Introduction
Human breast epithelial cells are constantly exposed
to polypeptide growth factors and steroid hormones as part of their
physiological control. The levels of epidermal growth factor (EGF)
and estrogen have an impact on cell physiology, including cell
proliferation rate, differentiation and migration (1–5).
The exposure of cells to elevated levels of EGF and estradiol for a
prolonged period of time may irreversibly affect their physiology,
which may consequently impact on cell carcinogenic
transformation.
Tumorigenesis is a complex process involving
alterations of multiple genes, proteins and regulatory pathways
(6,7). In breast cancer, the overexpression
of epidermal growth factor receptor (EGFR) in the primary tumor
correlates with increased metastatic dissemination and aggressive
tumor progression (8). In total,
>70% of breast cancer tumors express high levels of estrogen
receptor-α (ERα), and a large number of these tumors require
estrogen to support cancer cell proliferation and tumor progression
(9). As the EGFR and estrogen
signaling pathways are closely associated with the development of
breast cancer, they are targets for the treatment of breast cancer
(10,11).
The EGF and estrogen signaling pathways share a
number of intracellular signaling mechanisms due to crosstalk
(10–12). The inverse correlation observed
between the expression levels of EGFR and ERα has been explained by
compensatory mechanisms, which are activated in malignant cells in
order to maintain a high proliferative status. When one of the
above receptors is upregulated or down-regulated, the expression of
the other receptor compensates for this alteration (12). It has been reported that EGF- and
ERα-dependent transcription may operate in parallel, although with
a marked overlap in the affected genes (13). Estrogen may also control the
downregulation of EGFR (10).
However, the exact molecular mechanisms of the crosstalk remain to
be fully elucidated. In addition, how long-term exposure to EGF and
estrogen may affect the carcinogenic properties of cells remains
unclear.
Breast cancer stem cells are defined as
self-renewing cells required to initiate a tumor and drive tumor
growth when transplanted into mice (14,15). In human breast cancer, cancer stem
cells show a cluster of differentiation
(CD)44+/CD24− pattern of surface markers
(16). This population of cells
exhibits the ability to form three-dimensional mammospheres under
low-adherence conditions and exhibit increased resistance to
chemotherapeutic compounds (14–17). The EGFR signaling pathway has been
implicated in the self-renewal of breast cancer stem cells
(18). It has been reported that
the EGFR tyrosine kinase inhibitor Iressa significantly decreases
the formation of mammospheres by cells derived from a ductal
carcinoma in situ (19). A
number of studies have reported that estrogen treatment may expand
the pool of breast cancer stem cells (20,21). The present study hypothesized
that, if prolonged exposure to EGF and estradiol changes the
physiology of breast cancer cells, then it may also modulate cell
responsiveness to anticancer drugs. The results revealed that
sustained exposure of conditionally tumorigenic MCF7 human breast
adenocarcinoma cells to EGF and 17β-estradiol led to the generation
of cells with increased proliferation rate, increased
CD44+/CD24− cell fraction population and a
different pattern of response to therapeutic drugs, including
Iressa, tamoxifen and the transforming growth factor (TGF)β type I
receptor kinase inhibitor SB431542, compared with non-exposed
control cells. The mouse xenograft experiments revealed that these
changes in cell physiology were not sufficient to ensure additional
tumor development in immunocompromised mice upon withdrawal of
treatment.
Materials and methods
Cell culture
The MCF7 human breast adenocarcinoma cell line was
purchased from the American Type Culture Collection (Rockville, MD,
USA; HTB-22™) and cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Merck
KGaA, Darmstadt, Germany), 100 U/ml penicillin, 100 µg/ml
streptomycin and 2 mM glutamine. The cells were maintained in a
humidified atmosphere with 5% CO2 at 37°C. To generate
cell clones exposed to EGF, estrogen and EGF + estrogen, the MCF7
cells at an initial density of 1×106 cells/plate were
grown on agarose-coated culture dishes with 5 ng/ml of EGF
(Sigma-Aldrich; Merck KGaA), 5 nM of 17β-estradiol (Sigma-Aldrich;
Merck KGaA) or 5 ng/ml of EGF + 5 nM 17β-estradiol. Agarose coating
prevented the attachment of cells to the plate. After 4 weeks, the
cells were transferred to 96-well plates coated with agarose. The
growing clones of cells were expanded, and 12 clones for each of
the treatment conditions were randomly selected for evaluation of
their proliferation status. For subsequent experiments, clones that
represented the average proliferation status of the initial clones
were selected, in addition to the 12 randomly selected clones (data
not shown). The total time of cell exposure to EGF and/or
17β-estradiol was 40–42 days, prior to the random selection of 12
initial clones, from which other clones were selected for the mouse
model and subsequent experiments. The selected clones were cultured
and analyzed on regular culture dishes without agarose coating.
