Introduction
Esophageal carcinoma, and in particular the
histological subtype esophageal squamous cell carcinoma (ESCC), is
frequently detected in developing countries including China
(1,2). At present, the main treatment
options available for ESCC include radical esophagectomy,
radiotherapy and chemotherapy. Despite advances in therapeutic
approaches, the 5-year survival rate of ESCC remains very low
(3,4). Therefore, it is of importance to
develop novel therapeutic modalities for ESCC.
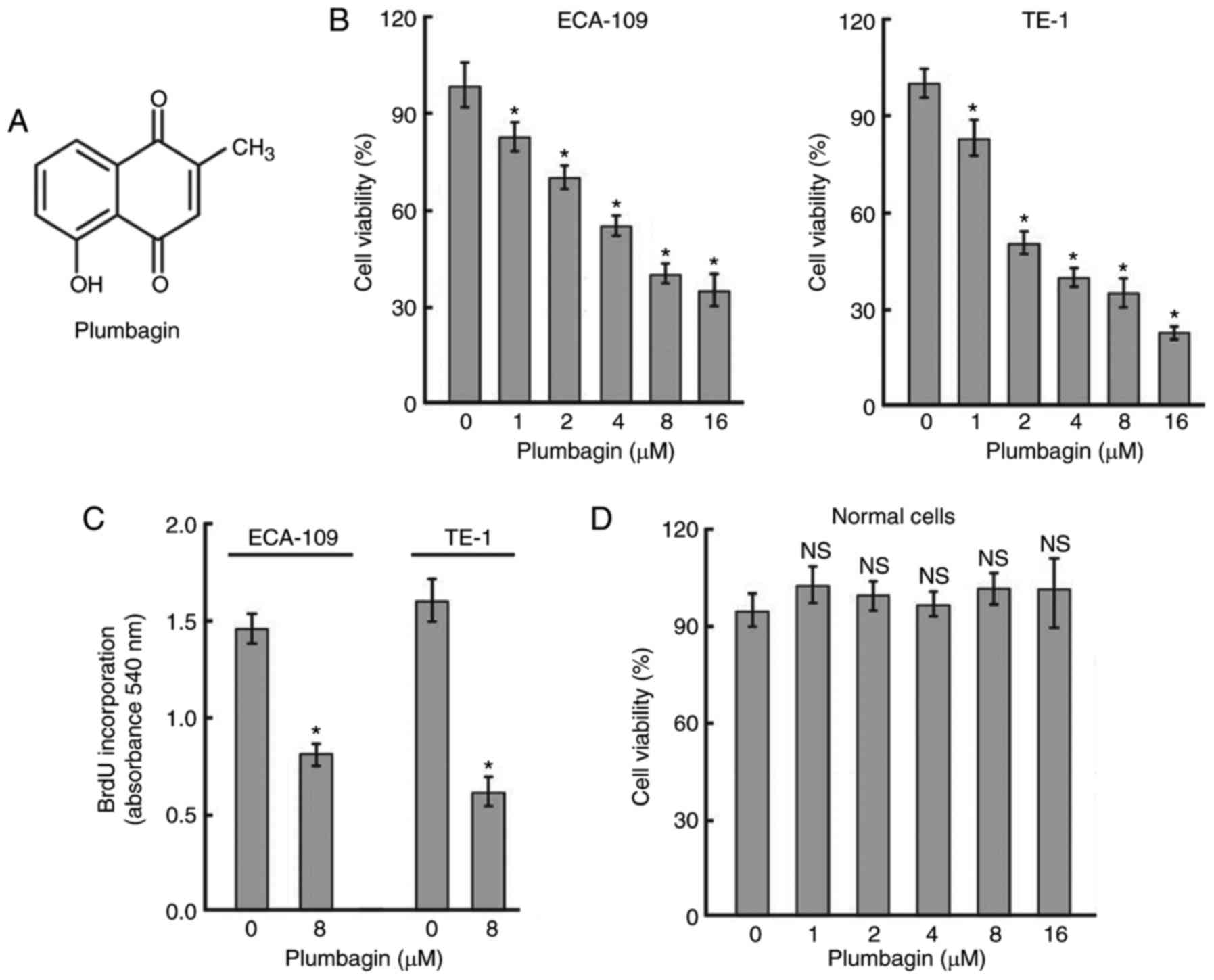
Plumbagin (Fig.
1A) is a natural naphthoquinone that is widely distributed in
the family of Plumbaginaceae. Plumbagin exhibits multiple
biological properties, including antibacterial (5), antimalarial (6), anti-inflammatory (7), and antitumor (8) activities. It has been documented
that plumbagin inhibits the invasion of breast cancer cells by
inactivating signal transducer and activator of transcription 3
(STAT3) signaling (8). Likewise,
plumbagin has been demonstrated to target the STAT3 pathway to
block the growth of pancreatic cancer cells (9). STAT3 is a key transcription
regulator that is aberrantly activated in ESCC (10,11). It has been reported that knockdown
of STAT3 reduces the proliferation of ESCC cells (11), indicating the requirement for
STAT3 activation in the progression of ESCC. Given the inhibitory
activity of plumbagin on STAT3 signaling, the present study
hypothesized that plumbagin may exert anticancer effects against
ESCC.
