain
reaction
Total RNA from NRK-52E cells was extracted using
TRIzol reagent (Invitrogen) according to the manufacturer's
protocol. After determining the purity and quality of the total
RNA, miR-29b was quantified by two step real-time PCR by using the
miScript-reverse transcription kit and the miRNA-SYBR-Green kit
(Qiagen, Valencia, CA, USA) according to the manufacturer's
recommendations. The PCR reaction was performed using the ABI 7500
Fast Real-Time PCR system (Applied Biosystems, Bedford, MA, USA).
The amplification program was 95°C for 15 min and then 40 cycles
consisting of 95°C for 10 sec and 60°C for 35 sec. The ABI Prism
7900HT Sequence Detection system (Applied Biosystems, Foster City,
CA, USA) was used to analyze the data, and the ΔΔCT method was used
to calculate the relative expression of the sample gene (14). The sequences of the primers, which
were designed using Primer Premier (v5.0) and were based on the
relevant sequences deposited in GenBank, were as follows:
rno-miR-29b, 5′-TAGCACCATTTGAAATCAGTGTT-3′; U6,
5′-CAAGGATGACACGCAAATTCG-3′. The relative amount of miR-29b was
normalized to that of the U6 RNA.
Transfection of miR-29 mimic or miR-29
inhibitors
miR-29b was either upregulated or downregulated in
the NRK-52E cells by transfection with miR-29b mimics or miR-29b
inhibitor, respectively. A single-cell suspension was prepared and
the cells were cultured in 6-well plates at the density of
1×105 cells/well 24 h prior to transfection. The NRK-52E
cells were transfected using Lipofectamine 2000 (Invitrogen)
according to the manufacturer's instructions. The cells were
divided into 5 groups: miR-29b inhibitor group (transfection with
miR-29b inhibitor), inhibitor control group (transfection with
miRNA synthesized randomly), the blank control group (no
transfection), miR-29b mimic group (transfection with miR-29
mimics), mimic control group (transfection with miRNA synthesized
randomly). All groups were treated with 100 nM Ang II 24 h
following transfection. The following experiments were carried out
after the cells were cultured at 37°C in an incubator containing 5%
CO2 for 24 or 48 h.
Dual-luciferase reporter activity
assay
miRNA targets were predicted by miRanda (www.microRNA.org) and TargetScan system (https://www.targetscan.org). The wild-type and
mutant-type seed region of PIK3R2 (PI3KR2, PI3K regulatory subunit
2, also known as p85-β) was synthesized and cloned into pMIR-REPORT
luciferase vector (Ambion, Inc., Grand Island, NY, USA) Either
miR-29b mimic or control were cotransfected with the constructed
wild-type or mutant-type luciferase reporter vector into NRK-52E
cells using Lipofectamine 2000 (Invitrogen). The cells were
harvested 48 h after transfection and luciferase activity was
assessed using the Dual-Luciferase Reporter assay system (Promega,
Madison, WI, USA).
Statistical analysis
Statistical analysis was performed by SPSS 20.0
software (IBM Corp., Armonk, NY, USA). The mean values were
analyzed for their normality by using the Shapiro-Wilk normality
test. All data passed the normality test and were tested for
significant difference by one-way analysis of variance, followed by
Student's t-test. The data are presented as the mean of SMD. All
experiments were performed in triplicate. P<0.05 was considered
statistically significant.
Results
Effects of Ang II on cell morphology
We first assessed the effect of Ang II on cell
morphology. Cell morphology was observed by light microscopy after
being incubated with 100 nmol/l Ang II for 24 h (Fig. 1A). The cells in Ang II group
showed obvious trans-differentiation with cell polarity
disappearing and shape changing from circular to elliptic,
suggesting that treatment with the Ang II can induce EMT from the
perspective of cell morphology.