MTT assay
Cell proliferation was measured using the CellTiter
96® Non-Radioactive Cell Proliferation assay (Promega
Biotech AB, Stockholm, Sweden). The MTT assay was performed
according to the manufacturer's protocol. In brief, 1,000 cells
were seeded per well in 96-well plates in triplicate, treated with
5 ng/ml EGF (Sigma-Aldrich; Merck KGaA), 5 nM 17β-estradiol
(Sigma-Aldrich; Merck KGaA) or 5 ng/ml EGF + 5 nM 17β-estradiol,
incubated in complete DMEM culture medium for 48 h, and then
subjected to the MTT assay. The formazan crystals were dissolved in
DMSO. The absorbance at 570 nm was recorded using a plate reader.
Statistical significance of observed differences was evaluated
using a one-way analysis of variance (ANOVA) with Tukey's honest
significant difference (HSD) test.
Colony formation assay
Colony formation assays were performed in 6-well
plates. Briefly, a bottom layer consisting of 0.5% agar in complete
culture medium was poured and, once solidified, was covered by a
layer containing 0.3% agar and 2,000 cells/well. Treatments were
performed by the addition of 5 ng/ml EGF (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), 5 nM 17β-estradiol (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) or 5 ng/ml EGF + 5 nM 17β-estradiol to
the medium in the top layer on experimental day 1, considering the
total volume of medium in the well. The plates were placed in the
incubator, and the colonies were counted under a light microscope
LeicaDHi1 (Leica Microsystems GmbH, Wetzlar, Germany) following 2
weeks of incubation. Treatments were introduced on day 1 of
experiment, and the cells were under treatment for the 2 weeks of
incubation. Colonies containing a minimum of 64 cells were counted.
The statistical significance of observed differences was evaluated
using a one-way ANOVA with Tukey's HSD test.
Mouse xenograft tumorigenesis assay
All experiments on mice were performed according to
Swedish and International guidelines (ethical approval no. C123/6,
granted by the Uppsala Animal Tests Committee of the Uppsala Court,
Uppsala, Sweden). Five severe combined immunodeficiency (SCID) mice
(females, 12 weeks of age, housed in pathogen-free conditions, at
25°C, 12-h day/night cycle, food and water ad libitum (free
access); Charles Rivers Laboratory, Worcester, MA, USA) were
injected subcutaneously in the flank with selected and tested cell
clones per condition, and with parental MCF7 cells, and six mice
were injected with wild-type cells. Each mouse received
5×106 cells/injection in a volume of 100 µl
suspended at a 1:1 ratio in PBS and Matrigel (BD Pharmingen, San
Diego, CA, USA) in the mouse flanks. The mice were monitored twice
a week for overall health and tumor formation. The tumor diameters
were measured with calipers, and the tumor volume in mm3
was calculated using the following formula: Volume=width2 × length ×0.5. The mice were sacrificed
at 29 weeks. All the tumors were excised, fixed in 4%
paraformaldehyde for 24 h at 4°C and embedded in paraffin for
analysis.
Immunohistochemistry
Paraffin-embedded sections of 5-µm were
deparaffinized, rehydrated and subjected to antigen retrieval by
incubation in citrate buffer (pH 6; Dako, Glostrup, Denmark) twice
for 7 min at 95°C. Quenching of endogenous peroxidase was performed
by incubation in 3% H2O2 in PBS for 10 min at
room temperature. Upon washing in PBS, the slides were incubated in
20% normal goat serum (cat. no. S-1000; Vector Laboratories, Inc.,
Burlingame, CA, USA) in PBST (PBS with 0.1% Tween-20) for blocking.
The tumor sections were then incubated with anti-human Ki-67 clone
MIB1 antibody (cat. no. M7240; Dako) diluted 1:250 and with
anti-von Willebrand factor (vWF) antibody (cat. no. ab6994; Abcam,
Cambridge, MA, USA) diluted 1:750 in blocking buffer overnight at
4°C. Following washing with PBST, a secondary biotinylated
universal antibody (horse anti-mouse/rabbit IgG; cat. no. BA-1400;
Vector Laboratories, Inc.) at dilution 1:50 was added and incubated
for 45 min in room temperature. The slides were stained using a
Vectastain Elite ABC kit (Vector Laboratories, Inc., Burlingame,
USA) following the manufacturer's protocol, and then
counter-stained with Mayer's hematoxylin, dehydrated and mounted
with Mountex (SouthernBiotech, Birmingham, AL, USA). Images of the
stained tissues were captured using a Leica DFC camera and images
were acquired with Leica QWin version 3 software (Leica
Microsystems Imaging Solutions, Ltd., Cambridge, UK). The
proliferative score was measured as the ratio of MIB1-stained cells
to the total number of cells. The vessel score was measured as the
number of vessels stained with anti-vWF antibody per tumor
section.
Flow cytometry
A total of 1×106 cells were used per
experimental condition. The cells were collected, washed twice in
cold PBS + 1% bovine serum albumin (BSA: Sigma-Aldrich; Merck KGaA)
and incubated at 4°C with either 1:25-diluted anti-CD44-fluoresein
isothiocyanate antibody (1:25; cat. no. 555478; clone G44-26; BD
Pharmingen) or anti-CD24-allophycocyanin antibody (1:25; cat. no.