To test this hypothesis, in the present study ESCC
cells were treated with different concentrations of plumbagin and
the effects of plumbagin on cell proliferation, cell cycle
progression and apoptosis were examined in vitro. In
addition, the in vivo effect of plumbagin on the growth of
ESCC xenograft tumors was investigated. Furthermore, the potential
involvement of STAT3 signaling in the activity of plumbagin in ESCC
cells was examined.
Materials and methods
Cell culture
Two human ESCC cell lines (ECA-109 and TE-1) were
purchased from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China) and cultured in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
at 37°C in a 5% CO2 atmosphere. Normal human esophageal
epithelial cells were obtained from ScienCell Research Laboratories
(San Diego, CA, USA) and maintained in Epithelial Cell Medium-2
(ScienCell Research Laboratories) containing 10% FBS.
Plumbagin treatment
Plumbagin (>95% in purity) was purchased from
Sigma-Aldrich (Merck KGaA) and dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich; Merck KGaA) to yield a stock solution of 20
mM. ECA-109 and TE-1 cells were exposed to different concentrations
of plumbagin (1-16 μM) (12) for
48 h and examined for viability, proliferation, cell cycle
distribution and apoptosis. If not stated otherwise, 8 μM of
plumbagin was used in in vitro experiments.
Cell viability assay
Cell viability was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. In brief, following plumbagin treatment, cells were
incubated with 0.5 mg/ml MTT (Sigma-Aldrich; Merck KGaA) at 37°C
for 4 h, followed by the addition of DMSO. The absorbance of the
MTT formazan reduction product was measured at 570 nm.
Cell proliferation assay
Cell proliferation was measured using
5-bromo-2′-deoxyuridine (BrdU) incorporation assay (13). In brief, cells were labeled with
BrdU (Roche Applied Science, Penzberg, Germany; 10 μM)
during the last 4 h of plumbagin treatment. For quantification of
BrdU incorporation, cells were incubated with anti-BrdU conjugated
to peroxidase (Roche Applied Science), followed by addition of
3,3′,5,5′-tetramethylbenzidine substrate solution (Roche Applied
Science). The absorbance was measured at 450 nm using a microplate
reader.
Cell cycle distribution and apoptosis
analysis by flow cytometry
For analysis of cell cycle distribution, cells were
fixed in ice-cold 70% ethanol and resuspended in staining solution
containing DNase-free RNase A (0.2 mg/ml) and propidium iodide (PI;
20 μg/ml), both from Sigma-Aldrich (Merck KGaA). Following
incubation for 15 min in the dark, cellular DNA content was
analyzed by flow cytometry. For measurement of apoptosis, cells
were fixed and stained with fluorescein isothiocyanate
(FITC)-conjugated Annexin V and PI using the Annexin V-FITC
Apoptosis Detection kit (R&D systems, Minneapolis, MN, USA).
Stained cells were subjected to flow cytometric analysis using
FlowJo software (version 10.0.4; Tree Star, Inc., Ashland, OR,
USA).
Western blot analysis
Cells were lysed in ice-cold
radio-immunoprecipitation assay (RIPA) buffer (Sigma-Aldrich; Merck
KGaA) containing a mixture of protease inhibitors (Pierce; Thermo
Fisher Scientific, Inc.). Protein concentrations were measured
using the BCA assay kit (Pierce; Thermo Fisher Scientific, Inc.).
Equal amounts of protein (40 μg) were separated by
SDS-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes. Following blocking with 5% fat-free milk,
membranes were incubated with primary antibodies overnight at 4°C
prior to incubation with horseradish peroxidase (HRP)-conjugated
goat anti-mouse IgG (1:2,000; cat. no. sc-2005; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or HRP-conjugated goat
anti-rabbit IgG (1:2,000; cat. no. sc-2030; Santa Cruz
Biotechnology, Inc.) for 1 h. The primary antibodies recognizing
tumor protein p53 (cat. no. ab131442; 1:500), cyclin-dependent
kinase inhibitor 1A (also known as p21; cat. no. ab109520; 1:500),
cyclin D1 (cat. no. ab134175; 1:500), cyclin-dependent kinase (CDK)
4 (cat. no. ab108357; 1:300), induced myeloid leukemia cell
differentiation protein Mcl-1 (Mcl-1; cat. no. ab32087; 1:500), and
β-actin (cat. no. ab8227; 1:2,000) were purchased from Abcam
(Cambridge, UK). The antibodies against phospho-STAT3 (Tyr705; cat.
no. 9131; 1:300), STAT3 (cat. no. 9132; 1:500), cleaved caspase-3
(cat. no. 9661; 1:300), and cleaved poly(ADP-ribose) polymerase
(PARP; cat. no. 5625; 1:300) were from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Protein signals were visualized using
enhanced chemiluminescence reagent (Amersham; GE Healthcare,
Chicago, IL, USA) and quantified using Quantity One software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Plasmids
A STAT3-responsive firefly luciferase plasmid
(pSTAT3-Luc) was purchased from Panomics (Redwood City, CA, USA). A
Renilla luciferase-expressing plasmid (pRL-TK) was purchased
from Promega Corporation (Madison, WI, USA). A construct expressing
constitutively active STAT3 mutant (pSTAT3-C) and empty vector were
obtained from Addgene (Cambridge, MA, USA).