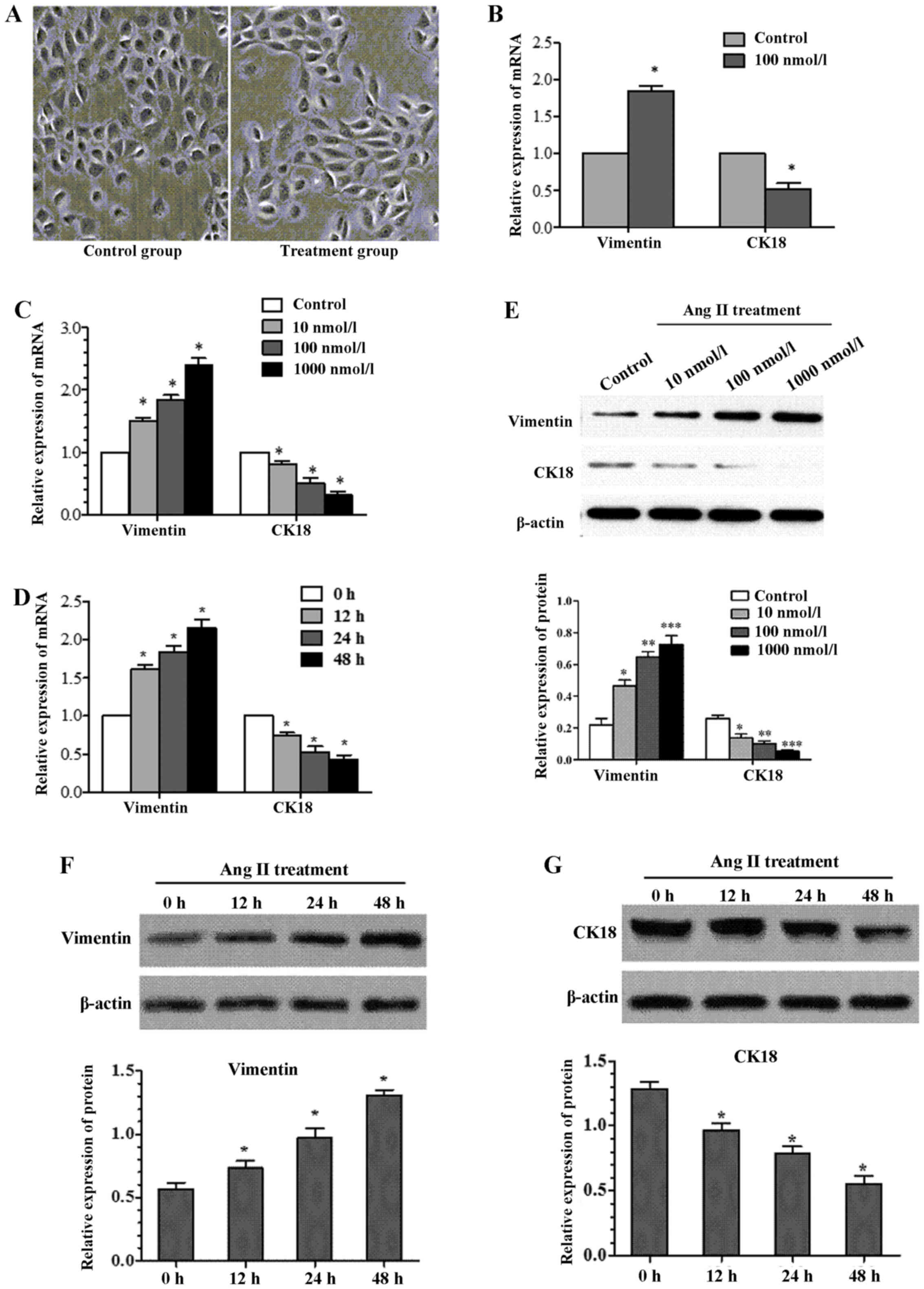 | Figure 1Effects of angiotensin II (Ang II) on
cell morphology and epithelial-mesenchymal transition (EMT)
molecular marker. (A) The cells in Ang II group showed obvious
trans-differentiation with cell polarity disappearing and shape
changing from circular to elliptic (magnification, ×400). (B)
NRK-52E cells were cultured in the presence of Ang II (100 nmol/l
for 24 h). After treatment with Ang II, vimentin gene was
significanttly increased by real-time quantitative PCR, whereas
cytokeratin 18 expression was significant decreased. (C) The
expression of cytokeratin 18 and vimentin as assessed by real-time
quantitative PCR, showed significant alteration after treatment
NRK-52E with Ang II (0, 10, 100 and 1,000 nmol/l) for 24 h. (D)
Treatment NRK-52E with Ang II (100 nmol/l) for 0, 12, 24 and 48 h,
resulted in a reduction in cytokeratin 18 expression and increase
in vimentin expression as assessed by real-time quantitative PCR
(*P<0.05 compared with control). (E) Western blotting
revealed a similar change in cytokeratin 18 and vimentin protein
expression after NRK-52E treatment with Ang II in doses 0, 10, 100
and 1,000 nmol/l for 24 h. After treatment with Ang II (100 nmol/l)
for 0, 12, 24 and 48 h, (F) vimentin protein was significantly
increased by western blotting, (G) whereas cytokeratin 18 protein
expression was significantly decreased (*P<0.05
compared with control). |
Effects of Ang II on EMT molecular
marker
Ang II treatment resulted in cell morphology change,
so we next examined the change of cell phenotype molecules
expressed by NRK-52E after Ang II treatment. By real-time PCR, the
mRNA expression level of cytokeratin 18, and vimentin present
significant discrepancy between experimental group (Ang II
treatment at concentration of 100 nmol/l for 24 h) and control
group (Fig. 1B). To detect
whether this alteration is time- or dose-dependent, the NRK-52E was
treated with Ang II (100 nmol/l) for 0, 12, 24 and 48 h,
respectively. Whilst, another batch of cells were incubated in Ang
II with different concentration (0, 10, 100 and 1,000 nmol/l) for
24 h. Cytokeratin 18 showed a time/dose-dependent decrease induced
by Ang II in NRK-52E (Fig. 1C and
D). In contrast, vimentin showed a time/concentration-dependent
increase (Fig. 1C and D). The
changes were confirmed by western blotting (Fig. 1E–G).