561646; clone ML5; BD Pharmingen) in PBS + 1% BSA for 30 min in the
dark. Subsequently, the cells were washed twice in cold PBS + 1%
BSA and resuspended in 400 µl cold PBS + 1% BSA for flow
cytometric analysis.
Mammosphere assays
The cells were trypsinized, mechanically separated
and passed through 40-µm strainers to obtain a single cell
suspension. Subsequently, the cells were plated at a density of
5,000 or 1,000 cells in 4 ml per well in super-low-attachment
plates. Treatments were added on day 1 upon seeding of cells, and
continued for 2 weeks as follows: EGF at 5 ng/ml, 17β-estradiol at
5 nM, TGFβ1 at 10 ng/ml, and Iressa, tamoxifen and SB431542 at 10
µM. The numbers of mammospheres formed were counted
following 2 weeks of incubation with these drugs.
Statistical analysis
Significant differences were calculated using a
one-way ANOVA with Tukey's HSD test. Data was analyzed using online
software (www.icalcu.com online test) and with IBM
SPSS software version 25 (IBM Corp., Armonk NY, USA). Results were
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Exposure to EGF and/or 17β-estradiol
enhances the proliferation rate of MCF7 cells
Following exposure of MCF7 cells to 17β-estradiol
and/or EGF for 40–42 days, 12 clones per experimental condition
were randomly collected. These clones were subjected to an MTT
proliferation assay, and representative clones were used for
subsequent experiments (data not shown). Prior to the proliferation
experiments, EGF and 17β-estradiol were removed, and the cells were
cultured in a standard culture medium without EGF or 17β-estradiol
for two passages (~1 week). The proliferation rate of the cells was
then measured. Control cells underwent the same manipulations as
the EGF and 17β-estradiol-exposed cells. To evaluate whether the
selection procedure by itself had an impact on the MCF7 cells,
parental MCF7 cells were also included in the analysis. The
parental cells were grown under standard culturing conditions as an
adherent culture, and were not subjected to a substrate-independent
selection.
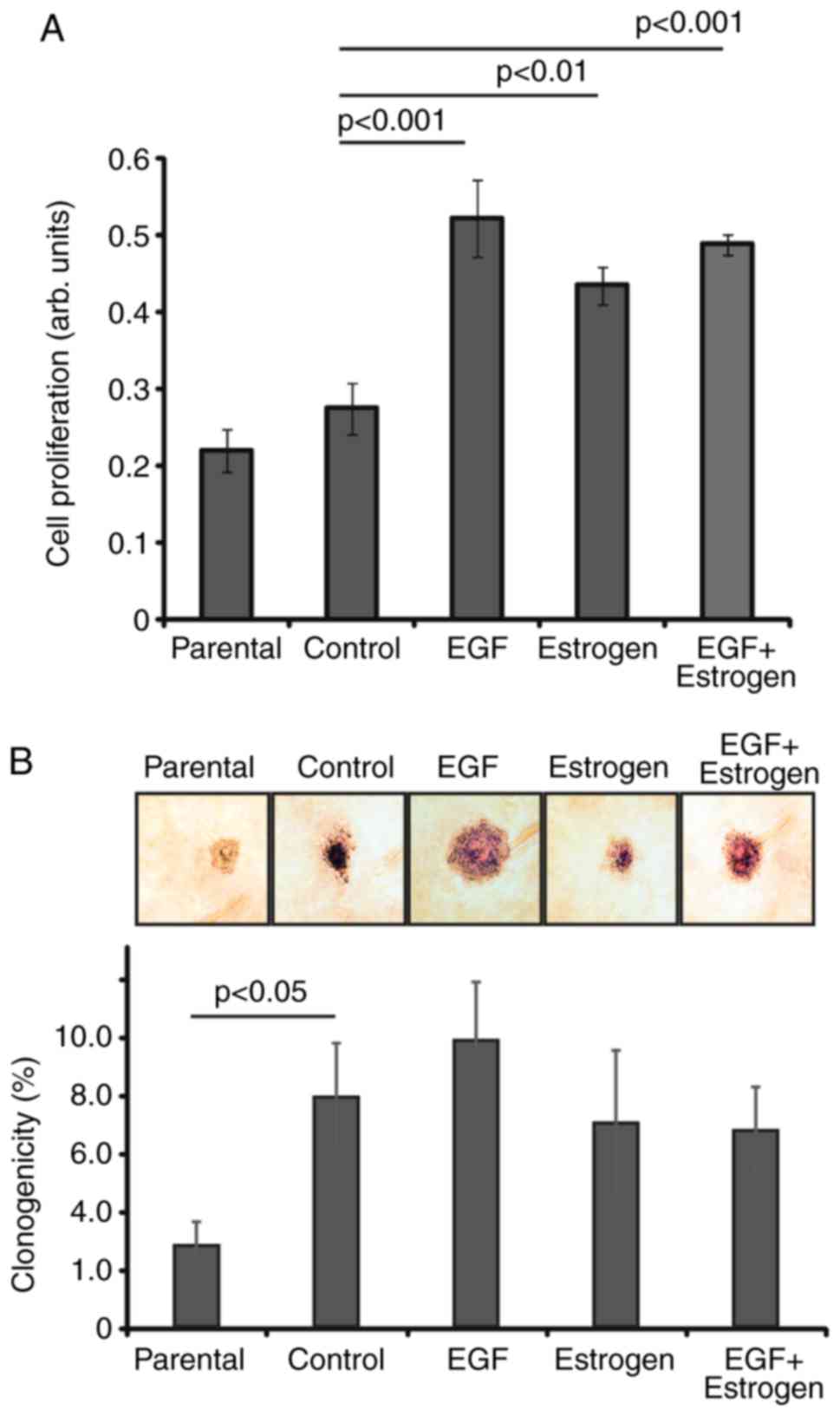
The results indicated that cells exposed to EGF
and/or 17β-estradiol exhibited a significantly enhanced rate of
cell proliferation, whereas the control cells only exhibited a
marginal increase in proliferation rate (Fig. 1A). These cell clones were used in
all subsequent experiments. The prolonged exposure to EGF and/or
17β-estradiol resulted in enhanced proliferation rates in
vitro.