Cell transfection and luciferase reporter
assay
For over-expression of constitutively active STAT3,
ESCC cells were seeded 12 h prior to transfection. At 80%
confluence, they were transfected with pSTAT3-C or empty vector
using Lipofectamine 2000 following the manufacturer's instructions
(Invitrogen; Thermo Fisher Scientific, Inc.). At 24 h after
transfection, cells were treated with plumbagin and subjected to
proliferation and apoptosis assays. For the luciferase reporter
assay, ESCC cells were co-transfected with pSTAT3-Luc (0.5 μg) and
pRL-TK (20 ng) 24 h prior to plumbagin treatment. Following
incubation for an additional 48 h, cells were lysed and luciferase
activities were measured using a luciferase assay kit (Promega
Corporation). The firefly luciferase activity was normalized to
that of Renilla luciferase.
Animal experiments
For xenograft studies, a total of 10 male nude mice
(aged 4-6 weeks) were used, which were purchased from the
Experimental Animal Center of Zhengzhou University (Zhengzhou,
China). Animals were housed in a laminar air flow hood under a
12/12 h light/dark cycle at 25°C and 50% humidity with free access
to food and water. ECA-109 and TE-1 cells (2×106
cells/mouse) were subcutaneously injected into the flanks of mice.
When xenograft tumors reached 100 mm3, the tumor-bearing
mice (n=5 for each group) were intraperitoneally administered with
plumbagin or vehicle (0.05% DMSO) at a dose of 2 mg/kg/day 3 times
per week for 4 weeks (14). Tumor
growth was monitored by measuring tumor volume. All animals were
sacrificed after 30 days of treatment. Tumors were removed and
processed for immunohistochemical staining using anti-Ki-67 (cat.
no. ab15580; 1:500; Abcam) and anti-phospho-STAT3 (1:100)
antibodies (15). Over 1,000
cells were counted from 5 random microscopic fields, and the % of
