Introduction
During immune response, indoleamine 2,3-dioxygenase
(IDO) is upregulated in antigen presenting cells (APCs) and
degrades L-tryptophan along the kynurenine pathway. Depletion of
L-tryptophan in the local microenvironment activates the amino-acid
sensor general control nonderepressible 2 kinase (GCN2K) in CD4+
T-cells (1–4). According to some, albeit not all,
investigators (1–5), L-tryptophan depletion also inhibits
the other amino-acid sensor, the mammalian target of rapamycin
complex 1 (mTORC1). In parallel, the produced kynurenine activates
the aryl hydrocarbon receptor (AhR) (6,7).
It is generally thought that through these pathways IDO suppresses
CD4+ T-cell function by inducing apoptosis, inhibiting
proliferation and promoting differentiation towards a regulatory
T-cell lineage (8).
Cell metabolism governs the fate of activated CD4+
T-cells. These cells rely on aerobic glycolysis and glutaminolysis
for energy and building blocks required for their survival,
proliferation and differentiation towards an effector cell lineage
(9–11). However, under conditions that
favor differentiation towards regulatory T-cells (Tregs), naïve
CD4+ T-cells rely on free fatty acid oxidation in order to cover
their energy demands. Notably, blockage of fatty acid oxidation
inhibits naïve CD4+ T-cells differentiation towards a regulatory
phenotype (11,12).
IDO affects the metabolism of CD4+ T-cells. By
upregulating tumor suppressor protein p53 (p53) and downregulating
MYC proto-oncogene (c-Myc), IDO alters the expression of key
enzymes resulting in suppressed utilization of glucose and
glutamine (3,13). In parallel, at least in part due
to energy deprivation, IDO induces CD4+ T-cell apoptosis and
inhibits their proliferation (2,3,13).
Nevertheless, a culture medium that was free of fatty acids was
used in the majority of these studies that evaluated the
immunosuppressive properties of IDO. Notably, in a study in which
free fatty acids were added in cell cultures, IDO increased free
fatty acid oxidation and although it promoted Treg differentiation,
it did not induce apoptosis or inhibited proliferation (7). Thus, contrary to what is generally
hypothesized, it is possible that the immunosuppressive properties
of IDO rely predominantly on the promotion of Treg differentiation,
and that, under physiological conditions where free fatty acids are
always present, IDO does not affect CD4+ T-cell apoptosis and
proliferation.
Since IDO is a potent immunosuppressive enzyme able
in experimental models to ameliorate autoimmune diseases (14,15), prevent allograft rejection
(16,17), protect the semiallogenic fetus
from the maternal immune system (18), and facilitate escape of cancer
from immunosurveillance (19), it
may serve as a useful pharmacological target in the future. Thus,
delineating the molecular pathways involved in the
immunosuppressive function of IDO is imperative. Accordingly, the
aim of the present study was to evaluate further the molecular
pathways implicated in IDO-induced alterations of glucose,
glutamine and free fatty acid metabolism in CD4+ T-cells, and how
they ultimately affect cell apoptosis and proliferation.
For this propose, two-way mixed lymphocyte reactions
(MLRs), a well-established model of alloreactivity (20), were performed in the presence or
not of the free fatty acid oleate and/or of the IDO inhibitor
1-methyl-DL-tryptophan (1-MT). 1-MT has been successfully used to
increase the activity of autoimmune diseases and break the immune
tolerance to semi-allogenic fetuses and allografts (15,16,18). In MLR-derived CD4+ T-cells, the
effect of both IDO-induced pathways, namely the activation of GCN2K
and AhR, can be evaluated. In addition, another system, free of
APCs, was generated. Isolated CD4+ T-cells, cultured in an
oleate-containing medium, were activated with anti-CD2/CD3/CD28
covered beads in the presence or not of the GCN2K activator
tryptophanol. Tryptophanol is known to activate GCN2K and inhibit
CD4+ T-cell proliferation and survival (1,3,4,13).
It is expected that in this system tryptophanol would activate only
GCN2K, leaving AhR unaffected. Comparing the results from these two
experimental systems, the effect of IDO-induced GCN2K activation
can be discriminated from the effect of AhR activation on CD4+
T-cells.
Materials and methods
Subjects
Blood samples were collected from 6 healthy
volunteers (4 males and 2 females; age, 35.83±5.78 years) in April
2017. A written informed consent was obtained from each individual
enrolled in the study. The Ethics Committee of the Faculty of
Medicine, University of Thessaly (Larisa, Greece) approved the
study protocol. The number of approval is 558/10-2-2017.
Peripheral blood mononuclear cells
(PBMCs) and CD4+ T-cell isolation and culture
PBMCs were isolated from whole blood by
Ficoll-Hypaque density gradient centrifugation (Histopaque 1077;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and counted by
optical microscopy on a Neubauer plaque. Cell viability was
assessed by trypan blue assay (Sigma-Aldrich; Merck KGaA).
PBMCs were resuspended in RPMI-1640 medium with
L-glutamine and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid (HEPES) (Sigma-Aldrich; Merck KGaA) and supplemented with 10%
fetal bovine serum (Sigma-Aldrich; Merck KGaA) and
antibiotic-antimycotic solution (Sigma-Aldrich; Merck KGaA). Then,
PBMCs from different individuals were seeded in couples in order to
set up ten different two-way MLRs. Unless otherwise stated, oleate
(Sigma-Aldrich; Merck KGaA) was added in all experiments to the
culture medium at a concentration of 0.8 mM. Oleate was selected
because this free fatty acid is not cytotoxic for human cells at a
concentration up to 1 mM (21).
In the case of experiments with the GCN2K activator
tryptophanol (Sigma-Aldrich; Merck KGaA), CD4+ T-cells were
isolated from fresh PMBCs using the Human CD4+ T-Cell Isolation kit
(Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Isolated CD4+
T-cells were cultured in the same medium as the PBMCs supplemented
with 0.8 mM oleate. The experiment was repeated six independent
times. All cultures were incubated at 37°C in a humidified
atmosphere containing 5% CO2.
L-tryptophan, glucose and oleate
consumption
MLRs were performed in 12-well plates for 7 days in
the presence or not of the IDO inhibitor 1-MT (100 μM;
Sigma-Aldrich; Merck KGaA). The concentration of 1-MT was selected
according to previous experiments (2,3,13).
The number of PBMCs of each member of the MLR couple was
5×105, bringing the total cell number to
1×106 cells/MLR. L-tryptophan concentration was assessed
in the supernatants using ELISA (cat. no. E01T0140; BlueGene
Biotech, Shanghai, China). The sensitivity of the above kit is 1
ng/ml. L-tryptophan consumption was calculated by subtracting the
results in the cell culture supernatants from the concentration
found in the supplemented culture medium. Similarly, glucose
consumption was assessed by using the Element blood glucose monitor
(Infopia, Inc., Titusville, FL, USA), and oleate consumption
colorimetrically through the Free Fatty Acid Quantification kit
(Abcam, Cambridge, UK). The detection limit of the above kit is 2
μM. Ten such MLRs were performed.
Glucose and oleate consumption were also assessed in
activated isolated CD+4 T-cells in the presence or not of
tryptophanol. For this propose, isolated CD4+ T-cells from fresh
PBMCs were cultured in 12-well plates (1×106 cells/well)
and activated for 72 h with anti-CD2/CD3/CD28-conjugated beads
using the T-Cell activation/expansion kit (Miltenyi Biotec GmbH) in
a bead to cell ratio of 1:2 and in the presence or not of 0.25 mM
tryptophanol. The above concentration of tryptophanol was selected
according to previous studies (1,3,4,13).
The experiment was repeated six independent times.
Cell proliferation mixed lymphocyte
reactions were performed in 96-well plates for 7 days in the
presence or not of 100 μΜ 1-MT
The number of PBMCs of each member of the MLR couple
was 5×104, bringing up the total cell number to
1×105 cells per MLR. In parallel, 1×105
resting PBMCs from each member were cultured in the same 96-well
plate, serving as controls. Cell proliferation was assessed with
the Cell Proliferation ELISA (cat. no. 11647229001; Roche
Diagnostics, Indianapolis, IN, USA) using bromodeoxyuridine
labeling and immunoenzymatic detection, according to the
manufacturer’s protocol. The proliferation index was calculated as
follows: Proliferation index = optical density (OD) derived from
each MLR/mean of the ODs derived from the control resting PBMCs
cultures of the two subjects that formed the specific MLR. Ten such
MLRs were performed, each in triplicate and the results refer to
the mean of the three measurements.
The proliferation of CD4+ T-cells retrieved from
freshly isolated PBMCs was also determined by Cell Proliferation
ELISA. Resting, stimulated with T-Cell activation/expansion kit or
stimulated in the presence of 0.25 mM tryptophanol CD4+ T-cells
were cultured in 96-well plates (1×105 cells/well) for
72 h. The proliferation index was calculated as follows:
Proliferation index=OD derived from activated CD4+ T-cells/ODs
derived from the respective resting CD4+ T-cells. T-cells were
derived from six individuals and the experiments were performed in
triplicates. The results refer to the mean of the three
measurements.
Assessment of proteins of interest in
MLR-derived CD4+ T-cells and in isolated CD4+ T-cells activated
with anti-CD2/CD3/CD28 covered beads
Ten MLRs were performed in 12-well plates in the
presence or not of 100 μΜ 1-MT, with a cellular number of
each PBMC population in the MLR context remaining the same as
before. At the end of the 7 day period that MLRs lasted, CD4+
T-cells were isolated from the MLRs by negative selection using the
Human CD4+ T-Cell Isolation kit. MLR-derived CD4+ T-cells were
counted via optical microscopy on a Neubauer plaque and cell
viability was determined by trypan blue assay. Equal numbers of
T-cells from each MLR were lysed using the T-PER tissue protein
extraction reagent (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with protease (Merck KGaA) and phosphatase (Roche
Diagnostics) inhibitors.
