Introduction
Epithelial-to-mesenchymal transition (EMT) is a
necessary condition associated with numerous cancer developmental
processes, including tumor invasion and metastasis, in various
types of epithelial cancer (1,2).
When EMT occurs, epithelial cells lose their cell adhesion and
apical-basal polarity. In addition, epithelial cell-matrix
interactions are altered due to reorganization of the actin
cytoskeleton, and cells gain a mesenchymal phenotype with high
mobility (3). During the EMT
process, EMT is frequently characterized by the reduced expression
of E-cadherin, N-cadherin and other epithelial markers, and by the
increased expression of Vimentin, Snail and other mesenchymal
markers (4). Mesenchymal cells
infiltrate blood vessels and lymph nodes, and may then migrate to
other tissues, resulting in tumor formation (5). Numerous studies have demonstrated
that activity of the phosphoinositide 3-kinase (PI3K)/protein
kinase B (AKT) pathway serves an important role in upregulating
cell proliferation, migration and EMT (6,7).
In addition to this pathway, the EMT process can promote the
activity of AMP-activated protein kinase (AMPK), which is an
important regulator of glucose uptake and energy balance (8). A previous study demonstrated that
activation of AMPK could induce migration by increasing the
expression levels of microRNA-451 in glioblastoma (9). The oxidative phosphorylation
(OXPHOS) process through the mitochondrial electron transport
system in cells is influenced by the intracellular energy metabolic
activity of AMPK.
Mitochondria serve a central role in the production
of cellular energy and in metabolism through the OXPHOS process of
the electron transport system via respiration (10). Mitochondrial dysfunction has been
reported to be associated with the development of various types of
human cancer, and it occurs through the production of reactive
oxygen species (ROS), which are substrates of oxidative
phosphorylation (11), through
the reduction and the activation of mitochondria respiration chain
complex. In addition, the high mutation rate of mitochondrial
(mt)DNA and nuclear gene-coded electron transport chain proteins
are known to influence mitochondrial dysfunction (12), and have been reported to regulate
apoptosis or programmed cell death (13). Oligomycin A is an inhibitor of ATP
synthase, and antimycin A is a specific inhibitor of mitochondrial
electron transport complex I, which previously led to an increase
in gastric cancer progression via the promotion of ROS (14). In addition, oligomycin A treatment
increased cytosolic Ca2+ and ROS in HepG2 cells, and
stimulated migration in SK-Hep-1 cells (15).
Although inhibitors of mitochondrial respiration,
including oligomycin A and antimycin A, have been reported to have
an effect on metastasis in various types of human cancer cells, the
molecular mechanism underlying mitochondrial respiration
inhibitor-mediated EMT in lung cancer cells remains to be
elucidated. Therefore, the present study aimed to determine the
effects of mitochondrial respiration inhibitors oligomycin A and
antimycin A on EMT, and on cell migration and invasion, in the A549
human lung cancer cell line. The results demonstrated that
treatment with mitochondrial respiration inhibitors may induce the
invasive phenotype, as well as cell migration and invasion.
Furthermore, treatment with mitochondrial respiration inhibitors
increased the protein expression levels of mesenchymal markers via
induction of the AKT and AMPK signaling pathways.
Materials and methods
Cells and materials
A549, H23 and H1793 lung carcinoma cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in RPMI 1640 (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
in an atmosphere containing 5% CO2 at 37°C. Oligomycin A
and antimycin A were obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). All chemicals, including N-acetyl-L-cysteine
(NAC; ROS scavenger), wortmannin (PI3K inhibitor) and Compound C
(AMPK inhibitor) were obtained from Sigma-Aldrich; Merck KGaA,
unless otherwise indicated. Densitometric analysis was performed on
scanned western blot and reverse transcription-polymerase chain
reaction (RT-PCR) images using ImageJ software (version 2018;
National Institutes of Health, Bethesda, MD, USA).
Cell viability assay
Human non-small cell lung cancer cell lines were
seeded in 96-well plates at 2×104 cells/well in RPMI
1640 media, and were allowed to adhere to the surface for 24 h.
Subsequently, fresh media containing various concentrations of
oligomycin A and antimycin A were added to the plates, and cells
were incubated for 24 h. Subsequently, 500 µg/ml MTT (Roche
Diagnostics, Indianapolis, IN, USA) was added to each well. The
amount of formazan deposits were dissolved in 100 µ1
dimethyl sulfoxide and absorbance was measured at 540 nm using a
plate reader (VersaMax™ Microplate Reader; Molecular Devices, LLC,
Sunnyvale, CA, USA), following a 4 h incubation with MTT in an
atmosphere containing 5% CO2 at 37°C.
Morphological analysis
Cells were cultured in a 60 mm dish and were
cultured until they reached 60% confluence. Subsequently, the media
were replaced with fresh RPMI 1640 media, and the cells were
incubated with 1 µg/ml oligomycin A or antimycin A for 24 h
in an atmosphere containing 5% CO2 at 37°C. Finally,
images of the cells were captured using amicroscope (Nikon Eclipse
TS100, Nikon Corporation, Tokyo, Japan).
Western blot analysis
Cells were seeded in a 60-mm dish at
1×106 cells/well in RPMI-1640 media, and were allowed to
adhere to the surface for 24 h. Subsequently, fresh media
containing various concentrations of oligomycin A and antimycin A
were added to the plates, and cells were incubated for 24 h. Cell
lysis, SDS-PAGE, transfer to a nitrocellulose membrane (EMD
Millipore, Billerica, MA, USA) and immunoblotting were performed as
previously described (16).
Briefly, cells were suspended in 0.1 ml lysis buffer containing 10
mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT,
0.5 mM phenylmethlysulfonyl fluoride, 2.0 µg/ml leupeptin
and 2.0 µg/ml aprotinin at 4°C for 30 min. Subsequently,
cells were centrifuged at 14,645 × g for 10 min. Protein
concentration was measured using the Bicinchoninic Acid (BCA) Assay
(Pierce™ BCA Protein Assay kit; Thermo Fisher Scientific, Inc.)
Total proteins (30 µg) were separated by 8–15% SDS-PAGE and
were transferred to nitrocellulose membranes. The membranes were
then blocked in 5% skim milk in Tris-buffered saline containing
0.1% Tween-20 (TBS-T) for 1 h at room temperature. The membranes
were then incubated with primary antibodies at 4°C overnight,
washed three times with TBS-T, and incubated with goat anti-mouse
immunoglobulin G (IgG; #31430) or goat anti-rabbit IgG (#65-6120)
secondary antibodies (Thermo Fisher Scientific, Inc.) at a dilution
of 1:2,000 for 1 h at room temperature, before being washed a
further three times with TBS-T. Signals were detected using
enhanced chemiluminescence (GE Healthcare, Chicago, IL, USA) and
ChemiDOC™ XRS (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
primary antibodies at a dilution of 1:1,000 used in the present
study were: E-cadherin (BD610182, BD Biosciences, Franklin Lakes,
NJ, USA); Snail (#3879), Slug (#9585), phosphorylated (p)-AMPKα
(#2531), p-acetyl-CoA carboxylase (ACC; #3661) and p-AKT (#4060)
(all from Cell Signaling Technology, Inc., Danvers, MA, USA);
Vimentin (sc-6260) and β-actin (sc-47778) (Santa Cruz
Biotechnology, Dallas, TX, USA).
