Introduction
Platelets are small, a nucleate blood cells, the
major role of which is in hemostasis and thrombosis owing to their
capacity to adhere to damaged blood vessels and to accumulate at
sites of injury (1). However,
platelets are also important contributors to thrombotic disorders,
including atherothrombosis, which are the final events complicating
cardiovascular diseases (2–4).
Upon vascular injury, platelets are exposed to the subendothelium,
and several agonists, including adenosine diphosphate (ADP) and
thrombin, are generated at the injury site, which can stimulate
platelet adhesion, activation and aggregation. Adherent, activated
platelets recruit additional platelets to the growing thrombus
(5,6). The uncontrolled progression of these
processes through a series of self-sustaining amplification loops
can initiate unrestrained platelet activation and aggregation, and
eventually lead to thromboembolic events (7,8).
It has been demonstrated clinically that the use of antiplatelet
agents to prevent and/or reverse platelet aggregation is a
successful strategy for the prevention of thrombosis (7,8).
However, due to their disturbance of the thromboregulatory balance,
existing antiplatelet drugs can cause severe side effects, the
majority of which limit the efficacy and safetyof these drugs.
Uncontrolled hemorrhage is the most frequent side effect of
antithrombotics/antiplatelets (9,10).
Therefore, understanding the molecular mechanisms of platelet
activation and identifying novel techniques for platelet inhibition
remain critically important.
Natural products remain a major resource and have
become increasingly important for novel drug identification. The
Callicarpa genus includes ~190 extant species (11). Among these, there are >10
medical herbs, and the majority of these have hemostasis-associated
usage (11). Callicarpa
nudiflora (C. nudiflora) Hook, which is one of the
medical herbs of Callicarpa with a long history, is used for
eliminating stasis in order to subdue swelling and hemostasis
(11). According to traditional
Chinese medicine theory, eliminating stasis to subdue swelling is
similar to the antithrombotic effect. Led by these concepts, the
present study hypothesized that antiplatelet activities may
contribute to the traditional usage of this plant. In our previous
study, hundreds of constituents were screened, including several
derivatives of luteolin. It was found that two novel triterpenoids,
extracted from the leaves of C. nudiflora, showed inhibitory
effects on ADP-induced platelet activation (12,13). In the present study, it was shown
that one of the derivatives of luteolin,
1,6-di-O-caffeoyl-β-D-glucopyranoside (LGP) exhibited potent
inhibitory effects on platelet activation, and it was demonstrated
that the effects of this natural compound may be mediated by dual
receptor antagonism on P2Y12 receptor and thromboxane
A2 (TXA2) receptor (TP).
Materials and methods
Drugs and chemicals
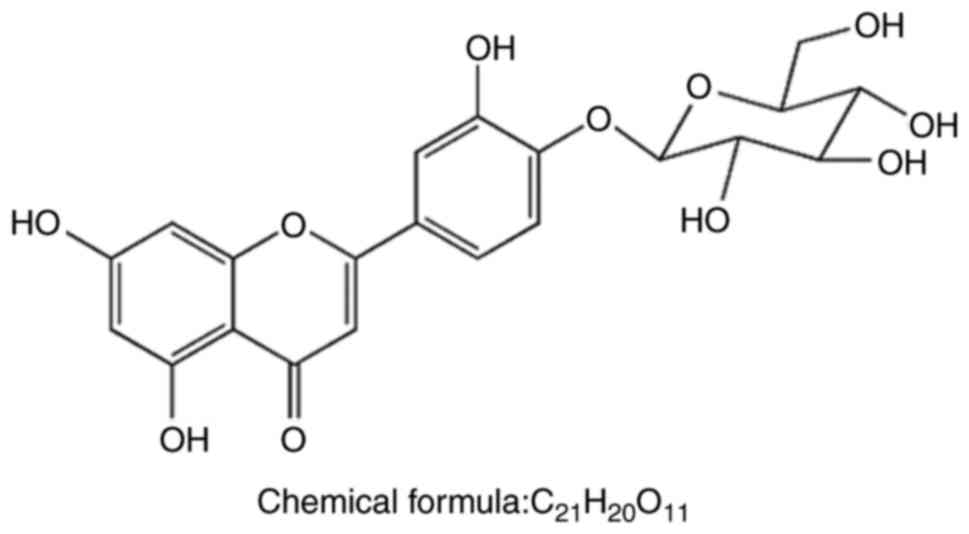
Luteolin-4′-O-β-D-glucopyranoside (LGP) was isolated
from the leaves of C. nudiflora Hook and its molecular
structure is shown in Fig. 1. The
purity of LGP was ≥95%, as determined by high-performance liquid
chromatography. A stock solution was prepared by dissolving the LGP
in 100% dimethyl sulfoxide (DMSO) and was used throughout the
investigation. The final DMSO concentration did not exceed 0.1%.
ADP, arachidonic acid (AA), ticagrelor (an antagonist of the
P2Y12 receptor) and BM-531 (an antagonist of the TP
receptor), were purchased from Sigma-Aldrich; EMD Millipore
(Billerica, MA, USA). U46619 was the product of Tocris Bioscience
(Bristol, UK). [3H]-2-methylthioadenosine diphosphate
([3H]-2-MeS-ADP) and [3H] SQ-29548 were
purchased from GE Healthcare Life Sciences (Chalfont, UK) and
PerkinElmer, Inc. (Waltham, MA, USA), respectively.
Animals
A total of 80 female Sprague-Dawley rats (aged 6–8
weeks old and weighing 180–220 g) were obtained from Vital River
Laboratories (Beijing, China) and maintained under pathogen-free
conditions in the Animal Center of Jiangxi University of
Traditional Chinese Medicine (Nanchang, China). All the animals
were maintained in a 12 h light/dark cycle at room temperature
(25±2°C) in 60% humidity. The animals were allowed water ad
libitum and were fed a standard laboratory diet. The protocol
used for animal experiments (JZAEC-2016-0031) was approved by the
Animal Ethics Committees of Jiangxi University of Traditional
Chinese Medicine, and all animal experiments were performed in
strict accordance with the requirements of this protocol.
Preparation of rat platelets
Blood was collected from the abdominal aorta of
anesthetized rats into a vacuum blood collection tube, which
allowed 10% blood volume with 3.8% sodium citrate as anticoagulant.
The citrated blood was then centrifuged (Allegra™ X-12R centrifuge;
Beckman Coulter, Inc., Brea, CA, USA) at 110 × g for 15 min at 4°C
to obtain platelet-rich plasma (PRP), and the quantity of platelets
in the PRP was determined using the automatic blood cell analyzer
(HEMAVET 950FS; Drew Scientific, Miami Lakes, FL, USA).
Platelet-poor plasma (PPP) was obtained by a second centrifugation
of the remaining blood (1,000 × g, 10 min, 4°C). The PRP was
adjusted to a platelet count of 400×109 platelets/l by
diluting in PPP.
Platelet aggregation
Platelet aggregation in 96-well plates was measured
using a modified light transmission method (14,15). Briefly, the PRP
(400×109 platelets/l) was incubated with 40, 80 and 160
μM of LGP, antagonists (positive controls) or dissolvent for
15 min at 37°C, respectively. The optical density (OD) was then
determined at 595 nm and marked as OD1. Platelet
aggregation was induced by the following agonists: ADP (10
μM), U46619 (1 μM) or AA (600 μM), and OD
(OD2) was determined again at 595 nm every 30 sec for 15
min, with 15 sec incubation and 15 sec shaking between readings. In
addition, the OD value at 595 nm was determined for the same volume
of PPP, and was marked as OD3. All experiments were
performed at least three times. The percentage of aggregation was
calculated using the absorbance of PRP without agonist as 0%
aggregation and the absorbance of PPP as 100% aggregation. The
relative aggregation was expressed using the following formula:
Relative aggregation
(%)=[(OD1−OD2)/(OD1−OD3)]
×100.