Substrate-independent growth is one of the key
features of transformed cells; these cells do not require
attachment to a substrate for proliferation. A soft agar colony
formation assay was used to analyze the ability of cells to grow
unattached to a surface. It was observed that the control and EGF-
and/or 17β-estradiol-exposed cells exhibited an enhanced ability to
form colonies, compared with the parental cells (Fig. 1B). The difference between the
parental and control cells may be due to the selection of control
cells in the absence of adherence, whereas the parental cells were
maintained as an adherent culture. Between the various selection
conditions, the observed differences were not significant
(P>0.05), the colonies did not differ significantly in their
shapes, and no significant spreading of cells from the colonies
observed (Fig. 1B). This higher
level of colony formation in semi-solid medium may have resulted
from the effect of the substrate-independent selection of cells
during drug exposure (Fig. 1B).
The proliferation-stimulating effect of the various treatments was
observed when the cells were grown as a two-dimensional culture
(Fig. 1A).
EGF- and/or 17β-estrogen-exposed cells do
not exhibit enhanced tumor formation ability in a xenograft mouse
model
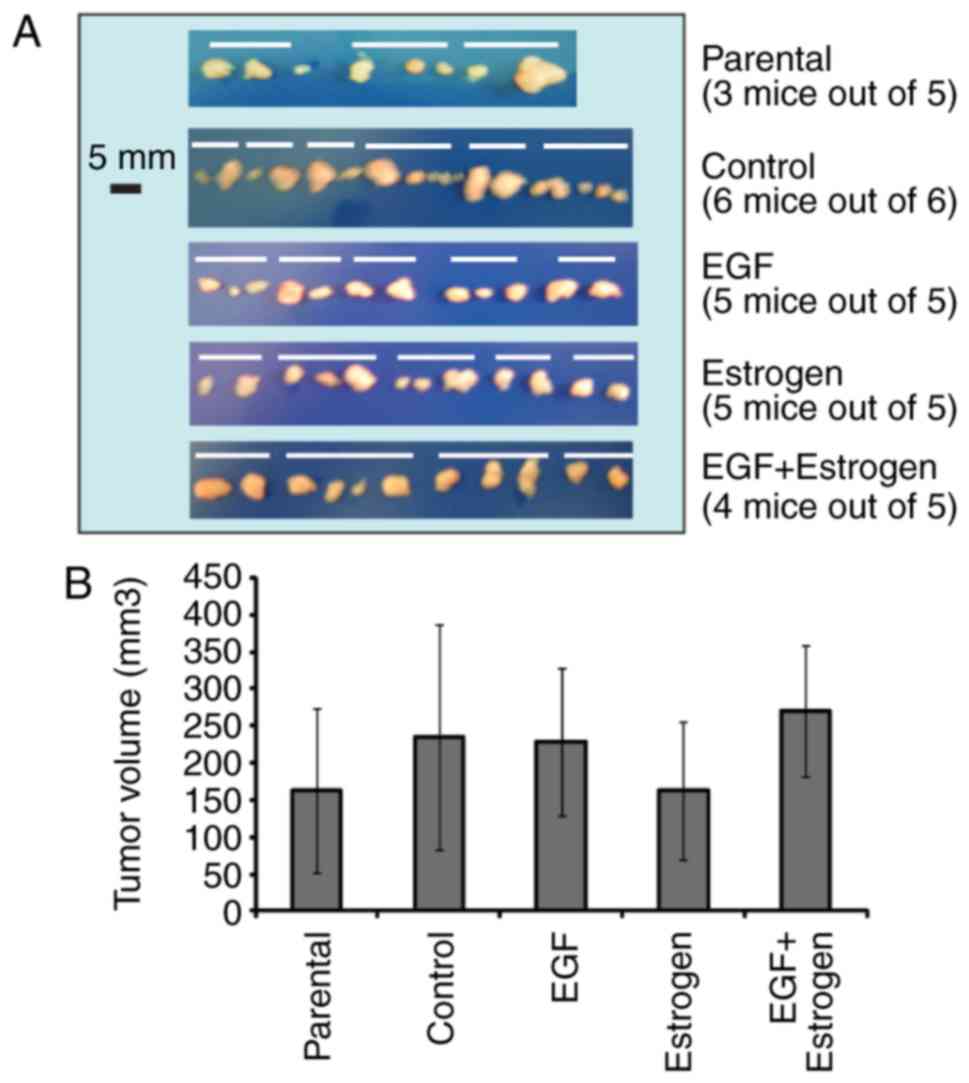
To investigate whether long-term exposure to EGF
and/or 17β-estradiol affects the ability of cells to form tumors
in vivo, 5×106 exposed cells were inoculated in
both flanks of each mouse (n=5 SCID mice per condition; n=6 mice
with non-exposed control cells). The tumor take, growth and the
overall state of the mice were monitored every second day for 29
weeks. It was observed that three of the five mice injected with
parental MCF7 cells developed tumors, whereas all six mice injected
with control cells, all five mice injected with cells exposed to
EGF or 17β-estradiol, and four of the five mice injected with cells
exposed to EGF + 17 β-estradiol presented with tumors (Fig. 2A). The volume of the tumors
collected from the injection sites was also measured (Fig. 2B). No significant difference in
volume was observed among the tumors derived from cells subjected
to different exposures.
To examine whether the molecular and functional
features of the cells in the xenograft tumors had changed, the
tumor sections were stained for proliferative status by Ki-67
staining, whereas the vessel density and apoptosis were evaluated
with the vascular marker vWF and a terminal deoxynucleotidyl
