Introduction
Orthotopic liver transplantation (OLT) is an
effective method of treating advanced liver disease; however,
immune rejection following transplantation remains a major clinical
problem (1). Patient outcomes
following liver transplantation (LT) have markedly improved due to
the extensive use of immunosuppressants. However, patients may
experience severe side effects and acute rejection (AR) following
LT, which may consequently reduce their quality of life (1,2).
It has been determined that AR following LT is a
local cellular immune responses primarily mediated by T lymphocytes
(3). Therefore, the number and
functional balance of T cell subsets [T helper (Th) 1/2; Th17 and T
regulatory (Treg) cells] is associated with the acquisition of
immune tolerance. T cell immunoglobulin and mucin domain (TIM)
genes, a novel family of co-stimulatory molecules, are critical in
determining the activation and differentiation of Th cells
(4). The TIM family of cell
surface proteins consists of eight members in mice (TIM 1–8) and
three in humans (TIM 1, 3 and 4) (5). All TIM family protein members are
type I cell surface glycoproteins and each possesses a common
immunoglobulin (Ig) V-like domain, a mucin-like domain, a single
transmembrane domain and a cytoplasmic region (5,6).
Previous studies have implicated TIM in the mediation of certain
immune responses, including autoimmunity, transplant tolerance,
allergic diseases and response to viral infections (5–7).
TIM-4 is the only non-T cell TIM protein and is solely expressed on
antigen presenting cells (APCs), particularly in macrophages and
cluster of differentiation (CD) 11c+ dendritic cells
(DCs). TIM-4 signals have been identified in activated human T
cells, including the CD4+ and CD8+ T cells
(8). TIM-4 was initially
identified as a phosphatidylserine (PS) receptor, functioning to
mediate the binding and engulfment of apoptotic bodies, which is a
crucial step in phagocytosis and innate immune reactions. By acting
as a TIM-1 ligand on T cells, TIM-4 may function as a
co-stimulatory molecule that regulates the adaptive immune response
(5,8,9).
It has been indicated that the interaction between TIM-4 and TIM-1
suppresses peripheral immune tolerance and activates T cells
(10). In addition, TIM-4 is able
to bind to naive CD4+ T cells in the absence of surface
TIM-4 expression (9,10), suggesting that an alternative
unidentified receptor is present on CD4+ T cells.
However, the specific effect of TIM-4 co-stimulation on T cell
activation in vivo remains unclear. In vitro studies
using TIM-4-immunoglobulin fusion proteins have presented
conflicting results: TIM-4 signaling increases the proliferation of
activated T cells but has the opposite effect on naive T cells
(9,11,12).
Kupffer cells (KCs) are the largest group of APCs
in vivo and account for 10-15% of total liver cells. They
also account for 80–90% of all monocyte-marophage cells and exhibit
high expression of TIM-4 (13).
KCs affect various processes, including antigen presentation, the
secretion of cytokines and immune regulation in patients following
LT (13). It has been
demonstrated that blocking TIM-4 expression in mice impairs the
intrinsic function of macrophages to phagocytose PS+
hepatic debris, which further mitigates toll like receptor
(TLR)-4-mediated inflammation in liver ischemia-reperfusion injury
(14). Blocking TIM-4 expression
in DCs initiates the production of induced (i) Tregs, which are
able to markedly prolong the survival of mice that have undergone a
skin allograft (15). Current
understanding regarding the action of TIM-4 is limited to its
involvement in immune tolerance; to the best of our knowledge, no
studies have assessed the effects of TIM-4 on rejection following
LT.
The present study demonstrated that OLT enhances
TIM-4 expression in liver KCs. It was assessed whether blocking
TIM-4 expression in KCs attenuates hepatic injury and inhibits the
inflammatory response. Additionally, naive CD4+ T cells
differentiation were directed to induce the generation of potent
and functionally suppressive iTregs by impeding interleukin
(IL)-4/signal transducer and activator of transcription 6 (STAT6)
signaling. It was also evaluated whether blocking TIM-4 expression
increases levels of transforming growth factor-β (TGF-β), which may
stimulate the expansion of iTregs. The results of the current study
demonstrated that blocking TIM-4 expression and administering
exogenous TGF-β following LT markedly increases the de novo
induction of iTregs from naive CD4+ T cells, thus
attenuating AR and promoting the survival of mice following LT.
Materials and methods
Experimental animals
A total of 40 8-10 week old wild-type female C57BL/6
mice weighing 16–22 g and 50 8–10 week female C3H mice weighing
16–22 g were purchased from the Animal Experimental Center of
Chongqing Medical University (Chongqing, China). All mice were
housed at a temperature of 23°C and humidity of 60% under a 12-h
light/dark cycle. Food and water were supplied ad libitum.
All protocols involving mice were conducted in accordance with the
Guide for the Care and Use of Laboratory Animals published by the
US National Institutes of Health (16) and all experiments were approved by
the Animal Care and Use Committee of Second Affiliated Hospital of
Chongqing Medical University (Chongqing, China).
OLT
An improved Kamada's two-cuff method with minor
modifications was used to perform OLT between female C57BL/6 donor
mice and inbred female C3H recipient mice, as previously described
(17). Donor and recipient mice
received 1.9% ethyl ether anesthesia via nostril cannula. The use
of ethyl ether was approved by the Animal Experimental Centre of
Chongqing Medical University (Chongqing, China). Warm ischemia,
cold ischemia and anhepatic times were set at 0 min, 1 h and 20 min
respectively. Only C3H recipient mice underwent the following
experiments. C3H mice that had undergone single LT from donor mice
but had not received subsequent treatments (n=5) were sacrificed
24, 48 and 72 h following surgery to determine KC activity and
TIM-4 expression in the liver. Recipient mice (n=30) were then
randomly divided into 3 treatment groups: A sham group (n=10) that
did not receive a transplant but still underwent abdominal cutting
and vascular exposures around the liver; a control mouse monoclonal
antibody (mAb) group (n=10; cat. no. GTX14149; 1:50 dilution; mAb
received from GeneTex, Inc., Irvine, CA, USA) treated with 0.35
mg/mouse mAb via portal vein injection to establish an LT model;
and a T cell immunoglobulin mucin protein 4 (TIM-4) mAb group
(n=10) that received 0.35 mg/mouse portal vein injections. The
indicated treatments and dosages in each group were administered
intravenously for 2 days following surgery at 24 h intervals. On
day 7 following surgery, all mice were anaesthetized via inhaled
1.9% ethyl ether and 0.5 ml/mouse blood samples were drawn from the
abdominal aorta. Mice were then sacrificed in a sealed-container
with an overdose of carbon dioxide (CO2 replacement
rate: 20% of the container volume/min). Murine liver tissues were
subsequently removed and examined and KCs were then isolated.
Another mouse model was then established. Donor mice
were injected with clodronate liposomes (CL; 10 mg/kg; cat. no.
40337ES05/10; Shanghai Yeasen Biotechnology Co., Ltd., Shanghai,
China) via the caudal vein to destroy liver KCs 24 h prior to
surgery. Following CL pretreatment, OLT or sham surgery was
performed (C57BL/6→C3H) and recipient C3H mice were administered
the aforementioned treatments. Recipient mice were randomly divided
into CL+sham, CL+control mAb and CL+TIM-4 mAb groups (all n=5). All
recipient mice were sacrificed and underwent tissue section
examination 7 days post-surgery.
Cell isolation and purification
OLT or sham surgery was performed and KCs were
isolated. A modified method of the in vitro type IV
collagenase digestion was used to dissociate liver tissue. Mice
were anaesthetized using inhaled 1.9% ethyl ether. Murine
heartbeats and thoracic breathing were monitored to ensure that
mice were anesthetized and not sacrificed. The liver was perfused
in situ with 10 ml PBS (3 ml/min) at 37°C to remove red
blood cells. Liver tissues were then dispersed in RPMI-1640 (Gibco;
Thermo Fisher Scientific Inc., Waltham, MA, USA) containing 0.5%
type IV collagenase (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
at 37°C for 30 min. Liver homogenate was filtered through a
200-mesh sieve to remove undigested tissue and non-parenchymal
cells of the liver were obtained using the gradient centrifugation
method. The cell suspension was centrifuged at 300 × g on an 5810R
machine (Eppendorf, Hamburg, Germany) for 5 min at 4°C. The top
aqueous phase was discarded and the cell sediment was reserved.
Cell sediments were then resuspended with 10 ml RPMI-1640 and
centrifuged at 300 × g for 5 min at 4°C, the top aqueous phase was
discarded and cell sediments were reserved. Cell sediments were
then resuspended with 10 ml RPMI-1640 and centrifuged at 50 × g for
3 min at 4°C. The top aqueous phase (cleared cell suspension) was
transferred into a new 10 ml centrifuge tube and centrifuged at 300
× g for 5 min at 4°C, the top aqueous phase was discarded and cell
sediments were reserved. To further purify KCs, the selective
adherence to plastic method was used following the protocol
described by Li et al (18). Cells were seeded in a 6-well plate
at a density of 1–3×107/well in Dulbecco's Modified
Eagle's medium supplemented with 10% fetal bovine serum (both
HyClone, GE Healthcare Life Sciences, Logan, UT, USA) and 100 U/ml
penicillin/streptomycin (Sigma-Aldrich; Merck KGaA) and incubated
for 2 h in a 5% CO2 atmosphere at 37°C. Following
washing with PBS, non-adherent cells were removed from the dish;
adherent cells were KCs.
Naive CD4+CD25− T cells were
isolated using the CD4+ T cell isolation kit II and CD25
microbeads (both Miltenyi Biotec GmbH, Bergisch Galdbach, Germany)
were utilized to select the CD25− population.
Immunohistochemistry
Liver tissues were fixed with 10% formaldehyde at
room temperature for 4 h and subsequently embedded in paraffin.