Activated αIIbβ3
integrin abundance
αIIbβ3 integrin is expressed
on the surface of platelets, which reflects platelet activation or
secretion from platelet granules. This was determined by the
measurement of fluorescent agent-labeled antibody binding, as
described previously (16,17).
For measurement of the expression of αIIbβ3
integrin, briefly, 50 μl activated platelets
(400×109 platelets/l) were pretreated for 15 min with
DMSO or the indicated concentrations (20, 40, 80 and 160 μM)
of LGP, and then fixed for 30 min in 0.5% paraformaldehyde.
Following washing once in incubation buffer, the fixed platelets
were added into 96-well plates, and incubated with Oregon
Green-labeled fibrinogen (Molecular Probes; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 15 min at room temperature
without shaking. The total fluorescence was determined by measuring
the fluorescence of the plate on a multi-label counter (VICTOR3™;
PerkinElmer, Inc.). Subsequently, the plates were centrifuged at
1,000 × g for 10 min at 4°C to remove platelets from the
supernatant. The supernatants were transferred to separate plates
and the fluorescence was determined (background or nonspecific).
Specific Oregon Green binding was determined by subtracting the
background fluorescence from the total fluorescence.
Measurement of serotonin (5-HT)
secretion
5-HT secretion was determined using a Serotonin
ELISA kit (Abcam, Cambridge, MA, USA; cat. no. ab133053) according
to the manufacturer’s protocol. The assay procedure is based on the
competition between an alkaline phosphatase-conjugated 5-HT
(supplied) and a non-labeled antigen (5-HT extracted from PRP) for
a fixed number of antibody binding sites on the micro-titer plate.
First, a curve of the OD405 of 5-HT, compared with its
concentration in the standard wells, was plotted. Subsequently, 50
μl platelets (400×109 platelets/l) were treated
with DMSO or the indicated concentrations (20, 40, 80 and 160
μM) LGP for 15 min, followed by 3 min incubation with
agonists (10 μM ADP or 1 μM U46619). The reaction was
terminated by snap freezing. Following thawing at room temperature,
the samples were centrifuged at 3,000 × g for 10 min at 4°C. The
supernatants were used for the measurement of 5-HT release. By
comparing the absorbance of the samples with the standard curve,
the 5-HT concentration in the unknown samples was determined, with
data representative of at least five independent experiments.
Measurement of TXA2
synthesis
In the present study, TXB2, the stable
metabolite of TXA2, was measured to reflect the level of
TXA2. PRP (400×109 platelets/l) was
pretreated with DMSO or various concentrations of LGP (20, 40, 80
and 160 μM) for 15 min at 37°C, and was stimulated with ADP
(10 μM) or U46619 (1 μM) at 37°C for 3 min whilst
stirring. The reaction was also terminated by snap freezing.
Following thawing at room temperature and centrifuging at 3,000 × g
for 10 min at 4°C, the supernatants were diluted (1:20) with the
assay buffer in the TXB2 ELISA kit (Cayman Chemical
Company, Ann Arbor, MI, USA). TXB2 was measured
according to the manufacturer’s protocol.
Western blot analysis
PRP (400×109 platelets/l), pretreated
with DMSO or various concentrations (40, 80 and 160 μM) LGP
for 15 min, were stimulated for 3 min with agonists, and the
reaction was terminated by rapid freezing of the sample in a dry
ice-ethanol bath. Following thawing at room temperature, the
samples were centrifuged at 3,000 × g for 10 min at 4°C. The
platelets were rinsed twice with PBS, and total proteins were
extracted with lysis buffer. Aliquots of each platelet lysate
containing equal quantities of protein (ranging between 500 and 750
μg between experiments) were added to SDS-PAGE gels (ranging
between 8 and 12%), and then transferred onto hybond nitroblotting
membranes and subjected to western blot analysis. Membranes were
blocked using 5% non-fat dried milk for 1 h at room temperature and
subsequently incubated with primary antibodies overnight at 4°C.
Following washing with 0.5% TBST three times, the membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies for 2 h at room temperature. The immunoreactive bands
were detected using an enhanced chemiluminescence kit (EMD
Millipore). β-actin (1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; cat. no. SC-130656) served as an internal control.
The signal intensities of the bands of interest were quantified and
normalized to β-actin using the Image-Pro Plus software version 6.0
(Media Cybernetics, Inc., Rockville, MD, USA). The primary
antibodies used in the present study were as follows: Anti-Rho
guanine nucleotide exchange factor 1 (ARHGEF1) antibody (1:1,000;
Abcam, cat. no. ab220892), anti-Ras homolog family member A (RhoA)
antibody (1:1,000; Abcam; cat. no. ab54835), anti-RhoA antibody
(1:1,000; phospho S188; Abcam; cat. no. ab41435),
anti-Rho-associated kinase 1 (ROCK1) antibody (1:1,000; Abcam; cat.
no. ab45171), anti-ROCK1 (phospho T455+S456) antibody (1:1,000;
Abcam; cat. no. ab203273), anti-phosphoinositide 3-kinase (PI3K)
p85 (phospho Y607) antibody (1:1,000; Abcam; cat. no. ab182651),
anti-pan-AKT antibody (1:1,000; Abcam; cat. no. ab8805),
anti-pan-AKT (phospho T308) antibody (1:1,000; Abcam; cat. no.
ab38449), anti-AKT1 (phospho S473) antibody (1:1,000; Abcam; cat.
no. ab81283) and anti-glycogen synthase kinase 3β (GSK3β) antibody
(1:1,000; phospho S9; Abcam; cat. no. ab75814). The secondary
antibodies used in the present study were as follows: Goat
polyclonal secondary antibody to mouse IgG (1:5,000; Abcam; cat.
no. ab6789) and goat anti-rabbit IgG (1:5,000; Abcam; cat. no.
ab6721).
Receptor-binding assay
The effects of LGP on P2Y12 ADP receptor
binding were determined by the binding of [3H]-2-MeS-ADP
to rat platelets with a filter technique to separate the free from
bound [3H]-2-MeS-ADP. [3H]SQ-29548
(PerkinElmer, Inc.) was also used in to assess the effects of LGP
on TXA2 receptor binding activities. Briefly, PRP
(1×109 platelets/ml) was incubated with
[3H]SQ-29548 (40 nM final concentration) in a total of
400 μl Tyrode’s buffer (pH 7.2) for 30 min at room
temperature. Subsequently, indicated concentrations of LGP were
added and incubated for 40 min at room temperature to compete
binding between agonists and their receptors. The binding assays
were terminated by rapid filtration on Packard GF-B filters
(Packard Instrument Co., Inc., Meriden, CT, USA). The filters were
then placed in plastic scintillation vials containing an
emulsion-type scintillation mixture (4 ml) and the radioactivity,
representing the binding of [3H]SQ-29548 to TP receptor
(B), was detected by Tri-Carb® Liquid Scintillation
(PerkinElmer, Inc.). The radioactivity of [3H]SQ-29548
(40 nM final concentration) in DMSO-treated platelets served as the
total binding (Bt) of [3H]SQ-29548 to the TP receptor.
Non-specific binding (Bns) was defined as the total radioactivity
measured in the presence of 100 μM (final concentration)
unlabeled SQ-29548. The specific binding rate (Bs) of
[3H]SQ-29548 to the TP receptor was calculated using the
following formula: Bs = (B − Bns)/Bt × 100.
For the P2Y12 ADP receptor binding assay,
a similar procedure to the TP receptor binding assay was used. PRP
(1×109 platelets/ml) was prepared, as previously
described, and incubated with [3H]-2-MeS-ADP (5 nM final
concentration) in a total of 400 μl Tyrode’s buffer (pH 7.2)
for 30 min at room temperature. Subsequently, indicated
concentrations of LGP, DMSO or 2-MeS-ADP (5 μM, final
concentration) were added for an additional 40 min at room
temperature. Following filtration on Packard GF-B filters,
radioactivity was detected by Tri-Carb® Liquid
Scintillation (PerkinElmer, Inc.) and the specific binding rate of
[3H]-2-MeS-ADP to the P2Y12 receptor was
calculated.
