Introduction
Atherosclerotic cardiovascular disease is the main
cause of high morbidity and mortality rates worldwide (1). It has been documented that
endothelial cell (EC) injury is an early step in the development of
cardiovascular diseases, including myocardial infarction, cardiac
failure and atherosclerosis (2).
The EC injury and dysfunctions are considered to be involved in the
recovery of the injured endothelium and human or animal
atherosclerotic lesions via regulating EC proliferation and
apoptosis, which is associated with the development of
atherosclerosis (3-5). Oxidized low-density lipoprotein
(ox-LDL) can cause an oxidative chain reaction and induce
endothelial dysfunction, which is an essential atherosclerotic risk
factor in the progression of atherosclerosis (6,7).
ox-LDL has been identified to bind with the lectin-like ox-LDL
receptor-1 on the endothelial cell surface, which induces apoptosis
and leads to reactive oxygen species (ROS) accumulation (7). Several studies have shown that EC
apoptosis is important in atherosclerosis via promoting
inflammatory cell infiltration (8), neointima formation (9), lipid transport (10) and plaque rupture (11). Therefore, it is critical to
protect endothelial cells against ox-LDL-induced apoptosis in the
treatment of atherosclerosis.
MicroRNAs (miRNAs) are small endogenous, non-coding
RNA molecules of 22-25 nucleotides, which function as unique gene
expression regulators at the post-transcriptional level by
repressing translation or promoting RNA degradation. Increasing
evidence has shown that miRNAs are able to modulate various
biological and pathological processes, including cellular
differentiation, proliferation, apoptosis and carcinogenesis
(12-14). A previous study demonstrated that
certain miRNAs function as essential regulators in atherosclerosis
by modulating crucial factors or key pathways (15). Sun et al (16) reported that the systemic delivery
of miR-181b attenuated atherosclerosis by inhibiting NF-κB
signaling in the vascular endothelium. miR-21 was shown to be
associated with the progression of atherosclerosis by suppressing
apoptosis and inducing proliferation in vascular smooth muscle
cells (17), and another study
reported that miR-513a-5p modulates tumor necrosis factor-α- and
lipopolysaccharide-induced apoptosis via downregulating X-linked
inhibitor of apoptotic protein in human umbilical vein endothelial
cells (HUVECs) (18). However,
whether miRNAs are also involved in ox-LDL- induced EC apoptosis
remains to be fully elucidated.
Several studies have shown that miR-34a has
pro-apoptotic effects on various cells, including vascular ECs
(19,20). A previous study revealed that
miR-34a was upregulated in atherosclerotic samples (21). In the present study, miRNA
microarray analysis from the Gene Expression Omnibus (GEO) database
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96621),
was obtained, and it was observed that miR-34a is upregulated in
atherosclerotic plaque tissues. Therefore, it was hypothesized that
miR-34a may be important in the development of atherosclerosis. In
the present study, the expression of miR-34a in atherosclerotic
patients was further verified, and the role of miR-34a in
ox-LDL-induced HUVEC apoptosis and the underlying mechanisms were
investigated.
Materials and methods
Atherosclerotic serum and plaque tissue
collection
A total of 25 patients with atherosclerosis and 25
healthy subjects who attended the Department of Cardiology, Huaihe
Hospital of Henan University (Henan, China) were recruited between
June 2015 and June 2016. Serum samples were obtained from the
patients with atherosclerosis and healthy subjects for the
subsequent experiments. Human advanced atherosclerotic plaques were
collected during carotid endarterectomy of patients from the
hospital. A total of six plaque tissues from different patients
were analyzed in the present study. In addition, ascending aortas
without visible atherosclerotic changes (control vessels) were
sampled as controls from six patients undergoing aortic valve
replacement surgery at Huaihe Hospital of Henan University. All
individuals provided informed consent for the use of human
specimens for clinical research. The present study was approved by
the Ethics Committees of Huaihe Hospital of Henan University.
Cell culture and treatment
HUVECs (HUVEC-CS; CRL-2873; American Type Culture
Collection, Manassas, VA, USA) were maintained in DMEM (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100
U/ml penicillin and 100 µg/ml streptomycin (Sigma; EMD
Millipore, Billerica, MA, USA). The cells were cultured at 37°C in
a humidified chamber containing 5% CO2. The HUVECs were
treated with different concentrations (10-200 µg/ml) of
ox-LDL (Union-Bio Technology, Beijing, China) and were collected at
24 h for further measurements.
Cell transfection
The miR-34a mimics/inhibitor and mimics/inhibitor
negative control (NC) were designed and synthesized by GenePharma
Co., Ltd. (Shanghai, China). B-cell lymphoma 2 small interfering
(si)RNA (siBcl-2; 5′-GGTACGATAACCGGGAGATAGTGAT-3) and siRNA NC
(si-Scramble; 5′-GGTTAGCAAGGCGAGATATGACGAT-3′) were synthesized by
BioMics Biotechnologies, Co., Ltd. (Nantong, China). The HUVECs
were seeded into six-well plates and transfected with miR-34a
mimics/inhibitor, mimics/inhibitor NC or si-Bcl-2 using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Following transfection
for 48 h, the cells were exposed to ox-LDL (80 µg/ml) for 24
h and collected for further measurements.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was isolated from atherosclerotic plaque
tissues and cells using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Subsequently, the miRNA and mRNA were reverse transcribed into cDNA
at 37°C for 1 h using the Reverse Transcriptase M-MLV kit (Takara
Biotechnology Co., Ltd., Dalian, China). RT-qPCR analysis was
performed on an Applied Biosystems 7500 Real-Time PCR machine with
miRNA-specific primers using a TaqMan Gene Expression Assay
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The RT-qPCR
reaction system (30 µl) contained 5 µl cDNA, 10
µl mix, 1 µl upstream primer, 1 µl downstream
primer and 13 µl double distilled H2O. The PCR
reaction consisted of an initial denaturation at 95°C for 3 min,
followed by 22 cycles of 95°C for 15 sec and 60°C for 30 sec. The
U6 gene served as an endogenous control for miR-34a and β-actin
served as a reference control for Bcl-2. The primers sequences were
as follows: miR-34a forward, 5′-GAGACAGCCAGGAGAAATCA-3′ and
reverse, 5′-CCTGTGGATGACTGAGTACC-3′; Bcl-2 forward,
5′-GGATTGTGGCCTTCTTTGAG-3′ and reverse, 5′-TAC
CCAGCCTCCGTTATCCT-3′; β-actin forward, 5′-GAGCGCGGCTACAGCTT-3′ and
reverse, 5′-TCCTTAATGTCACGCACGATT T-3′; U6 forward,
5′-CTCGCTTCGGCAGCA CA-3′ and reverse, 5′-AACGCTTCACGAATTTGC GT-3′.
The miR-34a relative expression was analyzed using the
2-ΔΔCq method (22).
All reactions were performed in triplicate.
Cell viability analysis
The
3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide (MTT)
assay was used to measure cell viability. Briefly, 1×104
cells were seeded into 96-well plates overnight. Following
transfection for 48 h, the cells were exposed to ox-LDL (80
µg/ml) for 24 h. Subsequently, 20 µl 5 mg/ml MTT
solution (Sigma; EMD Millipore) was supplemented into each well and
incubated for an additional 4 h. Subsequently, the supernatant was
removed and 150 µl dimethylsulfoxide was added to each well.
The absorbance was determined at 570 nm using a microplate reader
(Tecan Group, Ltd., Männedorf, Switzerland).
Analysis of caspase-3 activation
A fluorimetric assay kit (BioVision, Inc., Milpitas,
CA, USA) was used to measure the caspase-3 activity according to
the manufacturer's protocol. This assay is based on the principle
that activated caspases in apoptotic cells cleave the synthetic
substrates to release free chromophore p-nitroanilide (pNA),
which is measured at 405 nm. The pNA is generated following the
specific action of caspase-3 on tertrapeptide substrate DEVD-pNA.
In brief, the reaction mixture consisted of 50 µl of cell
extract protein, 50 µl of 2X reaction buffer containing 10
mM dithiothreitol, and 5 µl of 4 mM DEVD-pNA (for caspase-3)
substrate in a total volume of 105 µl. The reaction mixture
was incubated at 37°C for 1 h and the sample absorbance was
determined at 405 nm using a microplate reader (Model 680; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The results were determined
as fold changes compared with the control group.
Analysis of apoptosis
Following transfection for 48 h, the HUVECs were
treated with 80 µg/ml of ox-LDL for 24 h. Apoptosis was
measured using flow cytometry. Cells (1×106) were
collected by centrifugation at 1,000 × g for 10 min at 4°C,
followed by washing of the cell pellet twice with HEPES-buffered
saline. The cells were then fixed with 70% ice-cold methanol at 4°C
for 30 min. Following resus-pension with binding buffer, the cells
were added to 5 µl of AnnexinV-FITC and 1 µl of
propidium iodide (PI; 50 µg/ml) (BD Biosciences, Franklin
Lakes, NJ, USA) working solution in 100 µl cell suspension.