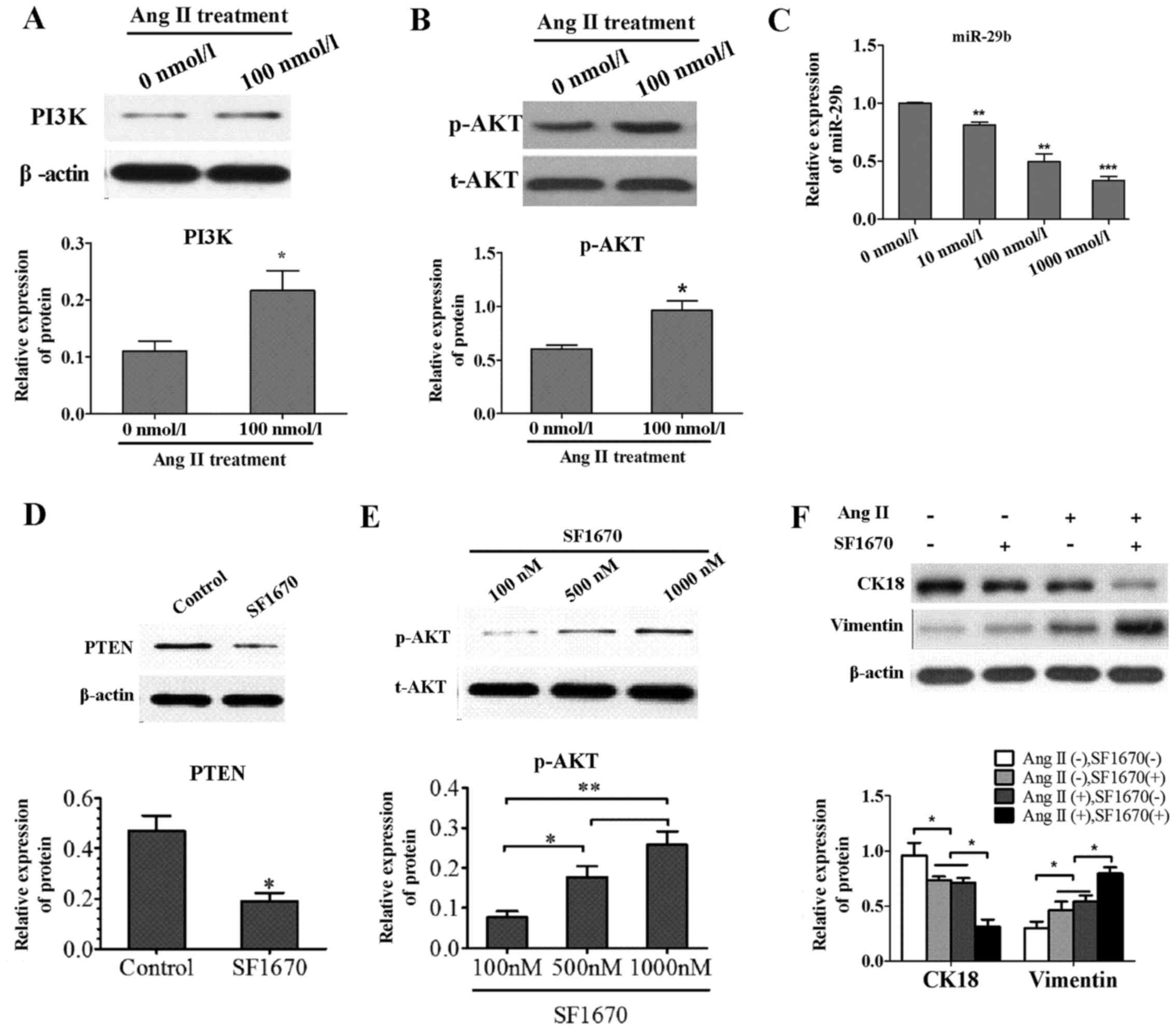
Effects of Ang II on PI3K/AKT signal
pathway and miR-29b
To explore the mechanism underlying the effect of
Ang II on PI3K/AKT signal pathway molecules, the PI3K, p-AKT and
total AKT were determined in the Ang II group and control group by
western blotting. Compared to control group, the expression level
of PI3K protein was increased in the Ang II group (Fig. 2A) and the phosphorylation rate of
AKT was remarkably elevated (Fig.
