Introduction
BCL2-like 12 (BCL2L12), a new member of BCL2 family,
is currently recognized as an apoptosis regulator (1). Its functional role appears
contradictory in different types of cancer (2,3).
In glioblastoma, BCL2L12 is highly expressed and associated with
antiapoptotic properties. The BCL2L12-mediated antiapoptotic
property is thought to be due to several mechanims: i) It
attenuates caspase-dependent apoptosis through direct interaction
with caspase-7 and indirect inactivation of caspase-3 activity via
upregulating crystallin αB (CRYAB) expression. ii) BCL2L12
neutralizes TP53's fucntion at the transcriptional and
translational levels as demonstrated by using
co-immunoprecipitation (4,5).
Downregulating BCL2L12 expression has been demonstrated to reduce
glioblastoma growth in animal models (6). An RNAi-based therapy using spherical
nucleic acid nanoparticle conjugates was reported to effectively
knock down endogenous BCL2L12 mRNA and protein levels and sensitize
glioma cells toward therapy-induced apoptosis by enhancing effector
caspase and p53 activity (6). To
investigate the antiapoptotic properties of BCL2L12, we previously
demonstrated that it is as a substrate of glycogen synthase kinase
(GSK) 3β, and that GSK3b-mediated phosphorylation at BCL2L12 Ser156
is crucial for anti-apoptosis (7). Conversely, treatment with LiCl, a
GSK3β pharmaceutical inhibitor that diminishes GSK3b-mediated
BCL2L12 phosphorylation, restored caspase activity and thus it
might have great potential in treating glioblastoma multiforme
(GBM) in addition to being a mood stabilizer in the clinic
(7). Furthermore, a previous
study using bioinformatics has demonstrated that in addition to
harboring a BH2 domain, BCL2L12 contains a BH3-like domain
(8). Our group further
characterized the function of this BH3-like domain and its
potential antiapoptotic properties. Exogenous expression of the
BCL2L12 mutant that was directly site-mutated at the -4 or 0
hydrophobic residue demonstrated an elevated apoptotic marker
expression when compared with the BCL2L12 wild-type group. In
addition, it was demonstrated that this BH3-like domain may be
associated with temozolomide (TMZ)-induced autophagy (9). Without this hydrophobic groove
region, TMZ is unlikely to induce effective activation of autophagy
markers.
In cell biology, cell proliferation, apoptosis and
autophagy are the predominant activities that determine cell fate.
The balance of these three cellular activities leads to different
outcomes in cancer treatment. One of the main options for GBM
treatment, TMZ, is usually co-administered with radiotherapy,
generally with good response. More in-depth pretreatment and
treatment evaluations are still needed for refining effective
personalized medicine approaches. Traditionally, status of p53 and
O6-methylguanine DNA methyltransferase (MGMT) is essential
reference in order to determine the TMZ effect in GBM treatment
(10,11). However, more advanced MGMT
activity/methylation status, p53 expression, reactive oxygen
species (ROS) production following chemotherapeutic intervention,
measurement of apoptosis induction, and unwanted autophagy level
(drug-induced pro-survival autophagy) are all critical indicators
in modern cancer therapy. A previous study has revealed that glioma
cells treated with high doses of TMZ may enhance ROS production and
induce autophagy rather than apoptosis (12,13). This TMZ-induced autophagy is
uncommon as a pre-death procedure but is linked to drug resistance
and remains a significant challenge in GBM treatment. The canonical
pathway of autophagy is governed by a subset of autophagy genes
(14). One of these genes,
Beclin-1 [also known as autophagy-related (Atg) 6, unlike the
others, is known to regulate either apoptosis or autophagy based on
its interactive partnerships; antiapoptotic when it interacts with
BCL2 apoptosis regulator (BCL2) and BCL-extra large (BCL-XL) via
the BH3 domain, or autophagy-inducing when it interacts with
phosphatidylinositol 3-kinase Vps34 (Vps34) and phosphoinositide
3-kinase (PI3K) (15,16). As aforementioned, BCL2L12 harbors
a BH3-like domain associated with TMZ-induced autophagy and
antiapoptotic properties in GBM (9). In addition, BCL2L12 and Beclin-1
share a similar binding pattern to BCL2 and BCL-XL (15,17). Ectopic expression of BCL2L12 also
enhanced Beclin-1 expression in glioma cell lines (9). Therefore, the present study
hypothesized that the BCL2L12 BH3-like domain might functionally
compensate for Beclin-1 in either regulating apoptosis or
autophagy.
Materials and methods
Cloning
BCL2L12, BCL-XL, BCL2, BCL2 associated X (Bax),
Beclin-1, induced myeloid leukemia cell differentiation protein
MCL-1 (Mcl-1), and GSK3β were cloned into pACT2 and pAS2-1 vectors
(Takara Bio Inc., Otsu, Japan) for yeast two-hybrid assay or into
the pEGFP-C1 vector for overexpression. Polymerase chain reaction
(PCR) technique was used to generate DNA fragments containing the
desired gene sequences utilizing primers designed to contain the
Xho I and Bam HI restriction enzyme recognition
sites. For gene-specific PCR, 100 ng genomic DNA was used as
template, and 2.5 µl 10X reaction buffer (Takara Bio Inc.),
4 µl 2.5 mM dNTPs (Takara Bio Inc.), 0.2 µl DNA
polymerase (5 U/µl; Takara Bio Inc.), and 1 µl of 10
µM primers were added into a 25 µl reaction mix. The
PCR was conducted on a GeneAmp 9,700 PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA), as
follows: 95°C for 30 sec, followed by 30 sec annealing, and
extension at 72°C for 35 cycles. The PCR products were separated on
2% agarose gel and visualized by staining with 0.5 µg/ml
ethidium bromide. The PCR products with correct sizes were further
purified using a PCR clean-up kit (GeneMark Technology Co., Ltd.,
Tainan, Taiwan) and subjected to restriction enzyme digestion.
Plasmid DNA was further confirmed by DNA sequencing (Mission
Biotech, Taipei, Taiwan).
Yeast two-hybrid assay
This assay is intended to screen an interaction
between two proteins called 'bait' and 'prey'. In the present
study, BCL2L12, Bax, Beclin-1, BCL2, BCL-XL, and Mcl-1, as well as
several BH3 mutants of BCL2L12, were included in the assay in order
to test their potential interactions. Standard techniques were used
to perform the yeast two-hybrid screening (18-20), using the MATCHMAKER Two-Hybrid
System 2 (Clontech Laboratories, Inc., Mountainview, CA, USA).
YRG-2 yeast host cells (MATa ura3-52 his3-200 ade2-101 lys2-801
trp1-901 leu2-3 112 gal4-542 gal80-538
LYS2::UASGAL1-TATAGAL1-HIS3
URA3::UASGAL4 17mers(×3)-TATACYC1-lacZ) were
purchased from Stratagene (Agilent Technologies, Inc., Santa Clara,
CA, USA). The yeast cells were cotransfected with the pAS2-1 and
pACT2 plasmids and then selected on G2 plates deficient in
tryptophan and leucine, and on G3 plates further deficient in
histidine. A positive interaction was determined by growth on G3
plates and by a visible blue color pattern in the subsequent colony
filter lift assay (19). In the
yeast two-hybrid system, YRG2 yeast cells were inoculated into 5 ml
YPD medium and grown at 28°C, 240 rpm for 20 h. YPD medium (45 ml)
was added to the original yeast culture to refresh their growth and
further incubated for 4 h. To detect whether gene A (encoding
'bait' protein) interacts with gene B (encoding 'prey' protein),
the pACT2 vector/gene A (1.2 µg) was premixed with pAS2-1
vector/gene B (1.2 µg), then further mixed with 10 µl
boiled salmon sperm DNA and 800 µl of the yeast solution.
The reaction was vortexed completely and incubated at 28°C, 7.7 × g
for 30 min, and supplemented with 80 µl DMSO (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). Then, the reaction was
heat-shocked at 42°C for 15 min and placed on ice for 2-5 min. The
reaction was further centrifuged at 25°C, 800 × g for 3 min, and
the supernatant was discarded. The transformed yeast pellet,
resuspended in sterilized water, was spread on nutritional
deficient plates G2 and G3, respectively. The G2 or G3 plates were
incubated at 28°C for colony growing. Afterwards, sterilized filter
papers were used to cover the G2/G3 plates. The filter papers, with
the yeast colonies attached and absorbed, were further dipped into
liquid nitrogen for 40 sec to break any enclosed yeast cell walls.
X-gal solution (1,600 µl) was added onto another filter and
incubated at 28°C to detect whether the 'bait' protein would
interact with 'prey' protein.
Site-directed mutagenesis
The specific primers for genes encoding the Bax L63A
mutation and the BCL2L12 BH3-like domain mutations h1, h2, h3,
charge, and h4, and the complementary sequences surrounding the
mutation sites, were designed and synthesized as previously
reported (19). Following PCR
amplification, 1.5 µl Dpn I (10 U/µl) was
added for digestion at 37°C for 10 min. Dpn I-treated DNA
was transferred into 45 µl of prepared competent cells.
Following incubation on ice for 30 min, heat shock was performed in
a 42°C water bath for 30 sec. The reaction mixture was kept on ice
for 2 min, and then 500 µl LB medium was added and incubated
at 37°C, 5.37 × g, for 1 h. Following incubation, bacterial
cultures were spread on selective plates containing antibiotics and
incubated at 37°C for 16-18 h. Several colonies were picked for
plasmid preparation, restriction enzyme digestion and sequencing to
confirm the desired mutations.
Molecular dynamics simulations of the
BCL2L12 phosphorylation at Ser156
The initial 3D model of BCL2L12 was obtained as
previously described (9). Next,
the Ser156 of BCL2L12 was phosphorylated with the AmberTools17 and
amber parameter database (21).
Then the model was inserted into the tip3p water box. The molecular
dynamics (MD) simulations were performed in the canonical ensemble
with a simulation temperature of 310 K, unless stated otherwise, by
using the Verlet integrator with an integration time step of 0.002
ps and SHAKE constraints (22) of
all covalent bonds involving hydrogen atoms. In the electrostatic
interactions, Atom-based truncation was performed using the PME
(23) method, and the switch van
der Waals function was used with a 1.80 nm cutoff for atom-pair
lists. The structure was minimized for 100,000 conjugate gradient
steps and was then subjected to a 100 s isothermal, constant volume
MD simulation. All the MD simulations were performed with Amber 16
(pmemd.cuda) program. The final structure was used in the BH3
domain comparisons (BCL2L12 and phosphorylation at Ser156 of
BCL2L12).
Cell culture, transfection, and
treatments
Human glioblastoma U87MG (cat. no. HTB-14), H4 (cat.
no. HTB-148) and T98G (cat. no. CRL-1690) cell lines were purchased
from American Type Culture Collection (Manassas, VA, USA). Cells
were cultured using MEM (Gibco; Thermo Fisher Scientific Inc.) and
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
antibiotics (penicillin and streptomycin, 100 IU/l) at 37°C, 5%
CO2 in air atmosphere. For transient transfections,
cells were seeded into 100 mm diameter culture dishes (BD
Biosciences, Franklin Lakes, NJ, USA) at a density of
2×106 cells per dish. DNA (1-2 µg/per well of 6
well cluster plate) was transfected into cells for 12-16 h using
Lipofectamine 2000 transfection reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). To induce apoptosis, staurosporine (STS; 0.5
µM; Sigma-Aldrich; Merck KGaA) or TMZ (Sigma-Aldrich; Merck
KGaA) (400 µM) was added to the culture for 24 and 48 h,
respectively. STS treatment was the positive control for apoptosis
induction. ABT-737 (Sigma-Aldrich; Merck KGaA) was applied alone or
in combination with TMZ at a concentration of 50 µM.