transferase dUTP nick end labeling (TUNEL) assay, respectively.
The Ki-67 protein is a nuclear marker of
proliferation (22). To
investigate the cell proliferation rates, immunohistochemistry was
performed by staining the collected mouse tumors with the
anti-Ki-67 antibody MIB1 (Fig.
2C). Cell proliferation was scored based on the intensity and
frequency of staining (Fig. 2D).
The anti-Ki-67 immunohistochemistry revealed higher overall
staining of cells in tumors derived from control and EGF-exposed
cells, and lower staining of cells in tumors derived from
17β-estradiol-exposed cells, compared with the staining of tumors
derived from parental MCF7 cells. The overall proliferation score
revealed that tumors from the control and EGF-exposed groups
exhibited the highest proliferation score, at 80 and 70%,
respectively. Tumors from the EGF and 17β-estradiol-exposed cells
exhibited marginally higher Ki-67 staining than the parental cells,
at 50, vs. 40%, respectively. However, tumors from the
17β-estradiol-exposed cells exhibited only 20% positive staining
for Ki-67 (Fig. 2D).
vWF is a glycoprotein that mediates platelet
adhesion to the sub-endothelium at sites of vascular injury, and
binds and stabilizes factor VIII in the blood (23,24). vWF appears to be expressed
exclusively in endothelial cells, where it exhibits a granular
pattern of reactivity. vWF is commonly used as an
immunohistochemical marker of endothelial cells (23,24). To evaluate the angiogenic status
of cells within the collected tumors, immunohistochemistry for vWF
was performed and vessel density was analyzed in the present study
(Fig. 2E). The tumors from the
control, EGF-exposed and EGF + 17β-estradiol-exposed cells
exhibited significantly enhanced angiogenesis compared with that of
the parental group, with an increase in vessel density of
>2.8-fold for the control and EGF-exposed cells, and a 3.5-fold
increase for the EGF + 17β-estradiol exposed cells. The tumors
derived from 17β-estradiol-exposed cells exhibited decreased
angiogenesis by 33% compared with that of tumors derived from
parental cells (Fig. 2F). No
significant cell death was observed in tumors stained with TUNEL
(data not shown). Therefore, the xenograft mouse study revealed
that tumor volume and tumor take did not differ among tumors formed
by cells exposed to EGF and/or 17β-estradiol. However, tumors
formed by cells exposed to 17β-estradiol exhibited decreased
expression of the proliferation marker Ki-67 and decreased vessel
formation.