Protein was also extracted from freshly isolated and
activated CD4+ T-cells. PBMCs were isolated from the blood of six
individuals. Following PBMCs isolation, CD4+ T-cells were isolated
with the Human CD4+ T-Cell Isolation Kit, cultured in 12-well
plates (1×106 cells/well) and activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or not of 0.25
mM tryptophanol for 72 h. Then they were counted via optical
microscopy on a Neubauer plaque, cell viability was determined by
trypan blue assay and equal numbers of cells were lysed using the
T-PER tissue protein extraction reagent (Thermo Fisher Scientific,
Inc.).
Protein was quantified via Bradford assay
(Sigma-Aldrich; Merck KGaA) and 10 μg from each sample was
used for western blotting. For this purpose, sodium dodecyl sulfate
(SDS) polyacrylamide 4–12% Bis-Tris gels (Thermo Fisher Scientific,
Inc.) and polyvinylidene fluoride (PVDF) membranes (Thermo Fisher
Scientific, Inc.) were used. Blots were incubated with the primary
antibody for 16 h at 4°C, followed by a 30 min incubation with the
secondary antibody at room temperature (anti-rabbit horseradish
peroxidase (HRP)-conjugated IgG; 1:1,000; cat. no. 7074; Cell
Signaling Technology, Inc., Danvers, MA, USA). For the primary
antibody against cytochrome P450 family 1 subfamily A polypeptide 1
(CYP1A1), a goat anti-mouse HRP-conjugated IgG secondary antibody
(1:1,000; cat. no. sc2005; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) was used. In case of reprobing the PVDF blots, the
previous primary and secondary antibody were removed with the
Restore Western Blot Stripping Buffer (Thermo Fisher Scientific,
Inc.) according to the manufacturer’s protocol. Analysis of the
western blots was performed using the ImageJ software (National
Institute of Health, Bethesda, MD, USA).
Primary antibodies targeting the following proteins
were used in western blotting: Eukaryotic initiation factor 2α
phosphorylated at serine 51 (p-eIF2α; 1:500; cat. no. 9721; Cell
Signaling Technology, Inc.) which is the substrate of GCN2K; 70 kDa
ribosomal protein S6 kinase phosphorylated at threonine 389
(p-p70S6K; 1:1,000; cat. no. 9234; Cell Signaling Technology, Inc.)
which is the substrate of mTORC1; the transcriptional target of
AhR, CYP1A1 (1:500; cat. no. sc-25304; Santa Cruz Biotechnology,
Inc.); the α subunit of activated AMP-activated protein kinase
phosphorylated at threonine 172 (p-AMPK; 1:1,000; cat. no. 2535;
Cell Signaling Technology, Inc.); the substrate of AMPK acetyl-CoA
carboxylase 2 phosphorylated at serine 221 (p-ACC2; 1:500; cat. no.
ab109540; Abcam); the activated cleaved caspase-3 at aspartate 175
(CC3; 1:500; cat. no. 9664; Cell Signaling Technology, Inc.);
glucose transporter-1 (GLUT1; 1:500; cat. no. sc-7903, Santa Cruz
Biotechnology, Inc.); hexokinase-II (HKII; 1:1,000; cat. no. 2867;
Cell Signaling Technology, Inc.); glutaminase 1 (GLS1; 1:100; cat.
no. AP18036PUN; Acris Antibodies GmbH: Origen Europe, Herford,
Germany); glutaminase2 (GLS2; 1:100; cat. no. AP17426PU-N; Acris
Antibodies GmbH; Origen Europe); carnitine palmitoyltransferase I
isoenzyme A (CPT1A; 1:1,000; cat. no. 12252S; Cell Signaling
Technology, Inc.); CPT1B (1:1,000; cat. no. ab134988; Abcam); and
CPT1C (1:250; cat. no. ab87498; Abcam). All western blot results
were normalized to β-actin (1:5,000; cat. no. 4967; Cell Signaling
Technology, Inc.).
Statistical analysis
The SPSS software (version 13; SPSS Inc., Chicago,
IL, USA) was used for statistical analysis. The normality of the
evaluated variables was assessed and confirmed by the one-sample
Kolmogorov-Smirnov test. For comparison of means, paired-sample
t-test was used. Results were expressed as mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of free fatty acids on the role of
IDO and GCN2K in T-cell proliferation
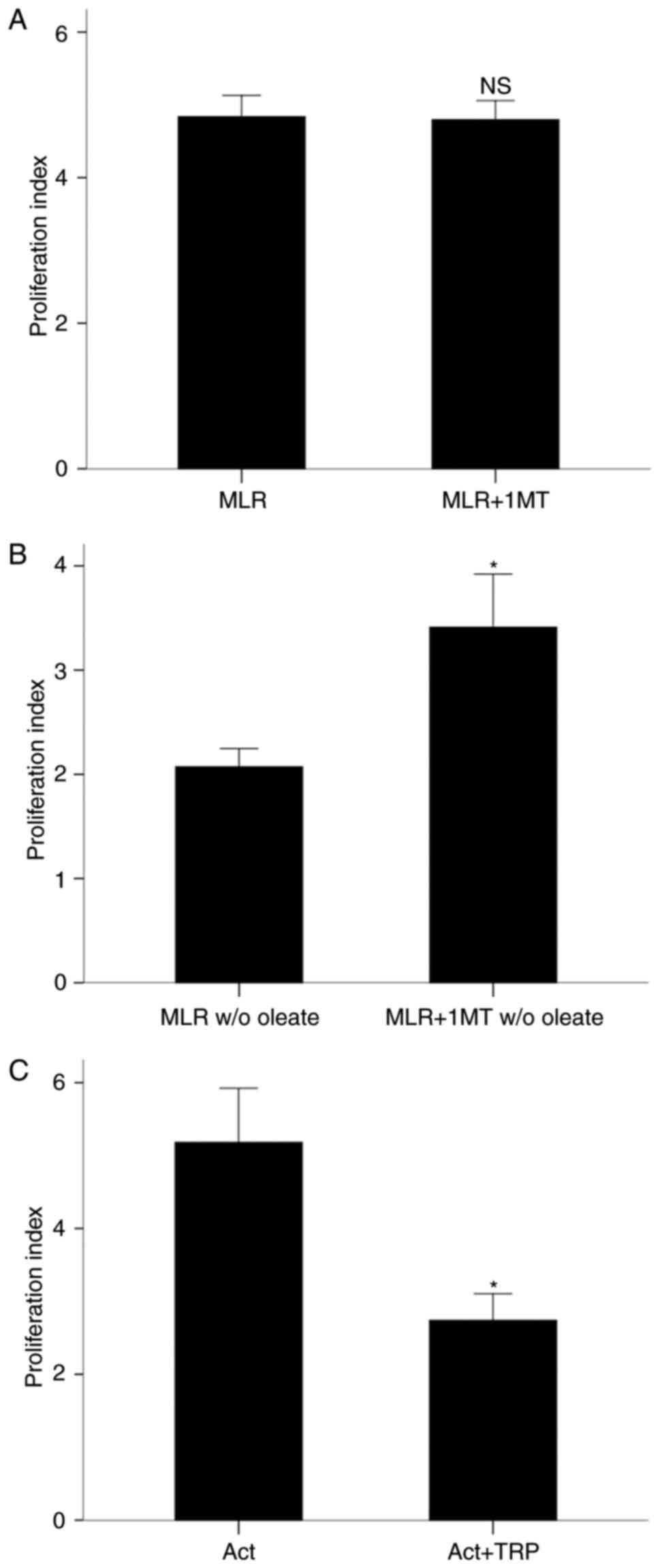
In the presence of oleate, the IDO inhibitor 1-MT
did not affect T-cell clonal expansion in MLRs. The proliferation
index was 4.84±0.29 in the absence of 1-MT and 4.80±0.26 with 1-MT
treatment (P=0.610; Fig. 1A). By
contrast, in the absence of oleate, 1-MT treatment increased the
proliferation index from 2.07±0.17 to 3.41±0.51 (P<0.001;
Fig. 1B). Of note, the T-cell
proliferation index was significantly higher in MLRs performed in
the presence of oleate compared with MLRs performed in the absence
of oleate (P<0.001; Fig. 1A and
B).
The GCN2K activator tryptophanol decreased
proliferation of isolated and activated with
anti-CD2/CD3/CD28-conjugated beads CD4+ T-cells despite the
presence of oleate. The proliferation index was 5.18±0.74 in the
absence of tryptophanol and 2.74±0.36 with tryptophanol treatment
(P<0.001; Fig. 1C). These
results demonstrated that IDO suppressed T-cell proliferation only
in the absence of free fatty acids, whereas the GCN2K activator
tryptophanol inhibited cell proliferation in activated CD4+ T-cells
despite the presence of free fatty acids.
Effect of free fatty acids on the role of
IDO and GCN2K in T-cell apoptosis
In MLR-derived CD4+ T-cells, and in the presence of
oleate, 1-MT treatment did not affect the protein levels of
activated cleaved caspase-3 compared with the untreated cells
(P=0.907; Fig. 2A and B). By
contrast, when oleate was absent from the culture medium, 1-MT
treatment resulted in a decrease of apoptosis in MLR-derived CD4+
T-cells (Fig. 2A), with the
levels of cleaved caspase-3 decreasing to 0.56±0.05-fold of the
levels in the untreated MLRs (Fig.
2C).