Western blotting
RIPA Lysis buffer (Protech Inc., Taipei, Taiwan) was
used to prepare the total lysates from cultured cells. For
detecting cytochrome c efflux, the cytosolic fraction was
prepared by a Mitochondria/Cytosol Fractionation kit (Merck
Millipore, Billerica, MA, USA), according to the manufacturer's
protocol. Protein concentration was determined using Protein Assay
reagent (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein
lysates (40 µg/lane) were separated on 12% SDS-PAGE and then
transferred onto methanol-treated polyvinylidene fluoride
membranes. The transferred membranes were blocked in TBST (TBS/0.1%
Tween-20) with 5% non-fat dry milk and was agitated for 1 h, and
then incubated with primary antibodies for 1 h. The primary
antibodies targeting microtubule associated protein 1 light chain 3
β (cat. no. 2775; LC3B; recognizing both LC3B-I and II), Atg12-Atg5
conjugates (cat. no. 2010), Beclin-1 (cat. no. 3738), full length
poly-ADP-ribose-polymerase (PARP; cat. no. 9542), cleaved PARP
(cat. no. 9542), cleaved caspase-9/-3 (cat. nos. 9502 and 9662),
cytochrome c (cat. no. 4272), BCL2 (cat. no. 2872), Bax
(cat. no. 2774), BCL-XL (cat. no. 2762), Mcl-1 (cat. no. 4572),
BCL2-like 11 (also known as Bim; cat. no. 2819), BCL2-related
ovarian killer protein (Bok; cat. no. 4521) and BCL2-binding
component 3 (also known as Puma; cat. no. 4976) were purchased from
Cell Signaling Technology, Inc. (Beverly, MA, USA). The primary
antobodies targeting green fluorescent protein (GFP, clone B-2;
cat. no. sc-9996), p53 (clone DO-1; cat. no. sc-126) and β-actin
(clone C4; cat. no. sc-47778) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Next, the membranes were
incubated with secondary antibodies conjugated with horseradish
peroxidase (HRP; Cell Signaling Technology, Inc.; anti-rabbit IgG,
HRP-linked antibody cat. no. 7074 and anti-mouse IgG, HRP-linked
antibody cat. no. 7076) for another 1 h. Both primary and secondary
antibodies were diluted in 1% non-fat dry milk or 5% BSA in TBST.
The protein signals were developed using enhanced chemiluminescence
reagent (GE Healthcare, Chicago, IL, USA) and recorded using Fuji
X-ray film Super RX (Fujifilm, Tokyo, Japan) for X-ray
autoradiography.
Statistical analysis
The western blot analyses were quantified with
ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Bar charts were generated using Sigma plot software version 12.3
(Systat Software Inc., Chicago, IL, USA). Data were expressed as
the mean ± standard deviation. All data were analyzed using the
SPSS for Windows 21.0 statistical software (IBM Corps., Armonk, NY,
USA). Statistical significance between groups was examined with
one-way analysis of variance for multiple comparisons followed by
Bonferroni correction for adjusting the P-value of multiple tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
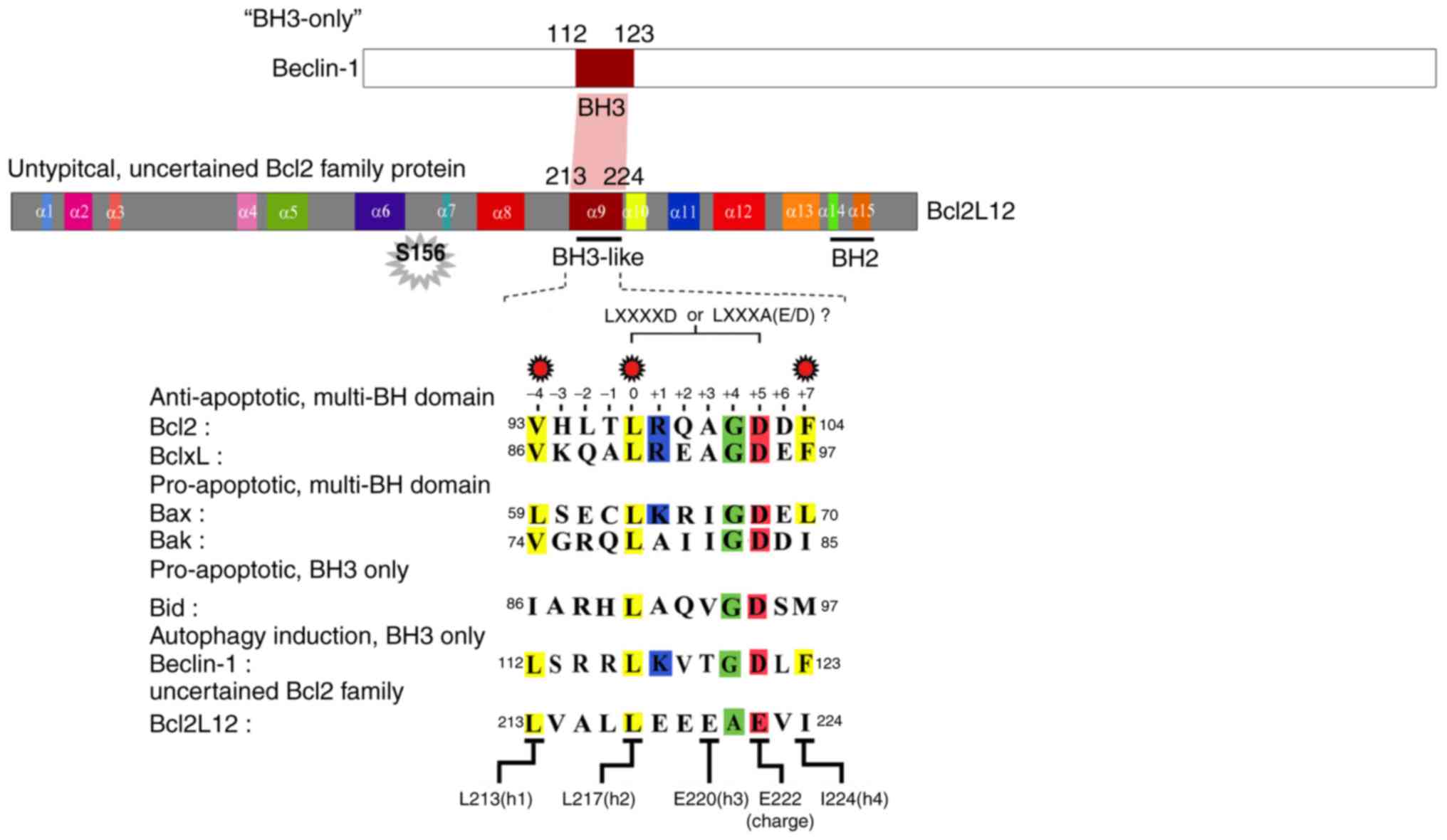
BCL2L12 contains a BH3-like domain on its
α-9 helix, and this 12-residue motif is conserved among the BCL2
family proteins
The structural similarity of the BH3-like domain of
BCL2L12 was compared to those of BCL2, BCL-XL, and Bax. As
illustrated in Fig. 1, the α-9
helix of BCL2L12 is structurally similar to the α-2 helix of
multiple BCL2 family proteins, including the antiapoptotic (BCL2
and BCL-XL) and proapoptotic (Bak and Bax) subgroups. To highlight
the structural/functional similarity among these BH3 domains, five
key amino acid residues were analyzed for their effects on the
interaction between BCL2L12, BCL2 and BCL-XL. As reported
previously, the L213 (-4), L217 (0) and I224 (+7) hydrophobic
residues are crucial for the BCL2L12 interaction with BCL2 and
BCL-XL in a yeast two-hybrid system (Fig. 1) (9). It was further determined that the
BH3 domain most likely consists of a 12-residue-long core motif of
LXXXAE/D in BCL2L12 instead of the canonical motif 'LXXXXD' in Bak
or other BCL2 family proteins. Since BCL2L12 interacts with BCL2
and BCL-XL, which shares similar interacting partnerships with Bax
and Beclin-1, it was hypothesized that the BCL2L12 BH3-like domain
may be necessary for both autophagy and apoptosis regulation.
Previously, it was reported that overexpressed BCL2L12 L213A and
L217A mutants resulted in reactivation of apoptotic markers with or
without STS treatment (9).
Therefore, the present study investigated L213A as a representative
BH3-like domain mutant in the subsequent cell-based assays.
BCL2L12 does not interact with Beclin-1,
but shares a similar binding partnership with Beclin-1 to interact
with BCL2 and BCL-XL, but not Mcl-1
The interactions between BCL2L12, Bax, Beclin-1 and
anti-apoptotic BCL2 family proteins, BCL2, BCL-XL and Mcl-1, were
investigated. GSK3β was used as positive control for its
interaction with BCL2L12. Several BH3 mutants of BCL2L12 (I209A,
L213A, L216A, L217A, E218A and E220A) (90-229, refers to the
BCL2L12 truncated fragment spanning between the 90th amino acid
residue to 229th residue. The plasmid harboring this BCL212
truncated fragment was further used as a parental vector to obtain
mutants by a site-direct mutagenesis technique) as well as the
binding-deficient mutant L63A of Bax (1-171), were tested in a
yeast two-hybrid system, in order to determine whether they could
abolish the interaction between BCL2L12, Bax, and their interacting
proteins. As listed in Table I,
both BCL2L12 (70-266) and Bax (1-171) could interact with BCL2
(1-211) and BCL-XL (1-209). Mutation on L213 of BCL2L12 (70-266)
and L63A of Bax (1-171) resulted in a loss of binding to either
BCL2 or BCL-XL. In addition, BCL2L12 (three different fragments
comprising 70-266, 70-334 and 90-229) could also interact with Bax
(1-171); however, BH3 mutants were unlikely to disrupt the binding
between BCL2L12 (90-229) fragments and Bax (1-171). BCL2L12
(70-266) failed to interact with Beclin-1 (1-450). Mcl-1 could not
interact with Beclin-1(1-450), Bax (1-171), BCL2L12 (70-266), and
BCL2L12 (70-334). Altogether, based on the binding partnership in
the present yeast two-hybrid system and the results from previous
studies, BCL2L12 and Bax shared similar binding partnerships
towards BCL2 and BCL-XL, probably through the BH3 domain; however,
their interaction (BCL2L12-Bax) was independent of the BH3-like
domain (Table I). BCL2L12 and
Beclin-1 share positive binding to BCL2 and BCL-XL and non-binding
to Mcl-1, even though they are unlikely to interact with each
other.
 | Table IYeast two-hybrid assay of BCL2L12 and
Beclin-1 interaction with BCL2 family proteins and GSK3β. |
Table I
Yeast two-hybrid assay of BCL2L12 and
Beclin-1 interaction with BCL2 family proteins and GSK3β.
| pGAL4-DBD
vector(pAS2-1) | pGAL4-AD vector
(pACT2)
|
|---|
|
BCL-XL(1-209) |
BCL2(1-211) |
Beclin-1(1-450) |
Bax(1-171) |
BCL2L12(70-266) |
BCL2L12(70-334) |
BCL2L12(90-229) |
BCL2L12(153-191) |
BCL2L12(90-229), I209Aa |
|---|
|
BCL2L12(70-266) | B+ | B+ | – | B+ | ND | ND | ND | ND | ND |
|
BCL2L12(70-266), L213A | – | – | ND | ND | ND | ND | ND | ND | ND |
|
Bax(1-171) | B+ | B+ | ND | ND | B+ | B+ | B+ | ND | B+ |
|
Bax(1-171), L63A | – | – | ND | ND | ND | ND | ND | ND | ND |
|
Mcl-1(1-327) | ND | ND | – | – | – | – | ND | ND | ND |
| GSK3β | ND | ND | – | ND | B+ | B+ | B+ | B+ | ND |
BH3-like domain L213A mutant restores
apoptotic marker activities in U87MG, but not T98G cells
In two glioma cell lines overexpressing GFP, BCL2L12
wt and L213A mutant, cleaved-PARP and cleaved-caspase-3 were
reactivated in the L213A overexpressing group compared with the
BCL2L12 wild-type group (Fig. 2A and
B). This phenomenon was evident in U87MG cells, but not in T98G
cells (Fig. 2A and B). STS
treatment further enhanced this effect (Fig. 2A and B). Anti-apoptotic BCL2
proteins, including BCL-XL, Mcl-1 and BCL2, were highly expressed
in U87MG cells compared with T98G cells. Overexpression of the
L213A mutant did not alter the expression levels of these
antiapoptotic BCL2 proteins (Fig.