Data presentation and statistical
analysis
Data are presented as the mean ± standard error of
the mean; n represents the number of independent experiments.
Statistical significance was determined using one-way analysis of
variance. Dose-response curves were generated using GraphPad Prism
software (version 4.0; GraphPad Software, Inc., La Jolla, CA, USA).
The IC50 value for each agent was determined from three
different concentrations of the agent using Schild analysis using
GraphPad Prism software. P<0.05 was considered to indicate a
statistically significant difference.
Results
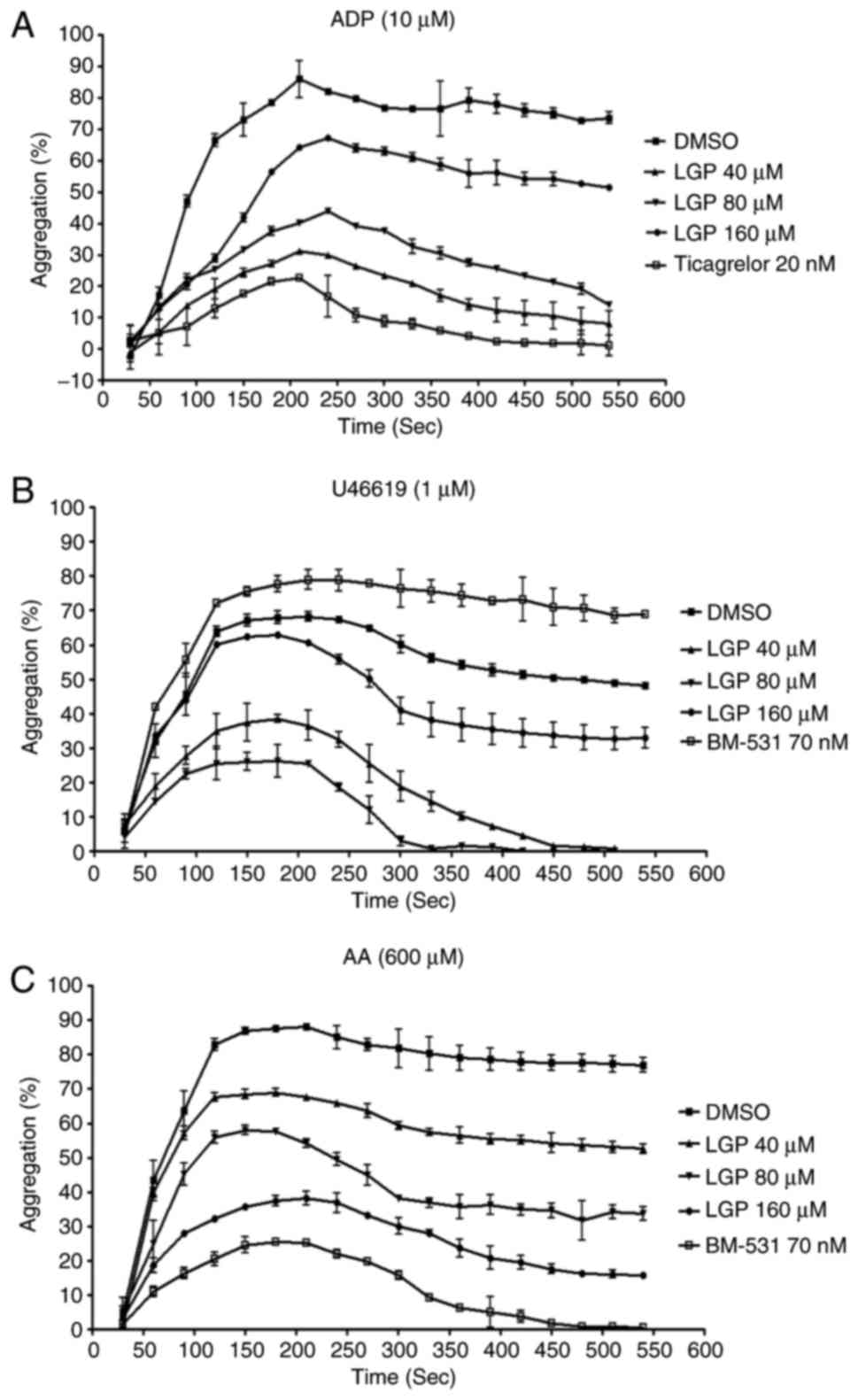
LGP inhibits ADP-, U46619- and AA-induced
platelet aggregation
The initial experiment defined the effects of LGP on
platelet aggregation induced by several agonists. Rat platelets
were isolated and platelet aggregation was observed. The platelets
were pretreated with LGP for 15 min and were incubated with 10
μM ADP. As shown in Fig.
2A, 10 μM ADP (to PRP) produced typical aggregation
curves. Ticagrelor and LGP significantly inhibited aggregation in a
dose-dependent manner, and the IC50 value of LGP at 540
sec was 74.9±1.6 μM. Similar results were obtained when
platelet aggregation was induced by U46619 (1 μM) and AA
(600 μM). LGP exhibited concentration-dependent inhibition
of aggregation induced by U46619 and AA with IC50 values
of 61.7±1.2 and 81.7±1.1 μM at 540 sec, respectively
(Fig. 2B and C).
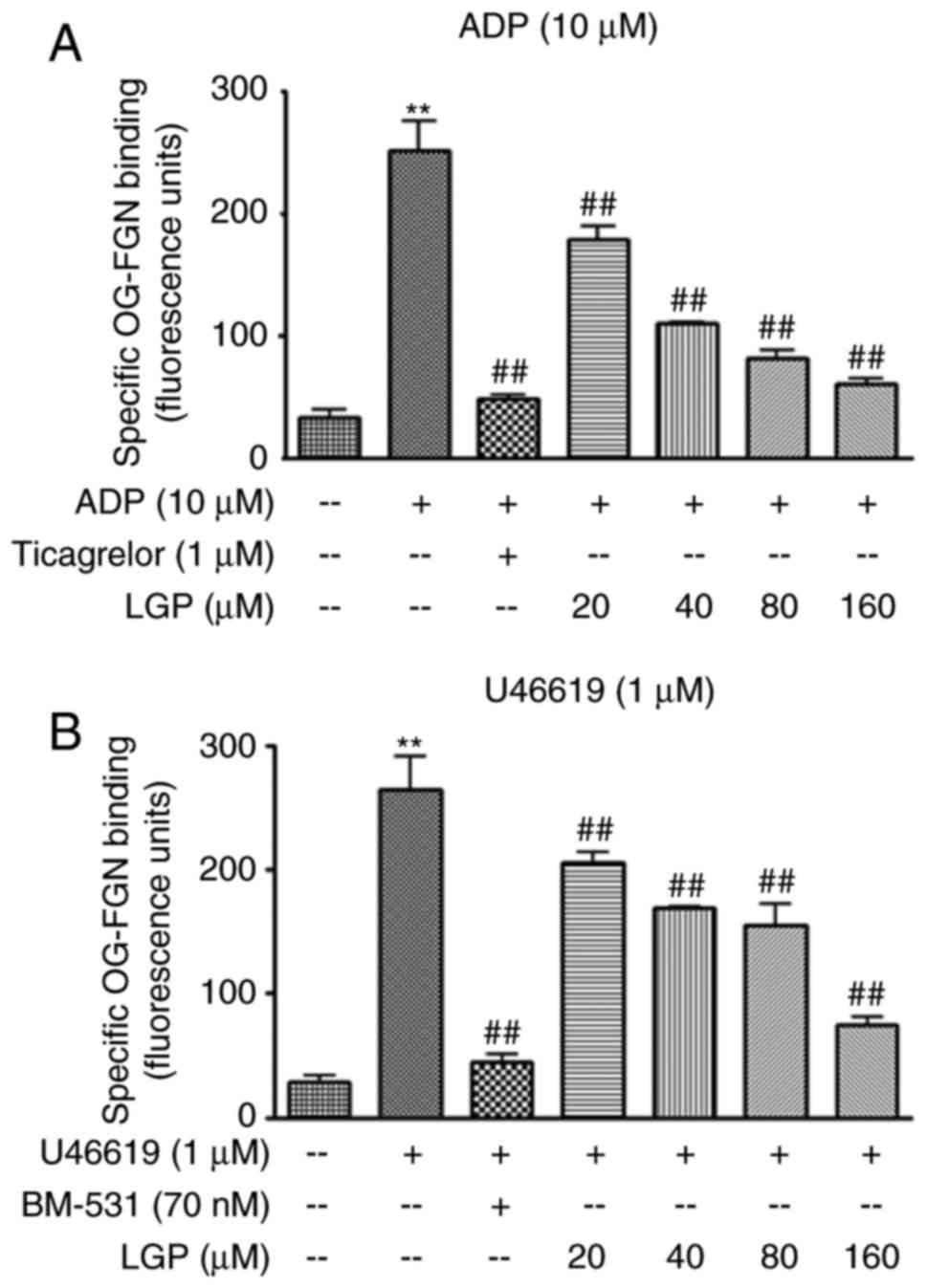
LGP suppresses ADP and U46619-mediated
αIIbβ3 integrin activation in rat
platelets
It is widely accepted that integrin
αIIbβ3-mediated outside-in signaling is the
most important amplifier of platelet activation. To confirm the