RT-PCR
Cells were seeded in a 60 mm dish at
1×106 cells/well in RPMI 1640 media, and were allowed to
adhere to the surface for 24 h. Subsequently, fresh media
containing 1 µg/ml oligomycin A and antimycin A were added
to the plates, and cells were incubated for 24 h. Total RNA was
extracted using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. For
RT-PCR, cDNA was synthesized from 1 µg total RNA using
AccuPower® RT PreMix (Bioneer Corporation, Daejeon,
Korea), according to the manufacturer's protocol. cDNA was
amplified by PCR (AccuPower® RT-PCR PreMix; Bioneer
Corporation) with the following primers: Snail, sense 5′-CAG CGA
GCT GCA GGA CTC TA-3′, antisense 5′-GTG GGA TGG CTG CCA GC-3′;
Slug, sense 5′-TGT GTG GAC TAC CGC TGC-3′, antisense 5′-TCC GGA AAG
AGG AGA GAG G-3′; and β-actin, sense 5′-CAA GAG ATG GCC ACG GCT
GCT-3′ and antisense 5′-TCC TTC TGC ATC CTG TCG GCA-3′. PCR
products were analyzed by 1.5% agarose gel electrophoresis and
visualized using ethidium bromide (E1510; Sigma-Aldrich; Merck
KGaA) and ChemiDOC™ XRS (Bio-Rad Laboratories, Inc.).
Transwell invasion assay
Matrigel-coated filter inserts (pore size, 8
µm; SPLInsert™ Hanging) that fit into 24-well migration
chambers were obtained from SPL Life Sciences (Pocheon, Korea).
Cells were plated at 2×104 cells/well in the upper
chamber. The lower chamber was filled with culture media containing
1 µg/ml oligomycin A or antimycin A. Cells were incubated in
the chamber for 24 h at 37°C; subsequently, cells that invaded
across the membrane surface were fixed with 100% methanol for 1 min
and stained with 0.5% crystal violet for 10 min at room
temperature. The cells that passed through the Matrigel-coated
inserts and were located on the underside of the filter were
counted. Random fields were counted by light microscopy (×200
magnification).
Wound-healing assay
The wound-healing assay was performed according to a
previously described procedure, with minor modifications (17). Briefly, cells were seeded in
6-well plates and were incubated until they reached 80% confluence.
Cell monolayers were then scratched with a 200-µl pipette
tip to create a wound, and cells were washed twice with serum-free
culture media to remove floating cells. Media were then replaced
with fresh serum-free media and cells were subjected to treatment
with 1 µg/ml oligomycin A or antimycin A for 24 h at 37°C;
images of the cells were captured after 24 h using a microscope
(Nikon Eclipse TS100; Nikon Corporation).
Immunofluorescence
For immunofluorescence, cells were cultured on cover
slips at 50% confluence, and were cultured with 1 µg/ml
oligomycin A or antimycin A for 24 h at 37°C. Cells on cover slips
were washed with PBS and were fixed in 100% methanol for 1 min and
permeabilized with 0.2% Triton X-100 in PBS for 5 min. Cells were
then blocked with 10% normal goat serum (#31872; Invitrogen; Thermo
Fisher Scientific, Inc.) in PBS for 1 h at room temperature and
were then incubated with primary antibodies against E-cadherin
(#3195; Cell Signaling Technology, Inc.) and Vimentin (sc-6260;
Santa Cruz Biotechnology, Inc.). The cells were washed three times
with PBS and were incubated with Alexa Fluor®
488-conjugated anti-mouse IgG (#R37120; Invitrogen; Thermo Fisher
Scientific, Inc.) and Flamma® 648-conjugated anti-rabbit
IgG (RSA1261a; BioActs, Incheon, Korea) secondary antibodies
diluted 1:200 in blocking buffer. Fluorescent signals were
visualized by confocal microscopy (Leica Microsystems GmbH,
Wetzlar, Germany).
Statistical analysis
All in vitro results were derived from at
least three independent experiments performed in triplicate. Data
were analyzed using GraphPad Prism 5 software (GraphPad Software,
Inc., La Jolla, CA, USA). The significance of differences between
experimental and control groups was determined using one-way
analysis of variance followed by Newman-Keuls multi-comparison test
(Prism 5. GraphPad Software, Inc., CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibitors of mitochondrial respiration
induce mesenchymal morphology in lung cancer cells
Prior to investigating the effects of oligomycin A
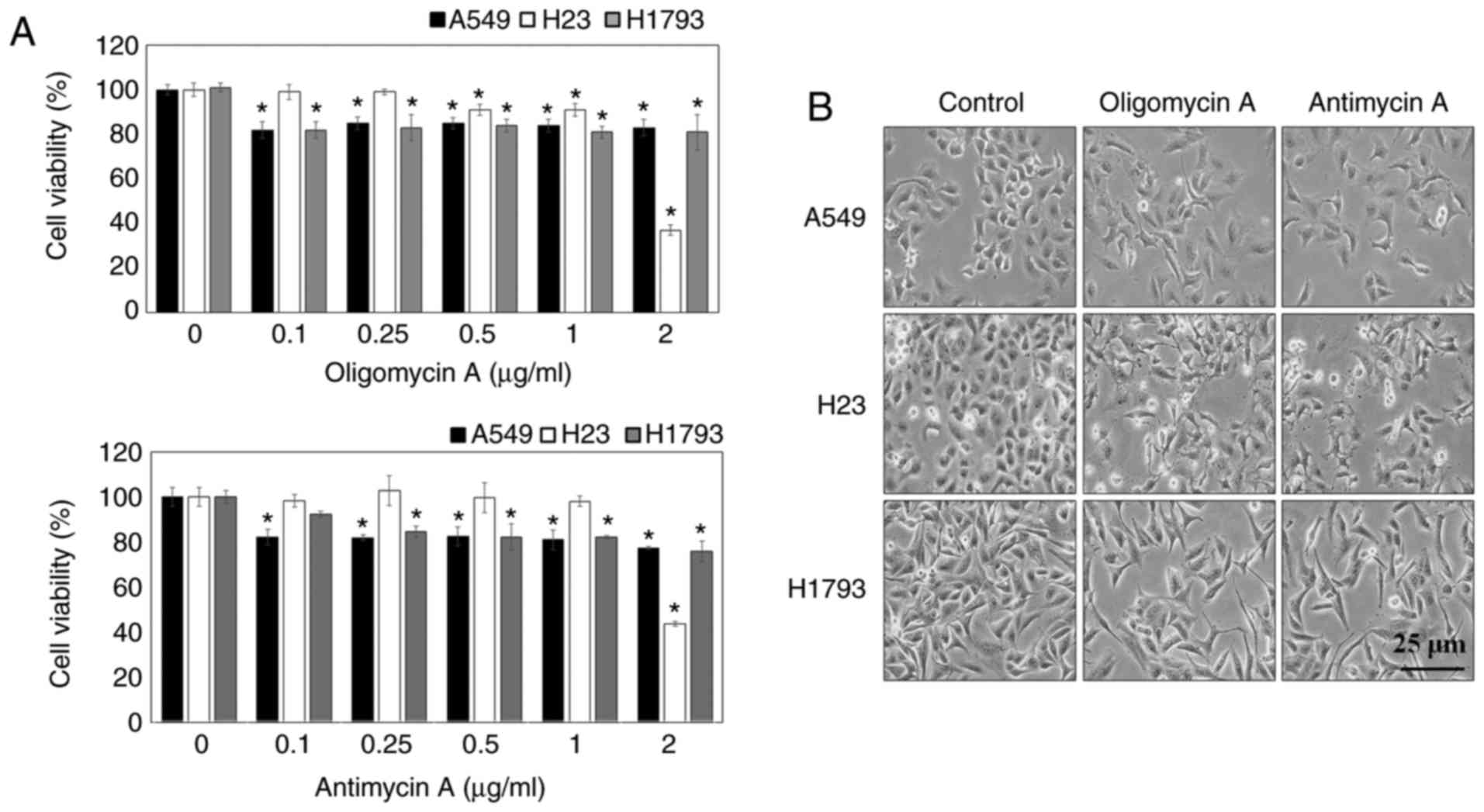
and antimycin A on EMT, the cytotoxic effects of oligomycin A and
antimycin A were examined using the MTT assay. The results
demonstrated that viability was >80% in A549, H23 and H1793
cells following treatment with oligomycin A and antimycin A at
doses <1 µg/ml. Cytotoxic effects of oligomycin A and
antimycin A were observed following treatment at 2 µg/ml in
A549 cells (Fig. 1A). The present
study aimed to determine whether the inhibitors of mitochondrial
respiration, oligomycin A and anti-mycin A, may induce EMT in human
epithelial lung cancer cell lines, including A549, H23 and H1793.