The stained cells were analyzed using flow cytometry (FACSCalibur;
BD Biosciences). The measurements were recorded at least three
times in individual experiments.
Western blot analysis
Total protein from the treated HUVECs was isolated
using RIPA buffer with protease inhibitor cocktail (Pierce; Thermo
Fisher Scientific, Inc.). The cytosolic and mitochondrial protein
extracts from the HUVECs were prepared using the
Mitochondria/Cytosol fractionation kit (Enzo Life Sciences,
Farmingdale, NY, USA) according to the manufacturer's protocol. The
BCA Protein Assay kit (CWBiotech, Beijing, China) was used to
measure the protein concentration. The protein samples (30-50
µg) were then resolved by 10 or 12% SDS-PAGE (Sigma-Aldrich;
EMD Millipore) and transferred onto polyvinylidene difluoride
membranes (BD Pharmingen, San Diego, CA, USA). Following blocking
with 5% skimmed milk at room temperature for 1 h, the membranes
were incubated with primary antibodies at 4°C overnight against
Bcl-2 (1:500; cat. no. SC-526; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA), cleaved-caspase-3 (1:1,000; cat. no. SC-5298;
Santa Cruz Biotechnology, Inc.), cleaved caspase-8 (1:1,000; cat.
no. 9496; CST Biological Reagents Co., Ltd., Shanghai, China)
cleaved caspase-9 (1:1,000; cat. no. ab185719; Abcam, Cambridge,
MA, USA), Cytochrome c (Cyt c; 1:200; cat. no.
SC-65396; Santa Cruz Biotechnology, Inc.), Bcl-2-associated ×
protein (Bax; 1:50; cat. no. SC-526; Santa Cruz Biotechnology,
Inc.), cleaved-poly (ADP-ribose) polymerase (PARP; 1:1,000; cat.
no. SC-56196; Santa Cruz Biotechnology, Inc.), gp91 (1:1,000; cat.
no. SC-5827; Santa Cruz Biotechnology, Inc.) and p22phox (1:200;
cat. no. SC-271968; Santa Cruz Biotechnology, Inc.) at 4°C
overnight. The β-actin (1:1,000; cat. no. SC-47778; Santa Cruz
Biotechnology, Inc.) and Cyt c oxidase (Cox) IV (1:1,000;
cat no. ab14744; Abcam) antibodies served as the internal controls
for protein loading. Following this, membranes were incubated with
goat anti-mouse IgG horseradish peroxidase secondary antibody
(1:500; cat. no. SC-2005; Santa Cruz Biotechnology, Inc.) for 1 h
at room temperature. Subsequently, the protein bands were scanned
on X-ray film using an enhanced chemiluminescence detection system
(PerkinElmer, Inc., Waltham, MA, USA). The relative intensity of
each band on the western blots was determined using Alpha Imager
2200 software version 3.1.2 (Alpha Innotech Corporation, San
Leandro, CA, USA). The measurements were taken five times on three
separate occasions.
Measurement of mitochondrial membrane
potential
The loss of mitochondrial membrane potential was
determined via the fluoroprobe (JC-1) as previously described
(23). The cultured cells were
incubated with 2.0 µg/ml of JC-1 fluoroprobe at 37°C for 30
min. Following washing with ice-cold PBS, the hepatocytes were
visualized on a Nikon Eclipse TE2000 inverted microscope (Nikon
Corporation, Tokyo, Japan; ×400 magnification). The mitochondrial
membrane potential drives the formation of red fluorescing JC-1
dimers. The ratio of red fluorescent dimers to green fluorescent
monomers was evaluated through comparing the red fluorescence
intensity (Ex/Em: 580/590 nm) to green fluorescence intensity
(Ex/Em: 510/527 nm) using a Bio-Tek fluorescent microplate reader
(Bio-Tek Instruments, Inc., Winooski, VT, USA).
Intracellular ROS measurement
The effects of miR-34a on intracellular ROS levels
were measured using a total ROS detection kit (BioVision, Inc.,
Milpitas, CA, USA) according to the manufacturer's protocol.
Briefly, the HUVECs were collected and washed with PBS.
Subsequently, the cells were incubated with 500 µl ROS
detection solution and stained at 37°C for 30 min in the dark. The
staining solution was replaced with PBS, and the samples were
analyzed by flow cytometry.
Luciferase reporter assay
The potential binding site between Bcl-2 and miR-34a
was searched using TargetScan (http://www.targetscan.org). The miR-34a
mimics/inhibitor and corresponding NC were synthesized by
GenePharma Co., Ltd. The fragment of the 3′-untranslated region
(UTR) of Bcl-2 containing the putative wild-type (wt) binding sites
and mutated (mut) binding sites for miR-34a was amplified and
cloned into the pMIR-REPORT luciferase vector (Ambion; Thermo
Fisher Scientific, Inc.). Site-directed mutagenesis of the Bcl-2
3′-UTR at the putative miR-34a binding site was performed using a
QuikChange kit (Qiagen, Inc., Valencia, CA, USA). Subsequently, the
HUVECs (2×105/well) were seeded into 24-well plates and
co-transfected with 0.8 µg of pMIR-Bcl-2-3′-UTR or
pMIR-Bcl-2-mut-3′-UTR, 50 nM miR-34a mimic/inhibitor or
corresponding NC using Lipofectamine 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). At 48 h post-transfection, the
luciferase activity was measured using a Dual-Light luminescent
reporter gene assay (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The ratio of Renilla luciferase to Firefly luciferase
was calculated for each well. Each experiment was performed at
least three times in individual experiments.
Statistical analysis
All statistical analyses were performed using SPSS
14.0 software (SPSS, Inc., Chicago, IL, USA). Each experiment was
performed five times on three separate occasions. Numerical data
are presented as the mean ± standard error of the mean. The results
were evaluated using Student's t test. P<0.05 was considered to
indicate a statistically significant difference.
Results
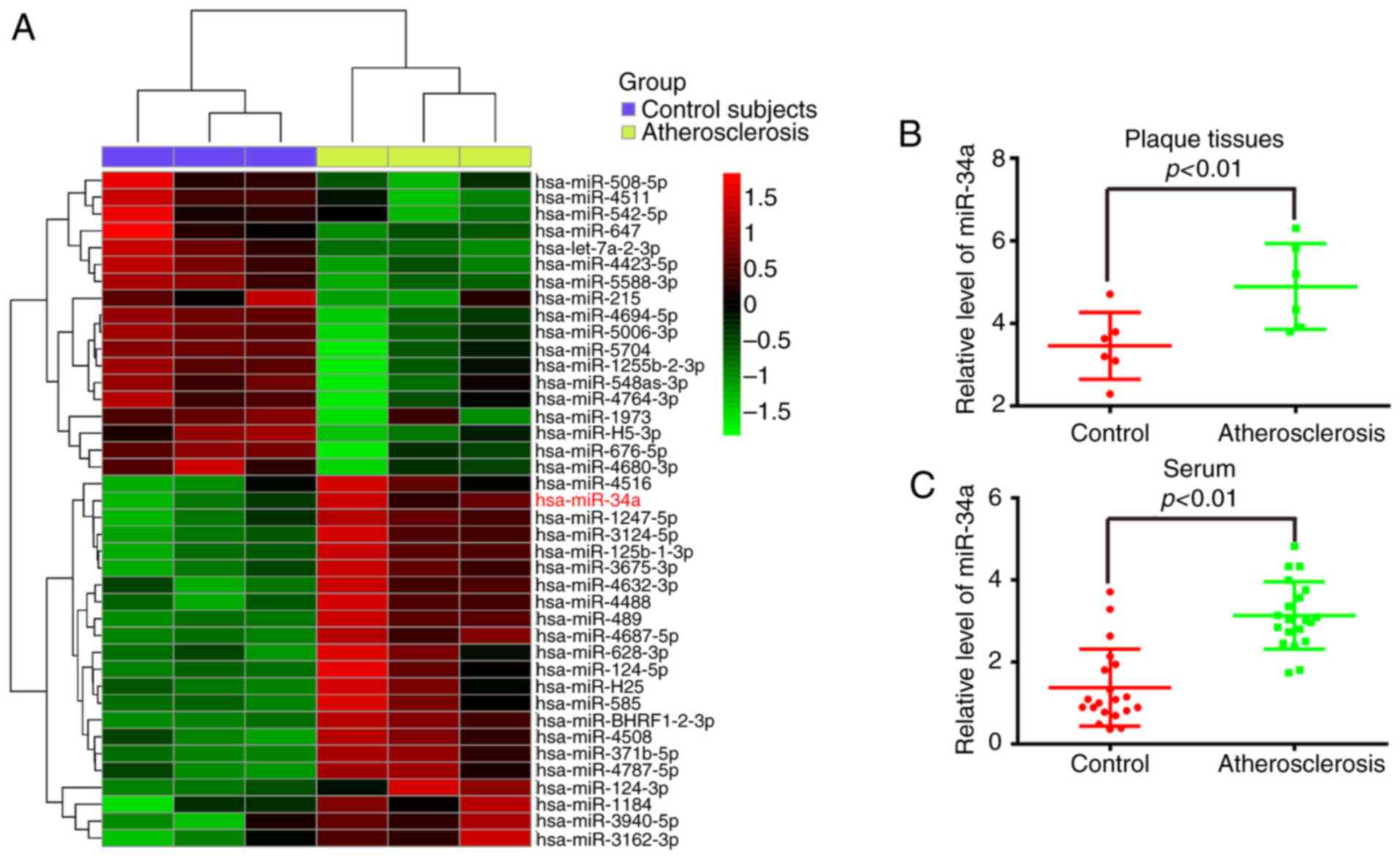
miR-34a is upregulated in human
atherosclerotic plaques and serums
To examine the potential involvement of miRNAs in
atherosclerosis, the miRNA expression profiles were obtained from
the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96621),
which exhibited the microarray profiles of atherosclerotic and
non-atherosclerotic serum of patients. Several miRNAs were altered
in the patients with atherosclerosis and miR-34a was one of the
miRNAs identified as being upregulated most markedly, compared with
the patients without atherosclerosis (Fig. 1A). A previous study also
identified miR-34a as being overexpressed in atherosclerotic
samples (21). To further
validate these results, atherosclerotic plaques and control vessels
were collected, and miR-34a was identified using RT-qPCR analysis.