immunoreactive cells was calculated. All experiments involving
animals were approved by the Experimental Animal Ethics Committee
of Shanghai Jiao Tong University School of Medicine (Shanghai,
China; Approval number: 2016-01-032).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical differences were determined using the Student's
t-test for comparison of two groups or one-way analysis of
variance followed by Tukey's post-hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Plumbagin inhibits the viability and
proliferation of ESCC cells in vitro
To assess the cytotoxicity of plumbagin, two ESCC
cell lines were exposed to different concentrations of plumbagin
for 48 h. As determined by MTT assay (Fig. 1B), plumbagin treatment resulted in
a concentration-dependent reduction in cell viability. The
IC50 values for plumbagin in ECA-109 and TE-1 cells were
6.2±0.5 and 2.4±0.6 μM, respectively. BrdU incorporation
assay was preformed to validate the antiproliferative activity of
plumbagin. The results demonstrated that treatment with 8 μM
plumbagin for 48 h decreased the proliferation of ECA-109 and TE-1
cells by 45.5 and 62.6%, respectively (Fig. 1C). However, plumbagin, up to the
concentration of 16 μM, had no significant impact on the
viability of normal esophageal epithelial cells following treatment
for 48 h (Fig. 1D).
Plumbagin induces cell cycle arrest at
the G0/G1 phase and triggers apoptosis in
ESCC cells
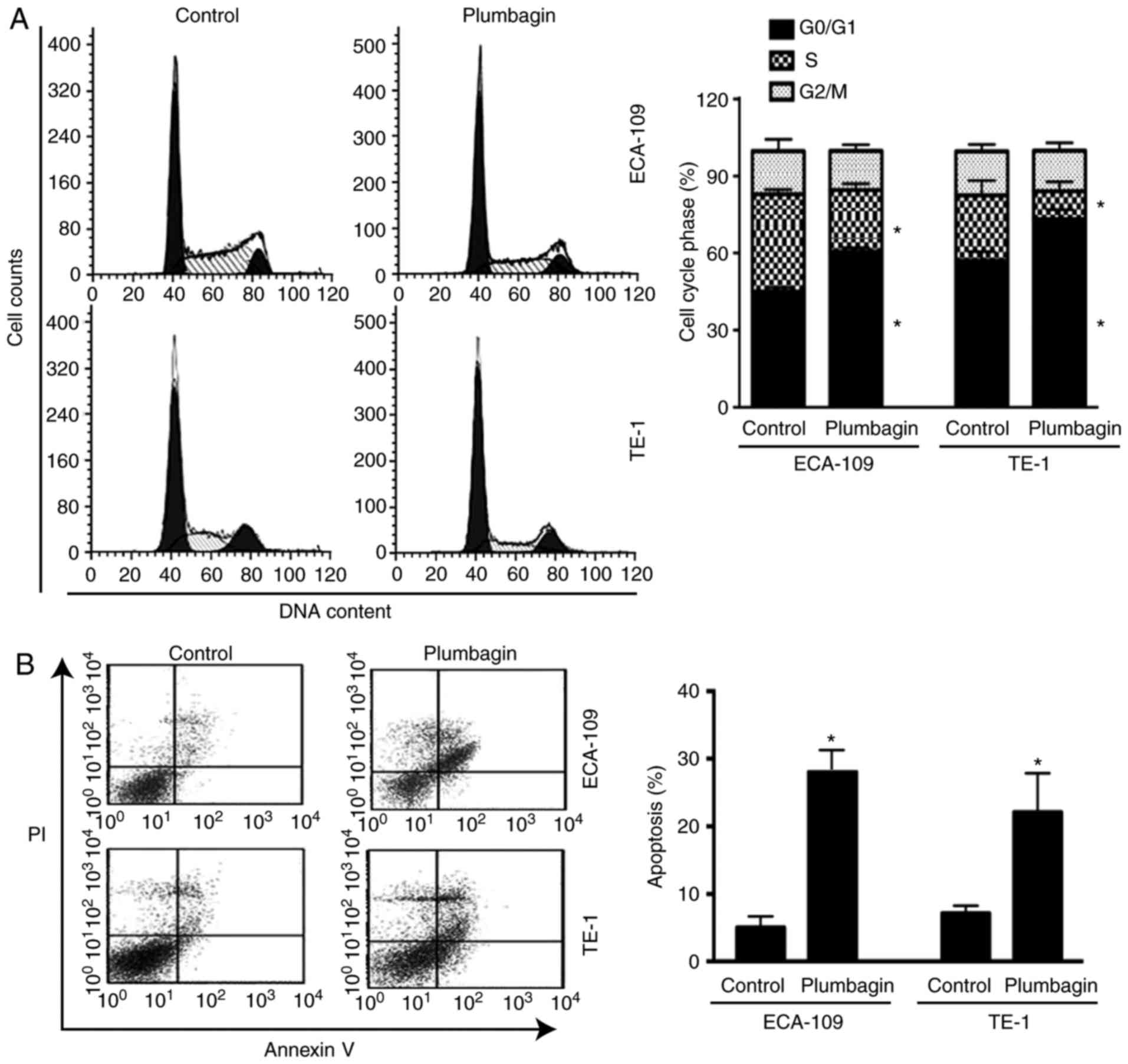
Flow cytometric analysis was performed to determine
the effect of plumbagin on cell cycle progression and apoptosis. As
presented in Fig. 2A, treatment
of ECA-109 cells with plumbagin for 48 h increased the proportion
of cells in the G0/G1 phase of the cell cycle
from 50 to 62% (P=0.001) and decreased the proportion of cells in