2B). Experiments were also conducted to determine the effect of
Ang II on miR-29b. After treatment NRK-52E with Ang II (0, 10, 100
and 1,000 nmol/l) for 24 h, miR-29b were significantly
downregulated by real-time quantitative PCR compared to control
(Fig. 2C).
Treatment with PTEN-specific inhibitor
SF1670 induced the activation of AKT pathway and occurrence of
EMT
To investigate whether the alteration of molecular
marker is regulated by PI3K/AKT pathway, the NRK-52E cells were
treated with SF1670, a specific inhibitor of PTEN. By inhibiting
the activity of endogenous PTEN with SF1670 (Fig. 2D), the AKT pathway was upregulated
via increasing the level of cellar PtdIns(3,4,5)P3
(Fig. 2E). Besides, cell
phenotype molecules were detected by WB and EMT occurred with the
upregulation of AKT pathway by treatment with PTEN-specific
inhibitor SF1670 (Fig. 2F). This
suggested that EMT is at least partly regulated by PI3K/AKT signal
pathway.
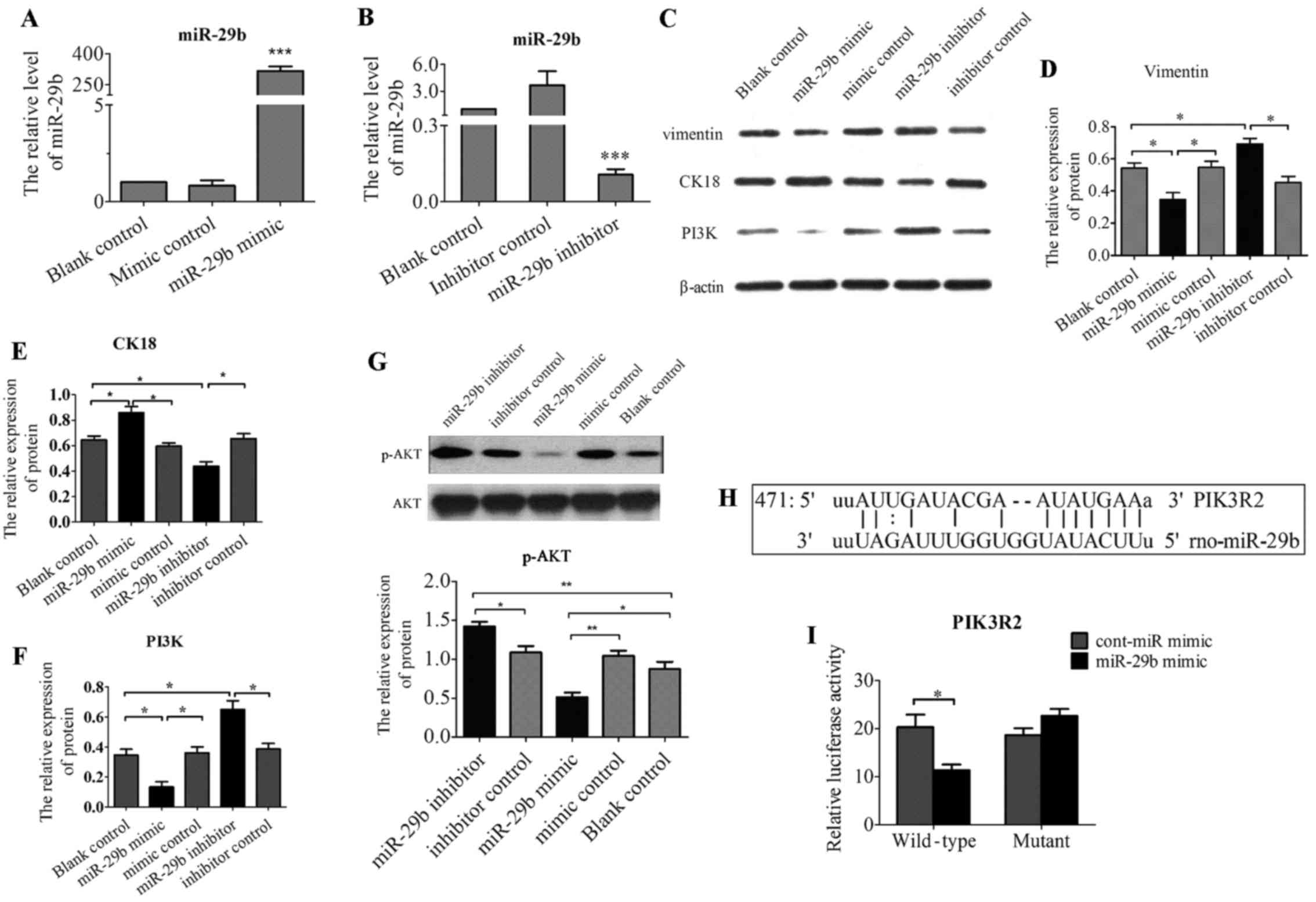
Overexpression of miR-29b suppressed
phenotype genes involved in EMT and inhibited PI3K and p-AKT
expression
Treatment with Ang II resulted in EMT-like
morphologic changes and the decrease of miR-29b. Then, in order to
determined whether overexpression of miR-29b could attune Ang
II-induced EMT, NRK-52E were transfected with double-stranded RNA
oligonucleotides containing the mature miR-29b sequence (miR-29b
mimic), control miRNAs (cont-miR-mimic), miR-29b inhibitor and
cont-miR-inhibitor. The efficacy of miR-29b transfection was
confirmed by real-time RT-PCR (Fig.