It was found that the level of miR-34a was significantly
upregulated in the atherosclerotic plaques, compared with that in
the control vessels (P<0.01; Fig.
1B). The results further verified that miR-34a was upregulated
in the serum of patients with atherosclerosis, compared with that
of the control (P<0.01; Fig.
1C). These data indicated that miR-34a may be important in
atherosclerotic progression and function as a biomarker in the
clinical diagnosis of atherosclerosis.
Knockdown of miR-34a prevents
ox-LDL-induced HUVEC apoptosis
As a model for investigations involving ECs, HUVECs
(HUVEC-CS) are widely used in the investigation of atherosclerosis
(18,24,25). To investigate the effect of
miR-34a on the development of atherosclerosis, the HUVEC model was
established, which was treated with different concentrations
(10-200 µg/ml) of ox-LDL for 24 h, and the level of miR-34a
was measured using RT-qPCR analysis. It was found that treatment
with 10-200 µg/ml of ox-LDL increased the levels of miR-34a
in a dose-dependent manner in the HUVECs (Fig. 2A). miR-34a was increased
273.92±23.07% at 80 µg/ml ox-LDL for 24 h, compared with
blank group. Therefore, this concentration was selected in
subsequent experiments. Firstly, the inhibition efficiency of
miR-34a inhibitor and overexpression efficiency of miR-34a mimic
were investigated in HUVECs transfected with miR-34a
mimic/inhibitor or mimic/inhibitor NC. As shown in Fig. 2B, transfection with miR-34a
mimic/inhibitor resulted in the upregulation and downregulation of
miR-34a, respectively, compared with the NC cells (P<0.01). To
determine whether miR-34a can influence the effects of
ox-LDL-mediated apoptosis on HUVECs, the HUVECs were treated with
ox-LDL (80 µg/ml) for 24 h following transfection with
miR-34a inhibitor or inhibitor NC, and an MTT assay was used to
measure cell viability. The results showed that ox-LDL treatment
markedly reduced the cell viability, compared with that in the
blank control group, however, the ox-LDL-induced reduction in cell
viability was reversed by the downregulation of miR-34a (P<0.01;
Fig. 2C). As shown in Fig. 2D and E, the knockdown of miR-34a
also significantly decreased the activity of caspase-3 and
proportion of apoptotic cells in the ox-LDL-treated HUVECs,
compared with the inhibitor NC cells (P<0.01). Additionally, to
evaluate the molecular mechanisms of miR-34a-mediated apoptosis,
western blot analysis was used to measure the expression levels of
apoptosis-related proteins in the ox-LDL-treated HUVECs following
transfection with miR-34a inhibitor or inhibitor NC. The data
revealed that ox-LDL treatment markedly increased the pro-apoptotic
proteins (cleaved-caspase-3, -8, -9, Bax and cleaved-PARP) and
decreased the protein expression of anti-apoptotic Bcl-2, compared
with the blank control group (P<0.01; Fig. 2F). However, the knockdown of
miR-34a significantly inhibited the expression of pro-apoptotic
proteins (cleaved-caspase-3, -8, -9, Bax and cleaved-PARP) and
increased the protein expression of anti-apoptotic Bcl-2, compared
with the inhibitor NC group (P<0.01; Fig. 2F).
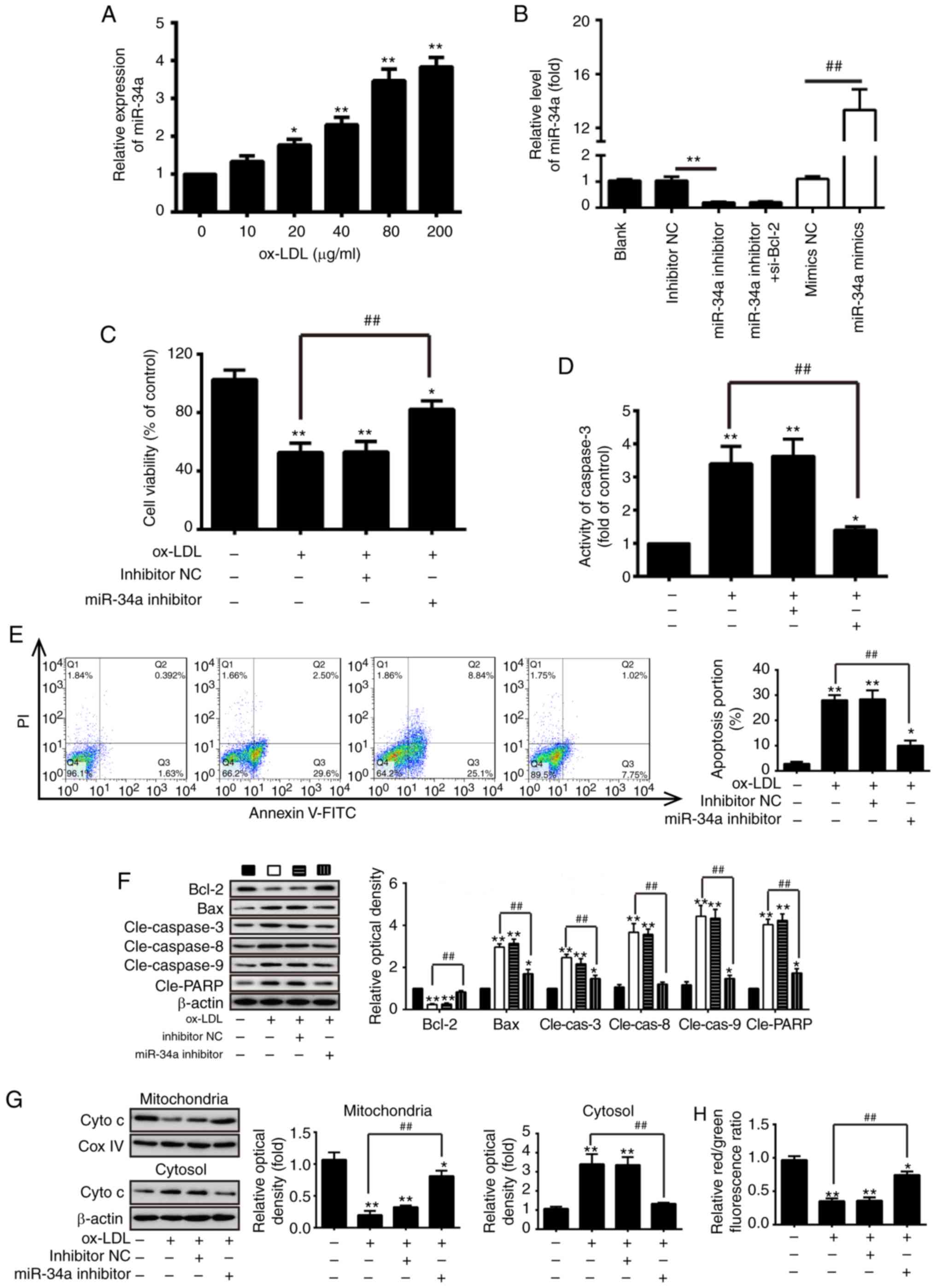 | Figure 2Knockdown of miR-34a inhibits
ox-LDL-induced HUVEC apoptosis. (A) RT-qPCR analysis was performed
to identify the expression of miR-34a in HUVECs following treatment
with different concentrations (10-200 µg/ml) of ox-LDL for
24 h. (B) HUVECs were transfected with miR-34a mimic/inhibitor or
mimic/inhibitor NC. The expression of miR-34a was measured using
RT-qPCR analysis (**P<0.01, vs. inhibitor NC;
##P<0.01, vs. mimic NC). (C) HUVECs were treated with
80 µg/ml of ox-LDL for 24 h following transfection with
miR-34a inhibitor or inhibitor NC, and an MTT assay was performed
to measure cell viability. Treated HUVECs were analyzed using a (D)
fluorimetric assay kit, (E) flow cytometry and (F) western blot
analyses to measure caspase-3 activity, apoptotic cells and
apoptosis-related proteins (Bcl-2, cleaved-caspase-3, -8, -9, Bax
and cleaved-PARP), respectively. (*P<0.05 and
**P<0.01, vs. blank group; ##P<0.01,
vs. ox-LDL-treated group). (G) Protein levels of Cyto c in
mitochondria and cytosol was measured using western blot analysis.