the S phase from 29 to 18% (P=0.002) relative to control. In
addition, plumbagin treatment resulted in a 4.7-fold (P=0.004)
increase in the % of Annexin V-positive apoptotic cells, compared
with vehicle-treated cells (Fig.
2B). Similar findings were observed in TE-1 cells following
treatment with plumbagin (Fig.
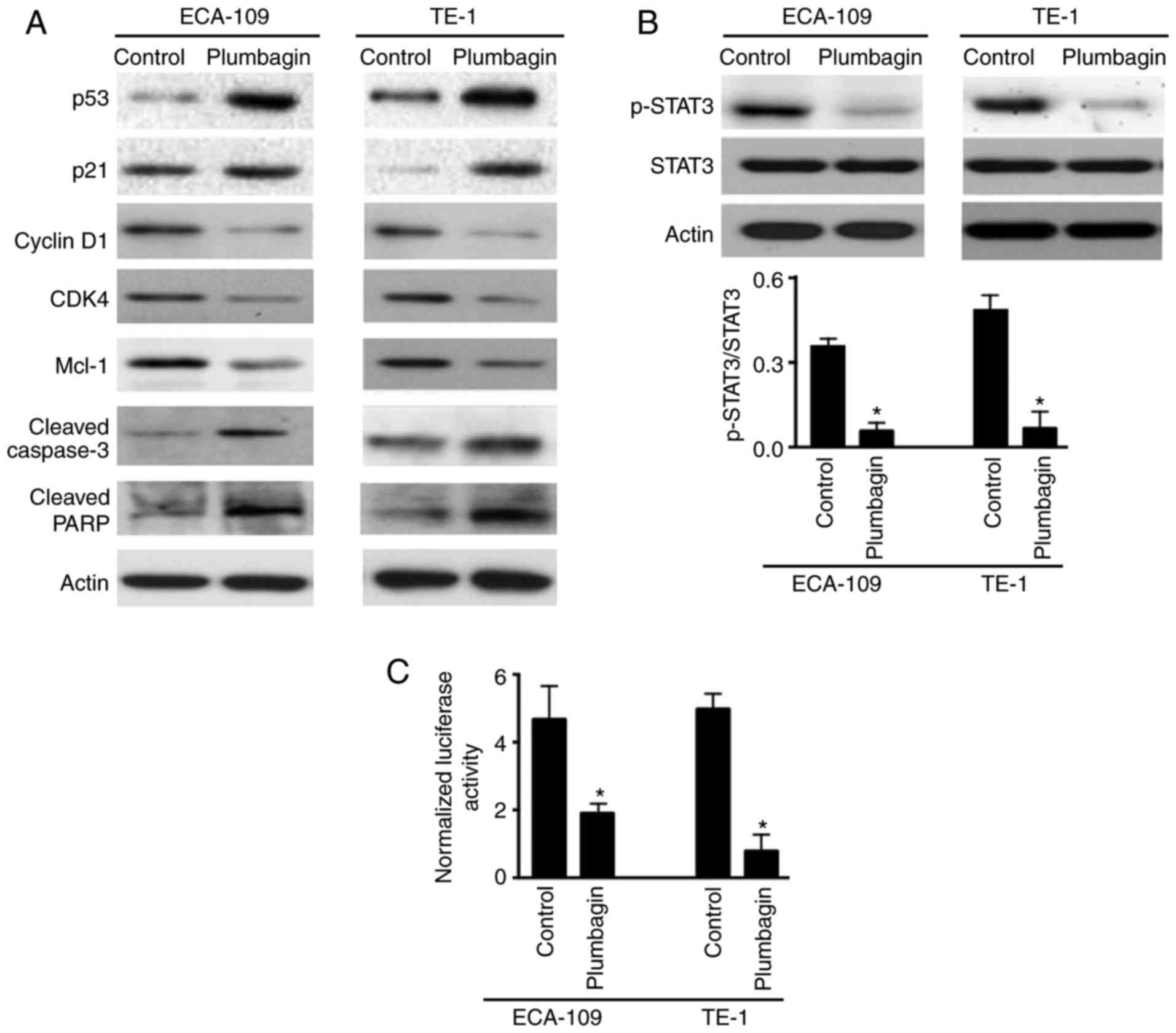
2). Western blot analysis of several key genes involved in cell
cycle progression and apoptosis confirmed that plumbagin treatment
resulted in a marked increase in p53 and p21 and decrease in cyclin
D1, CDK4 and Mcl-1 expression (Fig.
3A). In addition, cleavage of caspase-3 and PARP was markedly
enhanced in plumbagin-treated ECA-109 and TE-1 cells compared with
vehicle-treated cells (Fig.
3A).
Plumbagin inhibits the STAT3 signaling
pathway
Western blot analysis demonstrated that plumbagin
treatment led to a significant decrease in the levels of
phosphorylated STAT3, but not total STAT3 protein (Fig. 3B). Consistently, plumbagin-treated
ESCC cells displayed a 3- to 6-fold reduction of STAT3 reporter
gene activity, compared with control cells (Fig. 3C). These data indicated that
plumbagin had the capacity to interfere with STAT3 activation.
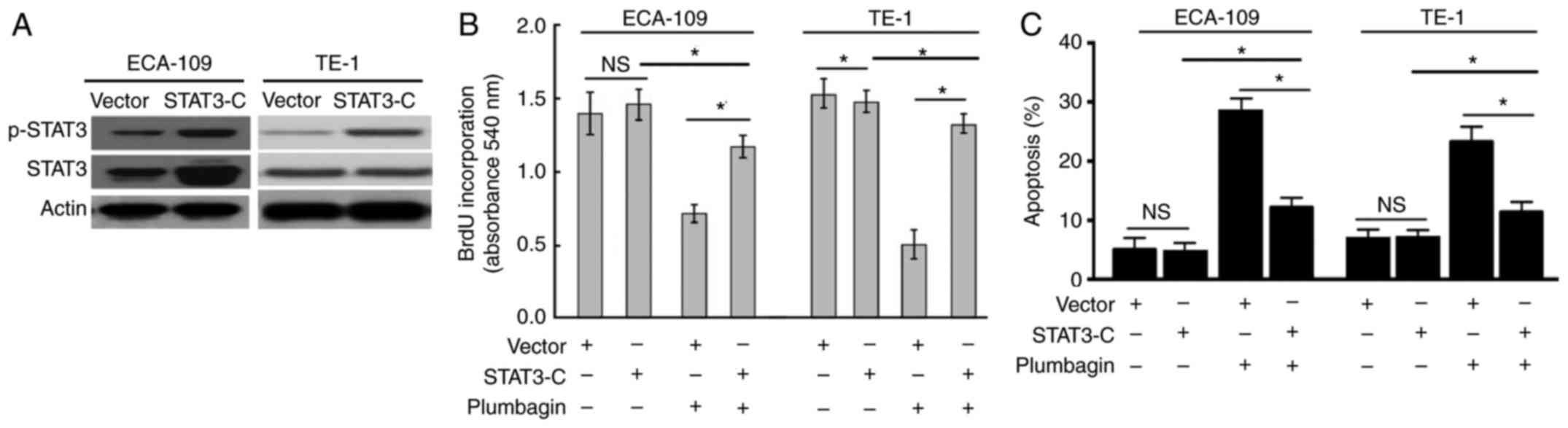
Overexpression of constitutively active
STAT3 reverses plumbagin-mediated growth suppression
Next, the hypothesis that the plumbagin-mediated
cytotoxicity in ESCC cells was directly associated with
inactivation of STAT3 signaling was examined. To this end, rescue
experiments were performed with a constitutively active STAT3
variant. The results demonstrated that ectopic expression of
constitutively active STAT3 (Fig.
4A) significantly attenuated the plumbagin-induced growth
suppression (Fig. 4B) and
apoptosis (Fig. 4C) in ESCC
cells.