3A and B). Introduction of miR-29b significantly reduced
protein expression of vimentin (Fig.
3C and D), and increased protein expression of cytokeratin 18
(Fig. 3C and E). In contrast, low
expression of miR-29b by transfecting miR-29b inhibitor resulted in
the increased expression of vimentin (Fig. 3C and D) and decreased expression
of cytokeratin 18 (Fig. 3C and
E). Lastly, to further assess the effect of miR-29b on
PI3K/AKT, the protein expression of PI3K, and p-AKT were examined
by western blot analysis after transfection of miR-29b-3p. As shown
in Fig. 3F and G, Ang II-treated
cells transfected with miR-29b mimic showed significant reduced
expression of PI3K and p-AKT compared to cont-miR-mimic or
untreated cells. In contrast, downregulation of miR-29b by
transfected miR-29b inhibitor resulted in the upregulation of
PI3K/AKT pathway.
miR-29b inhibits PI3K and AKT expression
by directly binding to their 3′-UTR regions
The above data suggested PI3K, AKT, vimentin,
cytokeratin 18 were regulated by miR29 in NRK-52E. However, the
regulation could be indirect. To investigate whether miR-29b
directly targets PI3K, AKT, vimentin, cytokeratin 18, we searched
for the targets of miR-29b using computational prediction analysis
in several miRNA target databases including TargetScan, Scan6.2,
miRANDA and miRDB. We found that miR-29b has one putative target
site in the 3′-UTR of PIK3R2 (PI3KR2, PI3K regulatory subunit 2,
also known as p85-β) (Fig. 3H),
but not AKT1, vimentin and cytokeratin 18. Moreover, these target
site sequences are highly conserved among human, mouse and rat,
demonstrating that miR-29b may act as a direct regulator of PIK3R2.
To validate whether the putative miR-29b target sequence in 3′-UTR
of PIK3R2 directly regulate gene expression, we constructed pMIR
report plasmids encoding a firefly luciferase transcript with
either wild-type or mutant 3′-UTR of PIK3R2 (wt-PIK3R2 and
mut-PIK3R2). We evaluated their respective luciferase reporter
activity after co-transfection with miR-29b mimic or cont-miR-mimic
in NRK-52E cells. The results showed that miR-29b mimic repressed
the reporter activity of the transcript containing wild-type 3′-UTR
of PIK3R2 (Fig. 3I), suggesting
that PIK3R2 is definite target of miR-29b.
Discussion
The important role of miRNAs as regulators of
physiologic and pathologic processes in human health and disease
has recently been recognized. The signal transduction pathway
within cells is a highly complex process mediating cell survival,
differentiation and metabolism. In this study, we demonstrated the
role of PI3K/AKT pathway that miR-29b plays in the regulation of
EMT induced by treatment of Ang II in NRK-52E.