β-actin and Cox IV were used as loading controls for the cytosolic
and mitochondrial fractions, respectively. (*P<0.05
and **P<0.01, vs. blank group;
##P<0.01, vs. ox-LDL-treated group). (H)
Mitochondrial membrane potential levels in the treated HUVECs were
analyzed using the JC-1 fluorescent probe. The relative red/green
fluorescence ratio was obtained by Microplate Multimode Reader
Modulus. Data are presented as the mean ± standard deviation of
three independent experiments (*P<0.05 and
**P<0.01, vs. blank group; ##P<0.01,
vs. ox-LDL-treated group). miR, microRNA; HUVECs, human umbilical
vein endothelial cells; ox-LDL, oxidized low density lipoprotein;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated × protein; PARP,
poly (ADP-ribose) polymerase; Cyt c, cytochrome c;
COX IV, Cyto c oxidase IV; NC, negative control; si-, small
interfering RNA; PI, propidium iodide; cle-, cleaved. |
The release of Cyto c from mitochondria into
the cytosol is a critical event in apoptosis. To further
investigate the effect of miR-34a on Cyto c release, the
HUVECs were treated with ox-LDL (80 µg/ml) for 24 h
following transfection with miR-34a inhibitor or inhibitor NC, and
the protein levels of Cyto c in the mitochondria and the
cytosol was measured using western blot analysis. The results
showed that ox-LDL treatment induced the release of Cyto c
from the mitochondria, and the knockdown of miR-34a attenuated the
ox-LDL-induced Cyto c release (P<0.01; Fig. 2G). Subsequently, mitochondrial
membrane potential was analyzed using the JC-1 fluorescent probe;
it was found that ox-LDL treatment significantly reduced the
red/green fluorescence ratio, compared with that in the blank
control cell (P<0.01; Fig.
2H). However, the knockdown of miR-34a significantly increased
the red/green fluorescence ratio in the ox-LDL-treated HUVECs
(P<0.01; Fig. 2H).
Collectively, these data suggested that the knockdown of miR-34a
protected HUVECs against ox-LDL-induced apoptosis via inhibiting
the mitochondrial apoptotic pathway.
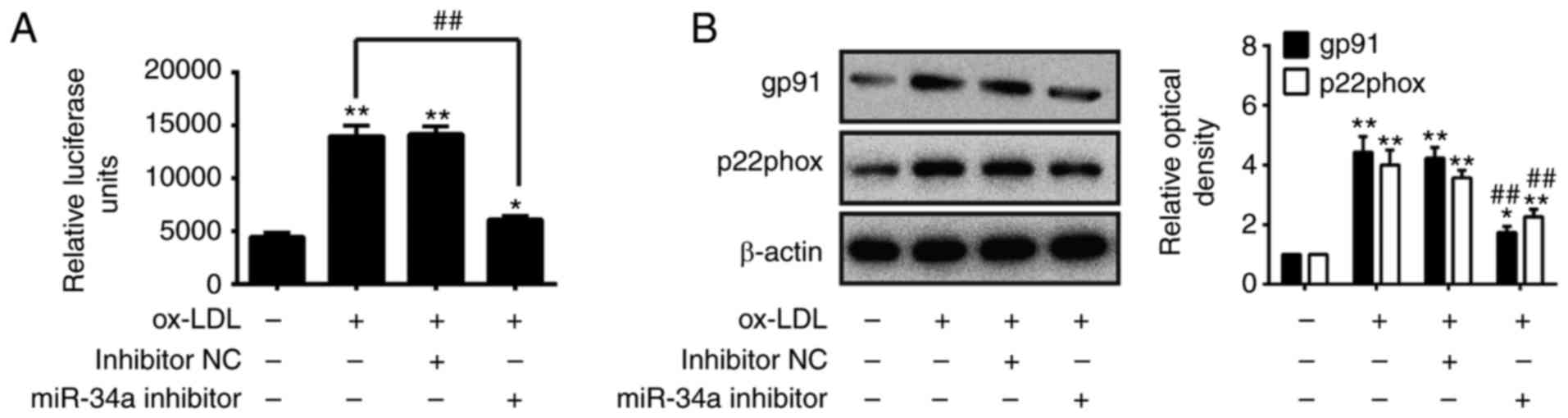
Knockdown of miR-34a protects against
ox-LDL-induced ROS
A previous study revealed that ROS is associated
with the initiation and progression stages of atherogenesis
(25). Therefore, the present
study investigated the effects of miR-34a on ROS induced by ox-LDL
in HUVECs. The HUVECs were treated with 80 µg/ml of ox-LDL
for 24 h following transfection with miR-34a inhibitor or inhibitor
NC, and the ROS levels were determined using a ROS detection kit.
As shown in Fig. 3A, ox-LDL
treatment markedly increased the ROS levels, compared with those in
the blank control group, however, the ROS levels were significantly
decreased by the knockdown of miR-34a, compared with the
ox-LDL-treated group (P<0.01; Fig.
3A). To confirm the anti-oxidative effects of the
downregulation of miR-34a, western blot analysis was performed to
detect the expression level of oxidative injury-related proteins
gp91 (NOX2) and p22phox (26). As
shown in Fig. 3B, the knockdown
of miR-34a markedly reduced the expression of gp91 and p22phox in
the ox-LDL-treated HUVECs (P<0.01; Fig. 3B). These results suggested that
the knockdown of miR-34a exerted anti-oxidative effects against
ox-LDL-induced ROS levels in the HUVECs.
miR-34a inhibits the expression of Bcl-2
by targeting its 3′-UTR in HUVECs
Previous studies have found that miR-34a exerts
pro-apoptotic effects by targeting anti-apoptotic protein Bcl-2 in
various types of cancer (27,28). Therefore, it was hypothesized that
ox-LDL-induced HUVEC apoptosis was regulated by miR-34a via
suppressing Bcl-2. The target gene of miR-34a was predicted using
bioinformatics analysis. It was found that miR-34a may regulate
Bcl-2 by binding with its 3′-UTR (Fig. 4A). As shown in Fig. 4B, compared with the mimic NC
group, miR-34a mimics markedly repressed the luciferase activity in
the presence of the wt 3′-UTR, however, the miR-34a inhibitor
markedly increased the luciferase activity (P<0.01; Fig. 4B), whereas no inhibition of
luciferase activity was found for the mut 3′UTR group (Fig. 4B). To verify that Bcl-2 was
negatively modulated by miR-34a, western blot and RT-qPCR analyses
were performed to detect the protein and mRNA levels of Bcl-2 in
HUVECs transfected with miR-34a mimic/inhibitor or corresponding
NC, respectively. The results demonstrated that the overexpression
of miR-34a markedly reduced Bcl-2 at the protein and mRNA levels,
whereas the knockdown of miR-34a led to a significant increase in
the protein and mRNA levels of Bcl-2, compared with those in the NC
group (P<0.01; Fig. 4C and D).
RT-qPCR analysis was used to detect the mRNA levels of Bcl-2 in
atherosclerotic plaque tissues (n=6), and it was found that the
expression of Bcl-2 was significantly decreased in the plaque
tissues, compared with that in the control (P<0.01; Fig. 4E). The results also verified that
the mRNA level of Bcl-2 was downregulated in the serum of patients
with atherosclerosis (n=25; P<0.01; Fig. 4F). The correlation analysis showed
a negative association between the expression of Bcl-2 and the
expression of miR-34a in serum samples from the patients with
atherosclerosis (r=-0.8404, P<0.001; Fig. 4G). Taken together, these data
indicated that miR-34a repressed Bcl-2 via targeting its 3′-UTR in
HUVECs, suggesting that miR-34a may induce HUVEC apoptosis in
atherosclerosis through targeting Bcl-2.