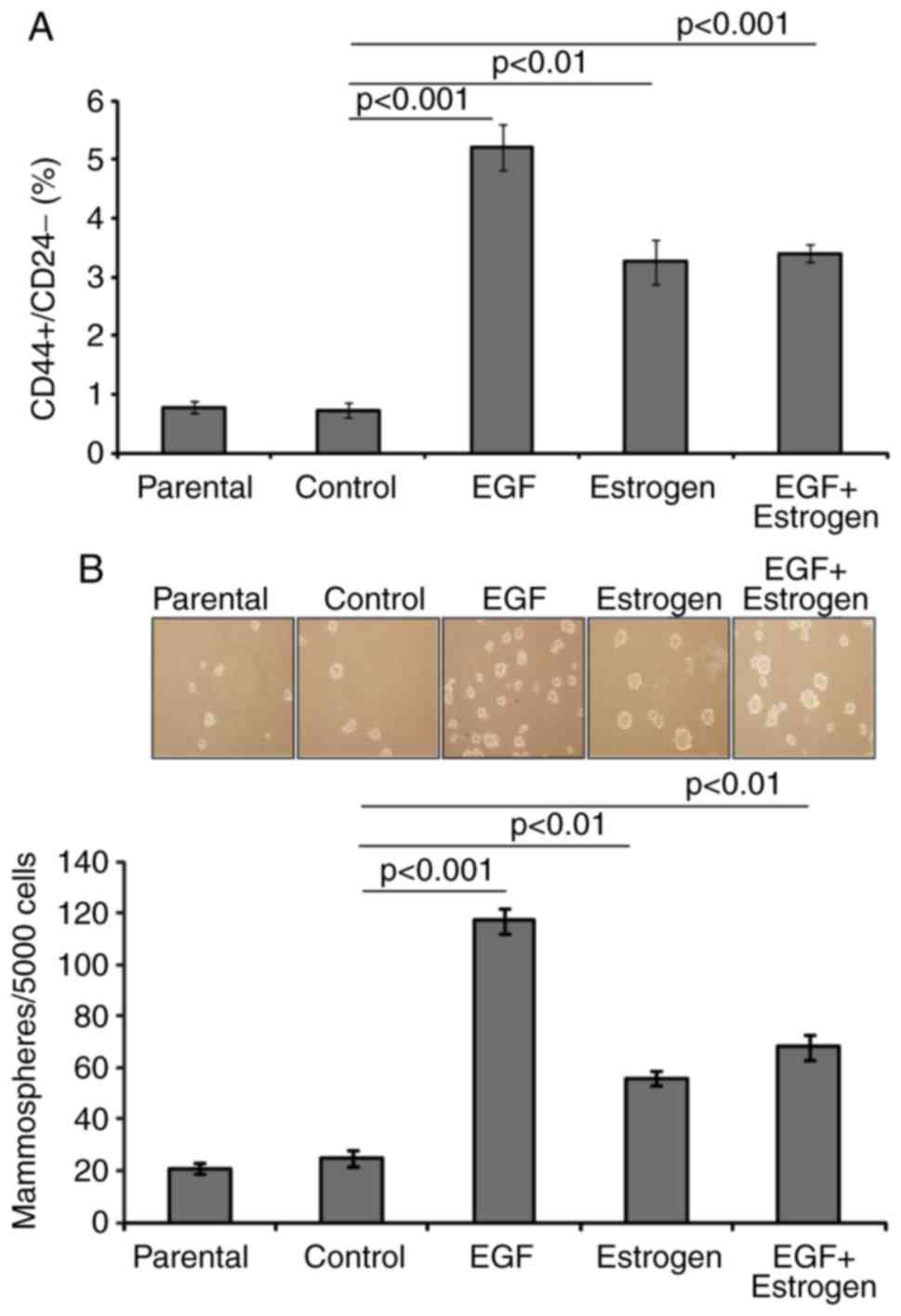
EGF and 17β-estradiol exposure increases
the breast cancer stem cell-like pool
To examine whether exposure to EGF and/or
17β-estradiol affects the number of breast cancer stem cells, the
proportion of stem-like cells was evaluated by flow cytometry and
mammosphere formation assays. It was observed that, upon EGF,
17β-estradiol, and EGF + 17β-estradiol exposure, the proportion of
CD44+/CD24− cancer stem-like cells was ~5-,
3- and 3-fold higher, respectively, than that of parental cells. No
significant change in the proportion of
CD44+/CD24− cells was detected in the control
cells compared with the parental cells (Fig. 3A). This suggested that the change
in expression of CD44+/CD24− markers was
attributed to the long-term treatments.
Mammosphere formation is indicative of cell
transformation and is associated with the degree of stemness
exhibited by the cells (25,26). The present study observed that the
EGF, 17β-estradiol and EGF + 17β-estradiol-exposed cells formed 6-,
3- and 3.5-fold higher numbers of mammospheres, respectively, than
the control cells (Fig. 3B). The
control cells exhibited the same low level of mammosphere formation
as the parental cells (Fig. 3B).
The enhanced proportion of CD44+/CD24− cells
and the increase in mammosphere formation suggested that exposure
to EGF and/or 17β-estradiol promoted an increase of cells with
cancer stem cell-like characteristics.
EGF and 17β-estradiol exposure modulates
mammosphere formation in response to treatments with Iressa,
tamoxifen and SB431542
As prolonged exposure to EGF and/or 17β-estradiol
altered the physiology of MCF7 cells, the effects of Iressa,
tamoxifen and SB431542 on the cells were further examined. These
compounds are inhibitory agents targeting EGF, estrogen, and TGFβ
signaling, respectively (27–30). The mammosphere formation capacity
of cells treated with EGF, 17β-estradiol, TGFβ1, and the inhibitors
of the corresponding signaling pathways, Iressa, tamoxifen and
SB431542, was evaluated (Fig.