Angiotensin II (Ang II) has been proven to plays an
important role in the occurrence and development of hypertension,
as well as in EMT (9,15), which is a crucial step in the
development of renal fibrosis. EMT is characterized by a phenotypic
transformation from epithelial cells to a fibroblast-like
morphology. During this process, there is acquisition of
mesenchymal markers, such as α-SMA and vimentin (16), and a loss of epithelium markers,
including E-cadherin and cytokeratin 18 (17), which is essential for the
structural integrity of the renal epithelium. In this study, we
found a significant decrease of cytokeratin 18 and increase of
vimentin in a concentration-dependent manner after treatment of
NRK-52E with Ang II. The change of cell morphology and phenotype
molecules observed in the study indicated that the cell underwent a
epithelial-to-mesenchymal transformation of tubular cells into a
myofibroblastic phenotype. These findings are consistent with the
previous study (9,14,18,19). In addition, treatment of NRK-52E
with Ang II resulted in the significant upregulation of PI3K/AKT
signal pathway just as reported by Yano et al (20) that Ang II can activate the
PI3K/AKT signal pathway through binding to AT1R. Besides,
inhibiting PTEN with SF1670 upregulated the level of p-AKT and
induced the occurrence of EMT which was also found following
treatment of Ang II.
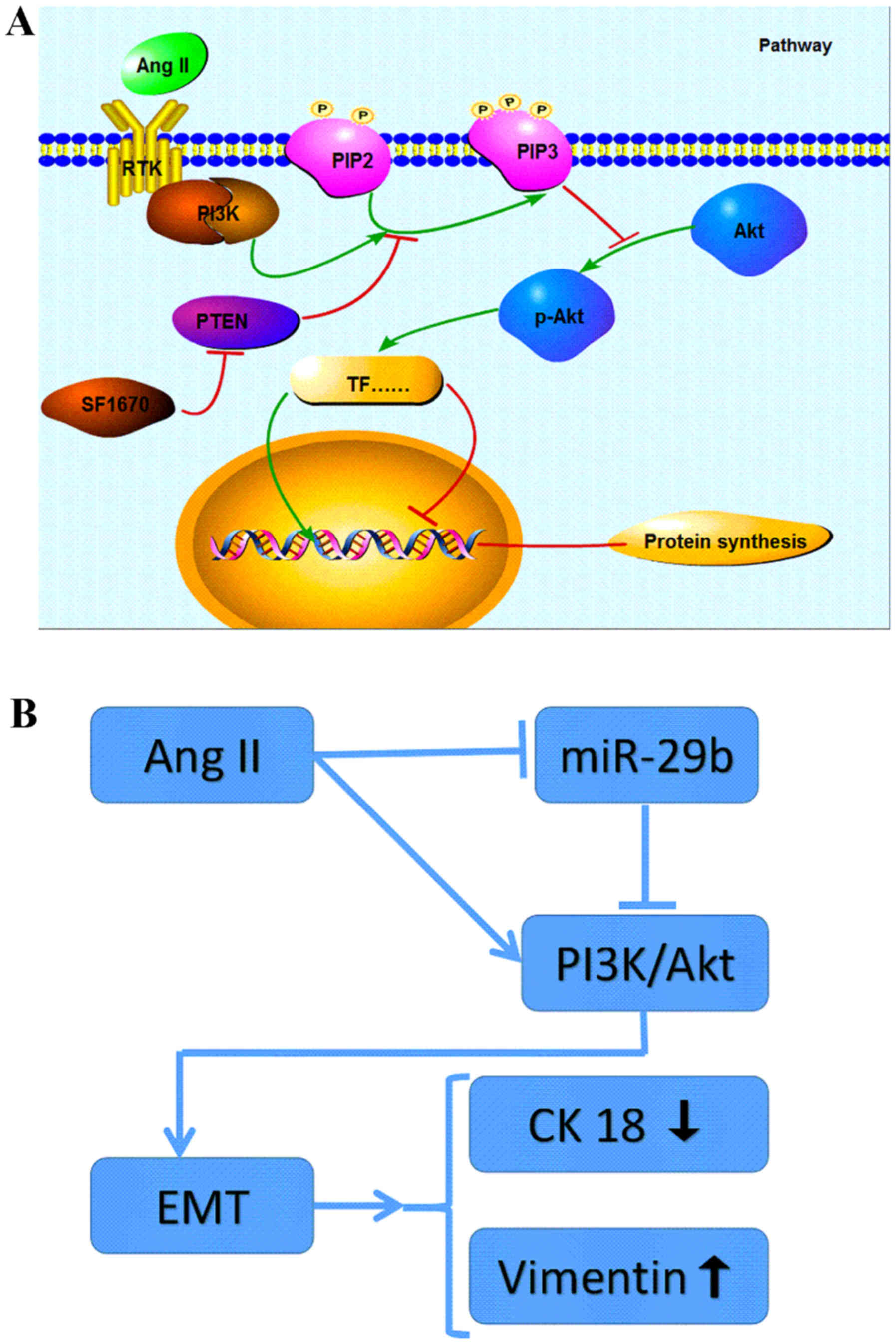
Together, these findings indicated that Ang II can
induce EMT through the signal pathway of PI3K/AKT in NRK-52E
(Fig. 4A). As a main regulator of
renal profibrotic factors (21),
the traditional roles of Ang II in the induction of EMT are mainly
mediated by directly activating TGF-β/Smad signalling pathway
(22–24), or by indirectly activating
TGF-β/Smad signaling via an ERK/p38 MAP kinase-Smad crosstalk
mechanism (25). This study
provided new insight into the intracellular signal transduction
involved in the EMT induced by Ang II treatment.