Cell morphology was observed by microscopy; the results
demonstrated that untreated lung cancer cells (control) exhibited
cobblestone-like cell morphology and cluster formation, which are
characteristic of epithelial cells (Fig. 1B). Notably, following treatment
with 1 µg/ml oligomycin A or antimycin A for 24 h, the cells
exhibited mesenchymal-like morphological features, including a
spindle shape and reduced intercellular contacts. The regulatory
effects of mitochondrial respiration inhibitors on the
EMT-associated markers E-cadherin and Vimentin were determined in
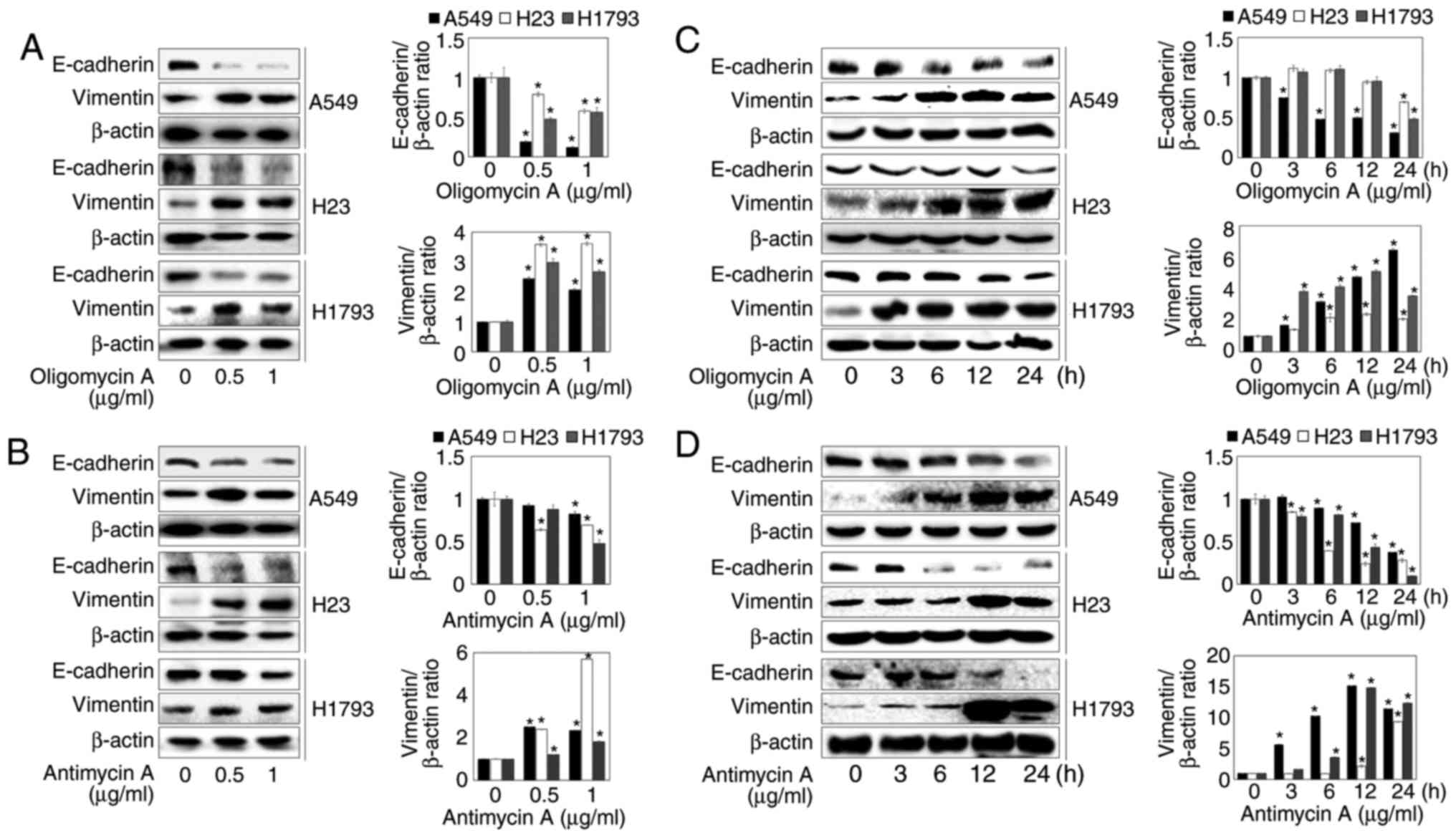
lung cancer cells using western blotting. As shown in Fig. 2A and B, the protein expression
levels of E-cadherin were significantly decreased by treatment with
oligomycin A and antimycin A at the indicated concentrations;
however, oligomycin A and antimycin A markedly increased Vimentin
protein expression. These results suggested that mitochondrial
respiration inhibitors may promote EMT. In addition, oligomycin A
and antimycin A enhanced the down-regulation of E-cadherin and the
upregulation of Vimentin in a time-dependent manner (Fig. 2C and D). Since mitochondrial
respiration inhibitor-induced EMT was most prominent in A549 cells
among the three types of epithelial lung cancer cells, subsequent
experiments were performed on the A549 cell line.