Paraffinized sections were placed in an oven at 58°C for 2 h,
dewaxed with xylene, placed in 100% ethanol and rehydrated using
90–70% ethanol for 3 min at each stage. Sections were washed with
PBS three times (3 min/wash), placed in a 0.01 M sodium citrate
antigen retrieval solution (pH 6.0, 95°C) for 15 min and then
cooled for 15 min at room temperature. Sections were washed with
PBS three times (3 min/wash), treated with 4.3%
H2O2 to block endogenous peroxidase for 20
min at room temperature. Sections were washed with PBS three times
(3 min/wash) and then blocked with 10% fetal calf serum (FCS; cat.
no. 25-01860; Shanghai Biomart Technology, Co., Ltd, Shanghai,
China) for 30 min at room temperature. Sections were washed with
PBS three times (3 min/wash) and incubated with anti-CD163 (EPR19;
cat. no. ab182422; 1:100 dilution; Abcam, Cambridge, MA, USA)
overnight at 4°C. Sections were washed with PBS three times (3
min/wash) and incubated with biotin-conjugated goat anti-rabbit and
goat anti-mouse immunoglobulin G secondary antibodies (cat. nos.,
BA1003 and BA1001, respectively; Wuhan Boster Biological
Technology, Ltd., Wuhan; China) at 37°C for 90 min. Sections were
washed with PBS three times (3 min/wash), treated with
3,3′-diaminobenzidine for 5 min at room temperature, stained with
hematoxylin for 30 sec at room temperature and treated with
hydrochloric acid for 1 sec. Sections were then placed in 70%
ethanol for 3 min and then treated with 80–100% ethanol for 3 min
each. Following dehydration, sections were mounted, observed using
a XSP-13C-LB microscope (Shanghai Precision & Scientific
Instrument Co., Ltd., Shanghai, China) at a magnification of ×400
and analyzed using Image pro plus 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA).
Immunostaining
KCs derived from recipient (sham, control mAb or
TIM-4 mAb mice) were collected and were fixed in 4%
paraformaldehyde for 1 h at room temperature. Slides were washed
with PBS three times (3 min/wash) and blocked with 30 min in 10%
FCS at room temperature. Slides were then washed with PBS three
times (3 min/wash) and incubated with TIM-4 antibodies (RMT4-53,
cat. no. GTX14149; 1:100 dilution; GeneTex Inc.) overnight at 4°C.
Slides were washed with PBS three times (3 min/wash) and incubated
with specific secondary antibodies labeled with
tetramethylrhodamine (red; 1:100 dilution; cat. no. GMS40036; Bio
Valley, Shanghai; China) or DAPI (blue) for 30 min at room
temperature. Slides were then washed with PBS three times (3
min/wash) and sealed with FluorFluoromount-G™ Slide Mounting medium
(Southern Biotech, Birmingham, AL, USA). Images were taken using a
Zeiss LSM 510 confocal microscope (magnification ×800; Zeiss AG,
Thornwood, NY, USA) and were analyzed using an Image Analysis
system, version 11.0 (Chang Heng Rong Technology Co., Ltd.,
Beijing; China).
Western blot analysis
Cells or liver homogenates were lysed in lysis
buffer (Gibco; Thermo Fisher Scientific, Inc.) and the
concentration of total protein was determined using a BCA Protein
assay kit (Sangon Biotech Co., Ltd., Shanghai, China). A total of
40 μg protein was loaded per lane, separated using 12%
SDS-PAGE gel and then electrotransferred onto polyvinylidene
difluoride membranes (Bio-Rad Laboratories Inc., Hercules, CA,
USA). Membranes were blocked using 5% non-fat milk in
Tris-buffered-saline with Tween at room temperature for 1 h and
incubated with primary antibodies overnight at 4°C. The primary
antibodies used included: TIM-4 (RMT4-53, cat. no. GTX14149; 1:100
dilution; GeneTex Inc.), tumor necrosis factor-α (TNF-α; 52B38,
cat. no. ab1793; 1:100 dilution; Abcam), interferon-γ (IFN-γ;
#37895R, cat. no. MAB485R; 1:100 dilution; R&D Systems Inc.,
Minneapolis, MN, USA), chemokine ligand 2 (CCL2; 2D8, cat. no.
GTX60582; 1:100 dilution; GeneTex Inc.), C-X-C motif chemokine
ligand 2 (CXCL2; cat. no. GTX74085; 1:200 dilution; GeneTex Inc.),
TGF-β (cat. no. ab92486; 1:100 dilution), phosphorylated (p)-signal
transducer and activator of transcription 6 (STAT6; phospho Y641,
cat. no. ab28829; 1:100 dilution), p-p65 (phospho S536, cat. no.
ab86299; 1:100 dilution), p-p38 (phospho Y182, cat. no. ab47363;
1:100 dilution), forkhead box P3 (Foxp3; 236A/E7, cat. no. ab20034;
1:100 dilution), transcription factor gata3 (Gata3; cat. no.
ab106625, 1:100 dilution) and β-actin (cat. no. ab8226; 1:100
dilution) (all from Abcam). Membranes were then washed with
TBS-Tween-20 and incubated with peroxidase-conjugated secondary
antibodies (cat. nos. A0545, A9044; 1:8,000 dilution;
Sigma-Aldrich; Merck KGaA) at room temperature for 1 h. An enhanced
chemiluminescence kit (GE Healthcare, Chicago, IL, USA) was used to
perform chemiluminescence and protein bands were visualized using
X-ray films. All images were analyzed using ImageJ software,
version 14.8 (National Institutes of Health, Bethesda, MD,
USA).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
RNAs were extracted from cell lysates using TRIzol
RNA-extraction reagent and reverse transcription was performed
using PrimeScript™ 1st Strand cDNA Synthesis kit (cat. no. 6110A)
(both from Takara Bio Inc., Otsu, Japan) following the
manufacturer's protocol. qPCR assays were performed using SYBR
Premix Ex Taq (Takara Bio Inc.) and a cDNA template on the Applied
Biosystems 7500 Real-time PCR System (Applied Biosystems; Thermo
Fisher Scientific Inc.). The primers used for qPCR amplification
were as follows: TIM-4; forward, 5′-CAATCGAGGTGACAGTGGG-3′ and
reverse, 5′-AAGGAGCCAGGTGTTGTTG-3′; β-actin; forward,
5′-GCTCTGGCTCCTAGCACCAT-3′ and reverse, 5′-GCCACCGATCCACACAGAGT-3′.
qPCR was conducted in a 50 μl reaction system, including 1
μl forward and 1 μl reverse primers, 0.5 μl
20x SYBR-Green I, 25 μl 2x PCR buffer, 2 μl cDNA, and
20.5 μl diethyl pyrocarbonate. The thermocycling conditions
used for qPCR were as follows: 94°C for 4 min, 30 cycles of 94°C
for 20 sec, 60°C for 30 sec, 72°C for 30 sec, and 72°C for 5 min.
Each individual sample was run in triplicate and the level of
expression was quantified using the comparative cycle threshold
(Cq) method. Results were normalized to β-actin expression and RNA
enrichments were calculated using the 2−ΔΔCq method
(19).
Carboxyfluorescein succinimidyl ester
(CFSE) assay
Splenic CD4+ T cells were purified from
recipient mice that had received single LT with no treatment (n=5).
Cells were labeled with 5 μM CFSE (cat. no. 65-0850-84;
eBioscience; Thermo Fisher Scientific, Inc.) for 20 min at 37°C in
the dark. Cold RPMI-1640 medium was then added and cells were
incubated on ice for 5 min. CD4+ T cells were then
co-cultured with TIM-4+ KCs (1:1). The following 5
experimental groups were constructed: The control group of cells
that were untreated; the control mAb group of cells that were
treated with control mAb (0.5 μg/ml, 500 μl); the
TGF-β group of cells that were treated with additional exogenous
TGF-β (1 ng/ml, 100 μl) following co-culture; the TIM-4 mAb
group of cells that were treated with TIM-4 mAb (0.5 μg/ml,
500 μl); and the TGF-β+TIM-4 mAb group of cells that were
treated with exogenous TGF-β and TIM-4 mAb. All cells were
co-cultured for 96 h at 37°C. Following washing, cells were
re-suspended in RPMI-1640 medium with 10% fetal bovine serum and
then blocked with 10% FCS for 30 min at room temperature. Cells
were then stained with APC-CD4 antibodies (cat. no. GK1.5, 1:100
dilution; eBioscience; Thermo Fisher Scientific, Inc.) in the dark
for 30 min at room temperature, centrifuged at 3,000 × g for 10 min
at 4°C and resuspended twice in PBS prior to fluorescence-activated
cell sorting (FACS) analysis. All experiments were performed in
triplicate.
Flow cytometry
Cells (KCs or T cells) were digested for 3 min in
0.25% trypsin at 37°C and collected following centrifugation at 50
× g for 5 min at room temperature. Cells were then rinsed in
pre-cooled PBS (4°C) twice. Cells were then resuspended in 400
μl binding buffer and the cell concentration was diluted to
1×106/ml. Fluorochrome-labeled mAbs, including
CD14-phycoerythrin (PE; cat. no. Sa2-8; 12-0141, dilution, 1:100),
CD163-APC (cat. no. GHI/61; 17-1639-42, dilution, 1:100), Foxp3-PE
(cat. no. 150D/E4; 12-477, dilution, 1:100), CD25-fluorescein
isothiocyanate (FITC; cat. no. CD25-4E3; 11-0257, dilution, 1:100),
were purchased from eBioscience; Thermo Fisher Scientific, Inc. KCs
were stained for CD14 and CD163 and T cells were stained for CD25
and Foxp3 for 1 h at 4°C in the dark. Flow cytometric cell sorting
was performed using FACSAria; flow cytometric data were acquired
using a FACSCalibur and analyzed using Cellquest version 5.1
software (BD Biosciences, Franklin Lakes, NJ, USA). Experiments
were performed in triplicate.