2C). By contrast, the proapoptotic and BH3-only BCL2 family
proteins Bax, Puma, Bok, and Bim, appeared more abundantly
expressed at the basal level and following STS treatment in T98G
compared with U87MG cells (Fig.
2D). However, these proapoptotic and BH3-only BCL2 family
protein expression levels are unlikely to be affected by BCL2L12 or
BCL2L12 L213A mutant expression. Since high expression of BCL2,
BCL-XL and Mcl-1 in cancer cells is reported to be associated with
acquired resistance to chemotherapy, the present study investigated
whether TMZ treatment caused any expression changes among these
three proteins in glioma cells. As illustrated in Fig. 3, TMZ treatment significantly
downregulated Mcl-1 expression, but not BCL-XL and BCL2 expression,
and this effect was independent of the expression status of BCL2L12
and its mutant (S156A and L213A).
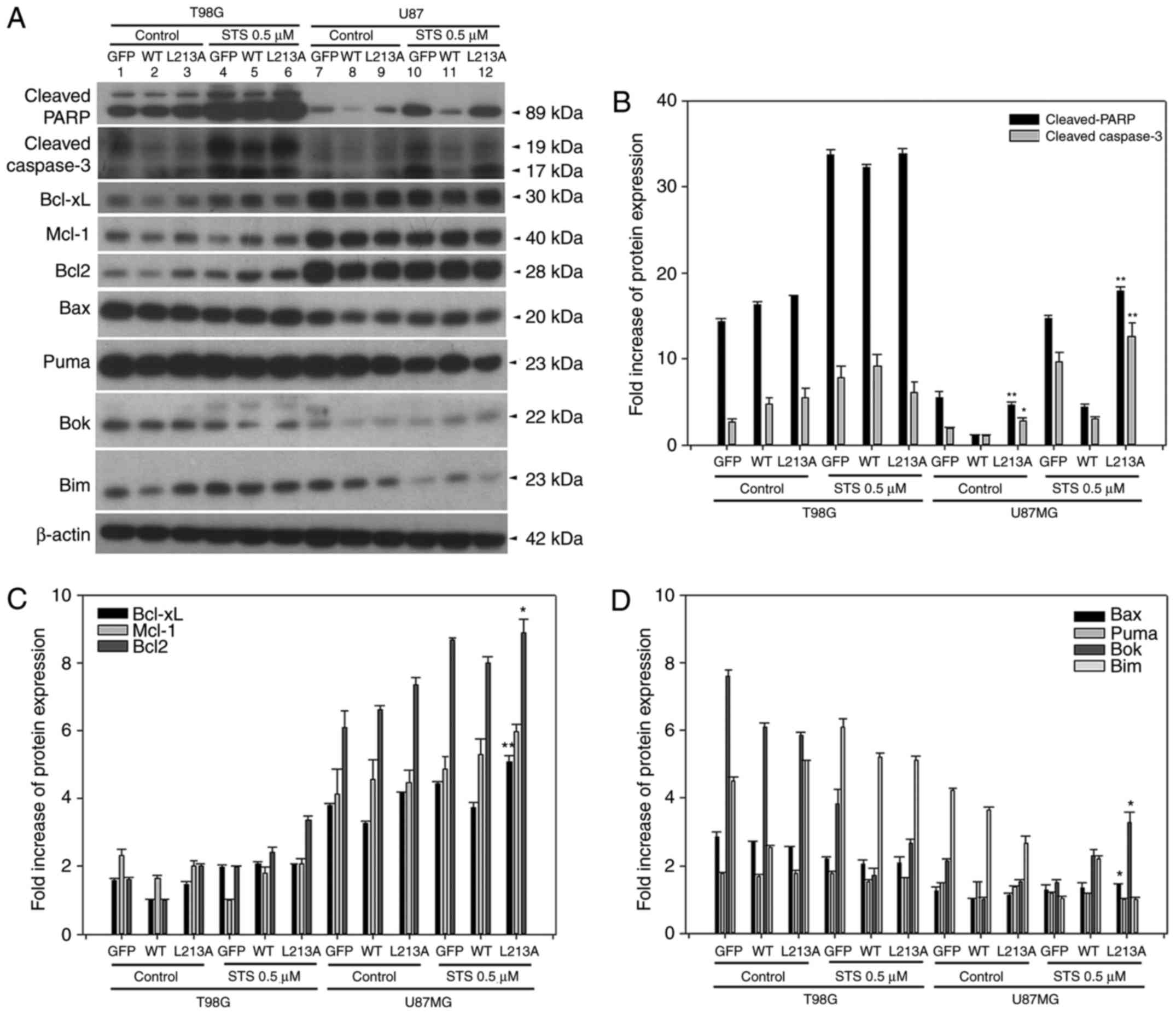 | Figure 2BCL2L12 L213A is associated with
reactivation of caspase-3 and the BCL2 family protein expression
profiles are different in T98G and U87MG cell lines. (A) The
protein expression levels of apoptotic markers and BCL2 family
proteins were detected by western blotting. (B) Quantitative plot
of cleaved PARP and cleaved caspase-3, (C) prosurvival proteins
BCL-XL, Mcl-1, and BCL2, and (D) proapoptotic BCL2 family proteins
expression in two glioma cell lines. All target protein expression
levels were normalized to β-actin expression prior to conversion
into fold increase measurements. *P<0.05 and
**P<0.01 compared with WT group. BCL2L12, BCL2-like
12; BCL2, BCL2 apoptosis regulator; PARP,
poly-ADP-ribose-polymerase; BCL-XL, BCL-extra large; Mcl-1, induced
myeloid leukemia cell differentiation protein MCL-1; WT, wild-type;
Bax, BCL2 associated X; Puma, BCL2-binding component 3; Bok,
BCL2-related ovarian killer protein; Bim, BCL2-like 11. |
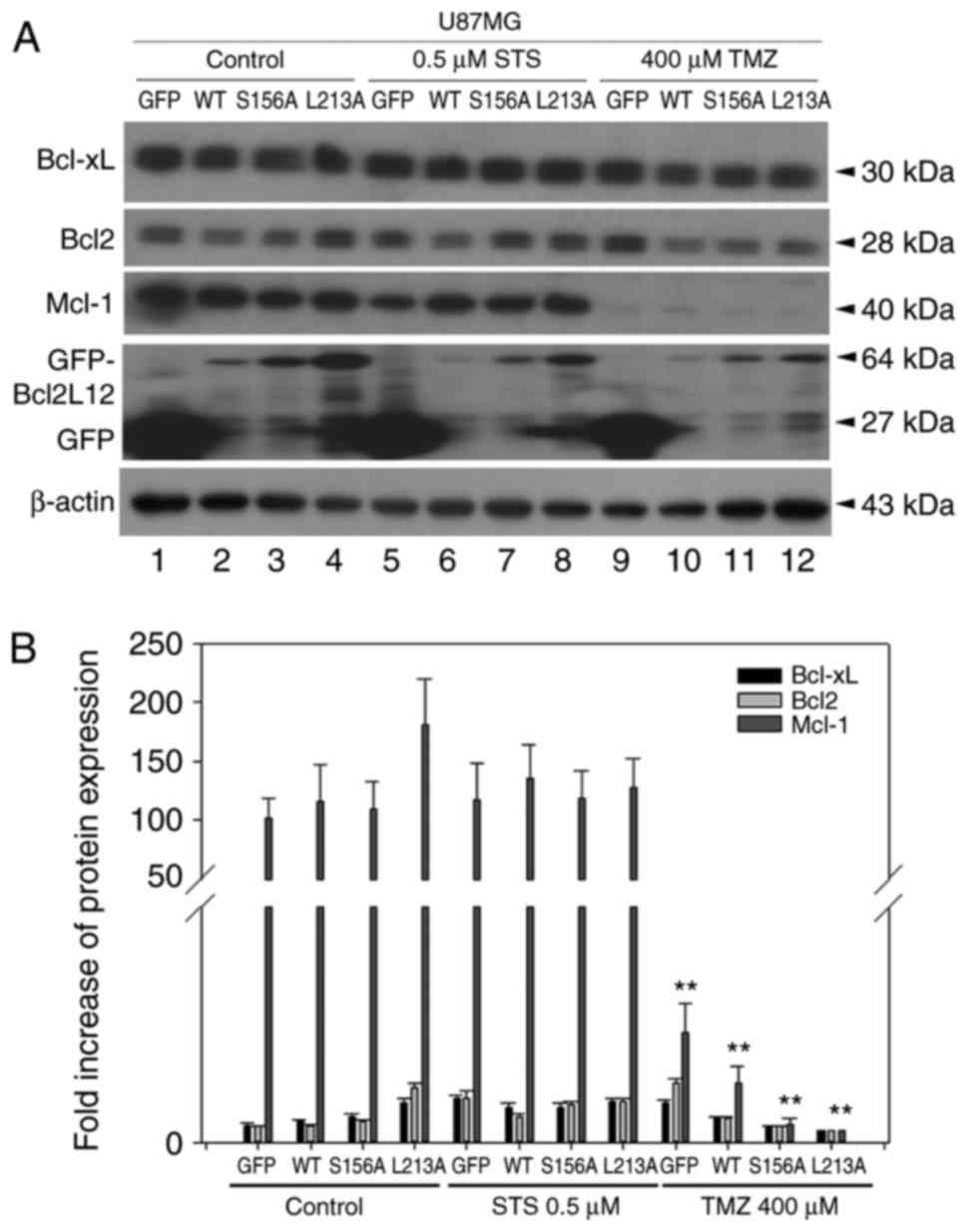 | Figure 3TMZ treatment downregulates Mcl-1,
creating a desirable microenvironment for applying ABT-737. (A) The
expression levels of prosurvival BCL2 family proteins were detected
in different experimental groups of U87MG cells by western
blotting. (B) Quantitative plot of BCL-XL, BCL2 and Mcl-1
expression in the different groups. **P<0.01 compared
with control. TMZ, temozolomide; Mcl-1, induced myeloid leukemia
cell differentiation protein MCL-1; BCL2, BCL2 apoptosis regulator;
BCL-XL, BCL-extra large; BCL2L12, BCL2-like 12; STS, staurosporine;
GFP, green fluorescent protein; WT, wild-type. |
Combination treatment with TMZ and
ABT-737 triggers enhanced apoptosis compared with each agent alone
in U87MG cells
Because of the high levels of BCL2, BCL-XL and
BCL2L12 expression in U87MG cells, it was postulated that BCL2L12
might interact with these antiapoptotic BCL2 family members and
lead to enhanced outer membrane integrity of mitochondria.
Therefore, BCL2L12 with BH3-like domain to trigger
mitochondria-dependent apoptosis was preceded by the treatment of
ABT-737, a mimetic agent of the BH3 domain of Bad, as well as a
selective inhibitor of BCL2, BCL-XL, and BCL-w, alone or in
combination with TMZ in U87MG cells. We previously reported that
both TMZ and ABT-737 resulted in moderate apoptosis compared with
an untreated GFP-alone group in U87MG cells, which express
wild-type p53 (associated with the effects of TMZ) and high levels
of BCL2 and BCL-XL. Bax was also expressed at an intermediate level
with ABT-737 treatment in U87MG cells. However, induction of
apoptosis by these two agents appears to be independent of BCL2L12
L217A expression, especially in combination use (9). When not treated with these two
agents, the L217A group demonstrated an increased level of cleaved
PARP like the L213A group compared with the untreated BCL2L12
wild-type and untreated GFP alone group in U87MG cells (9). Combination treatment with TMZ and
ABT-737 resulted in higher levels of cleaved PARP and cleaved
caspase-3 compared with each agent used alone (9). The induction level of apoptotic
protein expression by these two agents whether used alone or in
combination, the effect is increased compared with the effect
contributed by the exogenously expressed L217A mutant (9). Thus, combination use of these two
agents appears to be efficient in glioma cells with p53 wild-type,
high BCL2 and BCL-XL expression, moderate Bax expression and null
to low background MGMT activation. In addition, TMZ-induced
downregulation of Mcl-1 creates a desirable microenvironment for
the addition of ABT-737 in treating U87MG cells (Fig. 3), as mentioned in a previous study
(9).