Inhibitors of mitochondrial respiration
induce EMT-associated transcriptional activation by increasing
mesenchymal marker expression
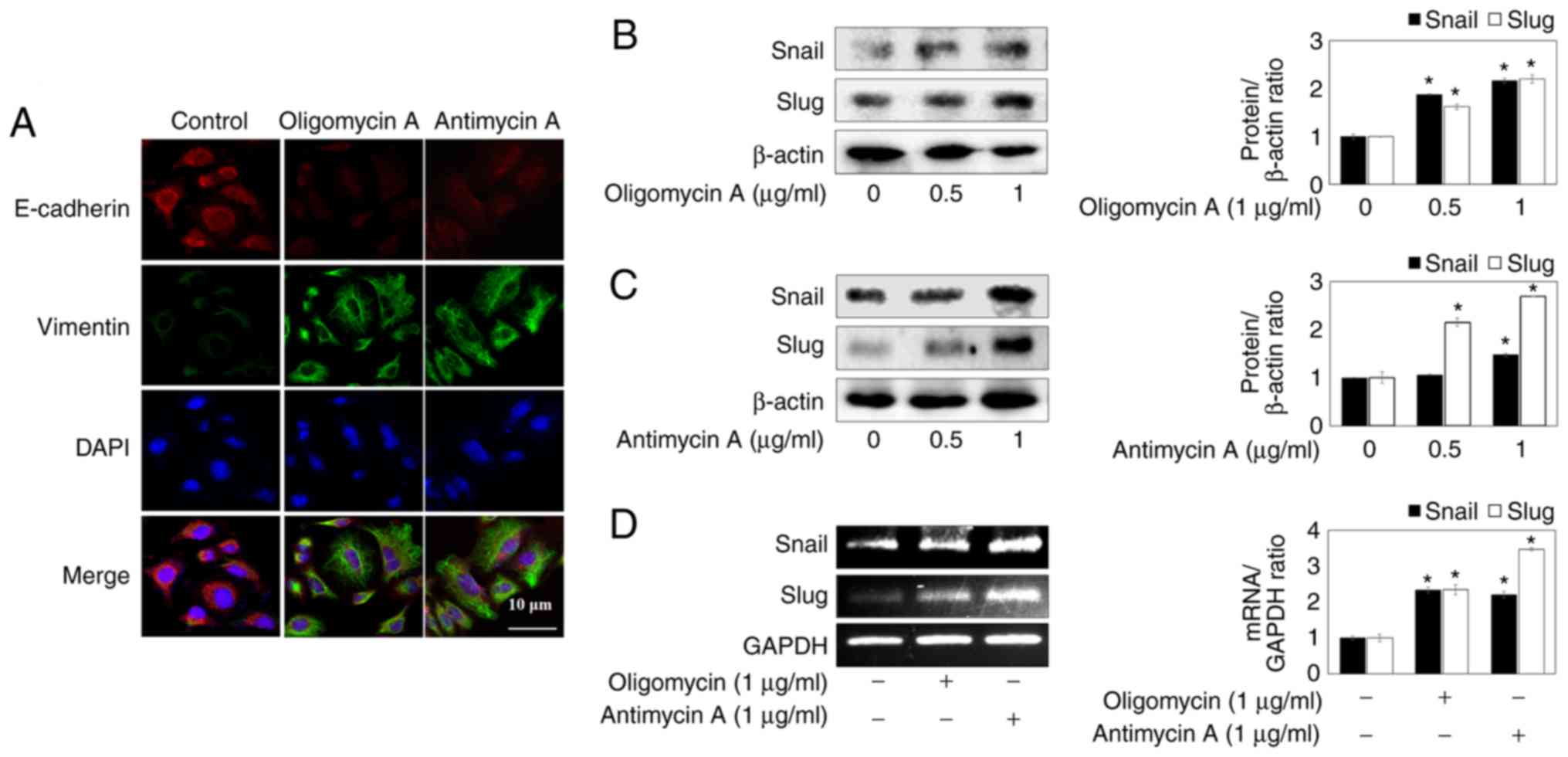
To confirm whether the oligomyicin A- and antimycin
A-induced mesenchymal-like morphology of lung cancer cells was
closely associated with EMT, immunofluorescence staining was
performed using E-cadherin and Vimentin antibodies. The results
detected a decrease in E-cadherin expression and an increase in
Vimentin expression upon oligomycin A (1 µg/ml) and
antimycin A (1 µg/ml) treatment of A549 lung cancer cells
(Fig. 3A).
Snail is a prominent inducer of EMT and strongly
suppresses E-cadherin expression. Snail and Slug are zinc-finger
transcriptional factors that downregulate E-cadherin expression and
upregulate Vimentin expression by binding E-boxes (18). Therefore, in order to determine if
the mitochondrial respiration inhibitors induce EMT by regulating
transcription factors, the protein expression levels of Snail and
Slug were detected. As shown in Fig.
3B and C, oligomycin A and antimycin A increased the protein
expression levels of Snail and Slug. In addition, the mRNA
expression levels of Snail and Slug were increased by oligomycin A
and antimycin A (Fig. 3D), thus
suggesting that mitochondrial respiration inhibitors may enhance
the down-regulation of E-cadherin and the upregulation of Vimentin
by promoting Snail and Slug expression in A549 lung cancer
cells.
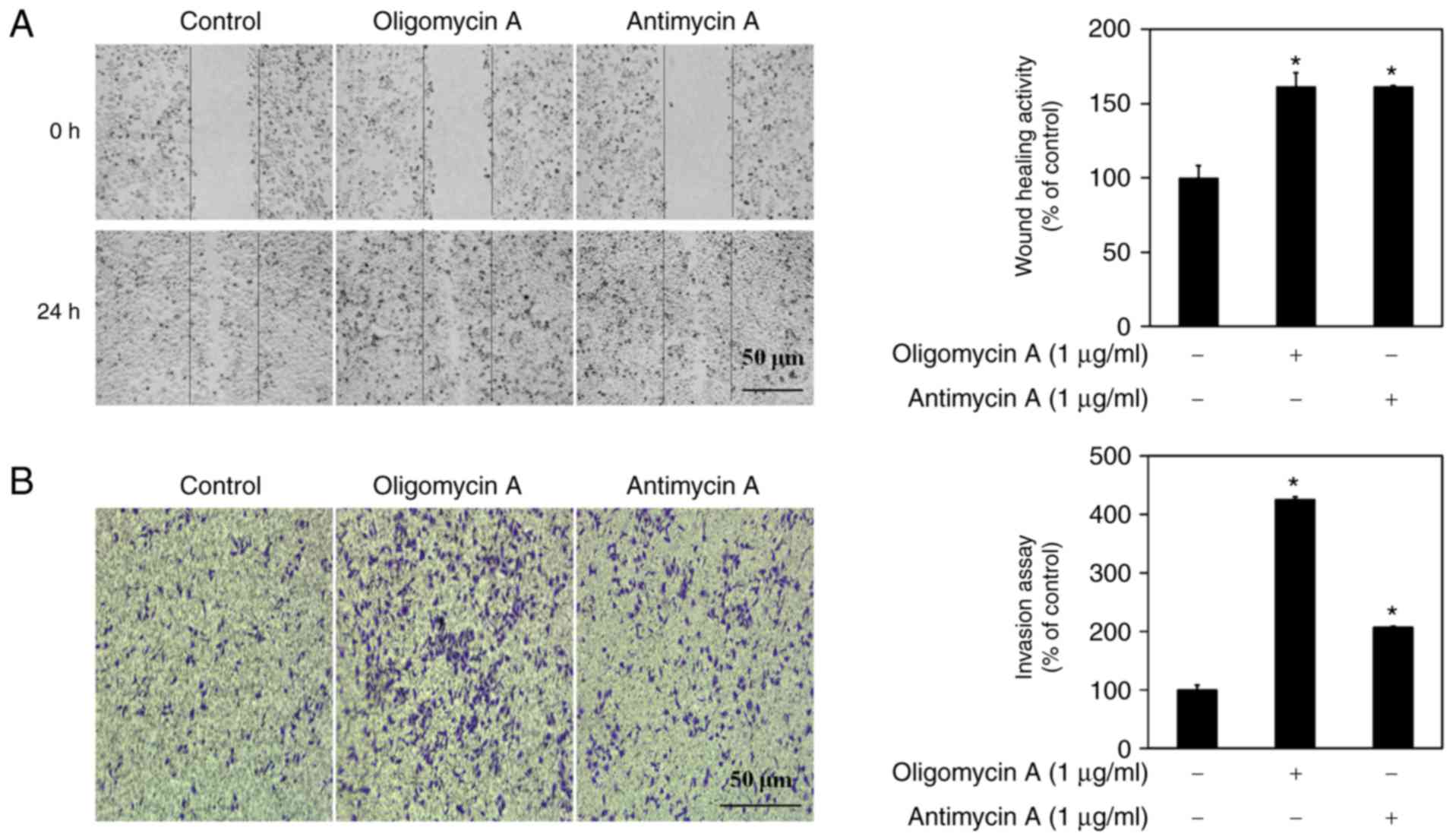
Inhibitors of mitochondrial respiration
increase cell migration and invasion of A549 lung cancer cells
EMT is an important process in epithelial-derived