effects of LGP on outside-in signal transduction, the active
integrin αIIbβ3 on the platelet surface was
assessed by the measurement of fibrinogen binding. As shown in
Fig. 3A, the level of integrin
αIIbβ3 was negligible at the surface of
resting platelets. There was a sharp increase in the level of
integrin αIIbβ3 following ADP (10 μM)
treatment, and a significant attenuation in the presence of LGP and
the positive control ticagrelor. Similarly, the level of integrin
αIIbβ3 was significantly increased by
treatment with U46619 (1 μM). Again, the effect was
significantly inhibited in the presence of LGP and BM-531 (Fig. 3B). These data indicated that LGP
inhibited the activation of integrin αIIbβ3
in a concentration-dependent manner when the platelets were
stimulated by ADP or U46619. These results are compatible with the
results of the aggregation assay.
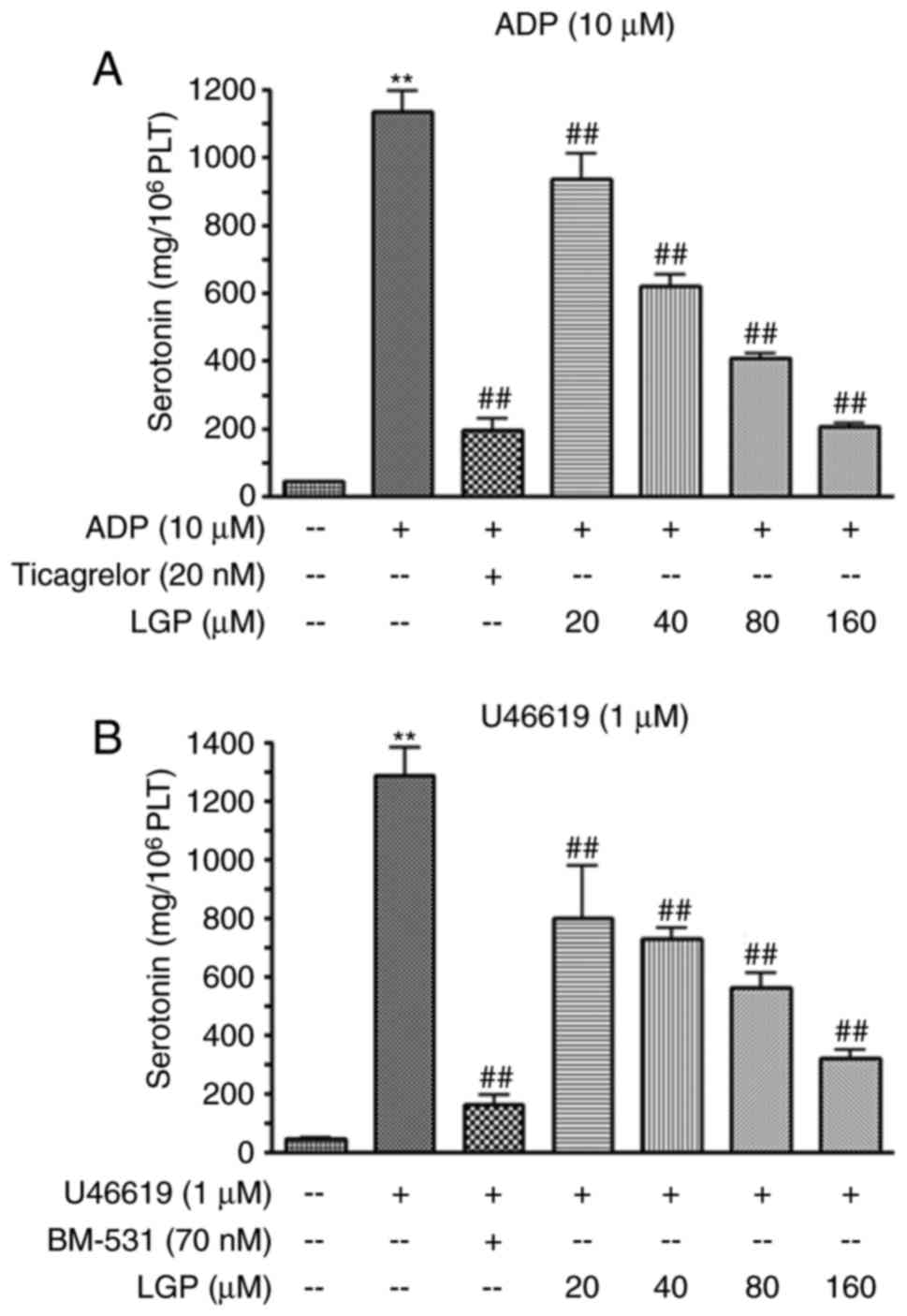
LGP inhibits5-HTrelease stimulated by ADP
and U46619
The platelets pretreated by LGP were incubated with
agonists to activate platelets, and the content of 5-HT in
supernatants was measured using a Serotonin ELISA kit. As shown in
Fig. 4A, the level of serotonin
was low when the platelets were treated with vehicle, whereas ADP
(10 μM) treatment induced a sharp increase in the level of
5-HT. The positive control (ticagrelor) significantly suppressed
the increase induced by ADP. LGP also caused a significant
reduction in the release of serotonin in a concentration-dependent
manner, with the inhibitory percentages of 22.55, 38.04, 47.71 and
65.95%, respectively. Similarly, LGP decreased the release of 5-HT
stimulated by U46619 (1 μM) in a concentration-dependent
manner (Fig. 4B).