GSK3b-mediated phosphorylation on Ser156
and the BH3-like domain contributes to the antiapoptotic property
of BCL2L12 and drug-induced autophagy in U87MG
Overexpressed BCL2L12 S156A and L213A increased the
levels of cleaved PARP and cleaved caspase-3, but not cleaved
caspase-9, compared with the BCL2L12 wild-type group (Fig. 4A and C). This effect was
strengthened in the two drug treatment groups, where cleaved PARP
and cleaved caspase-3 has dramatically altered expression levels.
For cleaved caspase-9, reactivation of apoptotic markers was only
observed in the TMZ-treated group (Fig. 4A and C). By contrast, p53 was
reactivated in S156A and L213A-overexpressing cells in both the
untreated and STS-treated group, but not in the TMZ-treated group
(Fig. 4B). In the drug-treated
group, overexpression of wild-type BCL2L12 resulted in an
activation of autophagic markers, including LC3B-II, Atg12-Atg5
conjugates, and Beclin-1 in U87MG cells (Fig. 4A and D). However, following
overexpression of the BCL2L12 S156A and L213A mutants, these
autophagic markers were no longer activated and were observed at
levels equal to the GFP group (Fig.
4A and D).
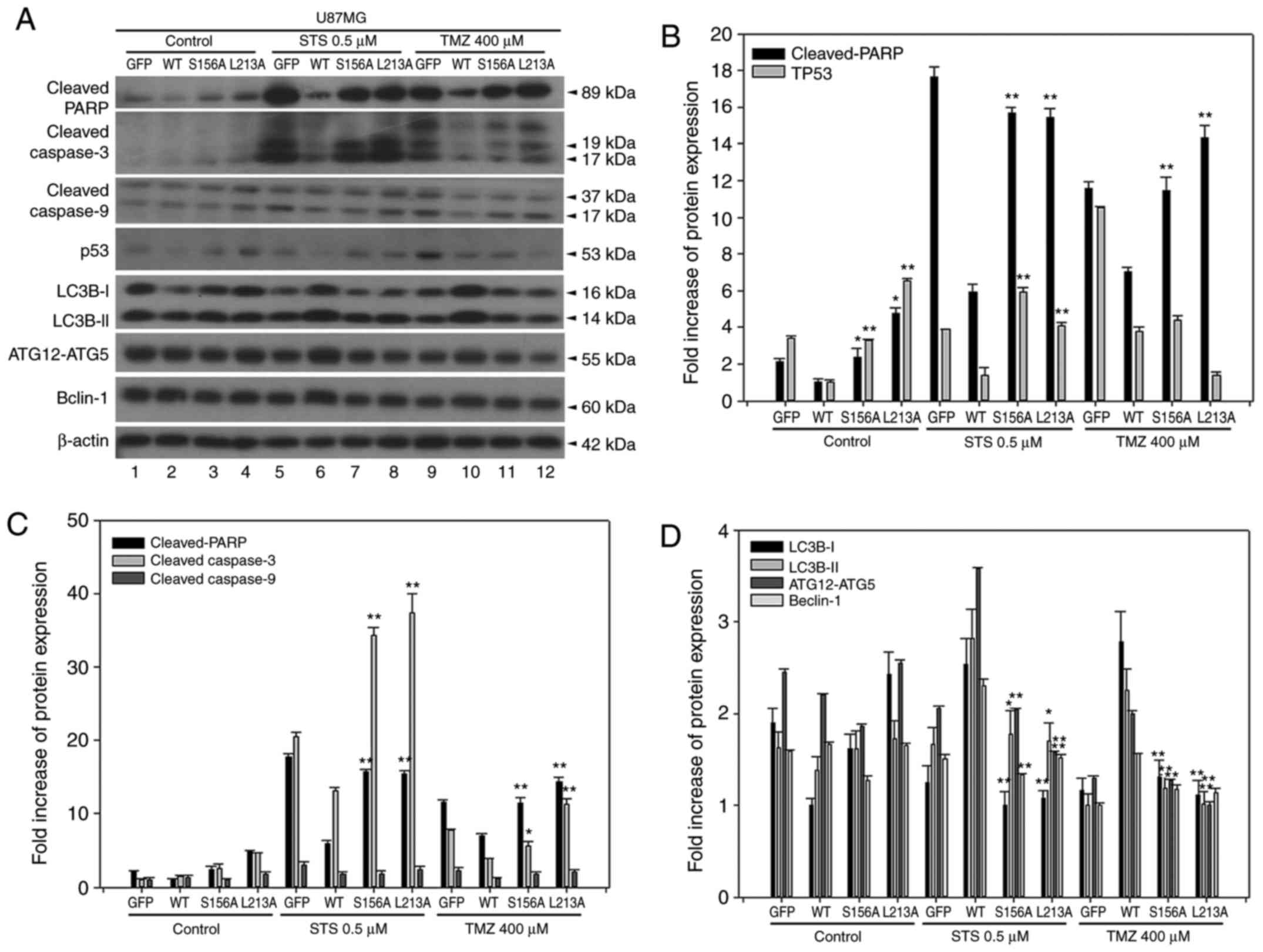 | Figure 4BCL2L12 S156A and L213A mutants are
involved in reactivation of apoptotic markers and drug-induced
autophagy. (A) The expression profiles of p53 and of various
apoptotic and autophagic markers were examined by western blotting.
(B) Quantitative plot of relative cleaved PARP and p53 expression
in U87MG cells with or without expression of BCL2L12 and its
mutants (S156A or L213A). (C) Quantitative plot of relative
expression of apoptotic markers cleaved PARP, cleaved caspase-3 and
cleaved caspase-9 in U87MG cells with or without expression of
BCL2L12 and its mutant proteins. (D) Quantitative plot of relative
expression of autophagy-related proteins LC3B-I, II, ATG12-ATG5
conjugates and Beclin-1 in U87MG cells with or without expression
of BCL2L12 and its mutant proteins. All target protein expression
levels were normalized to β-actin expression prior to conversion
into fold increase measurements. *P<0.05 and
**P<0.01 compared with WT group. BCL2L12, BCL2-like
12; p53, tumor protein p53; PARP, poly-ADP-ribose-polymerase; LC3B,
microtubule associated protein 1 light chain 3B; ATG,
autophagy-related gene; STS, staurosporine; TMZ, temozolomide; GFP,
green fluorescent protein; WT, wild-type. |
TMZ triggers an autophagy-apoptosis shift
event in a time-dependent manner in H4 cells
A previous study revealed that a high concentration
of TMZ may trigger autophagy, which is associated with ROS-mediated
activation of the extracellular signal-regulated kinase (ERK)
signaling pathway (12), as well
as acquired resistance to chemotherapeutics. These experiments were
repeated in the present study in three glioma cell lines in order
to determine whether high-dose TMZ treatment induces an autophagy
to apoptosis shift in a time-dependent manner. Similar to our
previously published work in U87MG cells (9), the present data for U87MG cells
demonstrated that cleaved PARP was activated at 24 h and gradually
reached a peak at 48 h following treatment with 400 µM TMZ
(9). Cleaved caspase-9 was
detected even in the untreated group, and it also increased from 8
to 48 h. Regarding autophagy markers, LC3B-II was detected in the
control group; following TMZ treatment its expression gradually
increased from 8 to 36 h and then dropped at 72 h. Beclin-1 was
also detected in the control group and progressively accumulated
from 8 to 36 h. The expression levels of Beclin-1 at 36 to 72 h
were not different between groups. The BCL-XL expression was
affected by TMZ treatment, gradually increasing from 8 to 72 h.
Altogether, the autophagy-apoptosis shift event in U87MG cells
treated with TMZ appears to be at a high point at 36 h, and
autophagy appears to be induced earlier than a subsequent apoptosis
event. In T98G cells, cleaved PARP and caspase-9/-3 activation was
observed in the untreated group from 8 to 72 h. However, in
contrast to apoptotic markers, expression levels concerning
LC3B-II, Beclin-1, and BCL-XL were unaltered by TMZ treatment
(9). In H4 cells, activation of
cleaved PARP and cleaved caspase-9 was detected in the untreated
group and gradually increased until 36 h (cleaved PARP) or 72 h
(cleaved caspase-9) following TMZ treatment (Fig. 5A and B). In addition, cytochrome
c was more abundantly expressed at 72 h (Fig. 5A and B). Autophagy markers LC3B-II
and Beclin-1 were consistently detected until 36 h and were
decreased at time points beyond 36 h (Fig. 5C). The BCL2 expression pattern was
highly similar to that of Beclin-1, and the BCL-XL expression
pattern was gradually accumulative until 96 h with a peak at 72 h
(Fig. 5D). Taken together,
treating H4 cells with TMZ triggered an autophagy-apoptosis shift
event similar to that observed in U87MG cells. Furthermore, TMZ
treatment enhanced the protein expression of antiapoptotic BCL2
family members, such as BCL2 (before 36 h) and BCL-XL (after 48 h;
Fig. 5D).
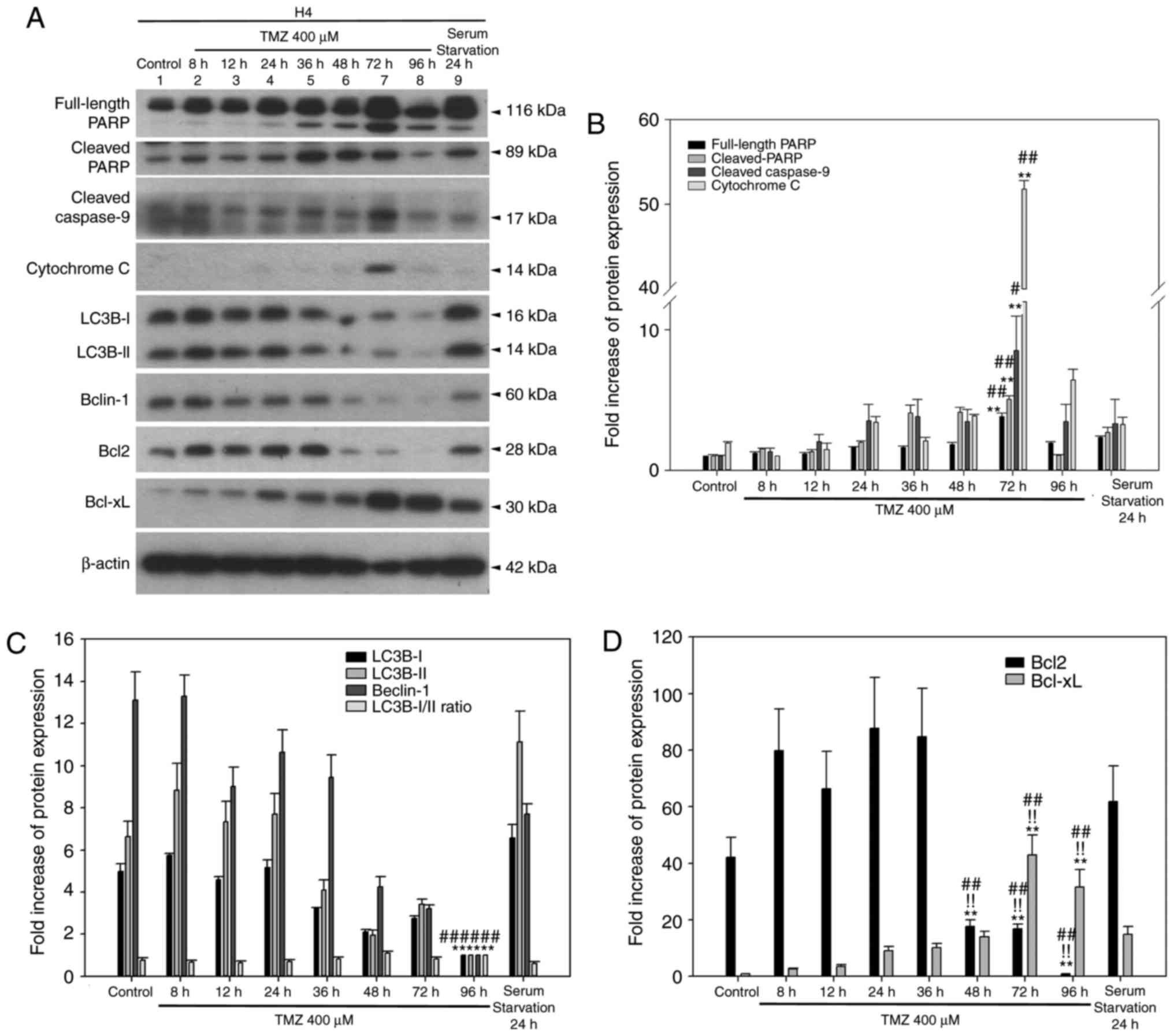 | Figure 5Time-dependent TMZ-induced autophagy
and apoptosis in H4 cells. (A) The expression profiles of apoptotic
and autophagic markers, BCL2 and BCL-XL were determined by western
blotting. (B) Quantitative plot of full-length PARP, cleaved-PARP,
cleaved caspase-9 and cytochrome c expression in H4 cells.