tumor malignancies, and is associated with cell adhesion. In
addition, a partial EMT step has been reported to upregulate wound
healing and the invasive activity of cancer cells (19,20). Therefore, the migratory effects of
mitochondrial respiration inhibitors were determined in A549 cells
using the wound-healing assay. Compared with in the control group,
treatment with 1 µg/ml mitochondrial respiration inhibitors
for 24 h significantly (1.5-fold) promoted the migration of A549
cells (Fig. 4A). In addition, the
effects of oligomycin A and antimycin A were determined on the
invasion of A549 cells using the invasion assay. After 24 h
treatment with oligomycin A and antimycin A, the invasion of A549
cells was increased 4.25- and 2.07-fold, respectively (Fig. 4B). The invasive effects of
oligomycin A were much higher than those of antimycin A. These
results suggested that mitochondrial respiration inhibitors may
enhance migration and invasion via induction of EMT in A549
cells.
Inhibitors of mitochondrial respiration
mediate EMT through activation of the AKT and AMPK pathways in A549
cells
It has previously been reported that EMT is induced
by factors, such as ROS, insulin receptor substrate-1 and hypoxia
(21–23). Mitochondrial dysfunction caused by
mitochondrial respiration inhibitors promotes cell migration
through the induction of intracellular ROS in SC-M1 cells (14). Therefore, the present study aimed
to determine whether oligomycin A and antimycin A may induce EMT
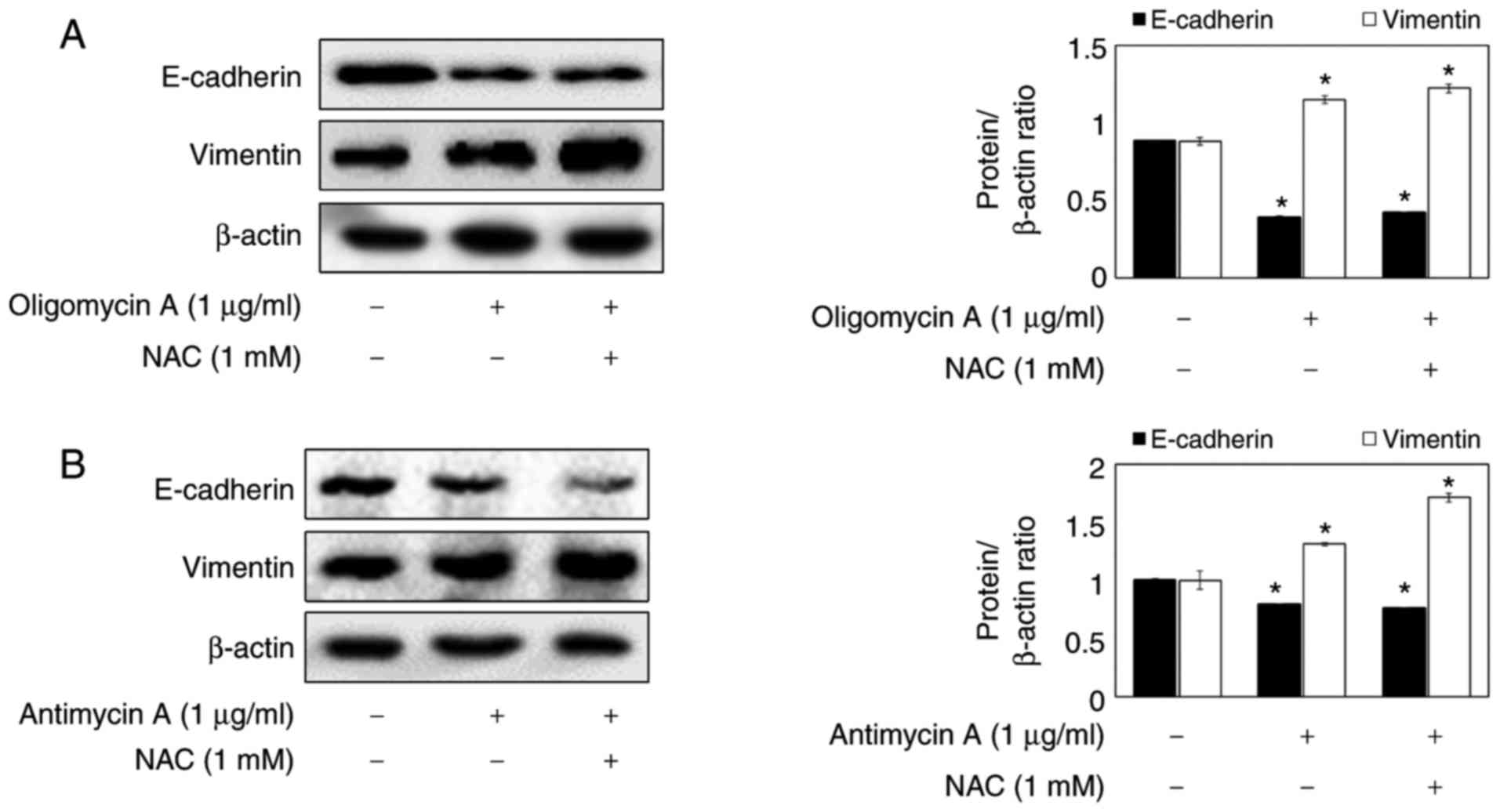
through ROS. As presented in Fig. 5A
and B, the ROS scavenger NAC had no effect on oligomycin
A/antimycin A-reduced E-cadherin and oligomycin A/antimycin
A-increased Vimentin.