Plumbagin suppresses the growth of ESCC
xenografts in mice
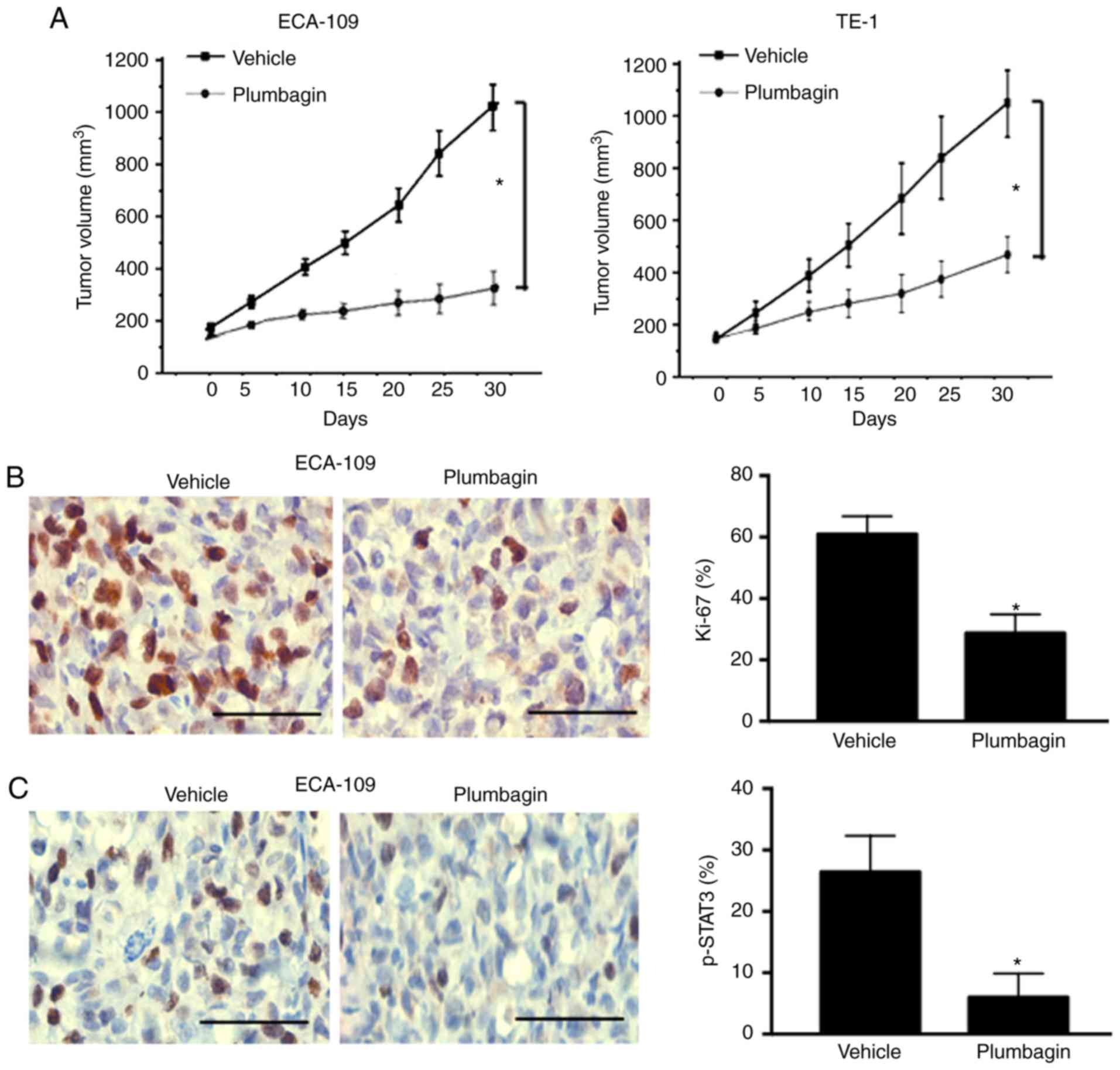
To determine the in vivo anticancer effect of
plumbagin, ECA-109 and TE-1 cells were injected into nude mice,
allowed to form xenograft tumors, and treated with plumbagin or
vehicle in vivo. After 30 days, plumbagin-treated ECA-109
and TE-1 xenograft tumors displayed 72 and 54% reduction in tumor
volume, respectively, relative to vehicle-treated tumors (Fig. 5A). Immunohistochemistry analysis
of tumor specimens confirmed that plumbagin-treated tumors
presented a significant reduction in the % of Ki-67-positive cells
(P=0.011; Fig. 5B). Additionally,
the levels of phosphorylated STAT3 were significantly lower in
plumbagin-treated tumors compared with vehicle-treated tumors
(P=0.001; Fig. 5C).
Discussion
Plumbagin has exhibited anticancer properties in
many types of cancer, including breast cancer (8), pancreatic cancer (9), prostate cancer (16), and gastric cancer (17). It has been reported that plumbagin
can block prostate carcinogenesis triggered by loss of phosphatase
and tensin homolog (PTEN) (16).