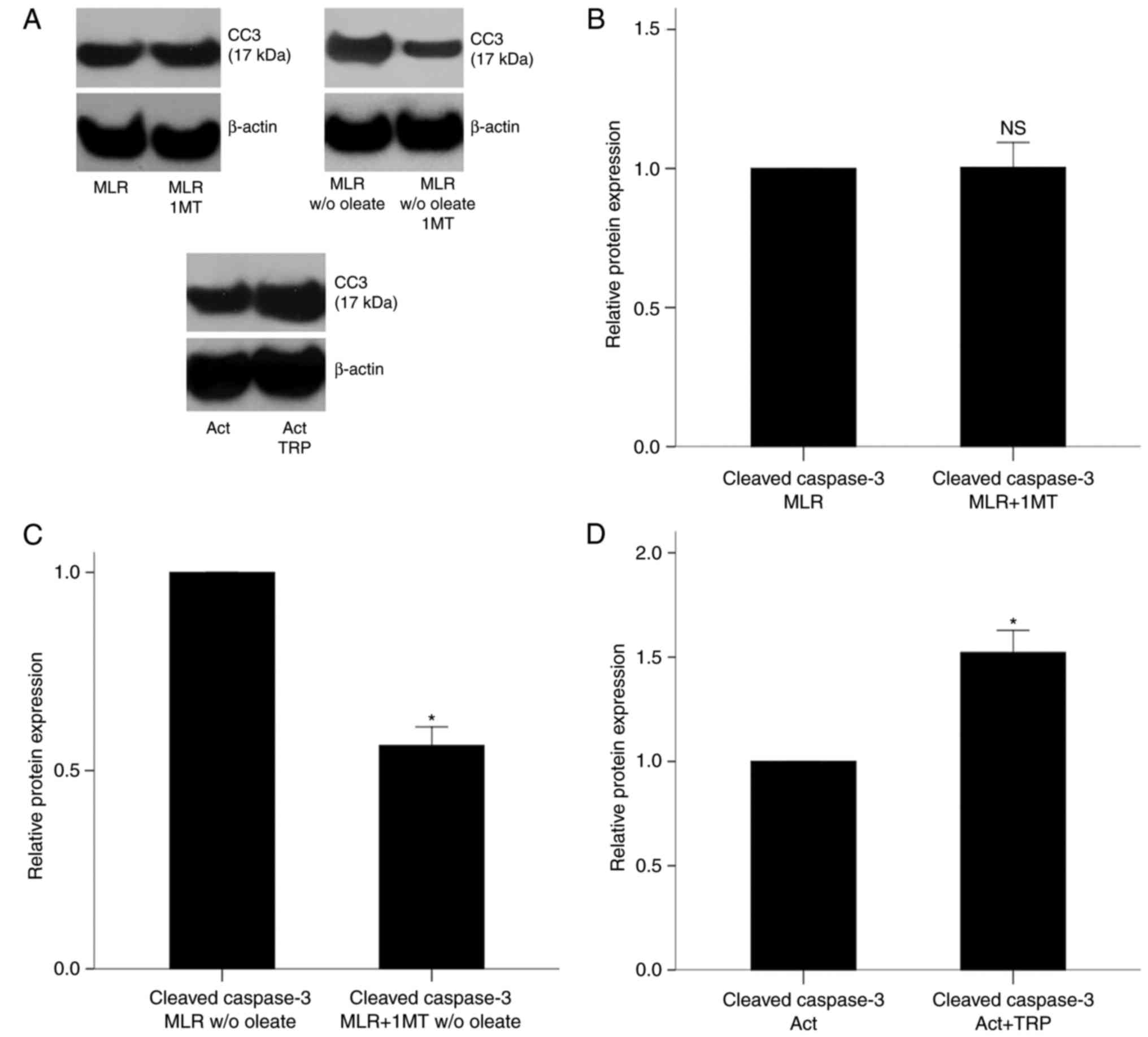 | Figure 2Effect of IDO inhibition and GCN2K
activation on cell apoptosis in MLR-derived CD4+ T-cells and
activated isolated CD4+ T-cells, respectively. (A) Representative
blots from western blot analysis for protein levels of cleaved
caspase-3 in experimental groups. (B) PBMCs from different
individuals were coupled in 10 different MLRs performed in the
presence or (C) absence of oleate and treated with the IDO
inhibitor 1-MT. Then, CD4+ T-cells were isolated from the MLRs and
protein was extracted for western blotting. 1-MT did not affect the
levels of cleaved caspase-3 in MLR-derived CD4+ T-cells cultured
with oleate but decreased the levels of cleaved caspase-3 in
MLR-derived CD4+ T-cells cultured without oleate. (D) CD4+ T-cells
were isolated from the PBMCs of 6 individuals, cultured in an
oleate-containing medium and activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or absence of
the GCN2K activator tryptophanol. Then, the protein was extracted
and western blotting revealed that tryptophanol increased cell
apoptosis. Results are presented as mean ± standard deviation.
*P<0.05. IDO, indoleamine 2,3-dioxygenase; GCN2K,
general control nonderepressible 2 kinase; MLR, mixed lymphocyte
reactions; PBMCs, peripheral blood mononuclear cells; CC3, cleaved
caspase-3; TRP, tryptophanol; NS, not significant. |
In isolated CD4+ T-cells cultured in an
oleate-containing medium and activated with
anti-CD2/CD3/CD28-conjugated beads, tryptophanol increased the
levels of cleaved caspase-3 by 1.52±0.11-fold compared with the
untreated cells (P<0.001; Fig. 2A
and D). These results demonstrated that, in MLR-derived CD4+
T-cells, IDO induced apoptosis only in the absence of free fatty
acids, whereas in activated isolated CD4+ T-cells the GCN2K
activator tryptophanol induced cell apoptosis despite the presence
of free fatty acids.
Effect of IDO and GCN2K on L-tryptophan
consumption and AhR activation
In MLRs performed in the presence of oleate, the IDO
inhibitor 1-MT decreased L-tryptophan consumption from 4.67±0.43
μg/ml in untreated cells to 1.16±0.73 μg/ml in
1-MT-treated cells (P<0.001; Fig.
3A). In CD4+ T-cells derived from the above 1-MT-treated MLRs,
GCN2K and AhR activity decreased, whereas mTORC1 activity remained
unaffected (Fig. 3B). More
precisely, 1-MT treatment decreased the levels of the GCN2K
substrate p-eIF2α and of the AhR transcriptional target CYP1A1 to
0.43±0.13 (P<0.001) and 0.46±0.18 (P<0.001) fold relative to
the levels in untreated MLRs, respectively (Fig. 3C). By contrast, the levels of the
mTORC1 substrate, p-p70S6K, were altered only by a factor of
1.05±0.14 (P=0.265; Fig. 3C).
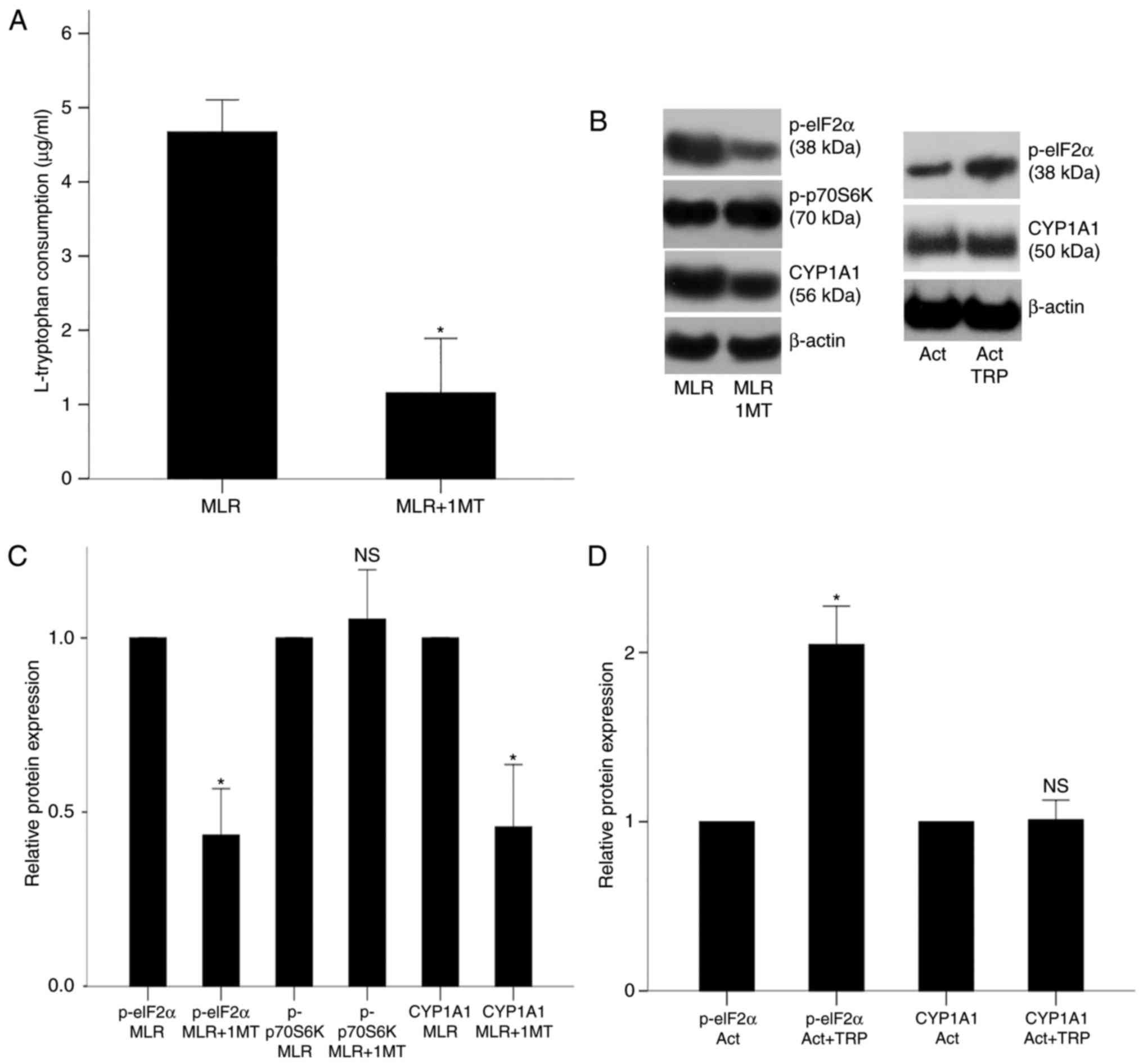 | Figure 3Effect of IDO on the activity of
GCN2K, mTORC1 and AhR in MLR-derived CD4+ T-cells and effect of
tryptophanol on the activity of GCN2K and AhR in activated isolated
CD4+ T-cells. (A) PBMCs from different individuals were coupled in
10 different MLRs performed in the presence of oleate and treated
with the IDO inhibitor 1-MT, then L-tryptophan consumption was
measured. (B) CD4+ T-cells were isolated from the MLRs and protein
was extracted for western blotting. Representative blots are shown
for expression of p-eIF2α, p-p70S6K and CYP1A1 in the experimental
groups. (C) Quantification of the western blot analysis results.