LGP inhibits ADP and U46619-induced
TXA2 synthesis
TXA2, which is produced by activated
platelets, serves to promote further platelet activation by binding
to TP receptor. The present study examined the effects of LGP on
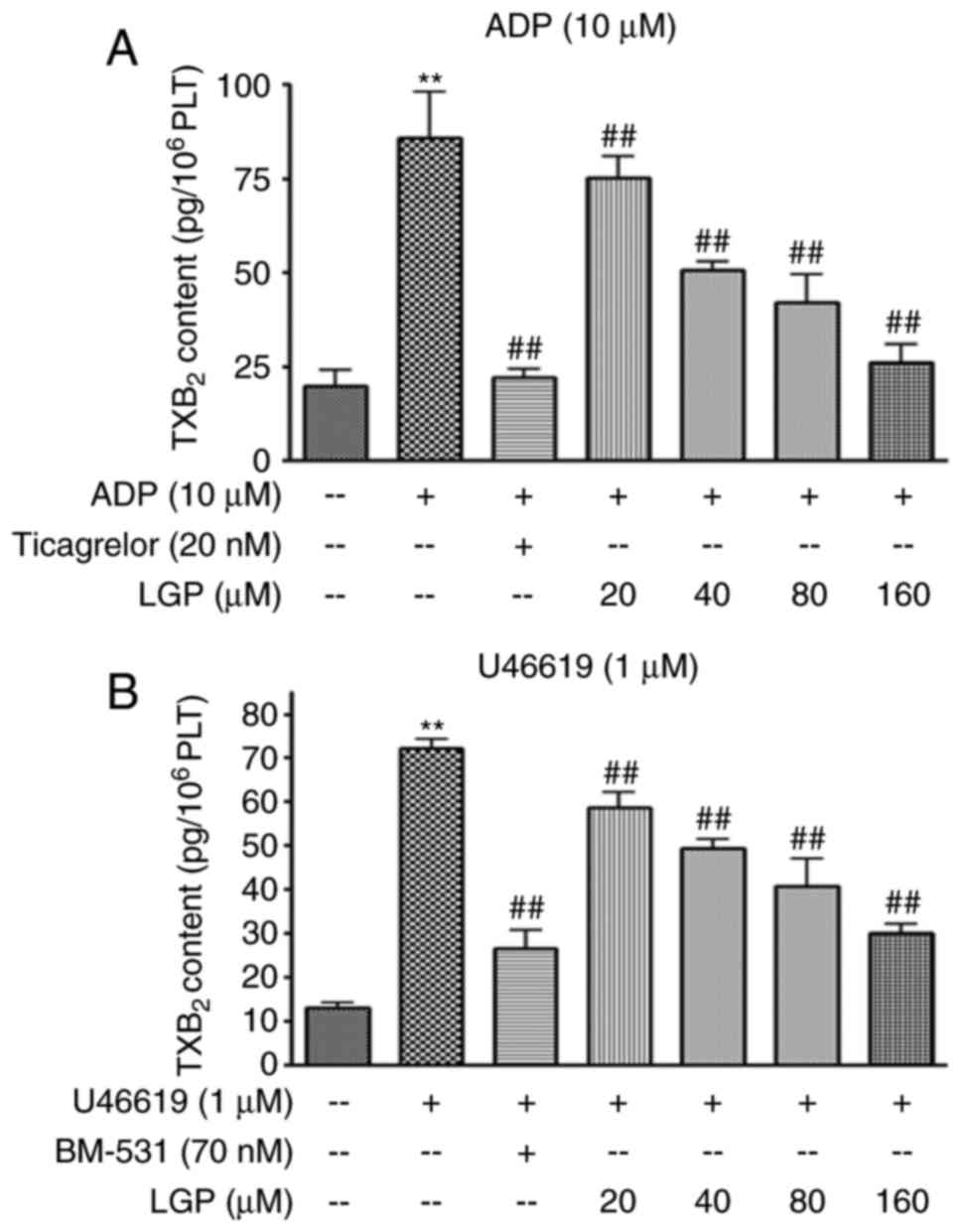
the content ofTXB2 in platelets treated with ADP and
U46619. As shown in Fig. 5A, ADP
markedly stimulated TXB2 release, whereas LGP and
ticagrelor caused a significant inhibition in the formation of
TXB2 in a dose-dependent manner. Similar results were
obtained with platelet activatorU46619 (Fig. 5B).
Effects of LGP on RhoA signaling induced
by U46619
One previous study with inhibitors and/or genetic
manipulations has demonstrated that RhoA signaling contributes to
TXA2-induced platelet activation by binding the TP
receptor (18). In order to
determine whether TP-mediated signal transduction is involved in
the inhibitory effects of LGP on platelet activation, the present
study examined the effects of LGP on the activation of RhoA
signaling transducers. As shown in Fig. 6A and B, LGP significantly reduced
the expression of phospho-RhoA in the presence of 1 μM
U46619, however, there was no significant change in the expression
of RhoA. Furthermore, it was found that the expression of ARHGEF1
(p115RhoGEF), an activator of RhoA, and phospho-ROCK1, an effecter
of RhoA, were decreased by LGP in a dose-dependent manner.
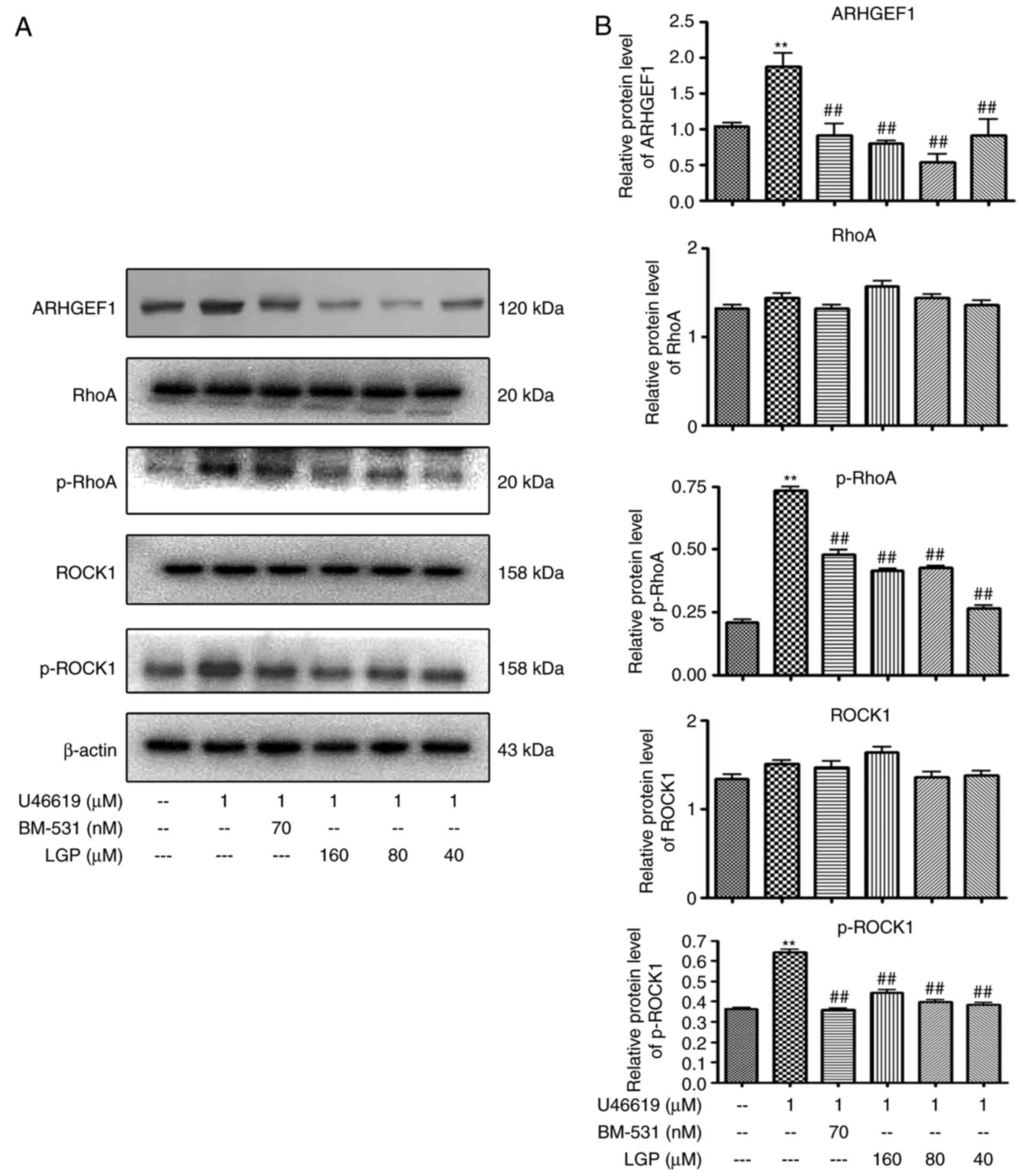 | Figure 6Effects of LGP on U46619-induced RhoA
signaling. (A) Western blot analysis of protein lysates with (B)
densitometric analysis of electrophoretic bands. Each assay was
performed in triplicate. **P<0.01, compared with
control; ##P<0.01, compared with U46619. Vehicle
(DMSO) was used as a control, and BM-531 served as the
positivecontrol. LGP, luteolin-4′-O-β-D-glucopyranoside;
ADP, adenosine diphosphate; RhoA, Ras homolog family member A;
ROCK1, Rho-associated kinase 1; ARHGEF1, Rho, guanine nucleotide
exchange factor 1; p-, phosphorylated. |
Effects of LGP on regulating
PI3K/Akt/GSK3β signal transduction stimulated by ADP
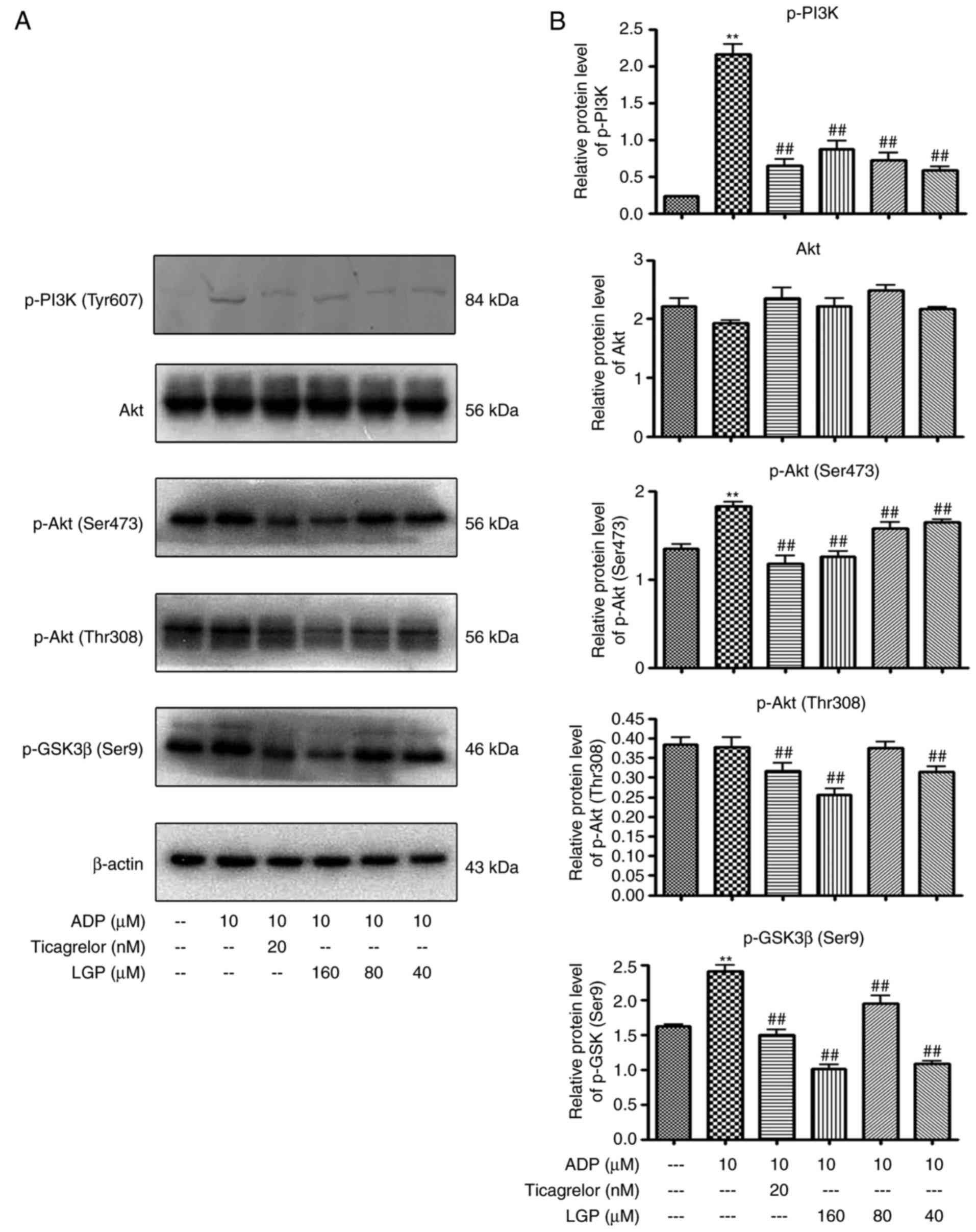
Subsequently, the present study examined the effects
of LGP on PI3K/Akt/GSK3β signal transduction stimulated by ADP.
Western blot analysis with platelet lysates revealed that
pre-incubation of the platelets with LGP (40, 80 and 160 μM)
attenuated the expression of phospo-PI3K in the presence of ADP.
Consistently, the phosphorylation of Akt and GSK3β were also
suppressed by LGP (40, 80 and 160 μM; Fig. 7A and B).
Effects of LGP on platelet
P2Y12 and TP receptor binding of
[3H]-2-MeS-ADP and [3H] SQ-29548
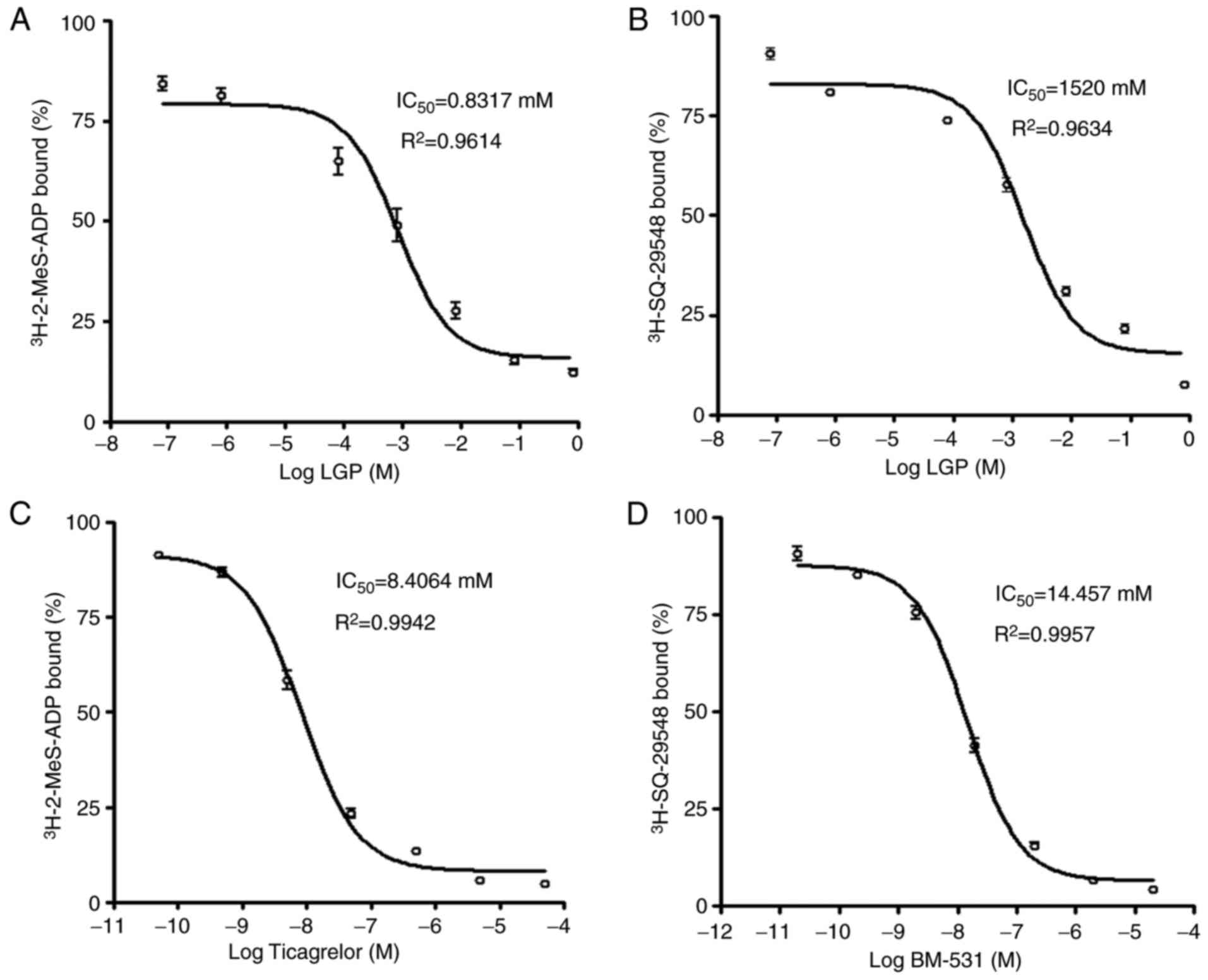
In order to further define whether the LGP-mediated
inhibitory effects on platelet activation are due to antagonism at
P2Y12 and TP receptors, the present study performed a
radiolabeled ligand binding assay using [3H]-2-MeS-ADP
and [3H]SQ-29548. LGP inhibited the binding of
[3H]-2-MeS-ADP to rat platelet membranes with
Ki=0.8317 mM as shown in Fig. 8A. LGP also exhibited apparent
competing effects on the TP receptor, which displaced
[3H] SQ-29548, a high affinity ligand of TP receptor
from rat platelet membranes with Ki=1.520 mM (Fig. 8B). In addition, ticagrelor, a
selective P2Y12 receptor antagonist, and BM-531, a
selective TP receptor antagonist, displaced
[3H]-2-MeS-ADP and [3H]SQ-29548 from their
respective receptors at concentrations in the nanomolar range
(Fig. 8C and D). From these data,
the dose-dependent displacement of [3H]-2-MeS-ADP and