Detection of liver function
Cardiac puncture blood samples were collected from
mice following euthanasia 7 days post-operation and allowed to
clot. Serum was obtained following centrifugation at 200 × g at 4°C
for 10 min. Levels of aspartate aminotransferase (AST), alanine
aminotransferase (ALT) and total bilirubin in serum (TBIL) were
determined using a Synchron CX7 analyzer (Beckman Coulter Inc.,
Brea, CA, USA) in the Clinical Biochemical Laboratory of Chongqing
Medical University. Experiments were performed in triplicate.
Enzyme-linked immunosorbent assay
(ELISA)
Liver tissues were removed from mice 7 days
following surgery. Then, liver tissues were sliced and washed
repeatedly with saline and formed into homogenates using a tissue
crusher. Liver homogenates were then centrifuged 3 times at 300 × g
at 4°C for 15 min to obtain supernatants. TIM-4+ KCs
were incubated with naive CD4+CD25- T cells
for 96 h at 37°C and were then centrifuged at 300 × g at 4°C for 10
min to obtain supernatants. Levels of inflammatory cytokines in
liver homogenates or supernatants were assessed using the following
ELISA kits: TNF-α (cat. no. MAT00B), IFN-γ (cat. no. MIF00), CCL2
(cat. no. MJE00), CXCL2 (cat. no. MM200), TGF-β (cat. no. MB100B),
IL-4 (cat. no. M4000B), IL-6 (cat. no. M6000B) and IL-13 (cat. no.
M1300CB; all R&D Systems, Inc.). All procedures were performed
following the manufacturer's protocols. Absorbance was read at 450
nm and experiments were performed in triplicate.
Histological examination
To assess hepatic injury, liver tissues were fixed
with 10% formaldehyde at room temperature for 4 h and then embedded
in paraffin. Tissues were sliced into sections 5-μm thick
prior to staining with hematoxylin for 10 min and eosin for 3 min
at room temperature. AR was classified into four types using the
Banff schema (20).
Terminal deoxynucleotidyl
transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end
labeling (TUNEL) assay
Apoptotic cells were detected using a commercial
TUNEL kit (cat. no. 11684817910; Roche Inc., Switzerland) following
the manufacturer's protocol. The TUNEL assay was performed for
paraffin-embedded sections fixed with 4% paraformaldehyde for 1 h
at room temperature and processed following the method described by
Kitamoto et al (21).
TUNEL + hepatocyte nuclei were quantified by calculating the mean
number of TUNEL + hepatocytes in five random fields of view per
animal with 100 nuclei counted per field. The mean percentage of
apoptotic cells was calculated.
Statistical analysis
All values are expressed as the mean ± standard
deviation. Data were analyzed using an unpaired two-tailed
Student's t-test or one-way analysis of variance with Tukey's
post-hoc test. P<0.05 was considered to indicate a statistically
significant result. Graft survival was assessed using the
Kaplan-Meier method and statistical differences were calculated
using the log rank test.
Results
AR injury following LT increases the
frequency of KC activation and TIM-4 expression
KCs are important APCs that serve bimodal roles in
the innate and adaptive immune response. It has been demonstrated
that liver KCs engulf and degrade pathogenic microorganisms and
remove endotoxins (22). To
assess the expression of TIM-4 in KCs following hepatic AR, flow
cytometry and immunohistochemistry were performed and the number of
activated KCs in transplanted livers were assessed. The number of
activated KCs in the LT group gradually increased with time
following surgery compared with the sham group (Fig. 1A and B). KCs were then isolated
from mice that had undergone LT and the expression of TIM-4 was
measured using western blotting. Mice that had undergone LT
exhibited significantly elevated TIM-4 expression 1, 3 and 7 days
post-surgery (P<0.05 vs. sham group; Fig. 1C). The expression of TIM-4 on day
3 following surgery increased to a peak and remained at a
relatively high level, indicating that the expression of TIM-4 is
promoted by activated KCs following LT.
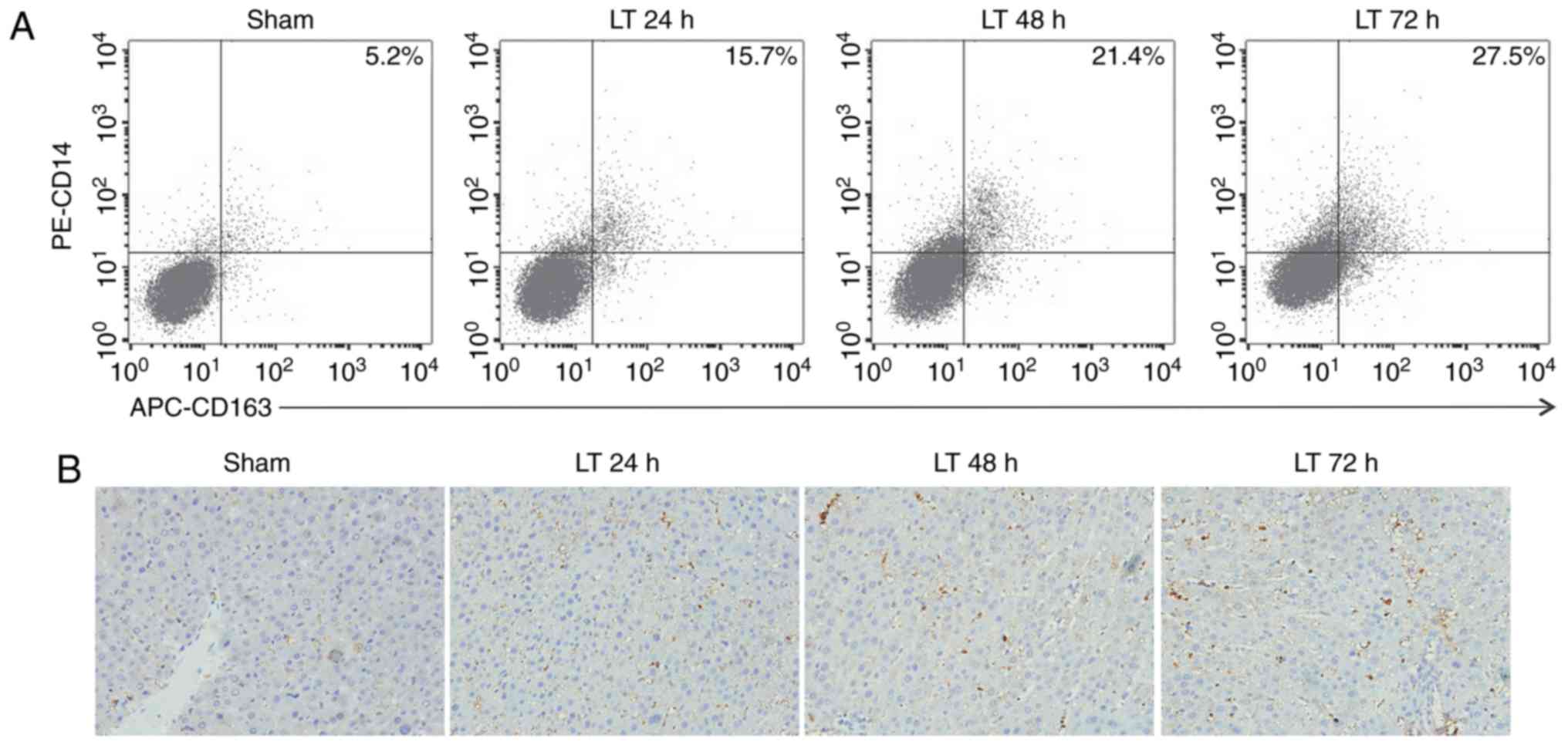 | Figure 1Orthotopic liver transplantation
increases KC activation and TIM-4 expression. (A) KCs were isolated
from mice in the sham and LT groups 24, 48 and 72 h following
surgery. Cells were double stained with PE-CD14 and APC-CD163 and
quantified using flow cytometry. (B) Liver samples were obtained
from mice in the sham and LT groups and assessed using
immunohistochemistry (magnification, ×400). Activated hepatic KCs
are stained brown. (C) KCs were isolated from mice in the sham and
LT groups 1, 3 and 7 days following surgery and TIM-4 expression
was analyzed using western blotting. Data are presented as the mean
± standard deviation. aP<0.05 vs. sham group. KC,
kupffer cells; TIM-4, T cell immunoglobulin-domain mucin-domain-4;
CD, cluster of differentiation; APC, antigen presenting cell; LT,
liver transplantation; PE, phycoerythrin; d, days. |
Blockade of TIM-4 in KCs ameliorates AR
and reduces the expression of inflammatory cytokines via the
nuclear factor (NF)-κB and p38 MAPK signaling pathways
TIM-4 was demonstrated to be present in activated
KCs at high levels; therefore, the role of TIM-4 signaling in the
function of KCs was assessed. The fluorescent expression of KCs was
markedly higher in the TIM-4 mAb group compared with the sham and
control mAb groups (Fig. 2A). The
effect of TIM-4 on KCs was determined using flow cytometric
analysis. TIM-4 expression in the TIM-4 mAb group was almost
entirely blocked compared with the control mAb group, indicating
that the expression of TIM-4 in KCs was successfully disrupted
(Fig. 2B).