Although the evidence suggesting a role for miRNAs
as biomarkers and novel targets for treatment in kidney disease has
been reported (26–28), the effect of known
nephroprotective treatments on the expression of miRNAs, or their
possible contribution as protective agents against renal fibrosis,
has yet to be fully established. In this context, we also evaluated
miR-29b expression in the Ang II-treated cells and we found that a
significant difference was displayed in the level of miR-29b
compared to the control group. Treatment with Ang II resulting in a
marked decrease in miR-29b expression apart from upregulation of
p-AKT and occurrence of EMT demonstrated that miR-29b may be
involved in PI3K/AKT signal pathway and regulate the EMT of NRK-52E
(Fig. 4B). This finding may
perfect the hypothesis of Pan et al (15) that at least two distinct
intracellular signaling pathways are involved in the low levels of
miR-29b expression after treatment of Ang II in NRK-52E.
If so, it would be important to establish that its
overexpression attenuated the severity of Ang II-induced EMT via
PI3K/AKT signal pathway. To confirm this, NRK-52E cells were
transfected with miR-29b mimics and then treated with Ang II for 24
h. The result of western blot analysis and immunoflorescence
staining demonstrated that overexpression of miR-29b displayed
downregulation of PI3K/AKT signal pathway and suppressed the Ang
II-induced upregulation of vimentin and downregulation of
cytokeratin 18. These results manifest that the overexpression of
miR-29b negatively modulates EMT by downregulation of PI3K/AKT
signal pathway. Moreover, transfection of NRK-52E with miR-29b
inhibitor was also conducted. Western blot analysis and
immunoflo-rescence staining revealed that miR-29b inhibitor induced
a marked increase of vimentin and decrease of cytokeratin 18 as
well as downregulation of PI3K/AKT signal pathway. The data
indicated that the treatment with Ang II can lower the level of
miR-29b, activate the PI3K/AKT signal pathway and eventually
trigger the occurrence of EMT,which can be negatively modulated by
overexpresson of miR-29b.
In conclusion, Ang II can induce EMT in NRK-52E via
the activation of PI3K/AKT signaling and elevated expression of
miR-29b may ameliorate Ang II-induced EMT by the suppression of
PI3K/AKT signaling. These results offer the possibility that
specific inhibitor of PI3K/AKT cascade or enhancing miR-29b level
by gene therapy may have beneficial effects in attenuating renal
interstitial fibrosis.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
Acknowledgments
The research was supported by the Research Center of
Zhongnan Hospital of Wuhan University.
References
|
1
|
Ballhause TM, Soldati R and Mertens PR:
Sources of myofibroblasts in kidney fibrosis: All answers are
correct, however to different extent! Int Urol Nephrol. 46:659–664.
2014. View Article : Google Scholar
|
|
2
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar
|
|
3
|
Cantrell DA: Phosphoinositide 3-kinase
signalling pathways. J Cell Sci. 114:1439–1445. 2001.PubMed/NCBI
|
|
4
|
Ye B, Jiang LL, Xu HT, Zhou DW and Li ZS:
Expression of PI3K/AKT pathway in gastric cancer and its blockade
suppresses tumor growth and metastasis. Int J Immunopathol
Pharmacol. 25:627–636. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hassan B, Akcakanat A, Holder AM and
Meric-Bernstam F: Targeting the PI3-kinase/Akt/mTOR signaling
pathway. Surg Oncol Clin N Am. 22:641–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang T, Liang Y, Lin Q, Liu J, Luo F, Li
X, Zhou H, Zhuang S and Zhang H: miR-29 mediates TGFβ1-induced
extracellular matrix synthesis through activation of PI3K-AKT
pathway in human lung fibroblasts. J Cell Biochem. 114:1336–1342.
2013. View Article : Google Scholar
|
|
7
|
Wang J, Chu ES, Chen HY, Man K, Go MY,
Huang XR, Lan HY, Sung JJ and Yu J: microRNA-29b prevents liver
fibrosis by attenuating hepatic stellate cell activation and
inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget.