[3H] SQ-29548 from their receptors by LGP was observed
in rat platelets; however, compared with the effects of selective
antagonists of these receptors, the Ki values
were higher than for the selective antagonists. These data
indicated that LGP exhibited weak dual receptor inhibitory effects
on P2Y12 and TP receptors.
Discussion
Platelets have a major role in thromboembolic
diseases, which are the final events complicating cardiovascular
diseases and peripheral vascular diseases (19). Therefore, antiplatelet therapy
remains crucial for patients with these diseases in treatment and
prophylaxis (19). However, the
multiple pathways of platelet activation limit the effects of
currently available antiplatelet agents, resulting in limited
clinical efficacy. The efficacy of existing antiplatelet therapies
cannot be dissociated from an increased risk of bleeding (20,21). Previous lessons have demonstrated
that, despite the implementation of existing treatments, the
incidence of side events remains high (20). Therefore, the development of
effective and safe methods to inhibit platelet function remains
critically important. Several medicinal herbs exhibit antiplatelet
effects, and these herbs are used in traditional Chinese medicine
for promoting blood circulation. In our previous study, the effects
of several derivatives of luteolin on ADP-induced platelet
aggregation were evaluated, and it was found that LGP exhibited
significant suppressive effects. The present study reported on the
anti-platelet effects of LGP, a flavonoid from C. nudiflora
which is used as a treatment for promoting blood circulation in
China.
Based on the screening results (data not shown), the
initial experiments performed in the present study were designed to
confirm the effects of LGP on platelet aggregation induced by
different agonists. It was found that LGP caused the
concentration-dependent inhibition of the platelet aggregation
induced by ADP, U46619 and AA. In general, the formation of a
stable platelet plug occurs in three distinct steps: Platelet
adhesion, platelet activation and platelet aggregation (22). Platelet adhesion to sites of
vascular injury is triggered by exposure of the subendothelial
extracellular matrix (ECM) following vascular injury, which is
mediated by platelet interactions with ECM components, particularly
Von Willebrand factor, collagen, fibronectin, thrombospondinand
laminin. The adhesion between platelets and ECM leads to the
deceleration of flowing platelets and capture of circulating
platelets to the vessel wall. At least two separate receptors on
platelet cell membranes, GpIb-V-IX complex and GpVI, function to
tether the platelet and initiate cellular activation,
simultaneously. Once the platelets have adhered to the damaged
vascular endothelium, they recruit additional platelets from the
circulation to augment the fragile platelet monolayer and
eventually form a stable plug (23). Following platelet activation, a
coordinated series of events is triggered, including a rapid
conformational change and the secretion of α,δ-granules and other
intracellular vesicles, including lysosomes, which provide a
positive feedback signal during platelet activation (19). Led by these concepts, the present
study examined the effects of LGP on the content of platelet
granule components to further confirm the effects of LGP on
platelet activation. It was found that the secretion of 5-HT, an
agonist of platelet activation stored in δ-granules, was
significantly decreased by LGP. TXA2, a labile