Similarly, plumbagin treatment resulted in a significant inhibition
of tumorigenesis and angiogenesis in ovarian cancer xenograft
models (14). In the present
study, plumbagin was also confirmed to exert growth suppressive
effects against ESCC cells. Treatment with plumbagin led to a
significant decline in cell viability and proliferation in ESCC
cells. Plumbagin-mediated toxicity appeared to be selective to
malignant cells, as the viability of a normal esophageal epithelial
cell line was not significantly changed following plumbagin
treatment. In agreement with the present findings, a previous study
has demonstrated that plumbagin was more effective at killing
melanoma cells than non-malignant cells in vitro and that it
displayed negligible toxicity to normal organs in vivo
(18).
To gain more insight into the growth suppression
induced by plumbagin, the effects of plumbagin on cell cycle
distribution and apoptosis were analyzed. Plumbagin-treated ESCC
cells displayed an accumulation of
G0/G1-phase cells and significant apoptotic
death, compared with vehicle-treated cells. At the molecular level,
multiple genes, including p53, p21, cyclin D1, CDK4 and Mcl-1 were
deregulated in response to plumbagin treatment. The protein p21 is
a well-defined CDK inhibitor that can bind to cyclin/CDK complexes
and inhibit their catalytic activities (19). p53 acts as a tumor suppressor and
has the ability to induce cell cycle arrest and apoptosis (20), while Mcl-1 serves an important
role in protecting cells from apoptosis (21). Taken together, plumbagin treatment
was demonstrated to modulate many key genes involved in the
regulation of cell cycle progression and apoptosis, consequently
leading to cell cycle arrest and apoptosis. Such effects were also
previously reported in breast cancer (22) and colon cancer cells (23). It should be mentioned that the
suppressive activity of plumbagin appears not to be
G1-phase specific, as this compound can cause
G2/M-phase arrest in A549 lung cancer cells (24).
Targeting STAT3 signaling represents an important
molecular mechanism for the anticancer activity of plumbagin in
melanoma (18), gastric cancer
(17), breast cancer (8), and prostate cancer (25). In line with these studies, the
present data demonstrated that plumbagin treatment significantly
suppressed STAT3 activation in ESCC cells. STAT3 has been reported
to regulate many genes involved in cell proliferation and survival,
including cyclin D1, Bcl-2, Mcl-1, and cellular inhibitor of
apoptosis 2 (c-IAP2) (26,27).
Consistent with the inactivation of STAT3, the expression of cyclin
D1 and Mcl-1 was downregulated in ESCC cells in response to
plumbagin treatment. Rescue experiments provided further evidence
for the involvement of STAT3 signaling in the anticancer activity
of plumbagin in ESCC. In vivo studies demonstrated that
plumbagin treatment significantly delayed the growth of established
ESCC xenograft tumors and suppressed STAT3 phosphorylation.
Collectively, plumbagin exerted its growth suppressive activity in
ESCC by targeting STAT3 signaling, which may provide an explanation
for selective inhibition of cancer cells. It was reported that
plumbagin can inhibit A549 lung cancer cell growth by inactivating
c-Jun N-terminal kinase signaling (24). Therefore, it is possible that
multiple signaling pathways other than STAT3 are involved in the
anticancer activity of plumbagin in ESCC.
In conclusion, the present data reinforce that
plumbagin has growth suppressive activity against ESCC cells, which
is, at least in part, mediated through inhibition of STAT3
activation. These findings suggest that plumbagin may be a
promising anticancer agent for ESCC.
Acknowledgments
Not applicable.
Funding
This work was funded by the Research Program of
Shanghai Health and Family Planning Commission of China (grant no.
20134036).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
YC and XY were responsible for study design, data
collection and analysis. YJ, BL and SW performed in vivo
studies and histological examination. MS performed in vitro
studies and statistical analysis. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All experiments involving animals were approved by
the Experimental Animal Ethics Committee of Shanghai Jiao Tong
University School of Medicine (Shanghai, China; Approval number,
2016-01-032).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shridhar R, Hoffe SE, Almhanna K, Weber
JM, Chuong MD, Karl RC and Meredith K: Lymph node harvest in
esophageal cancer after neoadjuvant chemoradiotherapy. Ann Surg
Oncol. 20:3038–3043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jackie Oh S, Han S, Lee W and Lockhart AC:
Emerging immuno therapy for the treatment of esophageal cancer.
Expert Opin Investig Drugs. 25:667–677. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Omosa LK, Midiwo JO, Mbaveng AT, Tankeo
SB, Seukep JA, Voukeng IK, Dzotam JK, Isemeki J, Derese S, Omolle
RA, et al: Antibacterial activities and structure-activity
relationships of a panel of 48 compounds from Kenyan plants against
multidrug resistant phenotypes. Springerplus. 5:9012016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sumsakul W, Chaijaroenkul W and
Na-Bangchang K: In vitro inhibitory effects of plumbagin, the
promising antimalarial candidate, on human cytochrome P450 enzymes.
Asian Pac J Trop Med. 8:914–918. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pile JE, Navalta JW, Davis CD and Sharma
NC: Interventional effects of plumbagin on experimental ulcerative
colitis in mice. J Nat Prod. 76:1001–1006. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan W, Tu B, Liu YY, Wang TY, Qiao H, Zhai
ZJ, Li HW and Tang TT: Suppressive effects of plumbagin on invasion
and migration of breast cancer cells via the inhibition of STAT3
signaling and down-regulation of inflammatory cytokine expressions.