**P<0.01 compared with control and
#P<0.05 and ##P<0.01 vs. the serum
starvation group. (C) Quantitative plot of autophagic proteins
LC3B-I, II, Beclin-1 expression and LC3B-I/II ratio.
**P<0.01 compared with control. (D) Quantitative plot
of BCL2 and BCL-XL expression. **P<0.01 indicate the
significance of comparisons between the TMZ-treated 48, 72, 96 h
group and control; !!P<0.01 vs. 8-36 h group;
##P<0.01 vs. the serum starvation. All target protein
expression levels were normalized to β-actin expression prior to
conversion into fold increase measurements. TMZ, temozolomide;
BCL2, BCL2 apoptosis regulator; BCL-XL, BCL-extra large; PARP,
poly-ADP-ribose-polymerase; LC3B, microtubule associated protein 1
light chain 3B. |
GSK3b-mediated phosphorylation on Ser156
influences the BH3-like domain and contributes to the antiapoptotic
property of BCL2L12
As described above, both GSK3b-mediated
phosphorylation on Ser156 and the BH3-like domain contribute to the
antiapoptotic properties of BCL2L12 and drug-induced autophagy.
Because of the prediction from the protein 3D structure of BCL2L12
containing 16 α-helixes that the BH3-like domain corresponded to
the residues of 211–226 (α-9 helix) (9), it was hypothesized that
phosphorylation at Ser156 may have a functional role on modulating
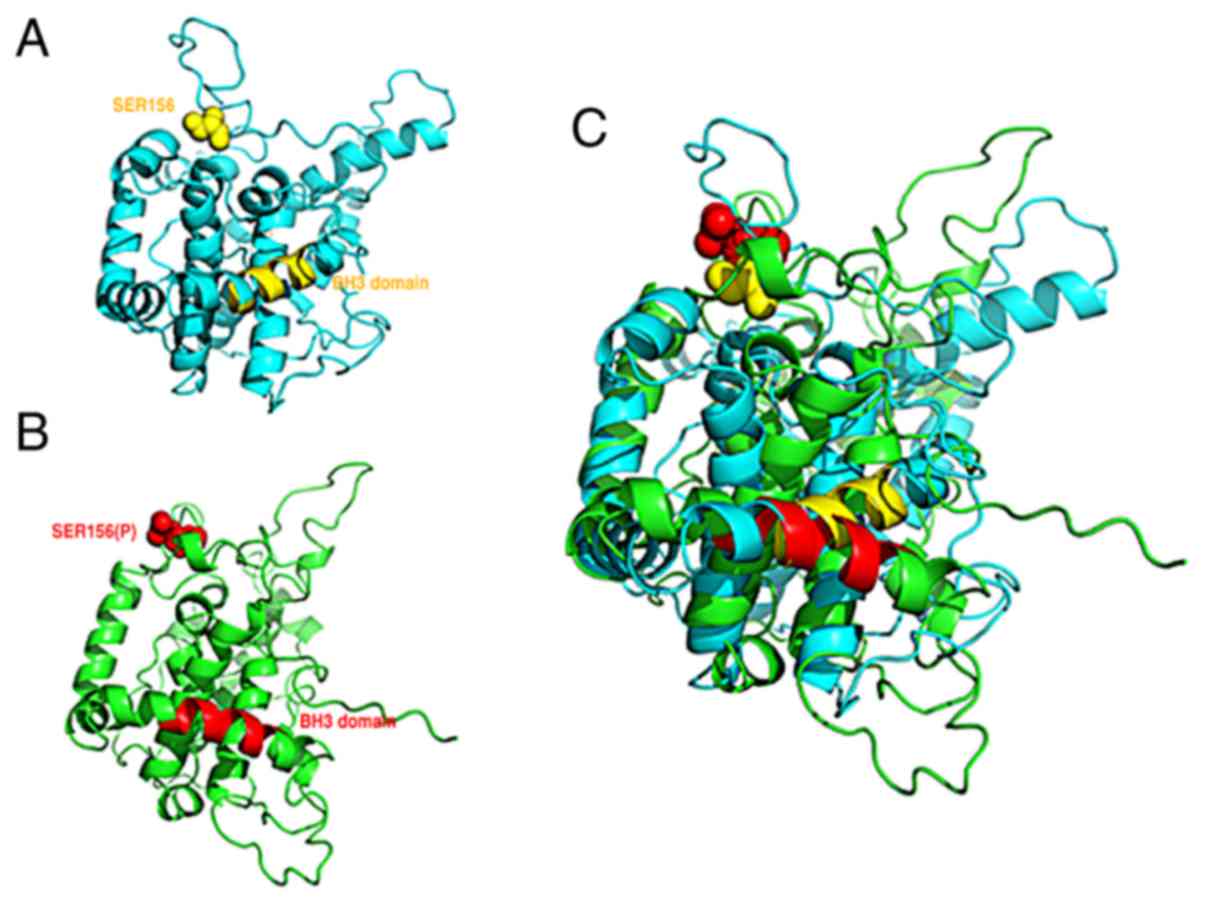
the BH3-like domain. Molecular dynamics simulation data
demonstrated that phosphorylation at Ser156 of BCL2L12 (within α-6
helix and α-7 helix) influences the BH3-like domain conformation
(α-9 helix), suggesting that GSK3β-mediated Ser156 phosphorylation
may function as an allosteric site to modulate the BH3-like domain
of BCL2L12 (Fig. 6). The present
results suggest that BCL2L12 participates in anti-apoptosis and
autophagy via the BH3-like domain and that this BH3-like
domain-mediated function is also further modulated by
GSK3β-mediated phosphorylation at Ser156.
Blockage of TMZ-induced autophagy leads
to a robust activation of apoptotic markers and p53 expression in
U87MG cells
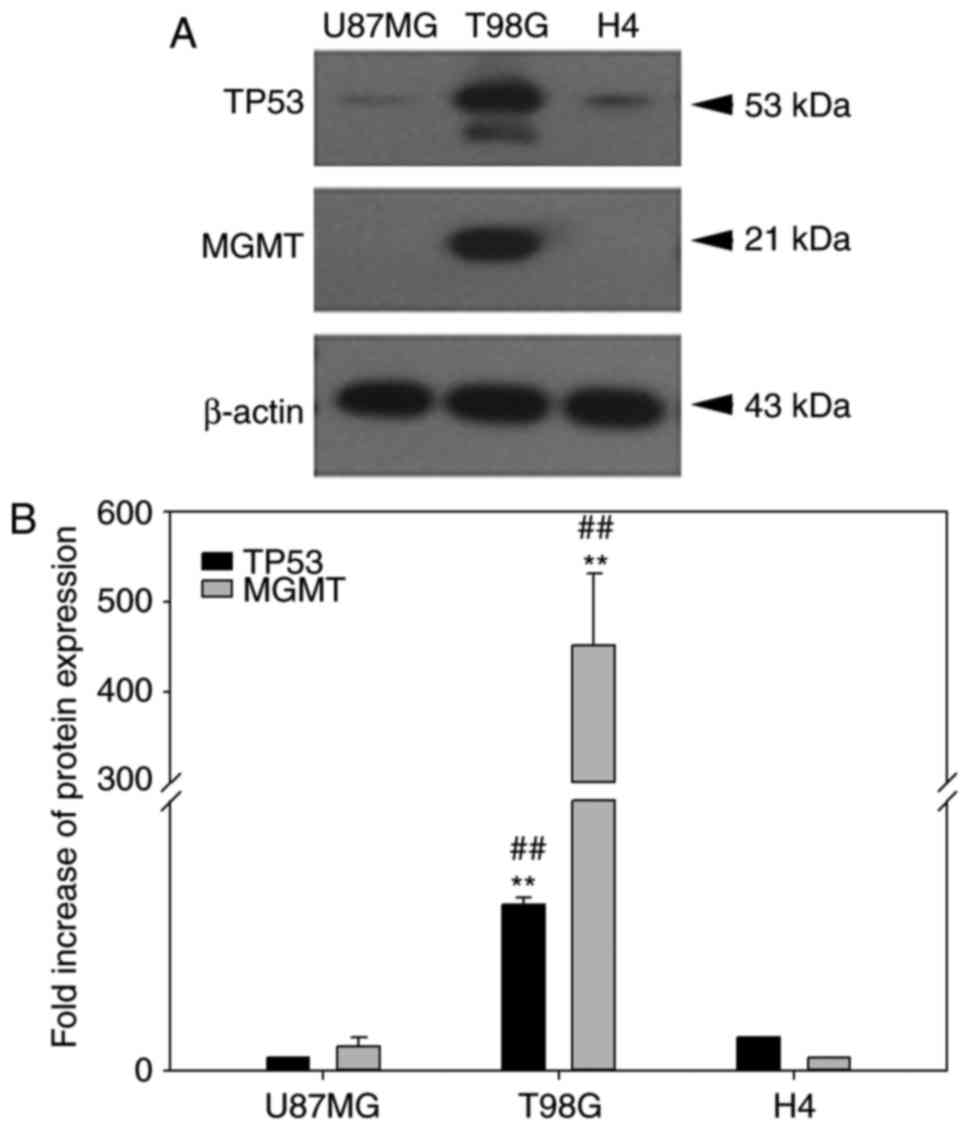
The null-MGMT expression of the U87MG glioma cell
line (24) was further examined
in the H4 cells (previously reported with a low MGMT mRNA
expression) (25), because TMZ is
better used in an MGMT-null background to determine its clinical
relevance. Furthermore, the p53 background and endogenous BCL2L12
expression can influence treatment outcome in glioma, due to their
reported physical interaction (2,26).
In addition to retrieving the p53 expression status using the IARC
p53 cell line database (http://p53.iarc.fr/CellLines.aspx), the endogenous
MGMT and p53 expression in the three glioma cell lines used in the
present study were investigated (Fig.