1-MT decreased the activities of GCN2K and AhR, assessed by the
phosphorylation of the GCN2K substrate eIF2α and the expression of
the AhR transcriptional target CYP1A1, respectively. By contrast,
1-MT did not affect the activity of mTORC1, assessed by the
phosphorylation of its substrate p70S6K. (D) CD4+ T-cells were
isolated from the PBMCs of 6 individuals, cultured in an
oleate-containing medium and activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or absence of
the GCN2K activator tryptophanol. Quantification of the western
blotting results from panel B. Tryptophanol increased GCN2K
activity, assessed by the level of p-eIF2α, but it did not alter
AhR activity, assessed by the expression of CYP1A1. Results are
presented as mean ± standard deviation. *P<0.05. IDO,
indoleamine 2,3-dioxygenase; GCN2K, general control
nonderepressible 2 kinase; mTORC1, mammalian target of rapamycin
complex 1; AhR, aryl hydrocarbon receptor; MLR, mixed lymphocyte
reactions; PBMCs, peripheral blood mononuclear cells; p-,
phosphorylated; eIF2α, eukaryotic initiation factor 2α; p70S6K, 70
kDa ribosomal protein S6 kinase; CYP1A1, cytochrome P450 family 1
subfamily A polypeptide 1; TRP, tryptophanol; NS, not
significant. |
In isolated CD4+ T-cells cultured in an
oleate-containing medium and activated with
anti-CD2/CD3/CD28-conjugated beads, tryptophanol, as expected,
increased the activity of GCN2K, but not of AhR (Fig. 3B). In specific, p-eIF2α levels
increased by a factor of 2.05±0.23 in the tryptophanol-treated
cells compared with untreated cells (P<0.001; Fig. 3D), while CYP1A1 expression was not
significantly altered by tryptophanol treatment (1.01±0.12-fold;
P=0.815; Fig. 3D). These results
demonstrated that IDO increased L-tryptophan consumption and
activated GCN2K and AhR, whereas the GCN2K activator tryptophanol
did not affect AhR activity in activated isolated CD4+ T-cells.
Effect of IDO and GCN2K on glucose
consumption
In MLRs performed in the presence of oleate, the IDO
inhibitor 1-MT increased glucose consumption from 105.60±37.39
mg/dl in untreated cells to 148.40±32.01 mg/dl in 1-MT-treated
cells (P<0.001; Fig. 4A). In
isolated CD4+ T-cells cultured in an oleate-containing medium and
activated with anti-CD2/CD3/CD28-conjugated beads, the GCN2K
activator tryptophanol decreased glucose consumption from
101.00±12.99 mg/dl in the untreated cells to 70.67±12.56 mg/dl in
the tryptophanol-treated cells (P<0.001; Fig. 4B).
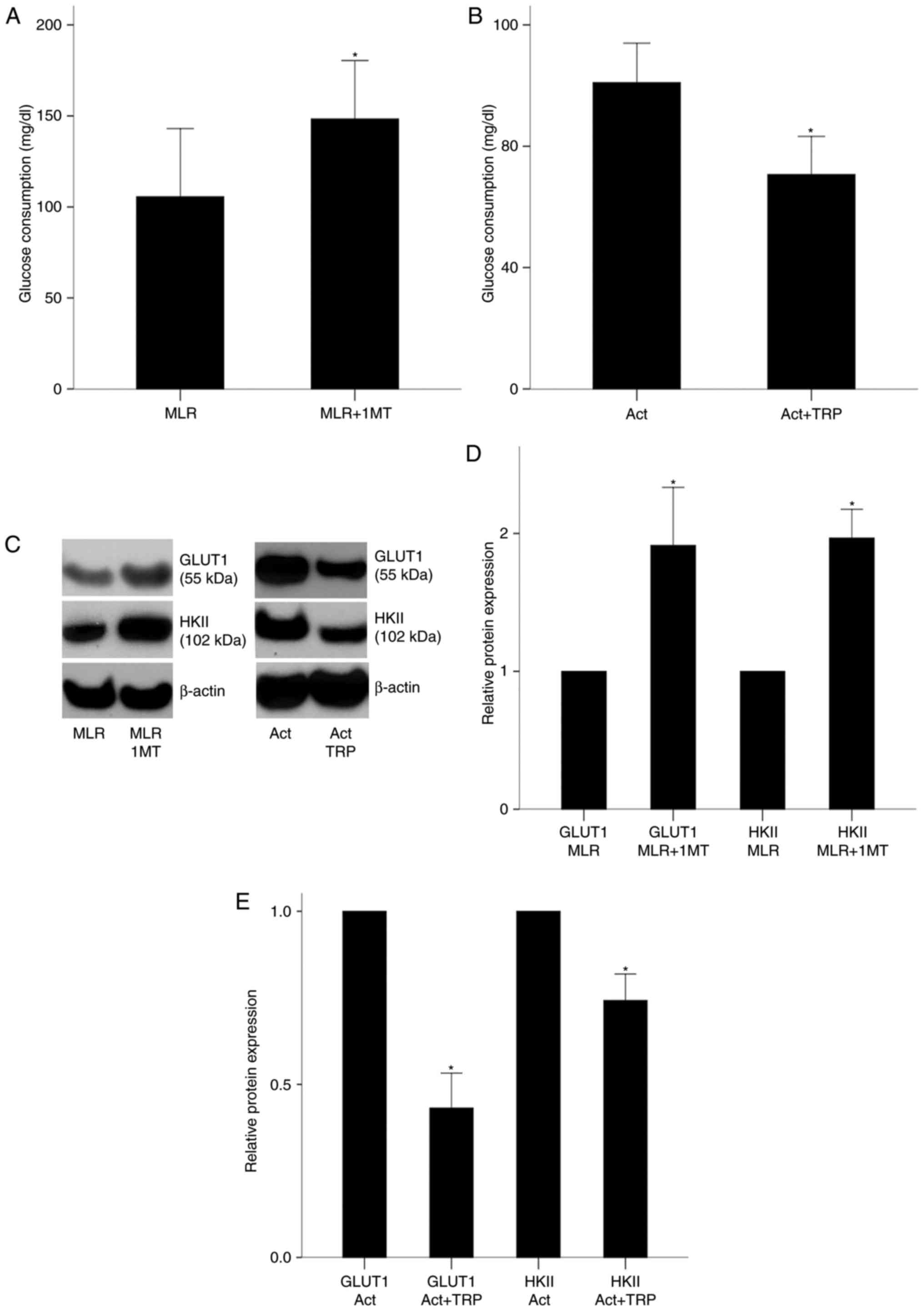 | Figure 4Effect of IDO on glucose consumption
in MLRs and on GLUT1 and HKII expression in MLR-derived CD4+
T-cells, and effect of GCN2K activation on glucose consumption,
GLUT1 and HKII expression in activated isolated CD4+ T-cells. (A)
PBMCs from different individuals were coupled in 10 different MLRs
performed in the presence of oleate, treated with the IDO inhibitor
1-MT, and their glucose consumption was measured. (B) CD4+ T-cells
were isolated from the PBMCs of 6 individuals cultured in an
oleate-containing medium, activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or absence of
the GCN2K activator tryptophanol, and their glucose consumption was
measured. (C) Representative blots of western blot analysis for
GLUT1 and HKII protein expression levels in the experimental
groups. (D) CD4+ T-cells were isolated from the MLRs and protein
was extracted for western blotting. Quantification of results from
panel C. 1-MT increased the expression of GLUT1 and HKII in
MLR-derived CD4+ T-cells. (E) Quantification of results from panel
C for the isolated and activated CD4+ T-cells. Tryptophanol
decreased GLUT1 and HKII expression. Results are presented as mean
± standard deviation. *P<0.05. IDO, indoleamine
2,3-dioxygenase; MLR, mixed lymphocyte reactions; GLUT1, glucose
transporter-1; HKII, hexokinase-II; GCN2K, general control
nonderepressible 2 kinase; PBMCs, peripheral blood mononuclear
cells; TRP, tryptophanol; NS, not significant. |
Next, the protein expression levels of GLUT1 and
HKII were examined, as a measure of glucolysis activity. In CD4+
T-cells derived from 1-MT-treated MLRs, GLUT1 and HKII expression
was increased by a factor of 1.91±0.42 (P<0.001) and 1.97±0.21
(P<0.001), respectively, compared with untreated T-cells
(Fig. 4C and D). In isolated,
cultured in the presence of oleate, and activated with
anti-CD2/CD3/CD28-conjugated beads CD4+ T-cells, tryptophanol
decreased GLUT1 and HKII expression to 0.43±0.10 (P<0.001) and
0.74±0.08 (P<0.001) fold relative to the levels of the untreated
activated CD4+ T-cells, respectively (Fig. 4C and E). These results
demonstrated that IDO and the GCN2K activator tryptophanol
decreased glucose consumption in CD4+ T-cells.