Since previous studies indicated that the AMPK and
AKT pathways control colony formation and cell proliferation,
migration and invasion in several types of cancer (24–26), the present study investigated
whether inhibitors of mitochondrial respiration may regulate the
activation of AMPK, its downstream signaling substrate ACC, and AKT
in A549 cells (Fig. 6). As shown
in Fig. 6A, B, D, and E,
oligomycin A and antimycin A significantly increased the expression
levels of p-AMPK, p-ACC and p-AKT. In H1793 and H23 cells, p-AMPK
and AKT expression levels were was significantly increased
following treatment with 2 µg/ml oligomycin A and antimycin
A (Fig. 6C and F). Therefore, in
order to determine whether the two pathways are associated with
oligomycin A/antimycin A-induced EMT, the effects of inhibitors of
AMPK (Compound C, 10 µM) and AKT (wortmannin, 200 µM)
were determined on EMT-associated proteins. Compound C suppressed
oligomycin A/antimycin A-induced E-cadherin downregulation and
oligomycin A/antimycin A-induced Vimentin upregulation, along with
p-AMPK expression (Fig. 6G).
Wortmannin also inhibited oligomycin A/antimycin A-regulated EMT
marker protein expression (Fig.
6H). These results suggested that the AMPK and AKT signaling
pathways may serve important roles in mitochondrial respiration
inhibitors-induced EMT in lung cancer cells.
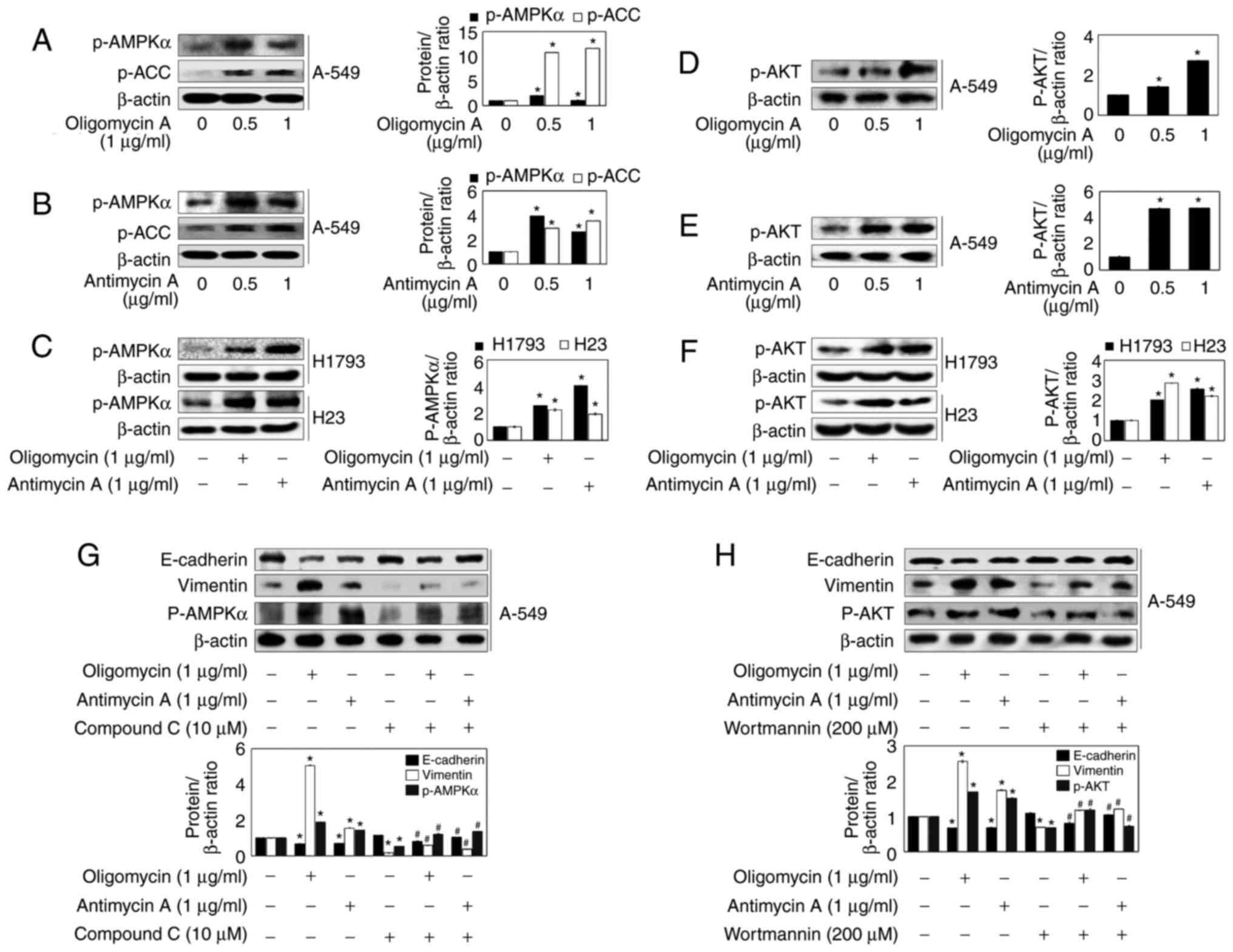 | Figure 6Inhibitors of mitochondrial
respiration modulate EMT through activation of AKT and AMPK
signaling in A549 cells. (A-F) A549, H1793 and H23 cells were
treated with the indicated concentrations of oligomycin A and
antimycin A for 24 h, and western blot analysis was conducted with
antibodies against p-AMPKα, p-ACC, p-AKT and β-actin. (G and H)
A549 cells were treated with 1 µg/ml oligomycin A or 1
µg/ml antimycin A in the absence or presence of 10 µM
Compound C or 200 µM wortmannin for 24 h. The expression
levels of E-cadherin, Vimentin, p-AMPKα and p-AKT were analyzed by
western blot analysis. β-actin was used as a control. Densitometric
analysis of each protein ratio was performed using ImageJ. Data are
presented as the means ± standard error of three independent
experiments. *P<0.05 vs. the control group,
#P<0.05 vs. the oligomycin A or antimycin A groups.
ACC, acetyl-CoA carboxylase; AKT, protein kinase; AMPK,
AMP-activated protein kinase; p-, phosphorylated. |
Discussion
Defective mitochondria were proposed by Otto Warbug
to explain why tumor cells undergo increased aerobic glycolysis,
namely the Warbug effect, unlike normal cells (27). Various mechanisms underlying
dysregulated energy generation in cancer cells are associated with
mitochondrial dysfunction induced by mtDNA mutations, mutations in
nuclear gene-encoded electron transport chain proteins and
treatment with inhibitors of mitochondrial electron transport
chains, including rotenone, oligomycin A and anti-mycin A (28–30). Previous studies have also
suggested that impaired mitochondria may serve critical roles in
tumor initiation and progression (1,31).
For example, OXPHOS dysfunction has been reported to be involved in
cell migration, invasion and metastasis (32). In addition, it was recently
demonstrated that mitochondrial dysfunction promotes EMT through
the transforming growth factor (TGF)-β/Smad/Snail signaling pathway
by activating c-Jun/activator protein-1 in hepatocellular carcinoma
cells (33). Another study also
demonstrated that mitochondrial dysfunction following TGFβ-1
exposure induces acquisition of a mesenchymal morphology and the
transfer from an epithelial to a mesenchymal phenotype in
pancreatic cancer (28). The
regulation of mitochondrial function by inhibitors of the
mitochondrial electron transport chain, and their effects on EMT,
cell migration and cell invasion, is critical but remains poorly
understood in lung cancer research.