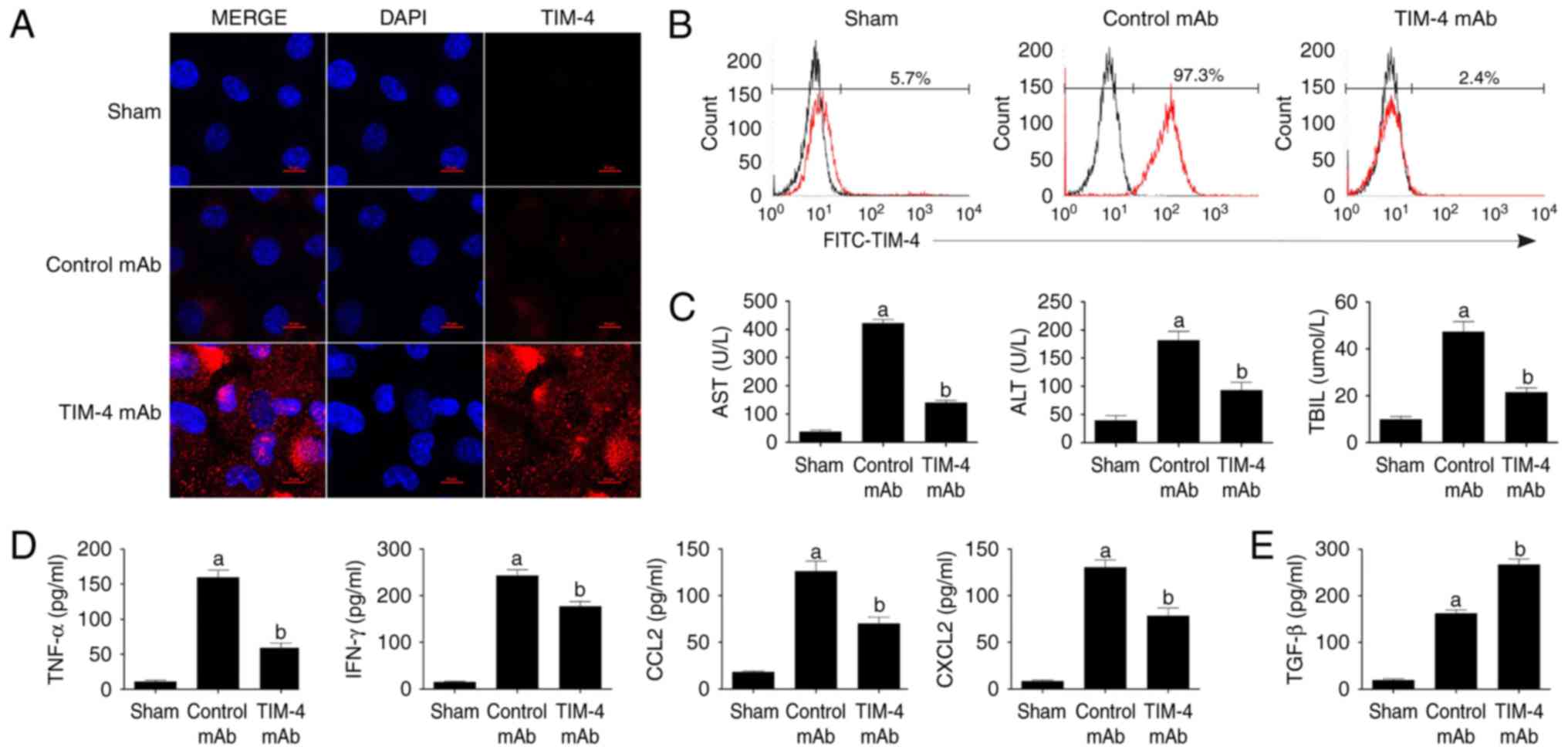 | Figure 2Blockade of TIM-4 improves hepatic
acute reaction response injury and reduces the secretion of
inflammatory cytokines. (A) KCs were isolated from mice in the
sham, control mAb and TIM-4 mAb groups on day 7 post-surgery. Cells
were stained with specific secondary antibodies labeled with
tetramethylrhodamine (red) and DAPI (blue) and then examined using
laser confocal microscopy (magnification ×800). (B) KCs were
isolated from each group and stained with FITC-TIM-4 mAb for flow
cytometry. (C) Blood samples were obtained (0.5 ml/mouse) from
murine abdominal aortas in each group on day 7 post-surgery. Levels
of AST, ALT and TBIL were determined in a clinical biochemical
laboratory. (D-F) Levels of inflammatory cytokines, including
TNF-α, IFN-γ, CCL2, CXCL2 and TGF-β were detected using ELISA and
western blotting. (G) Representative micrographs of the
pathological damage observed in each group post-transplantation
following staining with hematoxylin and eosin (magnification ×400).
(H) Donor mice received CL treatment to destroy KCs prior to
surgery. orthotopic liver transplantation or sham surgery was
subsequently performed and mice were treated either with or without
TIM-4 mAb. All mice underwent analysis to determine the RAI score
on day 7 following surgery. (I) The expression of key
phosphoproteins involved in the NF-κB and p38 MAPK signaling
pathways, including p65 and p38, respectively, were analyzed using
western blotting. Data are presented as the mean ± standard
deviation. aP<0.05 vs. sham group;
bP<0.05 vs. control mAb group. TIM-4, T cell
immunoglobulin-domain mucin-domain-4; mAb, monoclonal antibodies;
KCs, kupffer cells; FITC, fluorescein isothiocyanate; AST,
aspartate aminotransferase; ALT, alanine aminotransferase; TBIL,
total bilirubin in serum; TNF-α, tumor necrosis factor-α; IFN-γ,
interferon-γ; CCL2, chemokine ligand ; CXCL2, C-X-C motif chemokine
ligand 2; TGF-β, transforming growth factor-β; CL, clondronate
liposomes; NF-κB, nuclear factor-κB; MAPK, mitogen-activated
protein kinase. |
Compared with the control mAb group, the blockade of
KC TIM-4 expression significantly improved hepatic injury, as
determined by the liver function indices of AST, ALT and TBIL (each
P<0.05; Fig. 2C). Furthermore,
it was revealed that TIM-4 disruption significantly reduced levels
of the inflammatory cytokines TNF-α, IFN-γ, CCL2 and CXCL2 (each
P<0.05 vs. control mAb group; Fig.
2D and F) and significantly enhanced the levels of TGF-β
(P<0.05 vs. control mAb group; Fig. 2E) in the liver, thus reversing the
effects of AR. Additionally, visible focal necrosis and
vacuolization of liver parenchyma with lymphocyte infiltration was
observed in the control mAb group and classified as severe AR
according to the Banffe schema (20). However, tissues from mice in the
TIM-4 mAb group exhibited reduced infiltration of lymphocytes
surrounding the portal area. AR severity was determined following a
previously described protocol (20) and the rejection activity index
(RAI) was identified as being significantly lower than that of the
control mAb group (P<0.05; RAI score: 4.2±0.7 vs. 8.5±0.5;
Fig. 2G). These results suggest
that the blockade of TIM-4 by mAb contributes to AR amelioration
and inflammatory cytokine downregulation.
CL stimulates the depletion of macrophages in the
liver (23). To determine the
function of KCs in vivo, the livers of donor mice were
treated with CL to destroy hepatic KCs. The RAI score of liver
grafts treated with CL+TIM-4 mAb was associated with severe AR and
there were no significant differences in RAI scores between the
CL+TIM-4 mAb and CL+control mAb groups (Fig. 2H), suggesting that the disruption
of TIM-4 expressed on KCs is critical to the improvement of AR,
rather than the TIM-4 that is expressed on other hepatic APCs.
To analyze the possible pathways involved in this
process, the expression of key phosphorylated proteins involved in
the NF-κB and p38 MAPK signaling pathways, including p65 and p38
were assessed. A significant decrease in the expression of p-p65
and p-p38 was observed in mice following treatment with TIM-4 mAb
(P<0.05 vs. control mAb group; Fig. 2I). Therefore, the blockade of KC
TIM-4 may inhibit NF-κB and p38 MAPK signaling to improve liver
transplant injury.
Blockade of TIM-4 expressed by KCs with
exogenous TGF-β inhibits Th2 differentiation and promotes iTreg
generation in vitro
As aforementioned, it was determined that the
blockade of TIM-4 expressed by KCs enhances the secretion of TGF-β,
which is a protein that inhibits the polarization and facilitates
the proliferation of Th1/Th2 and Tregs, respectively. Therefore,
the effect of TIM-4 blockade (0.5 μg/ml) with exogenous
TGF-β (1 ng/ml) on the differentiation of naive CD4+ T
cells was assessed in vitro. TIM-4+ KCs were
isolated from model mice and purified using FACS. There was no
significant difference in the KC antigen-presenting ability between
the control mAb and TIM-4 mAb groups. However, compared with the
TIM-4 mAb group, the TGF-β+TIM-4 mAb group exhibited a significant
decrease in KC antigen-presenting ability (P<0.05; Fig. 3A). These results indicate that the
TIM-4 blockade does not decrease the antigen-presenting capacity of
KCs, however additional TGF-β treatment does. TIM-4+ KCs
were subsequently incubated with naive
CD4+CD25− T cells. It was revealed that the
blockade of TIM-4 with added TGF-β caused a significantly greater
reduction in CD4+CD25− T cell proliferation
compared with TGF-β and TIM-4 treatment alone (P<0.05; Fig. 3B). Furthermore, the measurement of
supernatants co-cultured cohorts demonstrated that TGF-β treatment
or TIM-4 blockade significantly decreased levels of the cytokines
IL-4, IL-6 and IL-13 (each P<0.05 vs. control mAb; Fig. 3C). The combination of TGF-β and
TIM-4 treatment further enhanced this effect (P<0.05; Fig. 3C), indicating that Th2
polarization was effectively inhibited. The proportion of Treg
(CD4+CD25+Foxp3+) cells was
significantly increased in the TGF-β and TIM-4 mAb groups compared
with the control mAb group (P<0.05; Fig. 3D) and the proportion of Treg cells
was significantly enhanced in the combined TGF-β+TIM-4 mAb group
(P<0.05; Fig. 3D). These
results suggest that the TIM-4 blockade serves a crucial role in
directing the conversion of iTreg and that the addition of TGF-β
treatment induces an even greater effect.