prostanoid synthesized by activated platelets, is referred to as a
second wave mediator of platelet activation (24). The synthesis of TXA2 is
mediated by a cascade of enzymes, including cyclooxygenase-1; this
enzyme is activated by elevated Ca2+, which induces
translocation to the plasma membrane and phosphorylation by the
stress kinase P38 and extracellular signal-regulated kinase 1/2.
Once synthesized, it diffuses across the platelet membrane and
causes conformational change, phosphoinositide hydrolysis,
Ca2+ mobilization, protein phosphorylation and
secretion, further amplifying the activation signaling (24,25). It was also observed in the present
study that LGP caused a significant decrease in the production of
TXA2. LGP also inhibited the activation of
αIIbβ3 integrin in a dose-dependent manner.
The activation of integrin, particularly
αIIbβ3 integrin, is considered the most
important step in platelet aggregation. Specific interactions of
agonists with their receptors generate inside-out signaling,
leading to the conformational activation of integrins, particularly
αIIbβ3 integrin, increasing their ligand
affinity. The binding of αIIbβ3 integrin to
its ligands, mainly fibrinogens, supports processes including the
close contact between aggregated platelets, and eventually promotes
platelet activation and aggregation. The present study demonstrated
that ADP, U46619 and AA induced platelet aggregation, and
α,δ-granule release and TXA2 synthesis were inhibited by
LGP in a dose-dependent manner. These data suggested that the
inhibitory effects of LGP on aggregation may be associated with its
suppression of platelet activation.
ADP is a critical mediator of platelet activation.
By binding to its receptor, this agonist leads to full activation
events, including platelet conformational change, Ca2+
influx, TXA2 synthesis and granule secretion.
Additionally, ADP is released from the δ-granules of activated
platelets and amplifies its own effects (26). Therefore, ADP, in addition to
TXA2 and 5-HY, are termed second wave mediators, which
are released from platelets and amplify effects of platelet
activation (26). There are two
distinct G-protein-coupled ADP receptors expressed on the surface
of human platelets, P2Y1 and P2Y12. Several
studies have suggested that P2Y12 is the major receptor
in the amplification of platelet activation, and the
PI3K/PDK1/Akt/GSK3β pathway, particularly p110β and p110γ PI3K
isoforms, has emerged as a major signaling axis regulating
P2Y12-mediated platelet activation (27–29). As mentioned above, TXA2
is another stimulator amplifying platelet activation, which is
synthesized by activated platelets. Once synthesized and diffused
from platelets, TXA2 activates and recruits platelets to
the growing platelet aggregation via the Gq- and
G12/13-coupled thromboxane-prostanoid receptors TPα and TPβ
(30). It is accepted that Rho
GTPase signaling is involved in TP-mediated platelet activation by
causing platelet conformational change and regulating platelet
secretion (31). Huang et
al found that RhoA was activated by ARHGEF1 when platelets were
stimulated by U46619, a mimetic of TXA2 (32). Therefore, in order to determine
whether the inhibitory effects of LGP on ADP-induced platelet
aggregation were due to P2Y12-mediated signaling
inhibition, the effects of LGP on the activities of
PI3K/PDK1/Akt/GSK3β were examined. LGP led to a dose-dependent
decrease in the expression of p-PI3K (Tyr607), p-AktSer473, Thr308)
and p-GSK3β (Ser9) (Fig. 7).