Bone Res. 1:362–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hafeez BB, Jamal MS, Fischer JW, Mustafa A
and Verma AK: Plumbagin, a plant derived natural agent inhibits the
growth of pancreatic cancer cells in in vitro and in vivo via
targeting EGFR, Stat3 and NF-κB signaling pathways. Int J Cancer.
131:2175–2186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Du XL, Wang CJ, Lin DC, Ruan X,
Feng YB, Huo YQ, Peng H, Cui JL, Zhang TT, et al: Reciprocal
activation between PLK1 and Stat3 contributes to survival and
proliferation of esophageal cancer cells. Gastroenterology.
142:521–530. 2012. View Article : Google Scholar
|
|
11
|
Timme S, Ihde S, Fichter CD, Waehle V,
Bogatyreva L, Atanasov K, Kohler I, Schöpflin A, Geddert H, Faller
G, et al: STAT3 expression, activity and functional consequences of
STAT3 inhibition in esophageal squamous cell carcinomas and
Barrett's adenocarcinomas. Oncogene. 33:3256–3266. 2014. View Article : Google Scholar
|
|
12
|
Niu M, Cai W, Liu H, Chong Y, Hu W, Gao S,
Shi Q, Zhou X, Liu X and Yu R: Plumbagin inhibits growth of gliomas
in vivo via suppression of FOXM1 expression. J Pharmacol Sci.
128:131–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Radestock Y, Hoang-Vu C and
Hombach-Klonisch S: Relaxin reduces xenograft tumour growth of
human MDA-MB-231 breast cancer cells. Breast Cancer Res.
10:R712008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sinha S, Pal K, Elkhanany A, Dutta S, Cao
Y, Mondal G, Iyer S, Somasundaram V, Couch FJ, Shridhar V, et al:
Plumbagin inhibits tumorigenesis and angiogenesis of ovarian cancer
cells in vivo. Int J Cancer. 132:1201–1212. 2013. View Article : Google Scholar
|
|
15
|
Liu C, Dai LH, Dou DQ, Ma LQ and Sun YX: A
natural food sweetener with anti-pancreatic cancer properties.
Oncogenesis. 5:e2172016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hafeez BB, Fischer JW, Singh A, Zhong W,
Mustafa A, Meske L, Sheikhani MO and Verma AK: Plumbagin inhibits
prostate carcinogenesis in intact and castrated PTEN knockout mice
via targeting PKCε, Stat3, and epithelial-to-mesenchymal transition
markers. Cancer Prev Res (Phila). 8:375–386. 2015. View Article : Google Scholar
|
|
17
|
Joo MK, Park JJ, Kim SH, Yoo HS, Lee BJ,
Chun HJ, Lee SW and Bak YT: Antitumorigenic effect of plumbagin by
induction of SH2-containing protein tyrosine phosphatase 1 in human
gastric cancer cells. Int J Oncol. 46:2380–2388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gowda R, Sharma A and Robertson GP:
Synergistic inhibitory effects of Celecoxib and Plumbagin on
melanoma tumor growth. Cancer Lett. 385:243–250. 2017. View Article : Google Scholar
|
|
19
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: A function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vousden KH and Lu X: Live or let die: The
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morciano G, Pedriali G, Sbano L, Iannitti
T, Giorgi C and Pinton P: Intersection of mitochondrial fission and
fusion machinery with apoptotic pathways: Role of Mcl-1. Biol Cell.
108:279–293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang XQ, Yang CY, Rao XF and Xiong JP:
Plumbagin shows anti-cancer activity in human breast cancer cells
by the up regulation of p53 and p21 and suppression of G1 cell
cycle regulators. Eur J Gynaecol Oncol. 37:30–35. 2016.
|
|
23
|
Eldhose B, Gunawan M, Rahman M, Latha MS
and Notario V: Plumbagin reduces human colon cancer cell survival
by inducing cell cycle arrest and mitochondria-mediated apoptosis.
Int J Oncol. 45:1913–1920. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hsu YL, Cho CY, Kuo PL, Huang YT and Lin
CC: Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces
apoptosis and cell cycle arrest in A549 cells through p53
accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation
at serine 15 in vitro and in vivo. J Pharmacol Exp Ther.
318:484–494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hafeez BB, Zhong W, Fischer JW, Mustafa A,
Shi X, Meske L, Hong H, Cai W, Havighurst T, Kim K and Verma AK:
Plumbagin, a medicinal plant (Plumbago zeylanica)-derived
1,4-naphthoquinone, inhibits growth and metastasis of human
prostate cancer PC-3M-luciferase cells in an orthotopic xenograft
mouse model. Mol Oncol. 7:428–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leslie K, Lang C, Devgan G, Azare J,
Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, et al:
Cyclin D1 is transcriptionally regulated by and required for
transformation by activated signal transducer and activator of
transcription 3. Cancer Res. 66:2544–2552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhattacharya S, Ray RM and Johnson LR:
STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents
apoptosis in polyamine-depleted cells. Biochem J. 392:335–344.
2005. View Article : Google Scholar : PubMed/NCBI
|