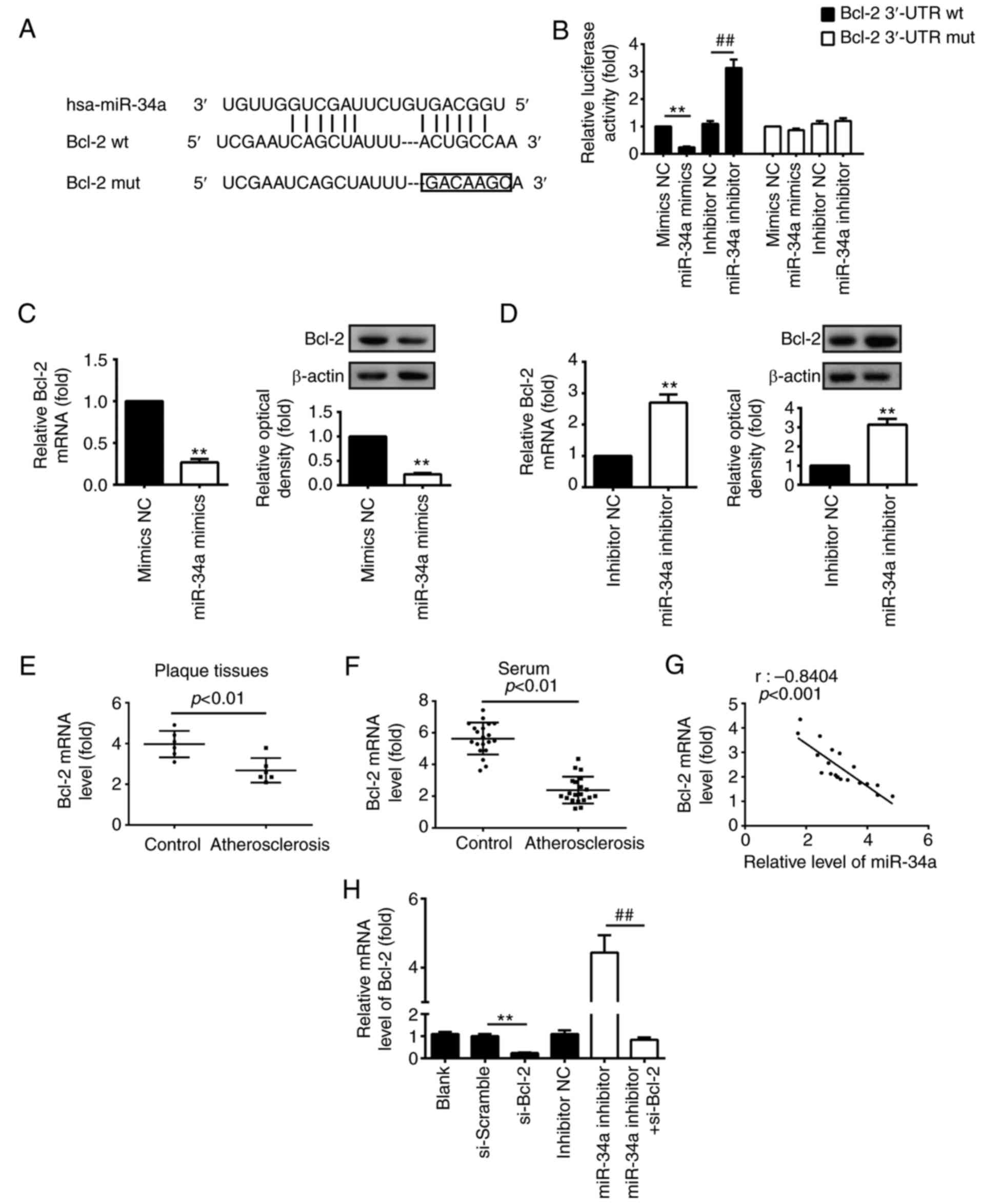 | Figure 4Bcl-2 is a direct target of miR-34a
in HUVECs. (A) Putative miR-34a binding sites on 3′-UTR of Bcl-2
mRNA were predicted using TargetScan. (B) Relative luciferase
activity of Bcl-2 wt or mut 3′-UTR in HUVECs following transfection
with the miR-34a mimic, inhibitor or corresponding NC
(**P<0.01, vs. mimic NC; ##P<0.01, vs.
inhibitor NC). HUVECs were transfected with (C) miR-34a mimic or
(D) inhibitor, or corresponding NC, the protein and mRNA levels of
Bcl-2 were measured using a western blot assay and RT-qPCR
analysis, respectively (**P<0.01, vs. corresponding
NC). β-actin was used as an internal control. (E) RT-qPCR analysis
was used to determine the mRNA expression of Bcl-2 in
atherosclerotic plaque tissues (n=6). (F) RT-qPCR analysis was used
to determine the mRNA expression of Bcl-2 in serum samples from
patients with atherosclerosis (n=20) and control subjects (n=20).
(G) Negative correlation between levels of Bcl-2 and miR-34a in
serum samples from patients with atherosclerosis and control
subjects (n=20; r=-0.8404, P<0.001). Data are presented as the
mean ± standard deviation of three individual experiments. (H)
HUVECs were transfected with si-Bcl-2 and si-Scramble, or
co-transfected with miR-34a inhibitor and si-Bcl-2. RT-qPCR
analysis was used to detect the mRNA level of Bcl-2 in HUVECs
(**P<0.01, vs. si-Scramble; ##P<0.01,
vs. miR-34a inhibitor). Data are presented as the means ± standard
deviation of three individual experiments. miR, microRNA; HUVECs,
human umbilical vein endothelial cells; UTR, untranslated region;
Bcl-2, B-cell lymphoma 2; si-, small interfering RNA; wt,
wild-type; mut, mutant; NC, negative control; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Silencing Bcl-2 inhibits the protective
effects of the downregu- lation of miR-34a on ox-LDL-induced HUVEC
apoptosis
The present study evaluated the interference
efficiency of si-Bcl-2 in HUVECs transfected with si-Bcl-2 or
si-Scramble, or co-transfected with miR-34a inhibitor and si-Bcl-2.
As shown in Fig. 4H, si-Bcl-2
interfered with the expression of Bcl-2, compared with that in the
si-Scramble group (P<0.01). In addition, the inhibition of
miR-34a significantly increased the expression of Bcl-2, whereas
silencing Bcl-2 eliminated the effect of the miR-34a
inhibitor-induced upregulation of Bcl-2 in HUVECs co-transfected
with miR-34a inhibitor and si-Bcl-2 (P<0.01; Fig. 4H). Subsequently, to verify whether
the protective effects of the downregulation of miR-34a on
ox-LDL-induced apoptosis is modulated by the expression of Bcl-2,
the HUVECs were co-transfected with miR-34a inhibitor and si-Bcl-2
or were transfected with either the miR-34a inhibitor or si-Bcl-2
prior to ox-LDL treatment. Western blot analysis, an MTT assay, a
fluorimetric assay, flow cytometric analysis, the JC-1 fluorescent
probe and a ROS detection kit were then used to measure the
expression of Bcl-2, cell viability, caspase-3 activity, apoptotic
cells, mitochondrial membrane potential and ROS levels,
respectively. Consistent with the above results, the knockdown of
miR-34a increased the protein expression of Bcl-2, cell viability
and the red/green fluorescence ratio, and decreased the activity of
caspase-3, apoptotic cells and ROS levels in the ox-LDL-treated
HUVECs. The effects of the downregulation of miR-34a on the
ox-LDL-treated HUVECs were eliminated by silencing Bcl-2
(P<0.01; Fig. 5A–F). These
results indicated that the protective effects of the downregulation
of miR-34a on ox-LDL-induced HUVEC apoptosis were weakened by
silencing Bcl-2.