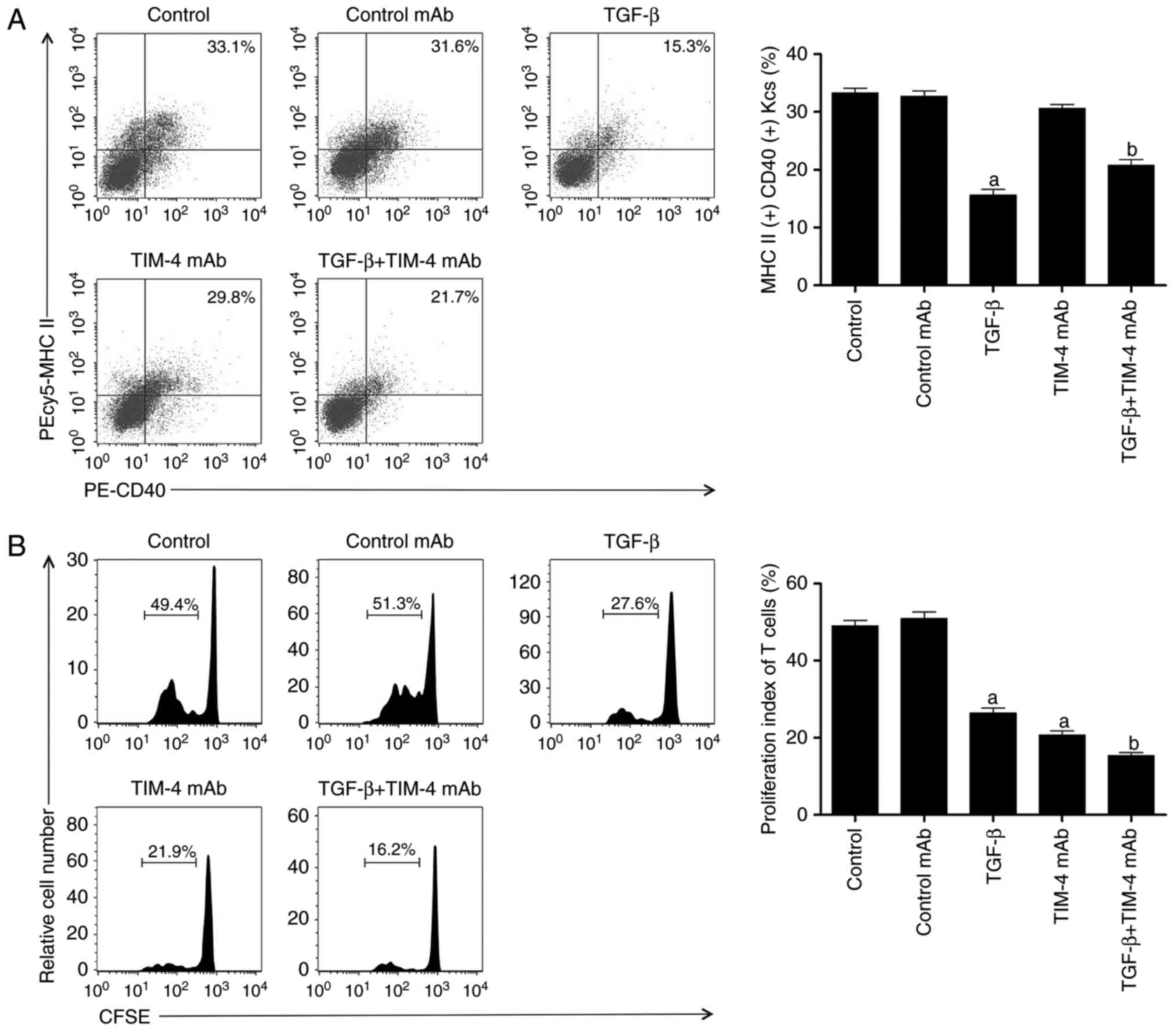 | Figure 3The effect of TIM-4 blockade with
addition of exogenous TGF-β on inhibiting T helper 2 cell
differentiation and inducing the conversion of
CD4+CD25+Foxp3+ T regulatory
cells. (A) TIM-4+ KCs were isolated from model mice and
purified using fluorescence activated cell sorting, followed by
addition of control mAb/TGF-β/TIM-4 mAb/TGF-β+TIM-4 mAb into medium
for incubation. Following washing and resuspension of cells, they
were stained with PEcy5-MHC II and PE-CD40 antibodies and acquired
for FACS analysis. (B) Spleen CD4+CD25− T
cells were purified from recipient mice and labeled with CFSE,
followed by co-culturing with KCs in condition described in A
(control as untreated). The proliferation of T cells was then
determined using a CFSE dilution gated on CD4+
populations. (C) The supernatants of the co-cultured system,
including the cytokines IL-4, IL-6 and IL-13, were detected using
enzyme-linked immunosorbent assay. (D) Splenic
CD4+CD25− T cells were purified from
recipient mice and co-cultured with KCs in conditions as described
in A. Following washing and re-suspension, the cells were stained
with PE-Foxp3 and FITC-CD25 antibodies and underwent FACS analysis.
Data are presented as the mean ± standard deviation.
aP<0.05 vs. control and control mAb groups;
bP<0.05 vs. TGF-β and TIM-4 mAb groups. TIM-4, T cell
immunoglobulin-domain mucin-domain-4; TGF-β, transforming growth
factor-β; KCs, kupffer cells; mAb, monoclonal antibodies; MHC II,
major histocompatibility complex II; CD, cluster of
differentiation; FACS, fluorescence-activated cell sorting; CFSE,
carboxyfluorescein succinimidyl ester; IL, interleukin; Foxp3,
forkhead box P3; FITC, fluorescein isothiocyanate; PE,
phycoerythrin; PEcy5, phycoerythrin-streptavidin. |
Blocking TIM-4 promotes iTreg expansion
by inhibiting the IL-4/STAT6/Gata3 signaling pathway
The TIM-4 blockade of KCs reduced the expression of
IL-4 and triggered the conversion of iTregs. The addition of
exogenous low-dose TGF-β amplified this effect. It has been
demonstrated that STAT6 signaling inhibits iTreg by binding to the
Foxp3 promoter, which directly interferes with the transcription of
Foxp3 (24). To determine the
role of the IL-4/STAT6/Gata3 pathway in the generation of iTregs,
fluorescent cell sorting was performed to attain TIM-4+
and TIM-4− KCs in the LT group. These were then
co-cultured with naive CD4+ T cells and the expression
of p-STAT6 in T cells was assessed. The results demonstrated that
levels of p-STAT6 in CD4+ T cells co-cultured with
TIM-4+ KCs were significantly higher than those in
CD4+ T cells co-cultured with TIM-4− KCs
(P<0.05; Fig. 4A).
TIM-4+/TIM-4− KCs were then co-cultured with
naive CD4+ T cells (which received either no
pretreatment or pretreatment with TIM-4 mAb) to detect levels of
p-STAT6. It was demonstrated that the addition of TIM-4 mAb
significantly blocked the expression of p-STAT6 in CD4+
T cells co-cultured with TIM-4+ KCs (P<0.05), but did
not affect p-STAT6 expression in CD4+ T cells
co-cultured with TIM-4− KCs (Fig. 4B). The exogenous addition of IL-4
to TIM-4+ KCs co-cultured with naive CD4+ T
cells demonstrated that the inhibition of p-STAT6 expression
induced by TIM-4 mAb was significantly reversed by IL-4 (P<0.05;
Fig. 4C). Furthermore, the
blockade of TIM-4 with additional TGF-β in the co-cultured model
significantly promoted the expression of Foxp3 (P<0.05) and had
a greater suppressive effect on Gata3 compared with TIM-4 blockade
alone (P<0.05; Fig. 4D).
Collectively, these data reveal that the generation of the iTreg
phenotype by TIM-4 blockade is dependent on the inhibition of the
IL-4/STAT6/Gata3 signaling pathway.
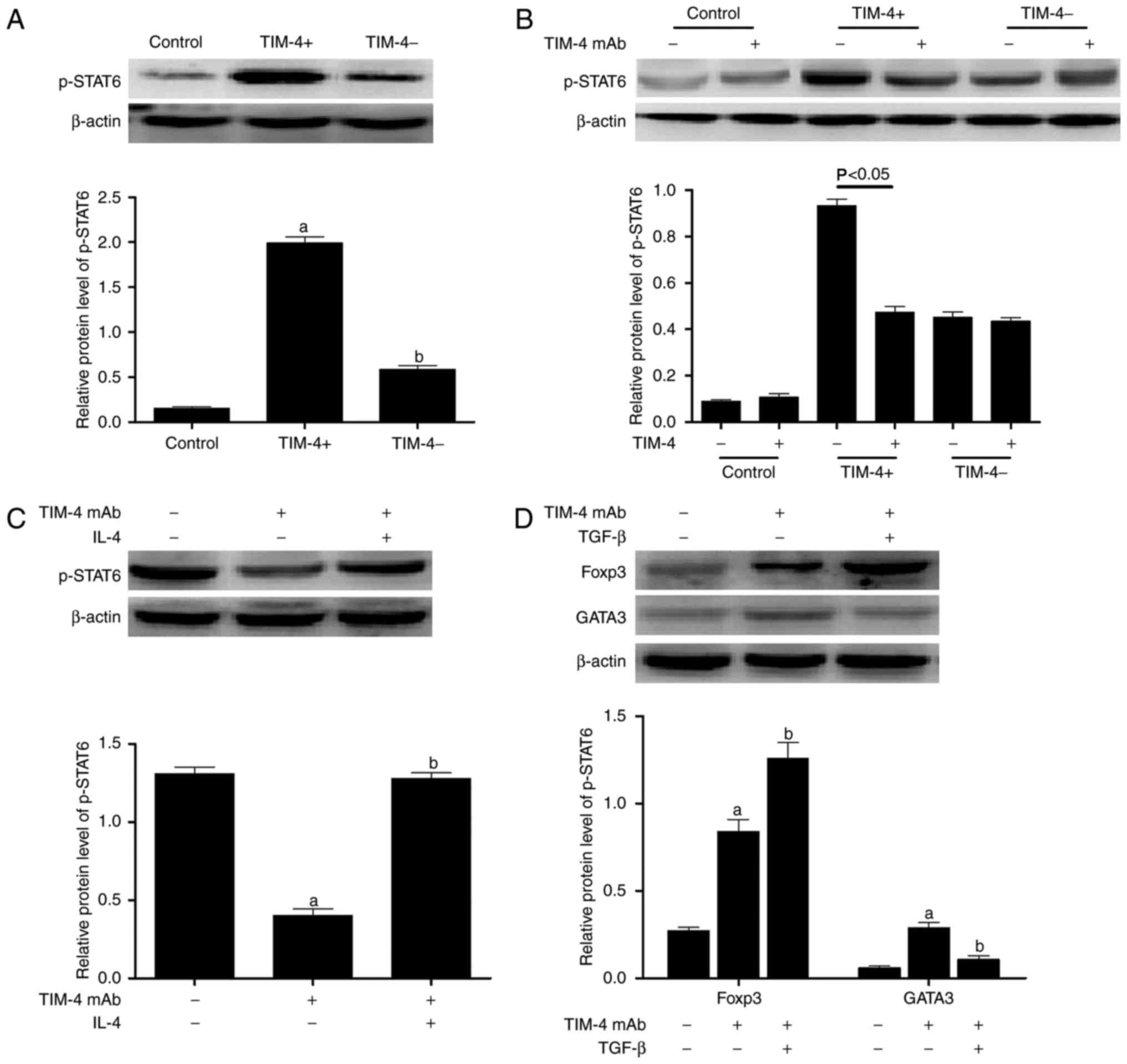 | Figure 4TIM-4 blockade stimulates the
differentiation of T cells into
CD4+CD25+Foxp3+ T regulatory cells
via the IL-4/STAT6/Gata3 pathway. (A) TIM-4+ and
TIM-4− KCs were obtained from mice following liver
transplantation and co-cultured with naive CD4+ T cells.