Effect of IDO and GCN2K on the expression
of key glutaminolysis-associated enzymes
In CD4+ T-cells derived from MLRs performed in an
oleate-containing medium and treated with 1-MT, GLS1 and GLS2
expression increased by a factor of 2.30±0.26 (P<0.001) and
1.40±0.29 (P<0.001), respectively, compared with untreated cells
(Fig. 5A and B). In isolated,
cultured with oleate, and activated with
anti-CD2/CD3/CD28-conjugated beads CD4+ T-cells, tryptophanol
treatment decreased GLS1 and GLS2 expression to 0.74±0.07
(P<0.001) and 0.58±0.11 (P<0.001) fold relative to the levels
of the untreated activated CD4+ T-cells, respectively (Fig. 5A and C). These results
demonstrated that IDO and the GCN2K activator tryptophanol
decreased the expression of key enzymes involved in
glutaminolysis.
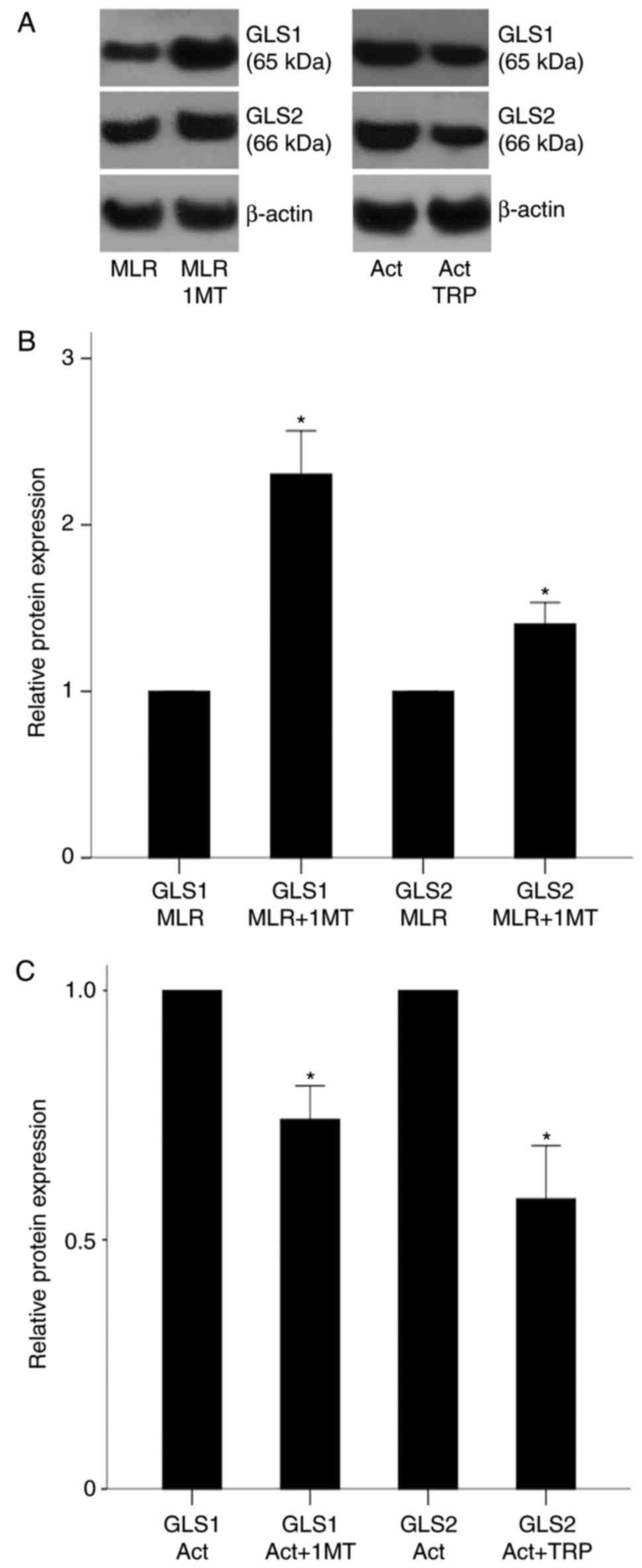 | Figure 5Effect of IDO inhibition and GCN2K
activation on GLS1 and GLS2 expression in MLR-derived CD4+ T-cells
and activated isolated CD4+ T-cells, respectively. (A)
Representative blots from western blot analysis for GLS1 and GLS2
protein expression levels in the experimental groups. (B)
Quantification of results from panel A. PBMCs from different
individuals were coupled in 10 different MLRs performed in the
presence of oleate and treated with the IDO inhibitor 1-MT. 1-MT
increased the expression of both GLS1 and GLS2 in MLR-derived CD4+
T-cells. (C) Quantification of results from panel A. CD4+ T-cells
were isolated from the PBMCs of 6 individuals, cultured in an
oleate-containing medium and activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or absence of
the GCN2K activator tryptophanol. Tryptophanol decreased GLS1 and
GLS2 expression. Results are presented as mean ± standard
deviation. *P<0.05. IDO, indoleamine 2,3-dioxygenase;
GCN2K, general control nonderepressible 2 kinase; GLS, glutaminase;
MLR, mixed lymphocyte reactions; PBMCs, peripheral blood
mononuclear cells; TRP, tryptophanol; NS, not significant. |
Effect of IDO and GCN2K on free fatty
acid consumption and expression of AMPK and CPT1
In MLRs performed in the presence of oleate, the IDO
inhibitor 1-MT decreased oleate consumption from 0.49±0.05 mM in
the untreated cells to 0.19±0.02 mM in the 1-MT-treated cells
(P<0.001; Fig. 6A). In
isolated CD4+ T-cells cultured in an oleate-containing medium and
activated with anti-CD2/CD3/CD28-conjugated beads, the GCN2K
activator tryptophanol did not significantly alter oleate
consumption, which remained low both in the absence (0.15±0.01 mM)
and in the presence (0.16±0.02) of the activator (P=0.158; Fig. 6B). These findings suggest that
although IDO increased free fatty acid consumption, the GCN2K
activator tryptophanol had not effect.