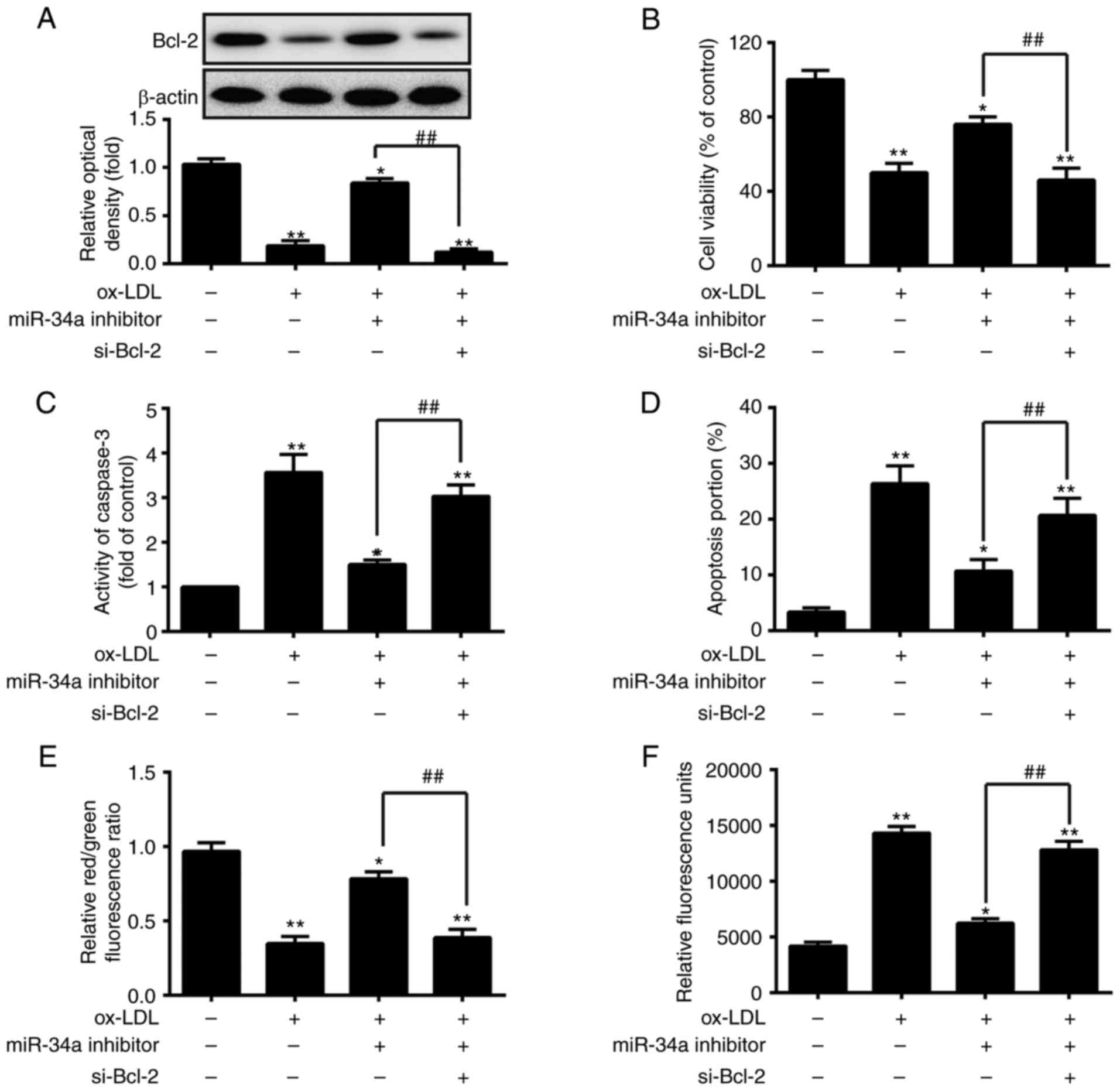 | Figure 5Silencing Bcl-2 suppresses the
protective effects of the downregulation of miR-34a on
ox-LDL-induced HUVEC apoptosis. The HUVECs were treated with 80
µg/ml of ox-LDL for 24 h following co-transfection with
miR-34a inhibitor and si-Bcl-2 or transfection with either miR-34a
inhibitor or si-Bcl-2. (A) Western blot analysis, an (B) MTT assay,
(C) fluorimetric assay, (D) flow cytometric analysis, (E) JC-1
fluorescent probe and a (F) ROS detection kit were used to measure
the expression of Bcl-2, cell viability, caspase-3 activity,
apoptotic cells, mitochondrial membrane potential and ROS levels,
respectively. Data are presented as the mean ± standard deviation
of three individual experiments (*P<0.05 and
**P<0.01, vs. blank group; ##P<0.01,
vs. ox-LDL-treated group). miR, microRNA; HUVECs, human umbilical
vein endothelial cells; Bcl-2, B-cell lymphoma 2; si-, small
interfering RNA; ox-LDL, oxidized low density lipoprotein; ROS,
reactive oxygen species; NC, negative control. |
Discussion
A previous study documented that certain specific
miRNAs are involved in the progression of atherosclerosis by
modulating the properties of vascular cells and different cellular
functions (29), and their
expression have been closely associated with the pathogenesis of
atherosclerosis (29). A previous
study reported that the upregulation of miR-876 induced EC
apoptosis in the development of atherosclerosis by repressing
anti-apoptotic protein Bcl-extra large (Bcl-xL) (30). Zhang et al (25) demonstrated that the inhibition of
miR-155 improved high glucose-induced EC injury by suppressing the
activation of nuclear factor-κB. miR-34a is upregulated in
atherosclerotic samples and is involved in the progression of
atherosclerosis (21), in
addition to possessing pro-apoptotic effects on ECs (20). Previous miRNA microarray analysis
confirmed that the levels of miR-34a are increased in the serum of
patients with atherosclerosis. However, the potential molecular
mechanism of miR-34a in atherosclerosis requires further
investigation to be fully elucidated. In the present study, the
expression of miR-34a was further validated in atherosclerotic
plaque tissues and the underlying mechanisms of miR-34a in
ox-LDL-induced HUVEC apoptosis were examined. It was found that
miR-34a was upregulated in the atherosclerotic plaque tissues. The
results also demonstrated that ox-LDL treatment increased the
levels of miR-34a in a dose-dependent manner, and that the
knockdown of miR-34a inhibited ox-LDL-induced apoptosis and ROS
levels in the HUVECs. In addition, Bcl-2 was identified as a direct
target of miR-34a, and silencing Bcl-2 abrogated the protective
effects of the downregulation of miR-34a on ox-LDL-induced
apoptosis in HUVECs.
It has been confirmed that ox-LDL is an important
factor in the initiation and progression of atherosclerosis
(31). Ox-LDL has also been
identified as being increased in diabetic patients, causing
atherogenesis and endothelial damage (32). Additionally, ox-LDL promotes the
recruitment of monocytes by vascular cells and triggers their
differentiation into macrophages (33). Ox-LDL is able to induce monocyte
adhesion, ROS generation and apoptosis in vascular ECs via binding
the lectin-like endothelial ox-LDL receptor (LOX-1) (34-36). However, the mechanism underlying
ox-LDL-mediated HUVEC apoptosis remain to be fully elucidated.
Chen et al (37) reported that miR-98 inhibits
proliferation and decreases ox-LDL-induced apoptosis through
targeting LOX-1 in HUVECs. miR-26a protects ECs from ox-LDL-induced
apoptosis in the setting of atherosclerosis by targeting transient
receptor potential cation channel subfamily C member 6 (38). The natural product
2,4,5-trihydroxybenz-aldehyde has been found to effectively inhibit
oxygen-glucose deprivation/reperfusion-induced EC injury via
suppressing miR-34a (20).
Therefore, it was hypothesized that miR-34a may modulate
ox-LDL-mediated apoptosis in HUVECs. RT-qPCR analysis was used to
measure the expression of miR-34a in HUVECs treated with different
concentration (10-200 µg/ml) of ox-LDL. The results
indicated that ox-LDL treatment increased the levels of miR-34a in
a dose-dependent manner, suggesting that miR-34a is important in
ox-LDL-induced apoptosis. It was also observed that ox-LDL
treatment reduced cell viability, and increased the activity of
caspase-3 and the number of apoptotic cells, whereas the
downregulation of miR-34a increased cell viability, and reduced the
activity of caspase-3 and apoptotic cells in the ox-LDL-treated
HUVECs. These data indicated that the downregulation of miR-34a
protected against ox-LDL-induced apoptosis in HUVECs.
Oxidative stress is a potent pathogenic mechanism in
atherosclerosis via inducing apoptosis, mitochondria dysfunction
and cellular injury (39,40). Oxidative stress is considered as
the increased bioactivity of ROS relative to antioxidant defenses
(41). ROS have been identified
to act as a major trigger of EC apoptosis (42). Zhang et al (43) revealed that
miR-34a/sirtuin-1/foxo3a exerts an important function in genistein
in preventing ox-LDL-induced oxidative damage in HUVECs. According
to these findings, the present study examined whether the knockdown
of miR-34a ameliorates ox-LDL-induced apoptosis via modulating ROS,
and examined the protective effects of the downregulation of
miR-34a on oxidative injury. The results demonstrated that the
knockdown of miR-34a reduced ox-LDL-induced ROS levels in the
HUVECs. Various enzymatic sources within the myocardium are capable
of producing excess ROS, including nicotinamide adenine
dinucleotide phosphate (NADPH). NADPH oxidases are multimeric
enzymes, which contain a membrane-bound core, comprising a
catalytic gp91phox subunit (NOX2) and a p22phox subunit (44). Li et al (45) revealed that miR-34a induced
apoptosis and enhanced the production of ROS in a human glioma cell
via targeting NOX2. In the present study, the results demonstrated
that the downregulation of miR-34a attenuated the expression of
gp91 and p22phox in the ox-LDL-treated HUVECs. However, further
investigation is required to determine the factors underlying the
possible pathway of miR-34a-regulated expression of gp91 and
p22phox in HUVECs. These findings suggested that the knockdown of
miR-34a exerted protective effects against ox-LDL-induced HUVEC
injury via suppressing the production of ROS.