The expression of p-STAT6 in T cells was determined using western
blotting. (B) TIM-4 mAb was added to co-cultured cohorts as
described in A, to analyze the expression of p-STAT6 in T cells.
(C) The exogenous addition of IL-4 to TIM-4+ KCs
co-cultured with naive CD4+ T cells (that received
either no pretreatment or pretreatment with TIM-4 mAb) was
performed and the expression of p-STAT6 in T cells was analyzed.
(D) TGF-β was added to TIM-4+ KCs co-cultured with naive
CD4+ T cells (that received either no pretreatment or
pretreatment with TIM-4 mAb) to determine the expression of Foxp3
and Gata3 protein in T cells. Data are presented as the mean ±
standard deviation of the mean. aP<0.05 vs. control;
bP<0.05 vs. TIM-4+ group. TIM-4, T cell
immunoglobulin-domain mucin-domain-4; IL-4, interleukin-4; p-,
phosphorylated; STAT6, signal transducer and activator of
transcription 6; Gata3, transcription factor gata3; CD, cluster of
differentiation; mAb, monoclonal antibodies; KCs, kupffer cells;
TGF-β, transforming growth factor-β; Foxp3, forkhead box P3. |
TIM-4 mAb combined with TGF-β produces a
lower AR and extends the survival time of mice following LT
The effect of TIM-4 blockade in combination with
TGF-β on AR was assessed in vivo. Recipient mice were
injected with either anti-TIM-4 mAb (0.35 mg/mouse), 0.5 ml TGF-β
(1 ng/ml) or the two in combination via murine portal veins to
establish a model of LT. Treatment was administered continuously at
the same dose for a total of 2 days following surgery. The results
of hepatic function and apoptotic tests indicated that TIM-4 mAb
combined with TGF-β induced a more significant improvement of liver
injury compared with the TGF-β or TIM-4 mAb groups (P<0.05;
Fig. 5A) and evidently
significantly ameliorates hepatocyte apoptosis in LT models on day
7 following surgery (P<0.05; Fig.
5B). As the present study demonstrated, TIM-4 mAb combined with
TGF-β induced the generation of significantly more
CD4+CD25+Foxp3+ T cells than in
mice treated with TGF-β or TIM-4 mAb alone (P<0.05; Fig. 5C). The mean survival time of mice
treated with TGF-β and TIM-4 mAb was markedly extended (>100
days) and was significantly longer than those that received TGF-β
or TIM-4 mAb alone (P<0.05; Fig.
5D). These results indicate that TIM-4 blockade combined with
additional TGF-β may ameliorate AR more effectively and potentially
induce the formation of immunosuppressive cells.
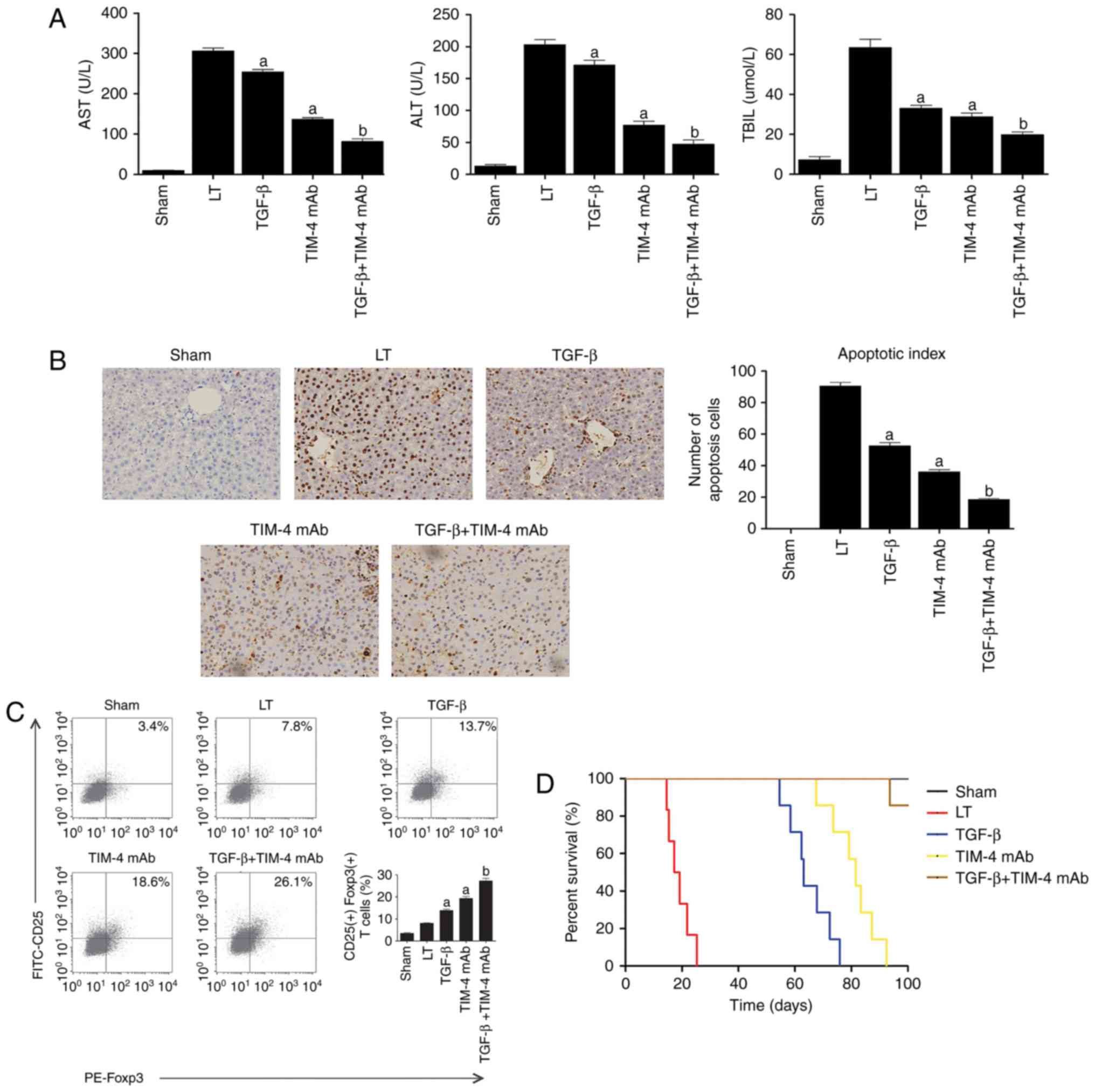 | Figure 5The effect of TIM-4 blockade with
exogenous TGF-β injection on acute reaction response in
vivo. (A) Mice were injected with either anti-TIM-4 mAb (0.35
mg/mouse), 0.5 ml TGF-β (1 ng/ml) or in combination via murine
portal veins to establish LT (LT group were treated with PBS as
control). Treatment in each group was administered continuously at
the same dose for a total of 2 days following surgery. Levels of
AST, ALT and TBIL were determined in a clinical biochemical
laboratory on day 7 following surgery. (B) Hepatocyte apoptosis was
detected using the terminal deoxynucleotidyl transferase-mediated
2′-deoxyuridine 5′-triphosphate nick-end labeling method
(magnification ×400). (C) T cells were purified from hepatic
tissue. Following washing and re-suspension, cells were stained
with PE-Foxp3 and FITC-CD25 antibodies and acquired for
fluorescence-activated cell sorting analysis. (D) Mice survival
time was observed and analyzed using the Log-rank test. Data are
presented as the mean ± standard deviation of the mean.
aP<0.05 vs. LT group; bP<0.05 vs. TGF-β
and TIM-4 mAb groups. TIM-4, T cell immunoglobulin-domain
mucin-domain-4; TGF-β, transforming growth factor-β; mAb,
monoclonal antibodies; LT, liver transplantation; AST, aspartate
aminotransferase; ALT, alanine aminotransferase; TBIL, total
bilirubin in serum; Foxp3, forkhead box P3; FITC, fluorescein
isothiocyanate; CD, cluster of differentiation; PE,
phycoerythrin. |
Discussion
The results of previous studies have demonstrated
that TIM-4 is expressed on mature APCs and engages TIM-1 ligands to
co-stimulate the activation of T cells in the adaptive immune
response (9,10,25). It has been demonstrated that the
interaction between TIM-4 and TIM-1 expressed on activated
CD4+ T cells increases the proliferation of T cells and
the polarization of Th2 (26).