EMT is a pathophysiological process that is
essential for the development of cancer cells, during which
epithelial cells lose their polarity and cell-cell adhesion is
disrupted, thus resulting in transformation into mesenchymal cells
through the reorganization of the cytoskeleton (34,35). In the present study, the effects
of two mitochondrial respiration inhibitors, oligomycin A and
antimycin A, on phenotypic alterations associated with EMT were
determined in H1793, H23 and A549 lung cancer cells. Oligomycin A
and antimycin A have previously been reported to increase cell
migration and expression of the mesenchymal protein marker Vimentin
in SC-M1 human gastric cancer cells (14). The damaged function of
mitochondrial OXPHOS has been demonstrated to initiate cell
migration, invasion and metastatic properties by regulating the
extracellular matrix (36,37).
As expected, the present study confirmed that oligomycin A and
antimycin A increased EMT, and induced the transformation of cells
into an invasive phenotype, thus suggesting that mitochondrial
respiration inhibition may be associated with the invasion and
migration of cancer cells.
During EMT, E-cadherin, which is an epithelial cell
marker that is located on the basolateral membrane of adherens
junctions, has been reported to be decreased due to the increase of
mesenchymal protein markers, including Vimentin and N-cadherin, and
of typical transcription factors, including Snail, Slug, TWIST and
zinc finger E-box-binding homeobox family proteins (38,39). In addition, increased expression
of Snail and Slug mRNA has a major role in EMT promotion in bladder
cancer compared with in normal cells and tissues (40). In the present study, treatment
with oligomycin A and anti-mycin A enhanced EMT by increasing the
expression of Vimentin and inhibiting the expression of E-cadherin
in H1793, H23 and A549 lung cancer cells. In addition, the protein
and mRNA expression levels of Snail and Slug were increased by
oligomycin A and antimycin A in A549 cells. These results indicated
that mitochondrial respiration inhibitors may promote regulation of
Vimentin and E-cadherin via induction of the transcription factors
Snail and Slug.
The present study also investigated the molecular
mechanisms through which oligomycin A and antimycin A operate as
oncogenic agents in lung cancer. AMPK is a key regulator involved
in maintaining energy homeostasis and regulating other
physiological functions in cells (41). Activation of AMPK is promoted by
various cellular environmental factors, including energy
deficiency, hypoxia and oxidative stress caused by non-functional
mitochondria (42). The present
study revealed that oligomycin A and antimycin A upregulated the
expression levels of p-AMPK. In addition, mitochondrial respiration
inhibitor-induced E-cadherin downregulation and Vimentin
upregulation were suppressed by Compound C (AMPK inhibitor) in A549
cells. Numerous studies have reported that ROS arising from
mitochondrial dysfunction mediate EMT and cell invasion in various
cancer cells via activation of the AMPK signaling pathway (29,42). Furthermore, mitochondrial
malfunction increases cell migration and invasion by increasing the
production of ROS in SKBR3 breast cancer cells (1). However, in the present study,
oligomycin A/antimycin A-regulated EMT marker protein expression
was not affected by treatment with a ROS scavenger in A549 cells.
This discrepancy between the present and previous studies may be
due to the different cell types used. These results suggested that
oligomycin A and antimycin A triggered EMT by activating AMPK
independent of ROS in A549 cells.
The AKT pathway is known to serve an important role
in intracellular processes, including glucose metabolism, cell
proliferation and metastasis. It is also highly activated in
various tumor cells (43). The
mitochondrial stress induced by mitochondrial respiration
inhibitors, including rotenone and oligomycin A, upregulates matrix
metalloproteinase-13 expression and invasion by activating AKT
pathway signaling in various cancer cells (44). In the present study, oligomycin A
and antimycin A upregulated the expression levels of p-AKT in three
types of lung cancer cells. In addition, inhibition of AKT
phosphorylation suppressed oligomycin A- and antimycin A-induced
E-cadherin downregulation and Vimentin upregulation, thus
suggesting that inhibitors of mitochondrial respiration may
initiate EMT by activating AKT pathway signaling in A549 cells.
In conclusion, the present study demonstrated that
oligomycin A and antimycin A may induce mesenchymal-like
morphological features, and cell migration and invasion, in various
human lung cancer cell lines. Oligomycin A and anti-mycin A
downregulated expression of the epithelial marker protein
E-cadherin, and upregulated expression of the mesenchymal marker
proteins Vimentin, Snail and Slug. In addition, oligomycin A and
antimycin A induced the protein expression levels of mesenchymal
markers via activation of the AKT and AMPK signaling pathways.
These results indicated that inhibition of mitochondrial
respiration may activate metastasis via EMT in lung cancer.
Therefore, control of mitochondrial respiration in patients with
lung cancer may be considered a novel approach for the prevention
of metastasis.
Acknowledgments
Not applicable.
Funding
This study was supported by the KIST intramural
research grant (grant no. 2Z04930) and also by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
NRF-2015R1D1A4A01019102).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YJJ and YCC designed the experiments. SYH and YJJ
performed the experiments. YC, SKH and YSB analyzed the data, and
contributed materials and analytical tools. SYH, YJJ and YCC wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ma J, Zhang Q, Chen S, Fang B, Yang Q,
Chen C, Miele L, Sarkar FH, Xia J and Wang Z: Mitochondrial
dysfunction promotes breast cancer cell migration and invasion
through HIF1α accumulation via increased production of reactive
oxygen species. PLoS One. 8:e694852013. View Article : Google Scholar
|
|
2
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji Q, Liu X, Han Z, Zhou L, Sui H, Yan L,
Jiang H, Ren J, Cai J and Li Q: Resveratrol suppresses
epithelial-to-mesenchymal transition in colorectal cancer through
TGF-β1/Smads signaling pathway mediated Snail/E-cadherin
expression. BMC Cancer. 15:972015. View Article : Google Scholar
|
|
6
|
Hou P, Zhao Y, Li Z, Yao R, Ma M, Gao Y,
Zhao L, Zhang Y, Huang B and Lu J: LincRNA-ROR induces
epithelial-to-mesenchymal transition and contributes to breast
cancer tumorigenesis and metastasis. Cell Death Dis. 5:e12872014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dumitriu IE, Dunbar DR, Howie SE, Sethi T
and Gregory CD: Human dendritic cells produce TGF-beta 1 under the
influence of lung carcinoma cells and prime the differentiation of
CD4+CD25+Foxp3+ regulatory T
cells. J Immunol. 182:2795–2807. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Godlewski J, Nowicki MO, Bronisz A, Nuovo
G, Palatini J, De Lay M, Van Brocklyn J, Ostrowski MC, Chiocca EA
and Lawler SE: MicroRNA-451 regulates LKB1/AMPK signaling and
allows adaptation to metabolic stress in glioma cells. Mol Cell.
37:620–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Godlewski J, Newton HB, Chiocca EA and
Lawler SE: MicroRNAs and glioblastoma; the stem cell connection.