Apoptosis, or programmed cell death, is important in
physiological and pathological conditions, and ox-LDL-induced EC
apoptosis is considered to be key in atherosclerosis (46). EC apoptosis is also involved in
atherosclerotic plaque development and progression (47). The balance of Bcl-2 family
members, including anti-apoptotic proteins Bcl-xL and Bcl-2, and
pro-apoptotic proteins Bcl-2-associated death promoter, Bcl-2
homologous antagonist/killer, and Bax, is disrupted, which is a
vital mechanism that contributes to apoptosis (48). A previous study demonstrated that
miR-1907 aggravates atherosclerosis-associated EC apoptosis by
inhibiting Bcl-2 (49). It has
been extensively reported that Bcl-2 is a direct target of miR-34a
in various cells (27,28,50,51). In the present study, the results
confirmed that miR-34a suppressed the expression of Bcl-2 by
directly targeting its 3′-UTR in HUVECs. Furthermore, silencing
Bcl-2 impaired the protective effects of the downregulation of
miR-34a on ox-LDL-induced apoptosis. Collectively, these data
suggested that miR-34a regulated ox-LDL-induced apoptosis by
targeting the anti-apoptotic protein Bcl-2 in HUVECs.
Mitochondria-mediated apoptosis, an intrinsic
pathway of apoptosis, is usually activated by loss of mitochondrial
membrane potential, and proceeds through the release of Cyto
c and ROS from the intermembraneous space of the
mitochondria to the cytosol (52). Mitochondrial dysfunction can
result in the release of Cyto c into the cytosol, where it
forms a complex with apoptotic peptidase activating factor 1, which
subsequently recruits and activates procaspase-9 into active
caspase-9 (53,54). Caspase-9 cleaves and activates
procaspase-3, a downstream executioner caspase protein, which
cleaves various 'death substrates' and thereby triggers the
apoptotic cell phenotype. A previous study demonstrated that the
knockdown of miR-34a altered the integrity of the mitochondrial
outer membrane and increased the release of Cyto c from
mitochondria, finally activating the caspase-mediated apoptotic
pathway in vascular ECs (20). In
the present study, the results showed that the knockdown of miR-34a
inhibited cleaved-caspase-3, -8, -9, Bax and cleaved-PARP, and
increased Bcl-2 in the ox-LDL-treated HUVECs. In addition, the
knockdown of miR-34a attenuated the ox-LDL-induced release of Cyto
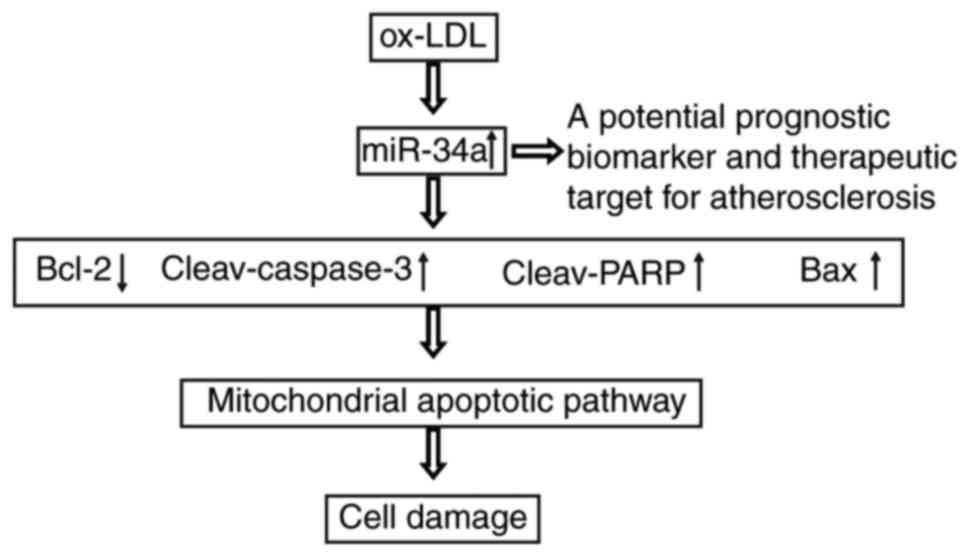
c and improved mitochondrial membrane potential. Taken
together, these findings indicated that the ox-LDL-mediated
upregulation of miR-34a induced HUVEC apoptosis via activating the
apoptotic pathway (Fig. 6),
suggesting it offers potential as a prognostic biomarker and
therapeutic target for atherosclerosis.
In conclusion, the results of the present study
demonstrated that miR-34a was upregulated in atherosclerotic plaque
tissues, and that the knockdown of miR-34a prevented ox-LDL-induced
HUVEC apoptosis by increasing cell viability, decreasing caspase-3
activity, and inhibiting apoptotic cells and ROS. In addition,
miR-34a modulated ox-LDL-induced apoptosis by repressing
anti-apoptotic protein Bcl-2 in the HUVECs. These findings
indicated that the knockdown of miR-34a protected against
ox-LDL-induced apoptosis and oxidative stress via inhibiting the
mitochondrial apoptotic pathway, suggesting that miR-34a serves as
a biomarker in the clinical diagnosis and treatment of
atherosclerosis.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XZ and PL and performed the experiments, contributed
to data analysis and wrote the paper. JL, RH and GC analyzed the
data. YL conceptualized the study design, contributed to data
analysis and experimental materials. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All individuals provided informed consent for the
use of human specimens for clinical research. The present study was
approved by Huaihe Hospital of Henan University Ethics
Committees.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Executive summary: Heart disease and stroke statistics-2014 update:
A report from the American Heart Association. Circulation.
129:399–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tabas I, Williams KJ and Borén J:
Subendothelial lipoprotein retention as the initiating process in
atherosclerosis. Circulation. 116:1832–1834. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Otsuka F, Finn AV, Yazdani SK, Nakano M,
Kolodgie FD and Virmani R: The importance of the endothelium in
atherothrombosis and coronary stenting. Nat Rev Cardiol. 9:439–453.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pober JS and Sessa WC: Evolving functions
of endothelial cells in inflammation. Nat Rev Immunol. 7:803–815.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stoneman VE and Bennett MR: Role of
apoptosis in atherosclerosis and its therapeutic implications. Clin
Sci. 107:3432004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sawamura T, Kume N, Aoyama T, Moriwaki H,
Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, et al: An
endothelial receptor for oxidized low-density lipoprotein. Nature.
386:73–77. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cominacini L, Rigoni A, Pasini AF, Garbin
U, Davoli A, Campagnola M, Pastorino AM, Lo Cascio V and Sawamura
T: The binding of oxidized low density lipoprotein (ox-LDL) to
ox-LDL receptor-1 reduces the intracellular concentration of nitric
oxide in endothelial cells through an increased production of
superoxide. J Biol Chem. 276:13750–13755. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Sivashanmugam P, Wu JH, Brian L,
Exum ST, Freedman NJ and Peppel K: Tumor necrosis factor receptor-2
signaling attenuates vein graft neointima formation by promoting
endothelial recovery. Arterioscler Thromb Vasc Biol. 28:284–289.
2008. View Article : Google Scholar
|
|
10
|
von Eckardstein A and Rohrer L:
Transendothelial lipoprotein transport and regulation of
endothelial permeability and integrity by lipoproteins. Curr Opin
Lipidol. 20:197–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kavurma MM, Bhindi R, Lowe HC, Chesterman
C and Khachigian LM: Vessel wall apoptosis and atherosclerotic
plaque instability. J Thromb Haemost. 3:465–472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
le Sage C and Agami R: Immense promises
for tiny molecules: Uncovering miRNA functions. Cell Cycle.
5:1415–1421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nature Rev Genet. 10:704–714.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Menghini R, Stöhr R and Federici M:
MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev.
17:68–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun X, He S, Wara AKM, Icli B, Shvartz E,
Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K
and Feinberg MW: Systemic delivery of microrna-181b inhibits
nuclear factor-kB activation, vascular inflammation, and
atherosclerosis in apolipoprotein E-deficient mice. Circ Res.
114:32–40. 2014. View Article : Google Scholar
|
|
17
|
Lin Y, Liu X, Cheng Y, Yang J, Huo Y and
Zhang C: Involvement of microRNAs in hydrogen peroxide-mediated
gene regulation and cellular injury response in vascular smooth
muscle cells. J Biol Chem. 284:7903–7913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shin S, Moon KC, Park KU and Ha E:
MicroRNA-513a-5p mediates TNF-α and LPS induced apoptosis via
downregulation of X-linked inhibitor of apoptotic protein in
endothelial cells. Biochimie. 94:1431–1436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li QL, Zhang HY, Qin YJ, Meng QL, Yao XL
and Guo HK: MicroRNA-34a promoting apoptosis of human lens
epithelial cells through down-regulation of B-cell lymphoma-2 and
silent information regulator. Int J Ophthalmol. 9:1555–1560.