However, a previous study demonstrated that TIM-4-Ig fusion
proteins inhibit naive CD4+ T cell activation via a
receptor other than TIM-1 in vitro (11). KCs are the largest type of APC and
function to mediate the balance between the effector and regulatory
T cell responses. KCs are activated following LT via various
pathways. Activated KCs possess a strong antigen presenting
capacity, demonstrated by their high expression of major
histocompatibility complex II and co-stimulatory molecules
(27). Furthermore, activated KCs
generate large quantities of Th1 cytokines, which activate
antigen-specific T cells to induce AR (13). However, activated KCs also
upregulate the expression of Fas ligand, which binds to
Fas+ T cells to induce apoptosis and promote the
secretion of Th2 cytokines, thereby inhibiting the immune response
(28). Therefore, the effect of
TIM-4 on KCs may serve a critical role in the immunological balance
associated with LT.
TIM-4 mediates co-stimulatory signaling and induces
the phagocytosis of apoptotic cells (29). It has been determined that TIM-4
expression increases in a dose-dependent manner in
CD11b+ monocyte macrophages treated with
lipopolysaccharide (9,30). By establishing a model of OLT, the
present study demonstrated that TIM-4 expression increases and is
maintained at high levels following the activation of pathological
inflammation-induced KCs. It has been demonstrated that the TIM-4
blockade inhibits the production of pro-inflammatory cytokines but
does not affect the phagocytosis of apoptotic bodies mediated by
DCs (15). Furthermore, it has
been demonstrated that the disruption of TIM-4 signaling inhibits
macrophage and neutrophil migration/function, diminishes the
phagocytosis of necrotic hepatocytes and suppresses the activation
of TLR2/4/9-dependent signaling (14). The present study disrupted
KC-expressed TIM-4 using anti-TIM-4 mAb and indicated that the
TIM-4 blockade contributed to the amelioration of hepatic injury
and reduced the release of inflammatory cytokines, including TNF-α,
IFN-γ, CCL2 and CXCL2. The degree of AR in the tissue sections of
mice that received TIM-4 mAb was markedly improved and was
exhibited only a mild rejection response. These results indicated
that TIM-4 serves an important role in LT injury and
inflammation.
Proteins in the NF-κB family act as transcription
factors and serve key roles during the regulation of inflammation
(31). TLR activation promotes
the activation of innate immune responses via the myeloid
differentiation primary response 88- or toll-interleukin
receptor-domain-containing adaptor-inducing IFN-β-dependent
pathways, which are instrumental for downstream NF-κB activation
and subsequent inflammatory reactions (14,32). In addition, it has been
demonstrated that the activation of KCs is closely associated with
p38 MAPK signaling in endoplasmic reticulum stress and ischemia
reperfusion injury (32). TIM-4
fragment crystallizable has been reported to inhibit the
proliferation and the differentiation of T cells, primarily via the
inhibition of MAPK signaling (12). The activation of NF-κB and MAPK
are considered to be important for the production of inflammatory
cytokines in KCs (32,33). The present study demonstrated that
the blockade of KC TIM-4 reduces the phosphorylation of p65 and
p38, which are key proteins of the NF-κB and p38 MAPK signaling
pathways. The results therefore demonstrated that TIM-4 disruption
in KCs may inhibit the NF-κB and p38 MAPK signaling pathways to
ameliorate AR damage in transplanted livers.
The present study also identified that TGF-β levels
were significantly increased in mice treated with TIM-4 mAb. TGF-β
is a key mediator of immune regulation and tolerance and serves a
functional role in the induction and inhibition of Foxp3 and
T-bet/Gata3 expression, respectively. However, the expression of
TGF-β is usually decreased following LT (34). The TGF-β-mediated transcription of
Foxp3 is associated with the Smad pathway (35,36). Distal to the Foxp3 gene is a
conservative enhancer, which associates with Smad3 and the nuclear
factor of activated T cells to synergistically induce the
transcription of Foxp3 (36). In
addition, TIM-4 signaling promotes Th2 differentiation, the
response to which markedly inhibits the induction of peripheral
Tregs from naive CD4+ T cells. The present study
therefore utilized a highly specific anti-TIM-4 mAb without
exogenous TGF-β and the results indicated that the antigen
presenting capacity of KCs that expressed TIM-4 following blocking
was not weakened; however, the reverse occurred following TIM-4
blockade combined with TGF-β treatment in vitro. The results
indicated that the addition of exogenous TGF-β favored negative
immune regulation in KCs, which is conducive to the effects of
immunosuppression. Furthermore, the proliferation of
CD4+ T cells in a co-culture system and Th2 cytokines,
including IL-4, IL-6 and IL-13 were significantly suppressed in the
presence of combined TIM-4 mAb and TGF-β, compared with individual
treatments administered alone. IL-4 suppresses the upregulation of
Foxp3 in CD4+ T cells and the suppression of Th2
differentiation is the central mechanism of Treg induction
(15,37,38). IL-4 may also block the generation
of TGF-β-mediated Treg differentiation and induce the population of
Th cells (15,24). The present study confirmed that
TIM-4 blockade with the addition of exogenous TGF-β exhibited a
greater effect on the induction of
CD4+CD25+Foxp3+ T cells. It was
further demonstrated that the TIM-4 blockade in TIM-4+
KCs attributes to iTreg conversion via IL-4/STAT6/Gata3 signaling
as the transcription of these proteins interfere with the
transcription of Foxp3. The differentiation of CD4+ T
cells is determined by the relative quantities of STAT6/Gata3 and
Foxp3.
A previous study demonstrated that the
administration of TGF-β in the presence of IL-6 promotes the
generation of pro-inflammatory Th17, causing chronic allograft
rejection (39). In addition, the
direct blockade of IL-4 also promotes the development of Th2-driven
diseases (40) and reduces the
long-term allograft survival of transplant models with a STAT6
deficiency (41). However, the
isolated impairment of Th2 differentiation is not sufficient to
promote immune tolerance. Based on these results, the present study
assessed the effect of TIM-4 blockade with the addition of
exogenous TGF-β on AR in vivo. The results demonstrated that
the combined treatment of anti-TIM-4 mAb and TGF-β was more
effective at protecting mice from AR injury that when either
reagent was used alone. It was also revealed that combined
treatment induced the generation of iTregs in liver allografts. The
survival time of mice treated with anti-TIM-4 mAb and TGF-β was
significantly longer than those in other treatment groups. TGF-β is
a primary pro-fibrogenic medium derived from KCs and hepatocytes in
inflammation. However, TGF-β-induced hepatic fibrosis resulting
from chronic hepatopathy with various etiologies, including HBV
infection and allograft rejection, is a long-term and reversible
process (42). The present study
utilized exogenous TGF-β (0.5 ml/mouse, 1 ng/ml) injections for 2
days following surgery to establish LT models. No marked changes in
hepatofibrosis pathophysiology were observed in any group.
Consequently, the blockade of KC-expressed TIM-4, in combination
with an exogenous TGF-β injection, exhibited synergistic effects on
the improvement of AR of LT.
In conclusion, the present study demonstrated that
LT may lead to the augmented expression of KC TIM-4 and that TIM-4
blockade markedly improves in vivo AR injury by inhibiting
NF-κB and p38 MAPK signaling. Additionally, TIM-4 blockade
increased TGF-β levels, inhibited Th2 differentiation and promoted
iTreg conversion by suppressing IL-4/STAT6/Gata3 signaling. These
effects were demonstrated more pronounced following the addition of
exogenous TGF-β. Therefore, the results of the present study
indicate that KC-expressed TIM-4 blockade in combination with an
exogenous TGF-β injection may be a useful strategy to treat AR and
improve the survival rates of mice that receive liver allografts.
However, further studies that utilize other animal models of liver
allografts are required to determine whether this may be the case
in humans too.
Abbreviations:
|
TIM-4
|
T cell immunoglobulin and mucin
domain-4
|
|
KCs
|
kupffer cells
|
|
APCs
|
antigen presenting cells
|
|
OLT
|
orthotopic liver transplantation
|
|
LT
|
liver transplantation
|
|
AR
|
acute rejection
|
|
Treg
|
T regulatory cell
|
|
NF-κB
|
nuclear factor-κB
|
|
MAPK
|
mitogen-activated protein kinase
|
|
TGF-β
|
transforming growth factor-β
|
|
IL-4
|
interleukin-4
|
|
STAT6
|
signal transducer and activator of
transcription 6
|
|
Gata3
|
transcription factor gata3
|
|
DCs
|
dendritic cells
|
|
AST
|
aspartate aminotransferase
|
|
ALT
|
alanine aminotransferase
|
|
TBIL
|
total bilirubin in serum
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IFN-γ
|
interferon-γ
|
Acknowledgments
Not applicable.
References
|
1
|
Knechtle SJ and Kwun J: Unique aspects of
rejection and tolerance in liver transplantation. Semin Liver Dis.
29:91–101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yoshida O, Dou L, Kimura S, Yokota S, Isse
K, Robson SC, Geller DA and Thomson AW: CD39 deficiency in murine
liver allografts promotes inflammatory injury and immune-mediated
rejection. Transpl Immunol. 32:76–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen ZY, Wu B, Liu T, Yang Y, Yin ML,
Zheng WP, Zhang BY and Song HL: Immunomodulatory effects of bone
marrow mesenchymal stem cells overexpressing heme oxygenase-1:
Protective effects on acute rejection following reduced-size liver
transplantation in a rat model. Cell Immunol. 313:10–24. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meyers JH, Sabatos CA, Chakravarti S and
Kuchroo VK: The TIM gene family regulates innate and adaptive
immunity. Trends Mol Med. 11:362–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Freeman GJ, Casasnovas JM, Umetsu DT and
DeKruyff RH: TIM genes: A family of cell surface phosphatidylserine
receptors that regulate innate and adaptive immunity. Immunol Rev.