7). In contrast to T98G cells, U87MG and H4 cells were similar
in MGMT and p53 expression, although the p53 expression in H4 cells
was slightly higher compared with the U87MG cells (Fig. 7). The high expression of MGMT in
T98G cells was consistent with a previous study (27). Therefore, the two major proteins
that may confound the therapeutic effect of temozolomide (28) are controlled in H4 and U87MG
cells. Similar results were reported in Figs. 4 and 5 for U87MG and H4 cell lines in the
present study, therefore reported issues regarding cell line
misidentification of U87MG ATCC when compared to U87MG Uppsala
(29) are unlikely to affect the
net results of the present study. More recently, a study also
demonstrated that the U87MG ATCC cell line has several typical
characteristics of glioblastoma through analyses of cell morphology
and gene expression profile, even though its origin is unknown
(30).
As illustrated in Fig.
8, apoptotic markers, including cleaved PARP, cleaved
caspase-3, cleaved caspase-9, cytochrome c, and p53 were
gradually activated from 12 to 48 h following TMZ treatment,
wheraeas Beclin-1 and BCL-XL levels were unchanged. Treatment with
3-methyladenine (3-MA), an autophagy inhibitor, for 2 or 4 h at 2
mM concentration, downregulated LC3B-II, while the apoptotic
markers p53, cleaved PARP and cleaved caspase-3 were activated
(Fig. 8). This enhancement was
more evident in the 4 h-treated group, indicating that inhibiting
TMZ-induced autophagy may, in turn, activate apoptotic genes and
may serve as a treatment option in GBM therapy.
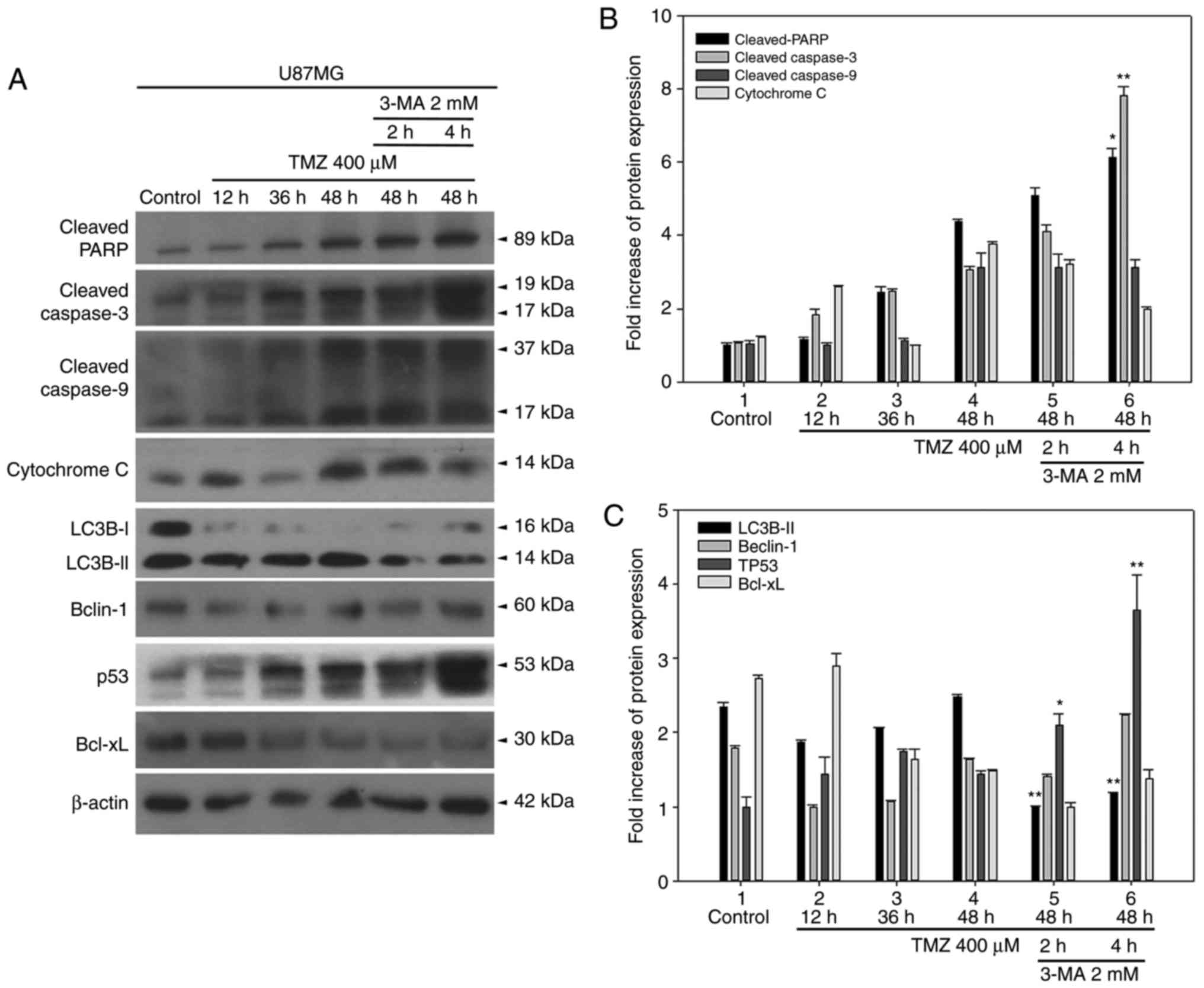 | Figure 8Inhibition of TMZ-induced autophagy
by 3-MA leads to enhanced activation of apoptotic markers and p53
expression in U87MG cells. (A) The expression levels of apoptotic
markers, autophagic markers, p53 and BCL-XL were detected by
western blotting. (B) Quantitative plot of cleaved PARP and cleaved
caspase-3 expression levels. (C) Quantitative plot of LC3B-II,
Beclin-1, p53, and BCL-XL expression levels. *P<0.05
and **P<0.01 compared with TMZ alone. TMZ,
temozolomide; 3-MA, 3-methyladenine; p53, tumor protein p53;
BCL-XL, BCL-extra large; PARP, poly-ADP-ribose-polymerase; LC3B,
microtubule associated protein 1 light chain 3B. |
Discussion
BCL2L12 is a relatively new member of the BCL2
family and only shares a low amino acid similarity to other BCL2
family proteins. It is reported to interact with BCL2, BCL-XL and
Bax (9), but not to interact with
Mcl-1 in yeast two-hybrid assays, and interacts with GSK3β
(7), p53 (27) and caspase-7 (26) in co-immunoprecipitation assays.
BCL2L12 was reported to have two functional variants in 2008
(31), which increased to 9
variants by 2012 (8). More
recently, using 3′RACE technique and new generation sequencing
approaches, another 50 transcripts have been confirmed, but their
functions have yet to be characterized (32). For the interaction between BCL2
proteins in regulating apoptosis, it is known that when BH3-only
proteins, such as Bim, Bad or Noxa, are activated, their
amphipathic α-helical BH3 domain inserts into the hydrophobic
groove of pro-survival BCL2 homologs. This fundamental interaction
initiates apoptosis, but cell death ensues only in cells that
express Bax and Bak (33), the
proapoptotic BCL2 members. When activated, Bax and Bak oligomerize
on the mitochondrial outer membrane and result in loss of outer
membrane integrity. The permeability alteration is accompanied by
the release of apoptogenic proteins and cytochrome c efflux.
These apoptogenic proteins promote activation of caspases, such as
caspase-9, caspase-7 and caspase-3. The BH3-only BCL2 family
proteins are effectors of canonical mitochondrial apoptosis. They
discharge their proapoptotic functions through BH1-3 proapoptotic
proteins, such as Bax and Bak, while their activity is suppressed
by BH1-4 antiapoptotic BCL2 family members, such as BCL2, BCL-XL
and Mcl-1. The precise mechanism by which BH3-only proteins mediate
apoptosis remains unresolved, but three mutually non-exclusive
models are proposed (34).
BCL2L12 contributes to apoptosis regulation, but has
different roles in different types of cancer type. It has yet to be
categorized in any distinct BCL2 protein subgroup, such as
antiapoptotic, proapoptotic, sensitizer, activator and effector. In
fact, as aforementioned, it shares only low amino acid similarity
to the majority of BCL2 family proteins based on full-length amino
acid sequence comparisons. However, its BH3-like domain does
conserve essential residue compositions and retains a similar helix
structure to BCL2, BCL-XL and Bax, as previously reported (9). BCL2L12 is relatively larger than
many BCL2 family proteins, and so is Beclin-1; these two proteins
are not as typical as their homologs. A BH3-only BCL2 protein like
Beclin-1 is unlike Bid, Bim, and Bak; it is 450 amino acids in
length and is suggested to not interact with Bax. Beclin-1 is a
novel BH3-only BCL2 member and autophagy effector. Anti-apoptotic
BCL2 homologs are known to downregulate autophagy through
interactions with Beclin-1, an essential autophagy effector, and
haploinsufficient tumor suppressor, through its BH3 domain, which
is required for BCL2-mediated inhibition of autophagy (16). On the other hand, BCL2L12 is 334
amino acids in length, and only known to have a conserved BH2
domain before 2009 (1,8). Of note, the BCL2L12 BH3-like domain
is located at the α-9 helix, while other BCL2 homolog BH3 domains
are located at the α-2 helix. BCL2L12 harbors a BH3-like domain
associated with TMZ-induced autophagy and antiapoptotic properties
in glioma cells. Beclin-1 also contains a BH3 domain interacting
with BCL2/BCL-XL in a context-specific dual role (autophagy and
anti-autophagy). A dual role (apoptosis/autophagy) is also reported
for another autophagy effector protein, autophagy and Beclin-1
regulator 1 (AMBRA1), which contains a BH3 domain at its C-terminus
that is necessary for AMBRA1 binding with the antiapoptotic factor
BCL2 for its anti-autophagy role. When the proapoptotic
amplification loop is turned on, the caspase-cleaved form of AMBRA1
is bound to antiapoptotic BCL2 proteins, such as Mcl-1, BCL2 and
BCL-XL, but not Bax and Bak (35). In addition to its BH3-like domain,
it is noted that the N-terminus of BCL2Ll2, especially the 1-70
residues, is unable to interact with either BCL2 or BCL-XL (data
not shown). A 3D simulation also reveals that BCL2L12 (1-173) does
not overlap with BCL2, BCL-XL or Bax. In addition, BCL2L12 (90-229)
interacts with Bax (1-171), and mutations on critical residues
within the BH3-like domain are unable to disrupt the interaction
between these two fragments in a yeast two-hybrid system (Table I). These findings indicate that
the N-terminus of BCL2L12 may have uncharacterized functions.
Whether BCL2L12 (70-153) serves as a Bax interacting region or has
any regulating role in Bax's biofunctions needs further
elucidation.
In the present study, overexpression of BCL2L12
Ser156A mutant resulted in reactivation of caspase activity and
downregulation of autophagy markers. It was postulated that
GSK3β-mediated phosphorylation at Ser156 of BCL2L12 may be related
to BH3-like domain functionality since this site is in the middle
of the α6 and α7 helix in proximity to the core motif LXXXAE/D of
the BCL2L12 BH3-like domain at the α9 helix. Phosphorylation at a
Serine site may cause conformational change and possibly more
significant exposure of its BH3-like domain for accessing the
hydrophobic groove of target proteins. We previously reported that
GSK3β-mediated phosphorylation at Ser156 of BCL2L12 is associated
with its antiapoptotic property. Addition of LiCl, a potent GSK3β
inhibitor, resulted in reactivation of caspase-3, -7 and -9, which
is identical to the process detected in U87MG cells over expressing
BCL2L12 Ser156A (7). In the
present study, the results demonstrated that following STS or TMZ
treatments, overexpression of the BCL2L12 L156A mutant reversed
autophagy, as the functional consequence of diminishing the
BH3-like domain via overexpression of the BCL2L12 L213A mutant.