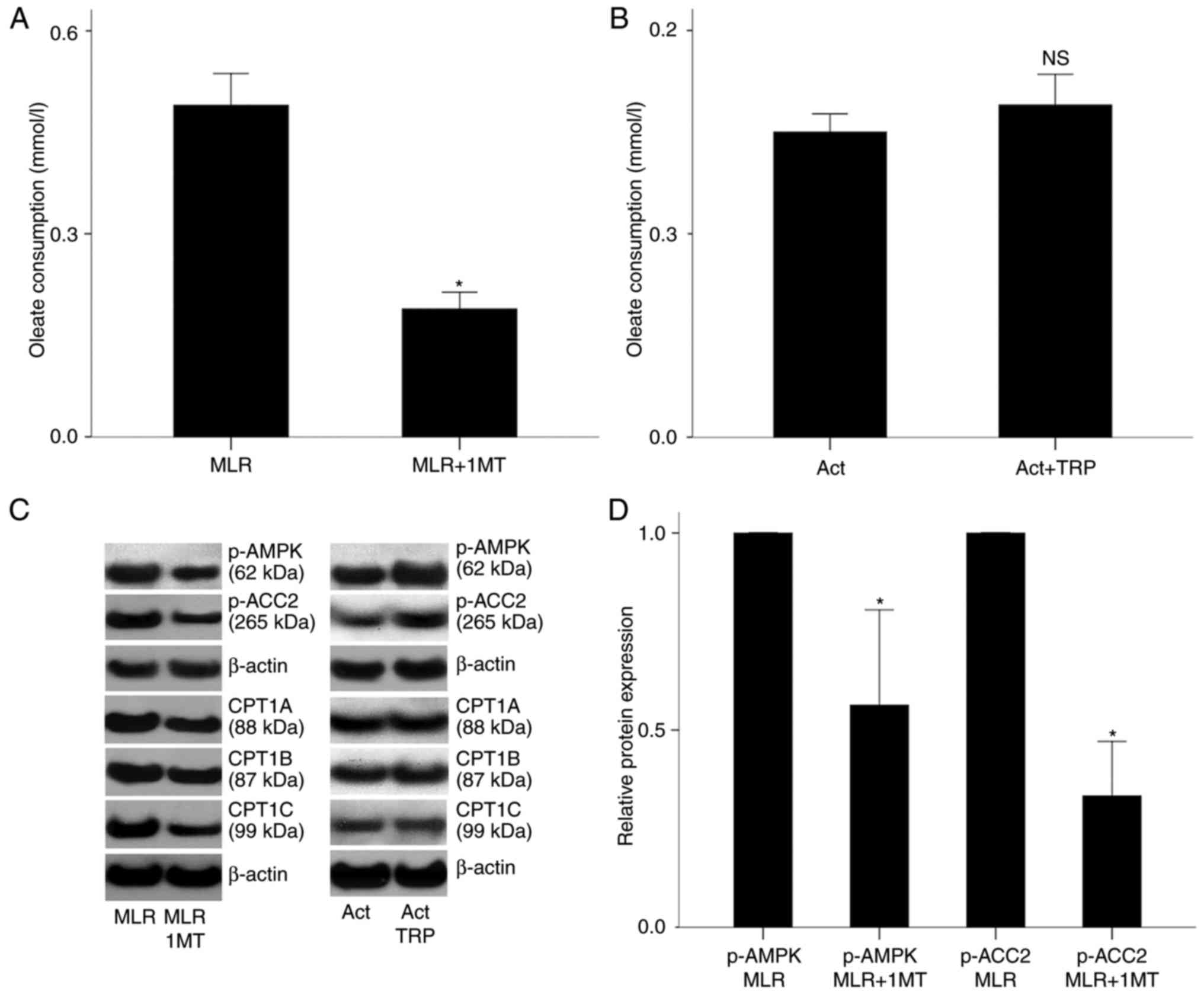 | Figure 6Effect of IDO on free fatty acid
consumption in MLRs and on p-AMPK, p-ACC2 and CPT1 isoenzymes in
MLR-derived CD4+ T-cells; and effect of GCN2K activation on free
fatty acid consumption, and on p-AMPK, p-ACC2 and CPT1 isoenzymes
in activated isolated CD4+ T-cells. (A) PBMCs from different
individuals were coupled in 10 different MLRs performed in the
presence of oleate, treated with the IDO inhibitor 1-MT, then their
free fatty acid consumption was measured. (B) CD4+ T-cells were
isolated from the PBMCs of 6 individuals cultured in an
oleate-containing medium, activated with
anti-CD2/CD3/CD28-conjugated beads in the presence or absence of
the GCN2K activator tryptophanol, and then their free fatty acid
consumption was measured. (C) Representative blots from western
blot analysis in all experimental groups. (D) Quantification of
results from panel C. In CD4+ T-cells isolated from the MLRs, 1-MT
decreased the activity of AMPK, assessed by the level of its
phosphorylated subunit α and the phosphorylation of its substrate
ACC2. (E) Quantification of results from panel C. In isolated and
activated CD4+ T-cells, tryptophanol increased p-AMPK and p-ACC2
levels. (F) Quantification of results from panel C. In CD4+ T-cells
isolated from the MLRs, 1-MT decreased the expression of all CPT1
isoenzymes. (G) Quantification of results from panel C. In isolated
and activated CD4+ T-cells, tryptophanol did not affect the
expression of CPT1 isoenzymes. *P<0.05. IDO,
indoleamine 2,3-dioxygenase; MLR, mixed lymphocyte reactions; p-,
phosphorylated; AMPK, AMP-activated protein kinase; ACC2,
acetyl-CoA carboxylase 2; CPT1, carnitine palmitoyltransferase I;
GCN2K, general control nonderepressible 2 kinase; PBMCs, peripheral
blood mononuclear cells; TRP, tryptophanol; NS, not
significant. |
In CD4+ T-cells derived from the above 1-MT-treated
MLRs, the levels of the activated phosphorylated form of AMPK, as
well as the levels of its substrate p-ACC2, decreased to 0.56±0.24
(P<0.001) and 0.33±0.14 (P<0.001) fold relative to the levels
of the untreated cells, respectively (Fig. 6C and D). In isolated, cultured
with oleate and activated with anti-CD2/CD3/CD28-conjugated beads
CD4+ T-cells, tryptophanol increased p-AMPK and p-ACC2 levels by
1.45±0.11 (P<0.001) and 1.46±0.10 (P<0.001) fold relative to
the untreated cells, respectively (Fig. 6C and E).
Finally, in CD4+ T-cells derived from MLRs performed
in an oleate-containing medium and treated with 1-MT, the
expression of all CPT1 isoenzymes was decreased compared with
untreated cells (Fig. 6C). The
protein expression levels of CPT1A decreased to 0.73±0.05
(P<0.001), CPT1B to 0.54±0.13 (P<0.001) and CPT1C to
0.42±0.12 (P<0.001) fold relative to the untreated cells
(Fig. 6C and F). By contrast, in
isolated, cultured with oleate and activated with
anti-CD2/CD3/CD28-conjugated beads CD4+ T-cells, tryptophanol did
not affect the expression of any of the CPT1 isoenzymes tested.
CPT1A protein expression levels were at 1.05±0.22 (P=0.598), CPT1B
at 0.99±0.17 (P=0.933) and CPT1C at 1.02±0.10 (P=0.706) fold
relative to the untreated cells (Fig.
6C and G). In conclusion, these results demonstrated that IDO
activated AMPK and increased CPT1 expression, whereas the GCN2K
activator tryptophanol, although it activated AMPK, did not
significantly alter CPT1 expression.
Discussion
Because IDO has an important role in immune
regulation, at least in part by altering T-cell metabolism
(3,4,7,8,13),
the present study evaluated the effect of different IDO-induced
pathways on glucose, glutamine, and free fatty acid metabolism and
ultimately on CD4+ T-cell survival and proliferation.
Regarding cell proliferation in MLRs, the present
study recapitulated the results of previous studies (1–4,7,13).
In MLRs performed in the commonly used culture medium, IDO
decreased T-cell proliferation; however, when a free fatty acid was
introduced in the culture medium, the IDO inhibitor 1-MT did not
affect T-cell clonal expansion. By contrast, as is demonstrated in
the present study for the first time, in activated isolated CD4+
T-cells, the presence of free fatty acids in the culture medium did
not abrogate the anti-proliferative effect of the GCN2K activator
tryptophanol. These results indicate that in the presence of free
fatty acids the IDO-induced GCN2K activation is not responsible for
the absence of the IDO antiproliferative effect on T-cells.
Similar results were obtained when apoptosis of CD4+
T-cells was assessed by examining the levels of activated cleaved
caspase-3, in which all the apoptotic pathways converge (22). IDO increased apoptosis in CD4+
T-cells derived from MLRs performed in the commonly used culture
medium, but when a free fatty acid was added into the culture
medium the IDO inhibitor 1-MT did not affect cell apoptosis. These
results are in agreement with the results of a previous study
(7). By contrast, as the present
study demonstrated for the first time, in activated isolated CD4+
T-cells, the presence of free fatty acids in the culture medium did
not abolish the apoptotic effect of the GCN2K activator
tryptophanol. These results indicate that other pathways, and not
the GCN2K activation IDO-induced pathway, may be responsible for
the absence of the IDO apoptotic effect on CD4+ T-cells when free
fatty acids are present.
During an immune response, IDO affects T-cells by
decreasing local L-tryptophan concentration, activating GCN2K and
possibly inhibiting mTORC1 (1–5),
as well as by producing kynurenine and activating AhR (6,7).
In the present study, experiments were performed to assay the
validity of our experimental systems regarding the above pathways
and to define whether there is a role for mTORC1. In MLRs performed
in the presence of oleate, IDO increased L-tryptophan consumption.
In addition, in MLR-derived CD4+ T-cells, IDO activated GCN2K,
assessed by the level of its phosphorylated substrate p-eIF2α
(23), but it did not affect
mTORC1 activity, assessed by the level of its phosphorylated
substrate p-p70S6K (24). This is
in accordance with previous studies in which cell cultures were
performed in the commonly used, fatty acid-free medium (1–4).
Thus, the presence of free fatty acids does not alter the signal
transduction pathways triggered by L-tryptophan depletion. No
effect on mTORC1 activity was detected in the present study, in
accordance with other studies reporting that mTORC1 is sensitive to
the depletion of the amino acids leucine, isoleucine, valine and
possibly arginine, but not of tryptophan (25). In addition to activation of GCN2K,
in MLR-derived CD4+ T-cells, IDO activated AhR, assessed by the
level of its transcriptional target CYP1A1 (26). As expected, in isolated CD4+
T-cells cultured and activated in an oleate-containing medium,
tryptophanol activated GCN2K, but not AhR.
Consistent with results obtained from MLRs in a
fatty acid-free medium (3,13),
the present study demonstrated that glycolysis was also decreased
by IDO in the presence of oleate. In addition, the expression of
GLUT1, the main glucose transporter in CD4+ T-cells (11), as well as of the first enzyme of
the glycolytic pathway HKII, decreased by IDO in MLR-derived CD4+
T-cells. These results were recapitulated by tryptophanol in
isolated CD4+ T-cells cultured with oleate and activated with
anti-CD2/CD3/CD28 beads. Thus, it is likely that IDO through
activation of GCN2K starves CD4+ T-cells from glucose.
In addition to glucose, another energy source for
activated CD4+ T-cells is glutamine (9,10).
However, as in MLRs performed in a fatty acid-free medium (3,4,13),
the present study demonstrated that, in the presence of oleate, IDO
decreased the expression of both GLS isoenzymes in MLR-derived CD4+
T-cells. This was also confirmed following tryptophanol treatment
in isolated CD4+ T-cells cultured and activated in an
oleate-containing medium. Hence, it can be hypothesized that IDO,
through activation of GCN2K, starves CD4+ T-cells of glutamine, as
well as glucose.
Regarding the third available energy source, the
free fatty acids, in MLRs, IDO increased oleate consumption, which
is in agreement with a previous study (7). By contrast, in isolated and
activated CD4+ T-cells, tryptophanol did not affect oleate
consumption, which remained at very low levels, precluding a role
for GCN2K in the IDO-induced increase in fatty acid consumption.
Therefore, the ability of IDO to increase free fatty acid
consumption in CD4+ T-cells could be attributed to the activation
of AhR. This alternative source of energy appears to compensate for
the decreased glucose and glutamine utilization and to provide the
required energy for cell survival and proliferation.