235:172–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Z, Ju Z and Frieri M: The T-cell
immunoglobulin and mucin domain (Tim) gene family in asthma,
allergy, and autoimmunity. Allergy Asthma Proc. 34:e21–e262013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yeung MY, McGrath M and Najafian N: The
emerging role of the TIM molecules in transplantation. Am J
Transplant. 11:2012–2119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baghdadi M, Yoneda A, Yamashina T, Nagao
H, Komohara Y, Nagai S, Akiba H, Foretz M, Yoshiyama H, Kinoshita
I, et al: TIM-4 glycoprotein-mediated degradation of dying tumor
cells by autophagy leads to reduced antigen presentation and
increased immune tolerance. Immunity. 39:1070–1081. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rodriguez-Manzanet R, Meyers JH,
Balasubramanian S, Slavik J, Kassam N, Dardalhon V, Greenfield EA,
Anderson AC, Sobel RA, Hafler DA, et al: TIM-4 expressed on APCs
induces T cell expansion and survival. J Immunol. 180:4706–4713.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meyers JH, Chakravarti S, Schlesinger D,
Illes Z, Waldner H, Umetsu SE, Kenny J, Zheng XX, Umetsu DT,
DeKruyff RH, et al: TIM-4 is the ligand for TIM-1 and the
TIM-1-TIM-4 interaction regulates T cell proliferation. Nat
Immunol. 6:455–464. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizui M, Shikina T, Arase H, Suzuki K,
Yasui T, Rennert PD, Kumanogoh A and Kikutani H: Bimodal regulation
of T cell-mediated immune responses by TIM-4. Int Immunol.
20:695–708. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao W, Ryan M, Buckley D, O'Connor R and
Clarkson MR: Tim-4 inhibition of T-cell activation and T helper
type 17 differentiation requires both the immunoglobulin V and
mucin domains and occurs via the mitogen-activated protein kinase
pathway. Immunology. 133:179–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Y, Liu Z, Liang S, Luan X, Long F,
Chen J, Peng Y, Yan L and Gong J: Role of Kupffer cells in the
induction of tolerance of orthotopic liver transplantation in rats.
Liver Transpl. 14:823–836. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ji H, Liu Y, Zhang Y, Shen XD, Gao F,
Busuttil RW, Kuchroo VK and Kupiec-Weglinski JW: T-Cell
immunoglobulin and mucin domain 4 (TIM-4) signaling in innate
immune-mediated liver ischemia-reperfusion injury. Hepatology.
60:2052–2064. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yeung MY, McGrath MM, Nakayama M, Shimizu
T, Boenisch O, Magee CN, Abdoli R, Akiba H, Ueno T, Turka LA and
Najafian N: Interruption of dendritic cell-mediated TIM-4 signaling
induces regulatory T cells and promotes skin allograft survival. J
Immunol. 191:4447–4455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Wasington, DC: 2011
|
|
17
|
Peng Y, Gong JP, Yan LN, Li SB and Li XH:
Improved two-cuff technique for orthotopic liver transplantation in
rat. Hepatobiliary Pancreat Dis Int. 3:33–37. 2004.PubMed/NCBI
|
|
18
|
Li PZ, Li JZ, Li M, Gong JP and He K: An
efficient method to isolate and culture mouse Kupffer cells.
Immunol Lett. 158:52–56. 2014. View Article : Google Scholar
|
|
19
|
Schmittgen TD: Real-time quantitative PCR.
Methods. 25:383–385. 2001. View Article : Google Scholar
|
|
20
|
No authors listed. Banff schema for
grading liver allograft rejection: An international consensus
document. Hepatology. 25:658–663. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitamoto K, Machida Y, Uchida J, Izumi Y,
Shiota M, Nakao T, Iwao H, Yukimura T, Nakatani T and Miura K:
Effects of liposome clodronate on renal leukocyte populations and
renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci.
111:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ikarashi M, Nakashima H, Kinoshita M, Sato
A, Nakashima M, Miyazaki H, Nishiyama K, Yamamoto J and Seki S:
Distinct development and functions of resident and recruited liver
Kupffer cells/macrophages. J Leukoc Biol. 94:1325–1337. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stienstra R, Saudale F, Duval C, Keshtkar
S, Groener JE, van Rooijen N, Staels B, Kersten S and Müller M:
Kupffer cells promote hepatic steatosis via
interleukin-1beta-dependent suppression ofperoxisome
proliferator-activated receptor alpha activity. Hepatology.
51:511–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takaki H, Ichiyama K, Kog K, Chinen T,
Takaesu G, Sugiyama Y, Kato S, Yoshimura A and Kobayashi T: STAT6
inhibits TGF-beta1-mediated Foxp3 induction through direct binding
to the Foxp3 promoter, which is reverted by retinoic acid receptor.
J Biol Chem. 283:14955–14962. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao CQ, Li TL, He SH, Chen X, An YF, Wu
WK, Zhou XH, Li P and Yang PC: Specific immunotherapy suppresses
Th2 responses via modulating TIM1/TIM4 interaction on dendritic
cells. Allergy. 65:986–995. 2010. View Article : Google Scholar
|
|
26
|
Rong S, Park JK, Kirsch T, Yagita H, Akiba
H, Boenisch O, Haller H, Najafian N and Habicht A: The TIM-1: TIM-4
pathway enhances renal ischemia-reperfusion injury. J Am Soc
Nephrol. 22:484–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang M, Xu S, Han Y and Cao X: Apoptotic
cells attenuate fulminant hepatitis by priming Kupffer cells to
produce interleukin-10 through membrane-bound TGF-β. Hepatology.
53:306–316. 2011. View Article : Google Scholar
|
|
28
|
Chen GS and Qi HZ: Effect of Kupffer cells
on immune tolerance in liver transplantation. Asian Pac J Trop Med.
5:970–972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tietjen GT, Gong Z, Chen CH, Vargas E,
Crooks JE, Cao KD, Heffern CT, Henderson JM, Meron M, Lin B, et al:
Molecular mechanism for differential recognition of membrane
phosphatidylserine by the immune regulatory receptor Tim4. Proc
Natl Acad Sci USA. 111:E1463–E1472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim HS, Lee CW and Chung DH: T cell Ig
domain and mucin 1 engagement on invariant NKT cells in the
presence of TCR stimulation enhances IL-4 production but inhibits
IFN-gamma production. J Immunol. 184:4095–4106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van Delft MA, Huitema LF and Tas SW: The
contribution of NF-κB signalling to immune regulation and
tolerance. Eur J Clin Invest. 45:529–539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu LM, Liang DY, Ye CG, Tu WJ and Zhu T:
The UII/UT system mediates upregulation of proinflammatory
cytokines through p38 MAPK and NF-κB pathway in LPS-stimulated
Kupffer cells. PLoS One. 10:e01213832015. View Article : Google Scholar
|
|
33
|
Gong XW, Xu YJ, Yang QH, Liang YJ, Zhang
YP, Wang GL and Li YY: Effects of soothing liver and invigorating
spleen recipes on the IKKβ-NF-κB signaling pathway in kupffer cells
of nonalcoholic steatohepatitis rats. Evid Based Complement
Alternat Med. 2015:6876902015. View Article : Google Scholar
|
|
34
|
Li J and Gong J, Li P, Li M, Liu Y, Liang
S and Gong J: Knockdown of microRNA-155 in Kupffer cells results in
immunosuppressive effects and prolongs survival of mouse liver
allografts. Transplantation. 97:626–635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li S, Fan Q, He S, Tang T, Liao Y and Xie
J: MicroRNA-21 negatively regulates Treg cells through a
TGF-β1/Smad-independent pathway in patients with coronary heart
disease. Cell Physiol Biochem. 37:866–878. 2015. View Article : Google Scholar
|
|
36
|
Tone Y, Furuuchi K, Kojima Y, Tykocinski
ML, Greene MI and Tone M: Smad3 and NFAT cooperate to induce Foxp3
expression through its enhancer. Nat Immunol. 9:194–202. 2008.
View Article : Google Scholar
|
|
37
|
Dardalhon V, Awasthi A, Kwon H, Galileos
G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et
al: IL-4 inhibits TGF-beta-induced Foxp3+ T cells and,
together with TGF-beta, generates IL-9+
IL-10+ Foxp3(−) effector T cells. Nat Immunol.
9:1347–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mantel PY, Kuipers H, Boyman O, Rhyner C,
Ouaked N, Rückert B, Karagiannidis C, Lambrecht BN, Hendriks RW,
Crameri R, et al: GATA3-driven Th2 responses inhibit
TGF-beta1-induced FOXP3 expression and the formation of regulatory
T cells. PLoS Biol. 5:e3292007. View Article : Google Scholar
|
|
39
|
Faust SM, Lu G, Marini BL, Zou W, Gordon
D, Iwakura Y, Laouar Y and Bishop DK: Role of T cell TGFbeta
signaling and IL-17 in allograft acceptance and fibrosis associated
with chronic rejection. J Immunol. 183:7297–7306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wei J, Duramad O, Perng OA, Reiner SL, Liu
YJ and Qin FX: Antagonistic nature of T helper 1/2 developmental
programs in opposing peripheral induction of Foxp3+ regulatory T
cells. Proc Natl Acad Sci USA. 104:18169–18174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou P, Szot GL, Guo Z, Kim O, He G, Wang
J, Grusby MJ, Newell KA, Thistlethwaite JR, Bluestone JA and Alegre
ML: Role of STAT4 and STAT6 signaling in allograft rejection and
CTLA4-Ig-mediated tolerance. J Immunol. 165:5580–5587. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang LY, Heller M, Meng Z, Yu LR, Tang Y,
Zhou M and Zhang YE: Transforming growth factor-β (TFG-β) directly
activates the JAK1-STAT3 axis to induce hepatic fibrosis in
coordination with SMAD pathway. J Biol Chem. 292:4302–4312. 2017.
View Article : Google Scholar : PubMed/NCBI
|