4A–C). It was observed that the parental and control cells had
similar pattern of responses, suggesting that the selection in
non-adherent conditions preserved the responsiveness mechanisms of
the cells. By contrast, the cells exposed to 17β-estradiol had a
lower amplitude of response to treatments compared with the other
cells. Tamoxifen was the only drug that exhibited a consistent
inhibitory effect in all the cells evaluated, which is consistent
with the ER-positive status of MCF7 breast cancer cells (31). Iressa inhibited mammosphere
formation in the EGF and EGF + 17β-estradiol-exposed cells.
Exposure to EGF resulted in an overall increase of cell
proliferation, which was inhibited upon treatment with
17β-estradiol, Iressa, tamoxifen and SB431542. Only TGFβ1
stimulated mammosphere formation in the EGF-treated cells. A
similar but less pronounced pattern of responsiveness was observed
for cells exposed to EGF and 17β-estradiol (Fig. 4).
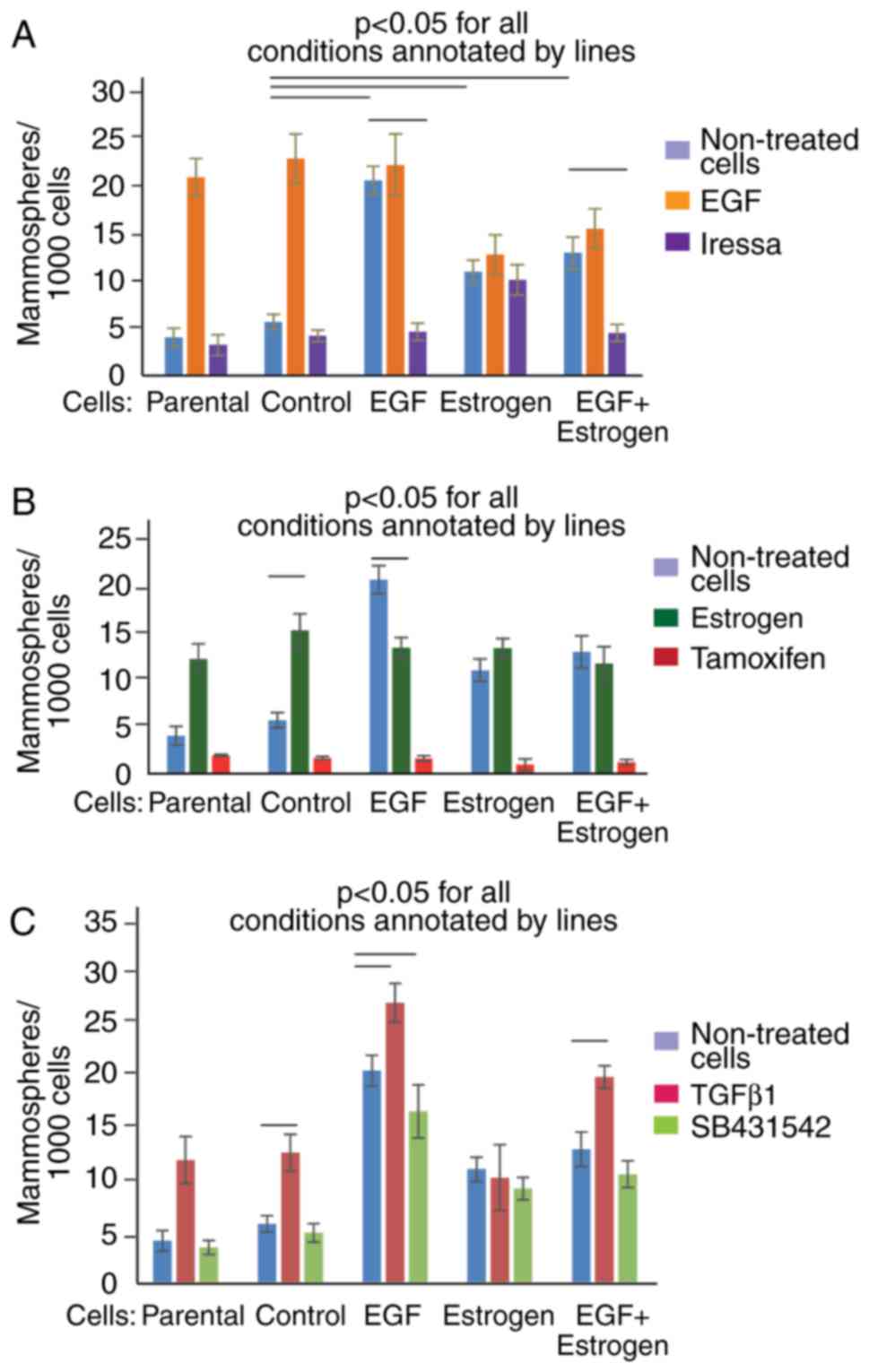 | Figure 4Exposure of cells to EGF and/or
17β-estradiol changes responsiveness of cells to treatments with
TGFb1, Iressa, tamoxifen, and SB431542. Cells groups comprised
Parental, Control, EGF, Estrogen (17β-estradiol), and EGF +
Estrogen (EGF + 17β-estradiol), as indicated. Cells were treated
with (A) EGF and Iressa, (B) estrogen and tamoxifen, and (C) TGFβ1
and SB431542. The statistical significance of differences was
evaluated using a one-way ANOVA with Tukey's HSD. Representative
results of three experiments performed are shown. EGF, epidermal
growth factor; TGFβ, transforming growth factor-β. |
The above results suggested that prolonged exposure
of human epithelial cells to EGF and/or 17β-estradiol altered the
response pattern of the cells to short-term treatments with EGF,
17β-estradiol, TGFβ1, and inhibitors of the corresponding signaling
pathways (Fig. 4).