Next, the molecular mechanisms that may be involved
in the IDO-induced increase in free fatty acid oxidation were
investigated in CD4+ T-cells. The IDO-induced and GCN2K-mediated
decrease in glucose and glutamine utilization would be expected to
decrease ATP production in CD4+ T-cells resulting in activation of
AMPK. Indeed, the present results demonstrated that IDO increased
the activated form of AMPK in MLR-derived CD4+ T-cells, and
tryptoph-anol had similar effect in isolated and activated CD4+
T-cells. Once activated, AMPK phosphorylates and inactivates ACC2,
which catalyzes the carboxylation of acetyl-CoA to the CPT1
inhibitor malonyl-CoA (27–29). Accordingly, the present results
demonstrated that IDO increased p-ACC2 levels in MLR-derived CD4+
T-cells, and tryptophanol had the same effect in isolated and
activated CD4+ T-cells. Since CPT1 controls free fatty acid entry
into the mitochondria for oxidation (28,29), it would be expected that
inactivation of ACC2 by AMPK would release CPT1 activity and
increase free fatty acid oxidation. However, although IDO activated
AMPK and inactivated ACC2 in MLR-derived CD4+ T-cells, similar to
tryptophanol in isolated, activated CD4+ T-cells, free fatty acid
consumption increased only in MLRs. Thus, an additional molecular
mechanism, specifically mediated by AhR may be responsible for the
IDO-induced increase in fatty acid oxidation.
Activation of AhR increases the expression of the
transcription factor peroxisome proliferator-activated receptor-α
(PPAR-α) (30), which is known to
upregulate the expression of all CPT1 isoenzymes (31–33). Therefore, the expression levels of
the isoenzymes CPT1A, CPT1B, and CPT1C were evaluated in the
present experimental systems. In MLR-derived CD4+ T-cells, IDO
increased the expression of all CPT1 isoenzymes, whereas, in
activated isolated CD4+ T-cells, tryptophanol did not alter their
expression. These results indicate that IDO may have increased CPT1
expression through activation of AhR. It is likely that through
this molecular mechanism IDO increases free fatty acid consumption
in CD4+ T-cells.
The present results support that free fatty acid
oxidation compensates for decreased glucose and glutamine
utilization in CD4+ T-cells providing the required energy for cell
survival and proliferation. On the other hand, IDO-induced and
AhR-mediated increase in free fatty acid oxidation may enhance CD4+
T-cell differentiation to Tregs. Inhibition of fatty acid oxidation
with the CPT1 inhibitor etomoxir abrogates differentiation of naïve
CD4+ T-cells to Tregs (12).
Notably, activation of AhR promotes Treg differentiation (6), activation of PPAR-α ameliorates the
course of autoimmune diseases (34–37), and PPAR-α activity is required for
Treg differentiation and suppressive function (38–40). Finally, it is possible that a
portion of free fatty acids is consumed by the expanded CD4+
T-cells as building blocks, since IDO by activating GCN2K decreases
fatty acid synthesis (4), which
is required for T-cell proliferation (41). This, along with the decreased
apoptosis, may contribute to the increased cell proliferation index
when MLRs were performed in a free fatty acid-containing culture
medium as observed in the present study.
Collectively, the present results propose the
following model regarding the effect of IDO on the utilization of
the main energy sources in activated CD4+ T-cells. As depicted in
Fig. 7, in the immune response
microenvironment, IDO, by degrading L-tryptophan along the
kynurenine pathway, activates GCN2K and AhR. Activation of GCN2K,
possibly by upregulating p53 and downregulating c-Myc, leads to
decreased glycolysis and glutaminolysis, two main energy sources in
activated CD4+ T-cells, resulting in reduced ATP production. The
latter activates AMPK, which phosphorylates and inactivates ACC2
resulting in decreased production of the CPT1 inhibitor
malonyl-CoA. In parallel, activation of AhR, possibly by
upregulating PPARα, increases the expression of all CPT1
isoenzymes. Since CPT1 controls free fatty acid oxidation, these
IDO-induced alterations promote free fatty acid oxidation as an
alternative fuel for ATP production, supplying the required energy
for CD4+ T-cell survival and proliferation.
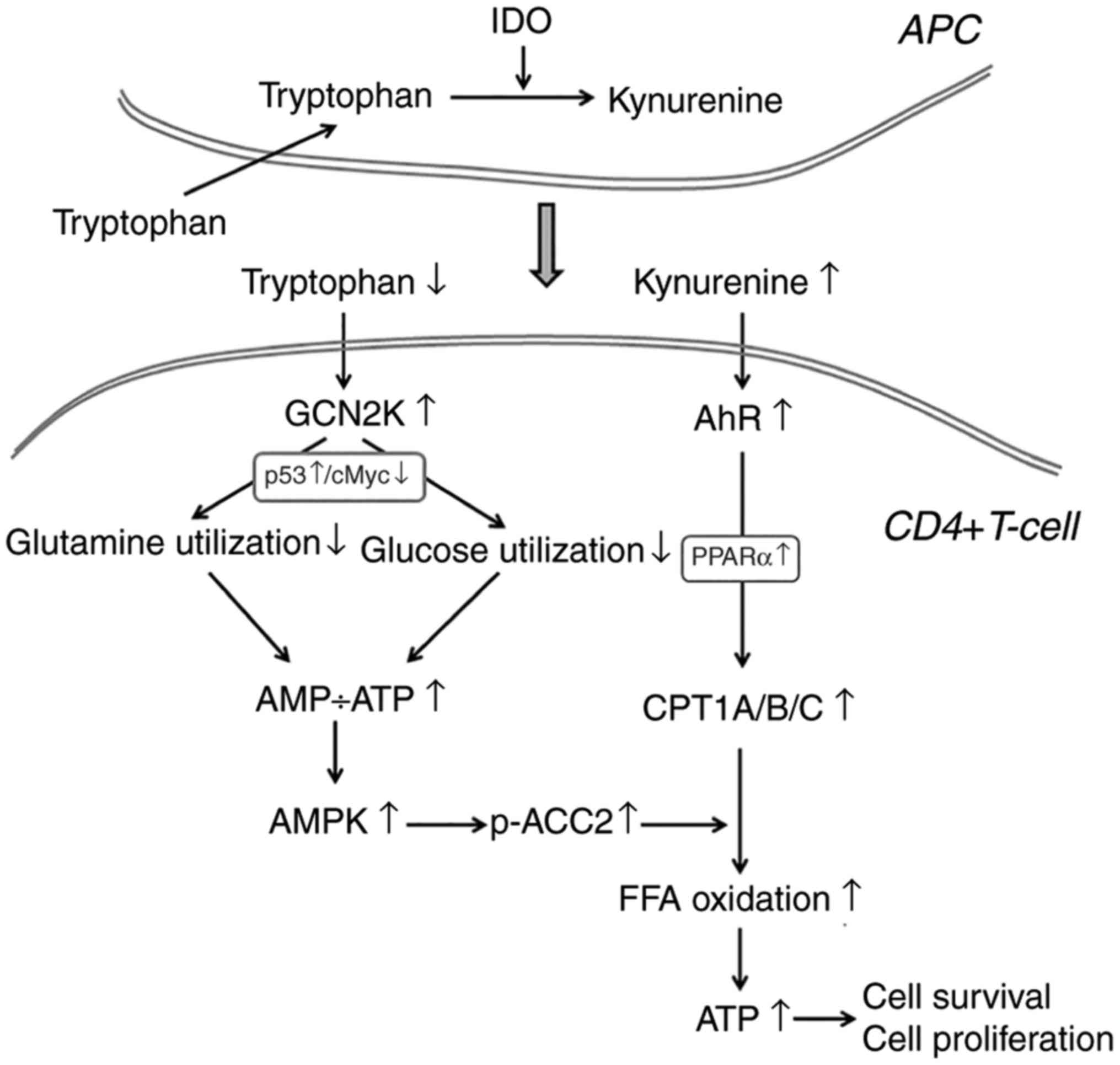 | Figure 7A proposed model regarding the effect
of IDO on the utilization of the main energy sources in activated
CD4+ T-cells. In the immune response microenvironment, IDO, by
degrading L-tryptophan along the kynurenine pathway, activates
GCN2K and AhR. Activation of GCN2K, possibly by upregulating p53
and downregulating c-Myc, leads to decreased utilization of glucose
and glutamine, two pivotal sources of energy in activated CD4+
T-cells, resulting in reduced ATP production. The latter activates
AMPK, which phosphorylates and inactivates ACC2, resulting in
decreased production of the CPT1 inhibitor malonyl-CoA. In
parallel, activation of AhR, possibly by upregulating PPARα,
increases the expression of all CPT1 isoenzymes. Since CPT1
controls free fatty acid oxidation, these IDO-induced alterations
promote free fatty acid oxidation as an alternative fuel for ATP
production, supplying the required energy for CD4+ T-cell survival
and proliferation. The steps in the present model that are based on
findings from previously published studies are noted in gray font.
IDO, indoleamine 2,3-dioxygenase; GCN2K, general control
nonderepressible 2 kinase; AhR, aryl hydrocarbon receptor; AMPK,
AMP-activated protein kinase; ACC2, acetyl-CoA carboxylase 2; CPT1,
carnitine palmitoyltransferase I; PPARα, peroxisome
proliferator-activated receptor-α. |
In conclusion, IDO decreases glycolysis and
glutaminolysis by activating GCN2K, while it increases free fatty
acid oxidation by activating AhR, providing the necessary energy
for CD4+ T-cell survival and proliferation. Thus, contrary to what
is generally hypothesized to date, in a normal environment that
contains fatty acids, the immunosuppressive effect of IDO cannot be
attributed to a decrease in CD4+ T-cell proliferation and
survival.