6:7325–7338. 2015.
|
|
8
|
Qin J, Xie YY, Huang L, Yuan QJ, Mei WJ,
Yuan XN, Hu GY, Cheng GJ, Tao LJ and Peng ZZ: Fluorofenidone
inhibits nicotinamide adenine dinucleotide phosphate oxidase via
PI3K/Akt pathway in the pathogenesis of renal interstitial
fibrosis. Nephrology (Carlton). 18:690–699. 2013.
|
|
9
|
Burns WC, Velkoska E, Dean R, Burrell LM
and Thomas MC: Angiotensin II mediates epithelial-to-mesenchymal
transformation in tubular cells by ANG 1-7/MAS-1-dependent
pathways. Am J Physiol Renal Physiol. 299:F585–F593. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan B, Guo Q, Fu FJ, Wang Z, Yin Z, Wei YB
and Yang JR: The role of miR-29b in cancer: Regulation, function,
and signaling. Onco Targets Ther. 8:539–548. 2015.PubMed/NCBI
|
|
11
|
Bayoumi AS, Sayed A, Broskova Z, Teoh JP,
Wilson J, Su H, Tang YL and Kim IM: Crosstalk between long
noncoding RNAs and microRNAs in health and disease. Int J Mol Sci.
17:E3562016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cushing L, Kuang PP, Qian J, Shao F, Wu J,
Little F, Thannickal VJ, Cardoso WV and Lü J: miR-29 is a major
regulator of genes associated with pulmonary fibrosis. Am J Respir
Cell Mol Biol. 45:287–294. 2011. View Article : Google Scholar :
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Pan J, Zhang J, Zhang X, Zhou X, Lu S,
Huang X, Shao J, Lou G, Yang D and Geng YJ: Role of microRNA-29b in
angiotensin II-induced epithelial-mesenchymal transition in renal
tubular epithelial cells. Int J Mol Med. 34:1381–1387. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okada H, Ban S, Nagao S, Takahashi H,
Suzuki H and Neilson EG: Progressive renal fibrosis in murine
polycystic kidney disease: An immunohistochemical observation.
Kidney Int. 58:587–597. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Djudjaj S, Papasotiriou M, Bülow RD,
Wagnerova A, Lindenmeyer MT, Cohen CD, Strnad P, Goumenos DS,
Floege J and Boor P: Keratins are novel markers of renal epithelial
cell injury. Kidney Int. 89:792–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen J, Chen JK and Harris RC: Angiotensin
II induces epithelial-to-mesenchymal transition in renal epithelial
cells through reactive oxygen species/Src/caveolin-mediated
activation of an epidermal growth factor receptor-extracellular
signal-regulated kinase signaling pathway. Mol Cell Biol.
32:981–991. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burns WC and Thomas MC: Angiotensin II and
its role in tubular epithelial to mesenchymal transition associated
with chronic kidney disease. Cells Tissues Organs. 193:74–84. 2011.
View Article : Google Scholar
|
|
20
|
Yano N, Suzuki D, Endoh M, Zhao TC,
Padbury JF and Tseng YT: A novel phosphoinositide
3-kinase-dependent pathway for angiotensin II/AT-1
receptor-mediated induction of collagen synthesis in MES-13
mesangial cells. J Biol Chem. 282:18819–18830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Macconi D, Remuzzi G and Benigni A: Key
fibrogenic mediators: Old players. Renin-angiotensin system. Kidney
Int Suppl (2011). 4:58–64. 2014. View Article : Google Scholar
|
|
22
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang F, Huang XR, Chung AC, Hou CC, Lai KN
and Lan HY: Essential role for Smad3 in angiotensin II-induced
tubular epithelial-mesenchymal transition. J Pathol. 221:390–401.
2010.PubMed/NCBI
|
|
24
|
Meng XM, Tang PM, Li J and Lan HY:
TGF-β/Smad signaling in renal fibrosis. Front Physiol. 6:822015.
View Article : Google Scholar
|
|
25
|
Rodríguez-Vita J, Sánchez-López E, Esteban
V, Rupérez M, Egido J and Ruiz-Ortega M: Angiotensin II activates
the Smad pathway in vascular smooth muscle cells by a transforming
growth factor-beta-independent mechanism. Circulation.
111:2509–2517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Denby L and Baker AH: Targeting non-coding
RNA for the therapy of renal disease. Curr Opin Pharmacol.
27:70–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simpson K, Wonnacott A, Fraser DJ and
Bowen T: MicroRNAs in diabetic nephropathy: From biomarkers to
therapy. Curr Diab Rep. 16:352016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gomez IG, Nakagawa N and Duffield JS:
MicroRNAs as novel therapeutic targets to treat kidney injury and
fibrosis. Am J Physiol Renal Physiol. 310:F931–F944. PubMed/NCBI
|