Cell Death Differ. 17:221–228. 2010. View Article : Google Scholar
|
|
11
|
Kowaltowski AJ, de Souza-Pinto NC,
Castilho RF and Vercesi AE: Mitochondria and reactive oxygen
species. Free Radic Biol Med. 47:333–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brandon M, Baldi P and Wallace DC:
Mitochondrial mutations in cancer. Oncogene. 25:4647–4662. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zamzami N, Susin SA, Marchetti P, Hirsch
T, Gómez-Monterrey I, Castedo M and Kroemer G: Mitochondrial
control of nuclear apoptosis. J Exp Med. 183:1533–1544. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hung WY, Huang KH, Wu CW, Chi CW, Kao HL,
Li AF, Yin PH and Lee HC: Mitochondrial dysfunction promotes cell
migration via reactive oxygen species-enhanced β5-integrin
expression in human gastric cancer SC-M1 cells. Biochim Biophys
Acta. 1820:1102–1110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang CJ, Yin PH, Yang DM, Wang CH, Hung
WY, Chi CW, Wei YH and Lee HC: Mitochondrial dysfunction-induced
amphiregulin upregulation mediates chemo-resistance and cell
migration in HepG2 cells. Cell Mol Life Sci. 66:1755–1765. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeong JH, Kang SS, Park KK, Chang HW,
Magae J and Chang YC: p53-independent induction of G1 arrest and
p21WAF1/CIP1 expression by ascofuranone, an isoprenoid antibiotic,
through downregulation of c-Myc. Mol Cancer Ther. 9:2102–2113.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin CW, Hou WC, Shen SC, Juan SH, Ko CH,
Wang LM and Chen YC: Quercetin inhibition of tumor invasion via
suppressing PKC delta/ERK/AP-1-dependent matrix metalloproteinase-9
activation in breast carcinoma cells. Carcinogenesis. 29:1807–1815.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yi BR, Kim TH, Kim YS and Choi KC:
Alteration of epithelial-mesenchymal transition markers in human
normal ovaries and neoplastic ovarian cancers. Int J Oncol.
46:272–280. 2015. View Article : Google Scholar
|
|
19
|
Cano A, Perez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blanco MJ, Barrallo-Gimeno A, Acloque H,
Reyes AE, Tada M, Allende ML, Mayor R and Nieto MA: Snail1a and
Snail1b cooperate in the anterior migration of the axial
mesendoderm in the zebrafish embryo. Development. 134:4073–4081.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, He K, Wang D, Yuan X, Liu Y, Ji H
and Song J: TMEPAI regulates EMT in lung cancer cells by modulating
the ROS and IRS-1 signaling pathways. Carcinogenesis. 34:1764–1772.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu X and Kang Y: Hypoxia and
hypoxia-inducible factors: Master regulators of metastasis. Clin
Cancer Res. 16:5928–5935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giannoni E, Parri M and Chiarugi P: EMT
and oxidative stress: A bidirectional interplay affecting tumor
malignancy. Antioxid Redox Signal. 16:1248–1263. 2012. View Article : Google Scholar
|
|
24
|
Han B, Cui H, Kang L, Zhang X, Jin Z, Lu L
and Fan Z: Metformin inhibits thyroid cancer cell growth,
migration, and EMT through the mTOR pathway. Tumour Biol.
36:6295–6304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu XL, Zhang XT, Meng J, Zhang HF, Zhao
Y, Li C, Sun Y, Mei QB, Zhang F and Zhang T: ING5 knockdown
enhances migration and invasion of lung cancer cells by inducing
EMT via EGFR/PI3K/Akt and IL-6/STAT3 signaling pathways.
Oncotarget. 8:54265–54276. 2017.PubMed/NCBI
|
|
27
|
Upadhyay M, Samal J, Kandpal M, Singh OV
and Vivekanandan P: The Warburg effect: Insights from the past
decade. Pharmacol Ther. 137:318–330. 2013. View Article : Google Scholar
|
|
28
|
Guo Q: Changes in mitochondrial function
during EMT induced by TGFβ-1 in pancreatic cancer. Oncol Lett.
13:1575–1580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Favre C, Zhdanov A, Leahy M, Papkovsky D
and O'Connor R: Mitochondrial pyrimidine nucleotide carrier (PNC1)
regulates mitochondrial biogenesis and the invasive phenotype of
cancer cells. Oncogene. 29:3964–3976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sequeira A, Martin MV, Rollins B, Moon EA,
Bunney WE, Macciardi F, Lupoli S, Smith EN, Kelsoe J, Magnan CN, et
al: Mitochondrial mutations and polymorphisms in psychiatric
disorders. Front Genet. 3:1032012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kwong JQ, Henning MS, Starkov AA and
Manfredi G: The mitochondrial respiratory chain is a modulator of
apoptosis. J Cell Biol. 179:1163–1177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nunes JB, Peixoto J, Soares P, Maximo V,
Carvalho S, Pinho SS, Vieira AF, Paredes J, Rego AC, Ferreira IL,
et al: OXPHOS dysfunction regulates integrin-β1 modifications and
enhances cell motility and migration. Hum Mol Genet. 24:1977–1990.
2015. View Article : Google Scholar
|
|
33
|
Yi EY, Park SY, Jung SY, Jang WJ and Kim
YJ: Mitochondrial dysfunction induces EMT through the
TGF-β/Smad/Snail signaling pathway in Hep3B hepatocellular
carcinoma cells. Int J Oncol. 47:1845–1853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pirozzi G, Tirino V, Camerlingo R, Franco
R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N and Rocco
G: Epithelial to mesenchymal transition by TGFβ-1 induction
increases stemness characteristics in primary non small cell lung
cancer cell line. PLoS One. 6:e215482011. View Article : Google Scholar
|
|
35
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S and Sarkar FH:
Metformin inhibits cell proliferation, migration and invasion by
attenuating CSC function mediated by deregulating miRNAs in
pancreatic cancer cells. Cancer Prev Res (Phila). 5:355–364. 2012.
View Article : Google Scholar
|
|
36
|
He X, Zhou A, Lu H, Chen Y, Huang G, Yue
X, Zhao P and Wu Y: Suppression of mitochondrial complex I
influences cell metastatic properties. PLoS One. 8:e616772013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Imanishi H, Hattori K, Wada R, Ishikawa K,
Fukuda S, Takenaga K, Nakada K and Hayashi J: Mitochondrial DNA
mutations regulate metastasis of human breast cancer cells. PLoS
One. 6:e234012011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Perez-Moreno M, Jamora C and Fuchs E:
Sticky business: Orchestrating cellular signals at adherens
junctions. Cell. 112:535–548. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Son H and Moon A: Epithelial-mesenchymal
transition and cell invasion. Toxicol Res. 26:245–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu M, Li J, Wang X, Meng S, Shen J, Wang
S, Xu X, Xie B, Liu B and Xie L: MiR-22 suppresses
epithelial-mesenchymal transition in bladder cancer by inhibiting
Snail and MAPK1/Slug/vimentin feedback loop. Cell Death Dis.
9:2092018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marshall S: Role of insulin, adipocyte
hormones, and nutrient-sensing pathways in regulating fuel
metabolism and energy homeostasis: A nutritional perspective of
diabetes, obesity, and cancer. Sci STKE. 2006:re72006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He K, Guo X, Liu Y, Li J, Hu Y, Wang D and
Song J: TUFM downregulation induces epithelial-mesenchymal
transition and invasion in lung cancer cells via a mechanism
involving AMPK-GSK3β signaling. Cell Mol Life Sci. 73:2105–2121.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li CH, Cheng YW, Liao PL, Yang YT and Kang
JJ: Chloramphenicol causes mitochondrial stress, decreases ATP
biosynthesis, induces matrix metalloproteinase-13 expression, and
solid-tumor cell invasion. Toxicol Sci. 116:140–150. 2010.
View Article : Google Scholar : PubMed/NCBI
|