Acknowledgments
Not applicable.
References
|
1
|
Munn DH, Sharma MD, Baban B, Harding HP,
Zhang Y, Ron D and Mellor AL: GCN2 kinase in T cells mediates
proliferative arrest and anergy induction in response to
indoleamine 2,3-dioxygenase. Immunity. 22:633–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eleftheriadis T, Pissas G, Yiannaki E,
Markala D, Arampatzis S, Antoniadi G, Liakopoulos V and Stefanidis
I: Inhibition of indoleamine 2,3-dioxygenase in mixed lymphocyte
reaction affects glucose influx and enzymes involved in aerobic
glycolysis and glutaminolysis in alloreactive T-cells. Hum Immunol.
74:1501–1509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eleftheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 levels in alloreactive human T cells,
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Indoleamine 2,3-dioxygenase
depletes tryptophan, activates general control non-derepressible 2
kinase and down-regulates key enzymes involved in fatty acid
synthesis in primary human CD4+ T cells. Immunology.
146:292–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cobbold SP, Adams E, Farquhar CA, Nolan
KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D and Waldmann H:
Infectious tolerance via the consumption of essential amino acids
and mTOR signaling. Proc Natl Acad Sci USA. 106:12055–12060. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mezrich JD, Fechner JH, Zhang X, Johnson
BP, Burlingham WJ and Bradfield CA: An interaction between
kynurenine and the aryl hydrocarbon receptor can generate
regulatory T cells. J Immunol. 185:3190–3198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eleftheriadis T, Pissas G, Sounidaki M,
Tsogka K, Antoniadis N, Antoniadi G, Liakopoulos V and Stefanidis
I: Indoleamine 2,3-dioxygenase, by degrading L-tryptophan, enhances
carnitine palmitoyltransferase I activity and fatty acid oxidation,
and exerts fatty acid-dependent effects in human alloreactive
CD4x T-cells. Int J Mol Med. 38:1605–1613. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Curti A, Trabanelli S, Salvestrini V,
Baccarani M and Lemoli RM: The role of indoleamine 2,3-dioxygenase
in the induction of immune tolerance: Focus on hematology. Blood.
113:2394–2401. 2009. View Article : Google Scholar
|
|
9
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J and Green DR: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caro-Maldonado A, Gerriets VA and Rathmell
JC: Matched and mismatched metabolic fuels in lymphocyte function.
Semin Immunol. 24:405–413. 2012. View Article : Google Scholar
|
|
11
|
Macintyre AN, Gerriets VA, Nichols AG,
Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen
BJ, Hale LP and Rathmell JC: The glucose transporter Glut1 is
selectively essential for CD4 T cell activation and effector
function. Cell Metab. 20:61–72. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Michalek RD, Gerriets VA, Jacobs SR,
Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG and
Rathmell JC: Cutting edge: Distinct glycolytic and lipid oxidative
metabolic programs are essential for effector and regulatory
CD4+ T cell subsets. J Immunol. 186:3299–3303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eleftheriadis T, Pissas G, Antoniadi G,
Tsogca K, Sounidaki M, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase downregulates T-cell receptor complex ζ-chain and
c-Myc, and reduces proliferation, lactate dehydrogenase levels and
mitochondrial glutaminase in human T-cells. Mol Med Rep.
13:925–932. 2015. View Article : Google Scholar
|
|
14
|
Gurtner GJ, Newberry RD, Schloemann SR,
McDonald KG and Stenson WF: Inhibition of indoleamine
2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in
mice. Gastroenterology. 125:1762–1773. 2003. View Article : Google Scholar
|
|
15
|
Kwidzinski E, Bunse J, Aktas O, Richter D,
Mutlu L, Zipp F, Nitsch R and Bechmann I: Indolamine
2,3-dioxygenase is expressed in the CNS and down-regulates
autoimmune inflammation. FASEB J. 19:1347–1349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alexander AM, Crawford M, Bertera S,
Rudert WA, Takikawa O, Robbins PD and Trucco M: Indoleamine
2,3-dioxygenase expression in transplanted NOD Islets prolongs
graft survival after adoptive transfer of diabetogenic splenocytes.
Diabetes. 51:356–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Tredget EE, Ghaffari A, Lin X,
Kilani RT and Ghahary A: Local expression of indoleamine
2,3-dioxygenase protects engraftment of xenogeneic skin substitute.
J Invest Dermatol. 126:128–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Munn DH, Zhou M, Attwood JT, Bondarev I,
Conway SJ, Marshall B, Brown C and Mellor AL: Prevention of
allogeneic fetal rejection by tryptophan catabolism. Science.
281:1191–1193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Munn DH and Mellor AL: Indoleamine
2,3-dioxygenase and tumor-induced tolerance. J Clin Invest.
117:1147–1154. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sato T, Deiwick A, Raddatz G, Koyama K and
Schlitt HJ: Interactions of allogeneic human mononuclear cells in
the two-way mixed leucocyte culture (MLC): Influence of cell
numbers, subpopulations and cyclosporin. Clin Exp Immunol.
115:301–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Staiger K, Staiger H, Weigert C, Haas C,
Häring HU and Kellerer M: Saturated, but not unsaturated, fatty
acids induce apoptosis of human coronary artery endothelial cells
via nuclear factor-kappaB activation. Diabetes. 55:3121–3126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castilho BA, Shanmugam R, Silva RC, Ramesh
R, Himme BM and Sattlegger E: Keeping the eIF2 alpha kinase Gcn2 in
check. Biochim Biophys Acta. 1843:1948–1968. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gallinetti J, Harputlugil E and Mitchell
JR: Amino acid sensing in dietary-restriction-mediated longevity:
Roles of signal-transducing kinases GCN2 and TOR. Biochem J.
449:1–10. 2013. View Article : Google Scholar :
|
|
26
|
Ma Q: Induction of CYP1A1. The AhR/DRE
paradigm: Transcription, receptor regulation, and expanding
biological roles. Curr Drug Metab. 2:149–164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schreurs M, Kuipers F and van der Leij FR:
Regulatory enzymes of mitochondrial beta-oxidation as targets for
treatment of the metabolic syndrome. Obes Rev. 11:380–388. 2010.
View Article : Google Scholar
|
|
30
|
Wang C, Xu CX, Krager SL, Bottum KM, Liao
DF and Tischkau SA: Aryl hydrocarbon receptor deficiency enhances
insulin sensitivity and reduces PPAR-α pathway activity in mice.
Environ Health Perspect. 119:1739–1744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song S, Attia RR, Connaughton S, Niesen
MI, Ness GC, Elam MB, Hori RT, Cook GA and Park EA: Peroxisome
proliferator activated receptor alpha (PPARalpha) and PPAR gamma
coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA
(CPT-1A) via independent gene elements. Mol Cell Endocrinol.
325:54–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kok BP, Dyck JR, Harris TE and Brindley
DN: Differential regulation of the expressions of the PGC-1α splice
variants, lipins, and PPARα in heart compared to liver. J Lipid
Res. 54:1662–1677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Y, Wang Y, Huang Y, Zeng H, Hu B,
Guan L, Zhang H, Yu AM, Johnson CH, Gonzalez FJ, et al: PPARα
regulates tumor cell proliferation and senescence via a novel
target gene carnitine palmitoyltransferase 1C. Carcinogenesis.
38:474–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dunn SE, Ousman SS, Sobel RA, Zuniga L,
Baranzini SE, Youssef S, Crowell A, Loh J, Oksenberg J and Steinman
L: Peroxisome proliferator-activated receptor (PPAR)alpha
expression in T cells mediates gender differences in development of
T cell-mediated autoimmunity. J Exp Med. 204:321–330. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JW, Bajwa PJ, Carson MJ, Jeske DR,
Cong Y, Elson CO, Lytle C and Straus DS: Fenofibrate represses
interleukin-17 and interferon-gamma expression and improves colitis
in interleukin-10-deficient mice. Gastroenterology. 133:108–123.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gocke AR, Hussain RZ, Yang Y, Peng H,
Weiner J, Ben LH, Drew PD, Stuve O, Lovett-Racke AE and Racke MK:
Transcriptional modulation of the immune response by peroxisome
proliferator-activated receptor-{alpha} agonists in autoimmune
disease. J Immunol. 182:4479–4487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Azuma YT, Nishiyama K, Matsuo Y, Kuwamura
M, Morioka A, Nakajima H and Takeuchi T: PPARα contributes to
colonic protection in mice with DSS-induced colitis. Int
Immunopharmacol. 10:1261–1267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim MS, Pyun HB and Hwang JK: Panduratin
A, an activator of PPAR-α/δ, suppresses the development of
oxazolone-induced atopic dermatitis-like symptoms in hairless mice.
Life Sci. 100:45–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lei J, Hasegawa H, Matsumoto T and
Yasukawa M: Peroxisome proliferator-activated receptor α and γ
agonists together with TGF-β convert human
CD4+CD25− T cells into functional Foxp3+
regulatory T cells. J Immunol. 185:7186–7198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hichami A, Yessoufou A, Ghiringhelli F,
Salvadori F, Moutairou K, Zwetyenga N and Khan NA: Peroxisome
proliferator-activated receptor alpha deficiency impairs regulatory
T cell functions: Possible application in the inhibition of
melanoma tumor growth in mice. Biochimie. 131:1–10. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Berod L, Friedrich C, Nandan A, Freitag J,
Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bähre H, et
al: De novo fatty acid synthesis controls the fate between
regulatory T and T helper 17 cells. Nat Med. 20:1327–1333. 2014.
View Article : Google Scholar : PubMed/NCBI
|