Similarly, it was found that LGP inhibited U46619-induced RhoA
signaling (Fig. 6). These data
indicated that the signal transduction mediated by P2Y12
and TP receptors was involved in the LGP-induced platelet
inhibition. To further assess this possibility, the effects of LGP
on P2Y12 and TP receptors were evaluated by a
radiolabeled ligand binding assay. As shown in Fig. 8, the dose-dependent displacement
of [3H]-2-MeS-ADP and [3H] SQ-29548 from
their receptors was caused by LGP in rat platelets, however, the
Ki values were higher compared with the selective
antagonists. These data confirmed the effects of LGP on the
activities of P2Y12 and TP receptors and downstream
signal transduction. However, how LGP affects the P2Y12
and TP receptors remains to be fully elucidated. Future
investigations will focus on the association between the chemical
structure of LGP and the P2Y12 and TP receptors, to
elucidate why and how this compound affects P2Y12 and TP
receptors.
In conclusion, the data presented in the present
study demonstrated that LGP, a natural compound from C.
nudiflora Hook, inhibited the development of platelet
aggregation and amplification of platelet activation. These
inhibitory effects may be associated with its dual-receptor
inhibition on P2Y12 and TP receptors.
Acknowledgments
The authors would like to thank Dr Xianghua Xu and
Dr Yaping Pan from Baylor College of Medicine (Houston, Texas) for
critically reading the manuscript.
References
|
1
|
Harrison P: Platelet function analysis.
Blood Rev. 19:111–123. 2005. View Article : Google Scholar
|
|
2
|
Ghoshal K and Bhattacharyya M: Overview of
platelet physiology: Its hemostatic and nonhemostatic role in
disease pathogenesis. ScientificWorldJournal. 2014:7818572014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weyrich AS: Platelets: More than a sack of
glue. Hematology Am Soc Hematol Educ Program. 2014:400–403.
2014.
|
|
4
|
Clemetson KJ: Platelets and primary
haemostasis. Thromb Res. 129:220–224. 2012. View Article : Google Scholar
|
|
5
|
Yun SH, Sim EH, Goh RY, Park JI and Han
JY: Platelet Activation: The mechanisms and potential biomarkers.
Biomed Res Int. 2016:90601432016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bye AP, Unsworth AJ and Gibbins JM:
Platelet signaling: A complex interplay between inhibitory and
activatory networks. J Thromb Haemost. 14:918–930. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Michelson AD: Antiplatelet therapies for
the treatment of cardiovascular disease. Nat Rev Drug Discov.
9:154–169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jackson SP and Schoenwaelder SM:
Antiplatelet therapy: In search of the ‘magic bullet’. Nat Rev Drug
Discov. 2:775–789. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sigalov AB: Novel mechanistic concept of
platelet inhibition. Expert Opin Ther Targets. 12:677–692. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Varon D and Spectre G: Antiplatelet
agents. Hematology Am Soc Hematol Educ Program. 267–272.
2009.PubMed/NCBI
|
|
11
|
Pei J and Chen SL: Flora of China.
Beijing: Science Press; 1982
|
|
12
|
Zhou Z, Wei X, Fu H and Luo Y: Chemical
constituents of Callicarpa nudiflora and their anti-platelet
aggregation activity. Fitoterapia. 88:91–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo YH, Ma SC, Hu SR, Fu HZ, Zhou ZQ and
Chen WK: Chemical constituents from callicarpa nudiflora. Zhong Yao
Cai. 38:2306–2310. 2015.In Chinese.
|
|
14
|
Del Turco S, Sartini S, Cigni G, Sentieri
C, Sbrana S, Battaglia D, Papa A, Da Settimo F, La Motta C and
Basta G: Synthetic analogues of flavonoids with improved activity
against platelet activation and aggregation as novel prototypes of
food supplements. Food Chem. 175:494–499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Armstrong PC, Truss NJ, Ali FY, Dhanji AA,
Vojnovic I, Zain ZN, Bishop-Bailey D, Paul-Clark MJ, Tucker AT,
Mitchell JA and Warner TD: Aspirin and the in vitro linear
relationship between thromboxane A2-mediated platelet
aggregation and platelet production of thromboxane A2. J
Thromb Haemost. 6:1933–1943. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schoenwaelder SM, Ono A, Sturgeon S, Chan
SM, Mangin P, Maxwell MJ, Turnbull S, Mulchandani M, Anderson K,
Kauffenstein G, et al: Identification of a unique co-operative
phosphoinositide 3-kinase signaling mechanism regulating integrin
alpha IIb beta 3 adhesive function in platelets. J Biol Chem.
282:28648–28658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao J and Shattil SJ: An enzyme-linked
immunosorbent assay to identify inhibitors of activation of
platelet integrin alpha IIb beta 3. J Immunol Methods. 181:55–64.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gratacap MP, Payrastre B, Nieswandt B and
Offermanns S: Differential regulation of Rho and Rac through
heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem.
276:47906–47913. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Angiolillo DJ, Ueno M and Goto S: Basic
principles of platelet biology and clinical implications. Circ J.
74:597–607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yousuf O and Bhatt DL: The evolution of
antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol.
8:547–559. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gachet C: Antiplatelet drugs: Which
targets for which treatments? J Thromb Haemost. 13(Suppl 1):
S313–S322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Meyer SF, Vanhoorelbeke K, Broos K,
Salles II and Deckmyn H: Antiplatelet drugs. Br J Haematol.
142:515–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shifrin MM and Widmar SB: Platelet
inhibitors. Nurs Clin North Am. 51:29–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakahata N: Thromboxane A2:
Physiology/pathophysiology, cellular signal transduction and
pharmacology. Pharmacol Ther. 118:18–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stegner D and Nieswandt B: Platelet
receptor signaling in thrombus formation. J Mol Med. 89:109–121.
2011. View Article : Google Scholar
|
|
26
|
Goggs R and Poole AW: Platelet signaling–a
primer. J Vet Emerg Crit Care. 22:5–29. 2012.
|
|
27
|
Gurbel PA, Kuliopulos A and Tantry US:
G-protein-coupled receptors signaling pathways in new antiplatelet
drug development. Arterioscler Thromb Vasc Biol. 35:500–512. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Von Kügelgen I and Hoffmann K:
Pharmacology and structure of P2Y receptors. Neuropharmacology.
104:50–61. 2016. View Article : Google Scholar
|
|
29
|
Cattaneo M: P2Y12 receptors: Structure and
function. J Thromb Haemost. 13(Suppl 1): S10–S16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang JS, Ramamurthy SK, Lin X and Le
Breton GC: Cell signalling through thromboxane A2
receptors. Cell Signal. 16:521–533. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goggs R, Williams CM, Mellor H and Poole
AW: Platelet Rho GTPases-a focus on novel players, roles and
relationships. Biochem J. 466:431–442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang JS, Dong L, Kozasa T and Le Breton
GC: Signaling through Gα13 switch region I is essential
for protease-activated receptor 1-mediated human platelet shape
change, aggregation, and secretion. J Biol Chem. 282:10210–10222.
2007. View Article : Google Scholar : PubMed/NCBI
|