Therefore, in U87MG cells, or in cells with wild-type p53, null or
low MGMT activation, high BCL2, BCL-XL and Mcl-1 expression,
drug-induced autophagy may be the initial step of anti-apoptosis
events. Thus, it can be suggested that cytoprotective autophagy
coordinated by both the BCL212 and Beclin-1 BH3 domain may
counteract p53-dependent apoptotic or BCL2-mediated anti-autophagic
signaling. Once the death signaling is irreversible, or the
pro-survival autophagy is overwhelmed, cells may undergo apoptosis.
When treated with TMZ or STS, a time-dependent autophagy-apoptosis
shift event occurs, and this pro-survival autophagy is linked to
acquired resistance to chemotherapeutics. Hence, inhibiting this
pro-survival process, or inhibiting drug-induced autophagy events,
may lead cancer cells to follow the fate of cell death (Fig. 9) (15,16,36).
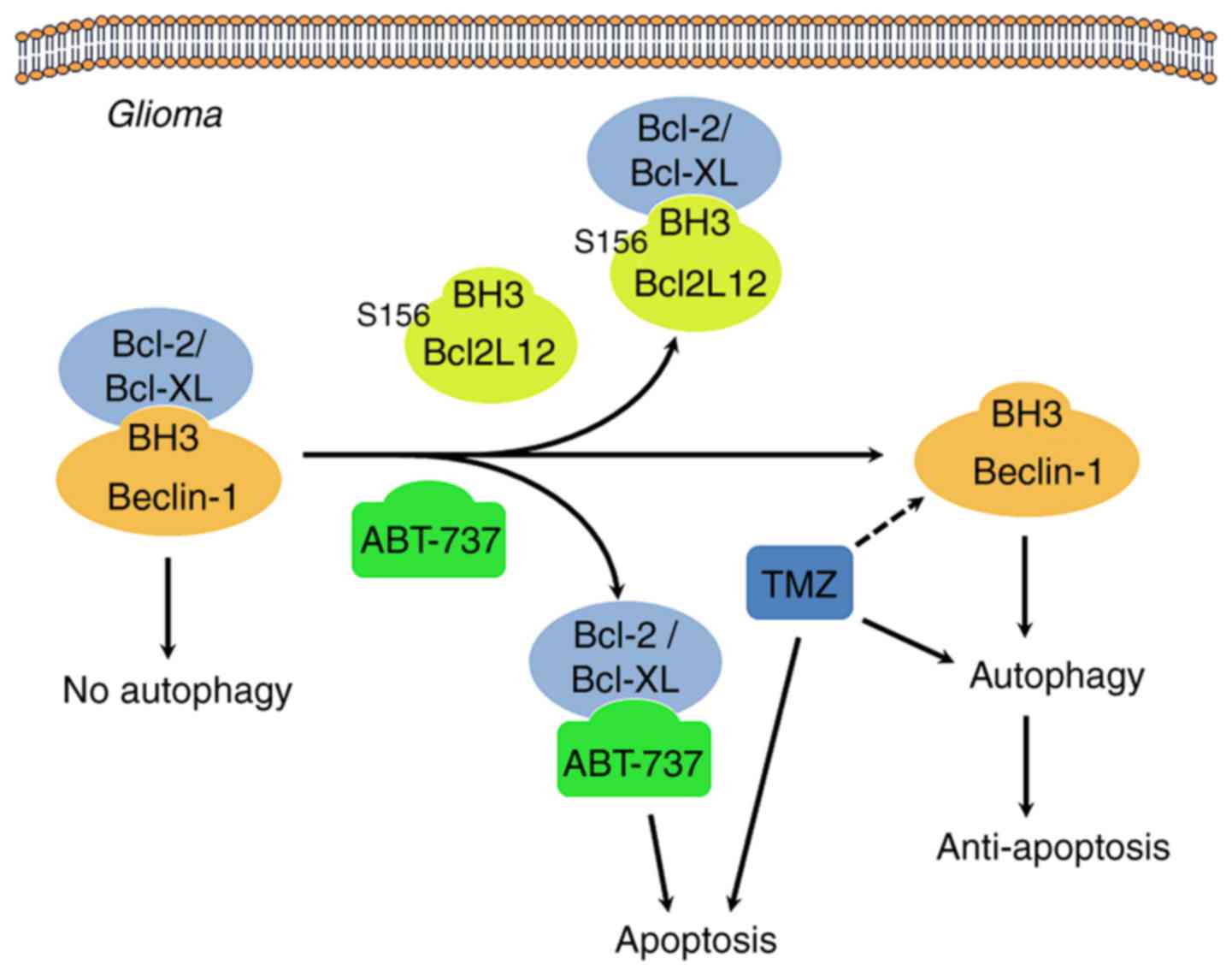 | Figure 9Proposed mechanism of Ser156
phosphorylation as an allosteric site to modulate a BH3-like domain
on BCL2L12 in glioma cells. When BCL2/BCL-XL interacts with the
Beclin-1 BH3 domain, autophagy is inhibited. Overexpression of
BCL2L12 may displace Beclin-1 in integrating with BCL2/BCL-XL via
its BH3-like domain, leading to release of Beclin-1 and initiation
of the autophagy process. In addition, since BCL2L12 occupies the
hydrophobic groove of BCL2/BCL-XL, BH3 only BCL2 activator or
sensitizer is unable to gain access, and the gross result of
anti-apoptosis is observed. The BH3 domain mimetic agent, ABT-737,
also binds to BCL2/BCL-XL, and hence competes and disrupts the
interaction between BCL2/BCL-XL and BCL2L12, making tumor cells
more vulnerable to apoptosis. Of note, GSK3β-mediated BCL2L12 S156
phosphorylation may affect BH3 domain function in glioma cells.
BCL2L12, BCL2-like 12; BCL2, BCL2 apoptosis regulator; BCL-XL,
BCL-extra large; GSK3β, glycogen synthase kinase 3β. |
By contrast, in neuronal cells, sphingosine kinase 2
(SPK2) interacts with BCL2 via its BH3 domain, thereby dissociating
it from Beclin-1 and activating autophagy. Tat-SPK2, a peptide
designed from the BH3 domain of SPK2, activates autophagy and
protects neural cells against oxygen-glucose deprivation injury. In
primary murine cortical neurons and HT22 hippocampal neuronal
cells, this cytoprotective autophagy appears beneficial to
prolonging cell survival (36).
Nevertheless, to further dissect the redundant role of BCL2L12 and
Beclin-1 beyond cytoprotective autophagy, other experimental
approaches are required. BCL2L12 may be tested in the future for
potential interactions with well-known interacting partners of
Beclin-1 in controlling autophagy, such as Vps34 PI3K (37) and UVRAG (38), in vitro and in vivo.
Ectopically expressed wild-type BCL2L12 and BCL2L12 S156A mutant,
but not BCL2L12A and BCL2L12 L213A mutants, restore the expression
of autophagic markers in Beclin-1 shRNA knockdown glioma cell lines
(unpublished data). This finding is more direct evidence that
BCL2L12 may be another Beclin-1 that regulates autophagy under
specific conditions.
Regarding GBM treatment, it may be necessary to
modulate multiple molecular mechanisms in order to reverse its
genetic antiapoptotic background and achieve better treatment
outcomes than traditional chemotherapeutics or radiotherapy.
Previous studies have pointed out that CRYAB and BCL2L12 are highly
expressed in GBM (5). Thus,
regardless of the expression of wild-type or mutant p53, p53
functions are compromised in GBM due to negative regulation by
BCL2L12 of p53 at the transcriptional and translational levels. The
first route of turning off p53 apoptotic events is directly binding
to p53 to prevent its nuclear translocation leading to activation
of downstream targets, such as Bax and Puma, an apoptosis modulator
(2). BCL2L12 may also use three
other routes to regulate apoptosis. First, it shuts down caspase-7
activation by directly binding and inactivating caspase-3 activity
through upregulating its negative regulator CRYAB (5). Second, it interacts through its
BH3-like domain with the hydrophobic groove region of BCL2 and
BCL-XL, similar to the BH3 domain of Bax; occupancy of the
hydrophobic groove of prosurvival BCL2 family proteins prevents
access of the BH3-only apoptotic activator and sensitizer to this
region to initiate apoptosis (9).
Finally, its 90-229 region may be responsible for binding with Bax,
scavenging active Bax to avoid Bax/Bak oligomer formation, thus
leading to a gross antiapoptosis effect. Therefore, downregulation
of BCL2L12 for treating GBM is a high priority goal (39). Previous studies also obtained good
inhibitory results on tumor growth in animal models via small
interfering (si)RNA directly against BCL2L12 (6) or SNA-miRNA-182 to downregulate
BCL2L12 expression (40). Using
TMZ is an option, but that involves p53 status and MGMT activation.
TMZ-induced apoptosis depends on the expression status of p53 and
MGMT, and TMZ-induced autophagy is linked to ERK
signaling-associated ROS production (12). In addition, TMZ-induced autophagy
occurs through mitochondrial damage and ER stress-dependent
mechanisms to protect glioma cells from apoptotic damage (13). This drug-induced pro-survival
autophagy is suggested to be an anti-apoptosis struggle with
apoptosis stimuli from chemotherapeutic (TMZ) and STS (bacterial
toxin, also protein kinase inhibitor) treatment (9).
ABT-737, a BH3 mimetic agent for treating cancer
cells with a high BCL2, BCL-XL expression and Mcl-1 neutralization,
may improve treatment outcome independent of p53 expression
(41). We previously applied
ABT-737 in combination with TMZ, since TMZ downregulates Mcl-1
expression in U87MG cells and thus creates a promising
microenvironment for ABT-737. ABT-737 may compete with the BCL2L12
BH3-like domain for binding with BCL2 and BCL-XL to selectively
inhibit their expression (9).
Inhibition of interaction between BCL2 and Bax is helpful for Bax
activation, forming Bax/Bak oligomerization to alter the outer
membrane integrity of mitochondria and activate caspases. Although
certain cell types lack Bax and Bak expression, ABT-737 may also
induce dissociation of BCL2-Beclin-1 and autophagy (42). LiCl was also successfully applied
to eliminate GSK3β-mediated phosphorylation at Ser156 of BCL2L12,
resulting in reactivation of caspase-dependent apoptosis (7). In the present study, blocking
TMZ-induced autophagy with 3-MA also revealed an enhancement of
apoptosis induction. Using agents targeting mitochondria or ER,
such as mitochondrial permeability transition pore inhibitor or
4-phenylbutyrate, an ER stress inhibitor, has also demonstrated
proper apoptosis induction with TMZ (13).
By integrating the findings from our previous work
and other studies, a combination of LiCl, siRNA of BCL2L12,
ABT-737, autophagy inhibitor and TMZ are suggested to be applicable
for GBM therapy. However, considering resistance to BCL2L12
targeting and ABT-737, as well as the low permeability of
blood-brain barrier, potential application of these drugs in the
clinic requires further investigation. To reach the goal of
individualized medicine, knowing the p53 and MGMT background is
essential for selecting which patients will respond to TMZ
treatment. A quick screening of the expression levels of BCL2,
BCL-XL, Mcl-1, Bax and Bak, in addition to p53 status and MGMT
activation, is suggested to be essential in order to determine
which patients will respond to the aforementioned combination
therapeutic approaches.
Acknowledgments
The authors would like to thank Gary Mawyer,
Managing Editor of Journal of the Wound, Ostomy and Continence
Nurses Society, for language editing.
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
TMZ
|
temozolomide
|
|
MGMT
|
O6-methylguanine DNA
methyltransferase
|
|
STS
|
staurosporine
|
|
MD
|
molecular dynamics
|
|
SPK2
|
sphingosine kinase 2
|
|
3-MA
|
3-methyladenine
|
Funding
The study was supported by a Ministry of Science and
Technology (MOST) grant (no. 104-2320-B-037-030-MY3) and other
funding sources from Kaohsiung Medical University, Kaohsiung
Medical University Teaching Hospital, and National Sun Yat-sen
University and Kaohsiung Medical University cooperative program
(grant nos. KMU-DK105004, KMTTH-105-003 and NSYSUKMU-106-P004).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
The study was conceived and designed by JKL and YRH.