2016.PubMed/NCBI
|
|
20
|
Liao LX, Zhao MB, Dong X, Jiang Y, Zeng KW
and Tu PF: TDB protects vascular endothelial cells against
oxygen-glucose deprivation/reperfusion-induced injury by targeting
miR-34a to increase Bcl-2 expression. Sci Rep. 6:379592016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Raitoharju E, Lyytikäinen LP, Levula M,
Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kähönen M,
Karhunen PJ, et al: miR-21, miR-210, miR-34a, and miR-146a/b are
up-regulated in human atherosclerotic plaques in the Tampere
Vascular Study. Atherosclerosis. 219:211–217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Nicholls DG: Fluorescence measurement of
mitochondrial membrane potential changes in cultured cells. Res
Gate. 810:119–133. 2012.
|
|
24
|
Zhang X, Xia S, Xu Q and Huang J: The
cytoprotective effects of Δ-17 fatty acid desaturase on injured
HUVECs and its underlying mechanism. Saudi Pharm J. 25:587–594.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Liu X, Li Y, Lai J, Zhang N, Ming
J, Ma X, Ji Q and Xing Y: Downregulation of microRNA-155
ameliorates high glucose-induced endothelial injury by inhibiting
NF-κB activation and promoting HO-1 and NO production. Biomed
Pharmacother. 88:1227–1234. 2017. View Article : Google Scholar
|
|
26
|
Lu J, Mitra S, Wang X, Khaidakov M and
Mehta JL: Oxidative stress and lectin-like Ox-LDL-receptor LOX-1 in
atherogenesis and tumorigenesis. Antioxid Redox Signal.
15:2301–2333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang F, Li QJ, Gong ZB, Zhou L, You N,
Wang S, Li XL, Li JJ, An JZ, Wang DS, et al: MicroRNA-34a targets
Bcl-2 and sensitizes human hepatocellular carcinoma cells to
sorafenib treatment. Technol Cancer Res Treat. 13:77–86. 2014.
View Article : Google Scholar
|
|
28
|
Li L, Yuan L, Luo J, Gao J, Guo J and Xie
X: MiR-34a inhibits proliferation and migration of breast cancer
through down-regulation of Bcl-2 and SIRT1. Clin Exp Med.
13:109–117. 2013. View Article : Google Scholar
|
|
29
|
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen
H, Dean DB and Zhang C: MicroRNA expression signature and
antisense- mediated depletion reveal an essential role of microrna
in vascular neointimal lesion formation. Circ Res. 100:1579–1588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu K, Liu P and Zhao Y: Upregulation of
microRNA-876 induces endothelial cell apoptosis by suppressing
Bcl-Xl in development of atherosclerosis. Cell Physiol Biochem.
42:1540–1549. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ehara S, Ueda M, Naruko T, Haze K, Itoh A,
Otsuka M, Komatsu R, Matsuo T, Itabe H, Takano T, et al: Elevated
levels of oxidized low density lipoprotein show a positive
relationship with the severity of acute coronary syndromes.
Circulation. 103:1955–1960. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jialal I and Devaraj S: The role of
oxidized low density lipopro-tein in atherogenesis. J Nutr. 126(4
Suppl): 1053S–1057S. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng H, Cui D, Quan X, Yang W, Li Y,
Zhang L and Liu E: Lp-PLA2 silencing protects against
ox-LDL-induced oxidative stress and cell apoptosis via Akt/mTOR
signaling pathway in human THP1 macrophages. Biochem Biophys Res
Commun. 477:1017–1023. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li D and Mehta JL: Antisense to LOX-1
inhibits oxidized LDL-mediated upregulation of monocyte
chemoattractant protein-1 and monocyte adhesion to human coronary
artery endothelial cells. Circulation. 101:2889–2895. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li D and Mehta JL: Upregulation of
endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and
implications in apoptosis of human coronary artery endothelial
cells: Evidence from use of antisense LOX-1 mRNA and chemical
inhibitors. Arterioscler Thromb Vasc Biol. 20:1116–1122. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen J, Mehta JL, Haider N, Zhang X,
Narula J and Li D: Role of caspases in Ox-LDL-induced apoptotic
cascade in human coronary artery endothelial cells. Circ Res.
94:370–376. 2004. View Article : Google Scholar
|
|
37
|
Chen Z, Wang M, He Q, Li Z, Zhao Y, Wang
W, Ma J, Li Y and Chang G: MicroRNA-98 rescues proliferation and
alleviates ox-LDL-induced apoptosis in HUVECs by targeting LOX-1.
Exp Ther Med. 13:1702–1710. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y,
Li X, Shen N, Xiao D, Zhang H, et al: MicroRNA-26a prevents
endothelial cell apoptosis by directly targeting TRPC6 in the
setting of atherosclerosis. Sci Rep. 5:94012015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stocker R and Keaney JF Jr: Role of
oxidative modifications in atherosclerosis. Physiol Rev.
84:1381–1478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Madamanchi NR and Runge MS: Mitochondrial
dysfunction in atherosclerosis. Circ Res. 100:460–473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kregel KC and Zhang HJ: An integrated view
of oxidative stress in aging: Basic mechanisms, functional effects,
and pathological considerations. Am J Physiol Regul Integr Comp
Physiol. 292:R18–R36. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wen H, Gwathmey JK and Xie LH: Oxidative
stress-mediated effects of angiotensin II in the cardiovascular
system. World J Hypertens. 2:34–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang H, Zhao Z, Pang X, Yang J, Yu H,
Zhang Y, Zhou H and Zhao J: MiR-34a/sirtuin-1/foxo3a is involved in
genistein protecting against ox-LDL-induced oxidative damage in
HUVECs. Toxicol Lett. 277:115–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun HN, Kim SU, Lee MS, Kim SK, Kim JM,
Yim M, Yu DY and Lee DS: Nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase-dependent activation of phosphoinositide
3-kinase and p38 mitogen-activated protein kinase signal pathways
is required for lipopolysaccharide-induced microglial phagocytosis.
Biol Pharm Bull. 31:1711–1715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li SZ, Hu YY, Zhao J, Zhao YB, Sun JD,
Yang YF, Ji CC, Liu ZB, Cao WD, Qu Y, et al: MicroRNA-34a induces
apoptosis in the human glioma cell line, A172, through enhanced ROS
production and NOX2 expression. Biochem Biophys Res Commun.
444:6–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qin B, Xiao B, Liang D, Xia J, Li Y and
Yang H: MicroRNAs expression in ox-LDL treated HUVECs: MiR-365
modulates apoptosis and Bcl-2 expression. Biochem Biophys Res
Commun. 410:127–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shin S, Chul K, Uk K and Ha E: Biochimie
MicroRNA-513a-5p mediates TNF-α and LPS induced apoptosis via
downregulation of X-linked inhibitor of apoptotic protein in
endothelial cells. Biochimie. 94:1431–1436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reuter S, Eifes S, Dicato M, Aggarwal BB
and Diederich M: Modulation of anti-apoptotic and survival pathways
by curcumin as a strategy to induce apoptosis in cancer cells.
Biochem Pharmacol. 76:1340–1351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao J, Ou SL, Wang WY, Yan C and Chi LX:
MicroRNA-1907 enhances atherosclerosis-associated endothelial cell
apoptosis by suppressing Bcl-2. Am J Transl Res. 9:3433–3442.
2017.PubMed/NCBI
|
|
50
|
Ji Q, Hao X, Meng Y, Zhang M, Desano J,
Fan D and Xu L: Restoration of tumor suppressor miR-34 inhibits
human p53-mutant gastric cancer tumorspheres. BMC Cancer.
8:2662008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bommer GT, Gerin I, Feng Y, Kaczorowski
AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al:
p53-mediated activation of miRNA34 candidate tumor-suppressor
genes. Curr Biol. 17:1298–1307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kapoor R, Rizvi F and Kakkar P: Naringenin
prevents high glucose-induced mitochondria-mediated apoptosis
involving AIF, Endo-G and caspases. Apoptosis. 18:9–27. 2013.
View Article : Google Scholar
|
|
54
|
Kim WH, Lee JW, Suh YH, Hong SH, Choi JS,
Lim JH, Song JH, Gao B and Jung MH: Exposure to chronic high
glucose induces β-cell apoptosis through decreased interaction of
glucokinase with mitochondria. Diabetes. 54:2602–2611. 2005.
View Article : Google Scholar : PubMed/NCBI
|