BXH and CHC performed the yeast two-hybrid system experiments.
Western blotting, Co-IP, and data analysis were performed by CWC,
CHC, WSH, MCY, SJC and JKL. YTW performed protein conformational
simulation. All other experiments were performed by CWC, BXH, and
CHC. MCY and YRH wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Scorilas A, Kyriakopoulou L, Yousef GM,
Ashworth LK, Kwamie A and Diamandis EP: Molecular cloning, physical
mapping, and expression analysis of a novel gene, BCL2L12, encoding
a proline-rich protein with a highly conserved BH2 domain of the
Bcl-2 family. Genomics. 72:217–221. 2001. View Article : Google Scholar
|
|
2
|
Stegh AH, Brennan C, Mahoney JA, Forloney
KL, Jenq HT, Luciano JP, Protopopov A, Chin L and Depinho RA:
Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes
Dev. 24:2194–2204. 2010. View Article : Google Scholar
|
|
3
|
Tzovaras A, Kladi-Skandali A, Michaelidou
K, Zografos GC, Missitzis I, Ardavanis A and Scorilas A: BCL2L12: A
promising molecular prognostic biomarker in breast cancer. Clin
Biochem. 47:257–262. 2014. View Article : Google Scholar
|
|
4
|
Stegh AH, Kim H, Bachoo RM, Forloney KL,
Zhang J, Schulze H, Park K, Hannon GJ, Yuan J, Louis DN, et al:
Bcl2L12 inhibits post-mitochondrial apoptosis signaling in
glioblastoma. Genes Dev. 21:98–111. 2007. View Article : Google Scholar
|
|
5
|
Stegh AH, Kesari S, Mahoney JE, Jenq HT,
Forloney KL, Protopopov A, Louis DN, Chin L and DePinho RA:
Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via
distinct mechanisms in glioblastoma. Proc Natl Acad Sci USA.
105:10703–10708. 2008. View Article : Google Scholar
|
|
6
|
Jensen SA, Day ES, Ko CH, Hurley LA,
Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI, et
al: Spherical nucleic acid nanoparticle conjugates as an RNAi-based
therapy for glioblastoma. Sci Transl Med. 5:209ra1522013.
View Article : Google Scholar
|
|
7
|
Chou CH, Chou AK, Lin CC, Chen WJ, Wei CC,
Yang MC, Hsu CM, Lung FW, Loh JK, Howng SL and Hong YR: GSK3β
regulates Bcl2L12 and Bcl2L12A anti-apoptosis signaling in
glioblastoma and is inhibited by LiCl. Cell Cycle. 11:532–542.
2012. View Article : Google Scholar
|
|
8
|
Kontos CK and Scorilas A: Molecular
cloning of novel alternatively spliced variants of BCL2L12, a new
member of the BCL2 gene family, and their expression analysis in
cancer cells. Gene. 505:153–166. 2012. View Article : Google Scholar
|
|
9
|
Yang MC, Loh JK, Li YY, Huang WS, Chou CH,
Cheng JT, Wang YT, Lieu AS, Howng SL, Hong YR and Chou AK: Bcl2L12
with a BH3-like domain in regulating apoptosis and TMZ-induced
autophagy: A prospective combination of ABT-737 and TMZ for
treating glioma. Int J Oncol. 46:1304–1316. 2015. View Article : Google Scholar
|
|
10
|
Blough MD, Beauchamp DC, Westgate MR,
Kelly JJ and Cairncross JG: Effect of aberrant p53 function on
temozolomide sensitivity of glioma cell lines and brain tumor
initiating cells from glioblastoma. J Neurooncol. 102:1–7. 2011.
View Article : Google Scholar
|
|
11
|
Srivastava A, Jain A, Jha P, Suri V,
Sharma MC, Mallick S, Puri T, Gupta DK, Gupta A and Sarkar C: MGMT
gene promoter methylation in pediatric glioblastomas. Childs Nerv
Syst. 26:1613–1618. 2010. View Article : Google Scholar
|
|
12
|
Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH,
Lin YF and Shih CM: Resveratrol enhances the therapeutic effect of
temozolomide against malignant glioma in vitro and in vivo by
inhibiting autophagy. Free Radic Biol Med. 52:377–391. 2012.
View Article : Google Scholar
|
|
13
|
Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH,
Chen TH and Shih CM: Inhibition of mitochondria- and endoplasmic
reticulum stress-mediated autophagy augments temozolomide-induced
apoptosis in glioma cells. PLoS One. 7:e387062012. View Article : Google Scholar
|
|
14
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar
|
|
15
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar
|
|
16
|
Sinha S and Levine B: The autophagy
effector Beclin 1: A novel BH3-only protein. Oncogene. 27(Suppl 1):
S137–S148. 2008. View Article : Google Scholar
|
|
17
|
Kim SY, Song X, Zhang L, Bartlett DL and
Lee YJ: Role of Bcl-xL/Beclin-1 in interplay between apoptosis and
autophagy in oxaliplatin and bortezomib-induced cell death. Biochem
Pharmacol. 88:178–188. 2014. View Article : Google Scholar
|
|
18
|
Fields S and Song O: A novel genetic
system to detect protein-protein interactions. Nature. 340:245–246.
1989. View Article : Google Scholar
|
|
19
|
Chien CT, Bartel PL, Sternglanz R and
Fields S: The two-hybrid system: A method to identify and clone
genes for proteins that interact with a protein of interest. Proc
Natl Acad Sci USA. 88:9578–9582. 1991. View Article : Google Scholar
|
|
20
|
Zhu L: Yeast GAL4 two-hybrid system. A
genetic system to identify proteins that interact with a target
protein. Methods Mol Biol. 63:173–196. 1997.
|
|
21
|
Homeyer N, Horn AH, Lanig H and Sticht H:
AMBER force-field parameters for phosphorylated amino acids in
different protonation states: Phosphoserine, phosphothreonine,
phosphotyrosine, and phosphohistidine. J Mol Model. 12:281–289.
2006. View Article : Google Scholar
|
|
22
|
Ryckaert JP, Ciccotti G and Berendsen HJC:
Numerical integration of the cartesian equations of motion of a
system with constraints: Molecular dynamics of n-alkanes. J Comput
Phys. 23:327–341. 1977. View Article : Google Scholar
|
|
23
|
Darden T, York D and Pedersen L: Particle
mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large
systems. J Chem Phys. 98:10089–10092. 1993. View Article : Google Scholar
|
|
24
|
Chahal M, Xu Y, Lesniak D, Graham K,
Famulski K, Christensen JG, Aghi M, Jacques A, Murray D, Sabri S
and Abdulkarim B: MGMT modulates glioblastoma angiogenesis and
response to the tyrosine kinase inhibitor sunitinib. Neuro Oncol.
12:822–833. 2010. View Article : Google Scholar
|
|
25
|
Malley DS, Hamoudi RA, Kocialkowski S,
Pearson DM, Collins VP and Ichimura K: A distinct region of the
MGMT CpG island critical for transcriptional regulation is
preferentially methylated in glioblastoma cells and xenografts.
Acta Neuropathol. 121:651–661. 2011. View Article : Google Scholar
|
|
26
|
Stegh AH and DePinho RA: Beyond effector
caspase inhibition: Bcl2L12 neutralizes p53 signaling in
glioblastoma. Cell Cycle. 10:33–38. 2011. View Article : Google Scholar
|
|
27
|
Tang JB, Svilar D, Trivedi RN, Wang XH,
Goellne EM, Moore B, Hamilton RL, Banze LA, Brown AR and Sobol RW:
N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER
inhibitor potentiation of glioma cells to temozolomide. Neuro
Oncol. 13:471–486. 2011. View Article : Google Scholar
|
|
28
|
Hermisson M, Klumpp A, Wick W, Wischhusen
J, Nagel G, Roos W, Kaina B and Weller M: O6-methylguanine DNA
meth-yltransferase and p53 status predict temozolomide sensitivity
in human malignant glioma cells. J Neurochem. 96:766–776. 2006.
View Article : Google Scholar
|
|
29
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar
|
|
30
|
Jia W, Jiang X, Liu W, Wang L, Zhu B, Zhu
H, Liu X, Zhong M, Xie D, Huang W, et al: Effects of
three-dimensional collagen scaffolds on the expression profiles and
biological functions of glioma cells. Int J Oncol. Mar 20–2018.Epub
ahead of print. View Article : Google Scholar
|
|
31
|
Hong Y, Yang J, Wu W, Wang W, Kong X, Wang
Y, Yun X, Zong H, Wei Y, Zhang S and Gu J: Knockdown of BCL2L12
leads to cisplatin resistance in MDA-MB-231 breast cancer cells.
Biochim Biophys Acta. 1782:649–657. 2008. View Article : Google Scholar
|
|
32
|
Adamopoulos PG, Kontos CK, Tsiakanikas P
and Scorilas A: Identification of novel alternative splice variants
of the BCL2L12 gene in human cancer cells using next-generation
sequencing methodology. Cancer Lett. 373:119–129. 2016. View Article : Google Scholar
|
|
33
|
Cheng EH, Wei MC, Weiler S, Flavell RA,
Mak TW, Lindsten T and Korsmeyer SJ: BCL-2, BCL-X(L) sequester BH3
domain-only molecules preventing BAX- and BAK-mediated
mitochondrial apoptosis. Mol Cell. 8:705–711. 2001. View Article : Google Scholar
|
|
34
|
Lomonosova E and Chinnadurai G: BH3-only
proteins in apop-tosis and beyond: An overview. Oncogene. 27(Suppl
1): S2–S19. 2008. View Article : Google Scholar
|
|
35
|
Di Rita A and Strappazzon F: AMBRA1, a
Novel BH3-like protein: New insights into the AMBRA1-BCL2-family
proteins relationship. Int Rev Cell Mol Biol. 330:85–113. 2017.
View Article : Google Scholar
|
|
36
|
Song DD, Zhang TT, Chen JL, Xia YF, Qin
ZH, Waeber C and Sheng R: Sphingosine kinase 2 activates autophagy
and protects neurons against ischemic injury through interaction
with Bcl-2 via its putative BH3 domain. Cell Death Dis.
8:e29122017. View Article : Google Scholar
|
|
37
|
Zeng X, Overmeyer JH and Maltese WA:
Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase
complex in macroautophagy versus endocytosis and lysosomal enzyme
trafficking. J Cell Sci. 119:259–270. 2006. View Article : Google Scholar
|
|
38
|
Liang C, Lee JS, Inn KS, Gack MU, Li Q,
Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C and Jung JU:
Beclin1-binding UVRAG targets the class C Vps complex to coordinate
autophagosome maturation and endocytic trafficking. Nat Cell Biol.
10:776–787. 2008. View Article : Google Scholar
|
|
39
|
Stegh AH, Chin L, Louis DN and DePinho RA:
What drives intense apoptosis resistance and propensity for
necrosis in glioblastoma? A role for Bcl2L12 as a multifunctional
cell death regulator. Cell Cycle. 7:2833–2839. 2008. View Article : Google Scholar
|
|
40
|
Kouri FM, Ritner C and Stegh AH: miRNA-182
and the regulation of the glioblastoma phenotype-toward miRNA-based
precision therapeutics. Cell Cycle. 14:3794–3800. 2015. View Article : Google Scholar
|
|
41
|
van Delft MF, Wei AH, Mason KD, Vandenberg
CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et
al: The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and
efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized.
Cancer Cell. 10:389–399. 2006. View Article : Google Scholar
|
|
42
|
Pedro JM, Wei Y, Sica V, Maiuri MC, Zou Z,
Kroemer G and Levine B: BAX and BAK1 are dispensable for
ABT-737-induced dissociation of the BCL2-BECN1 complex and
autophagy. Autophagy. 11:452–459. 2015. View Article : Google Scholar :
|