Discussion
The period of time that is required for EGF and
estrogen-exposed cells to acquire irreversible changes in
tumorigenesis-relevant physiology remains to be elucidated.
Short-term treatment, for hours or a few days, may not alter the
cells irreversibly, as cancer cells are known to be robust
(32,33). Therefore, the length of drug
exposure required to induce a sustainable change in cellular
physiology is of high relevance for understanding tumorigenesis.
The present study demonstrated that drug exposure of MCF7 cells for
40–42 days affected their proliferation rate and transformation
phenotype, but was not sufficient to affect tumor growth in mice
(Fig. 5).
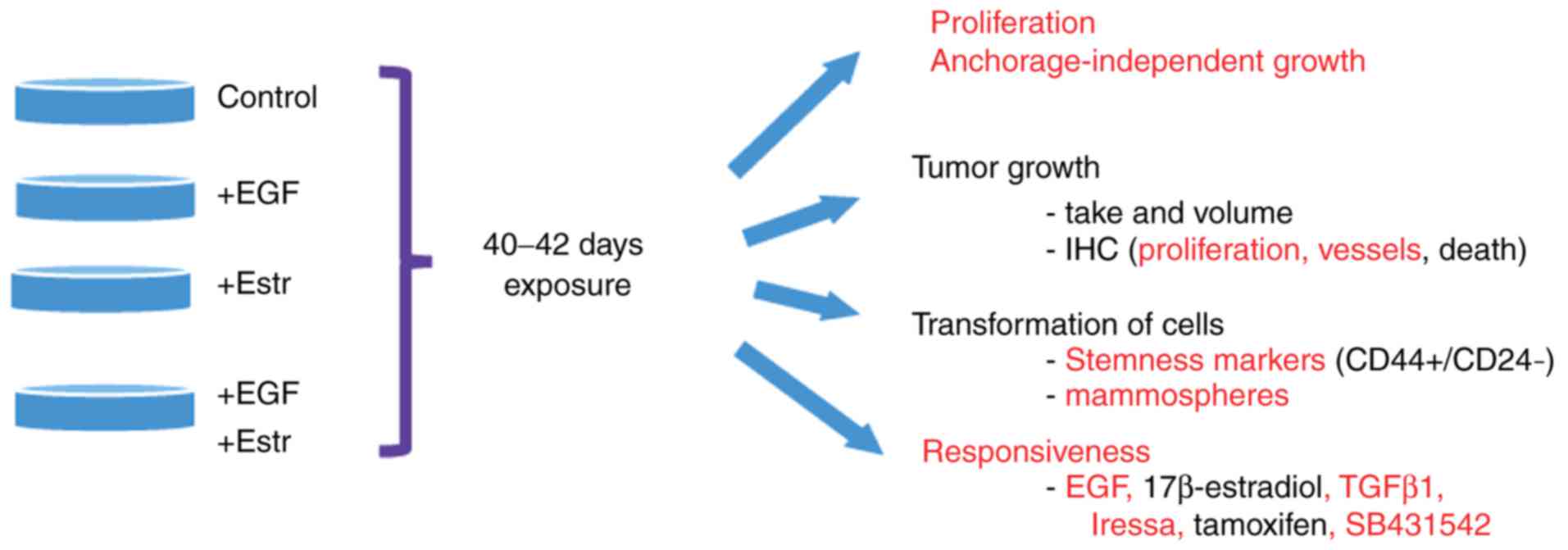 | Figure 5Long-term exposure of MCF7 cells to
EGF and/or 17β-estradiol alters cell proliferation, mammosphere
formation, expression of CD44+/CD24− markers,
and responsiveness to treatments, but is not sufficient to affect
tumor take or volume in a xenograft mice model. Cellular functions
affected by the exposure of cells to EGF and/or 17β-estradiol,
compared with parental or control cells, are annotated in red. EGF,
epidermal growth factor; Estr, 17β-estradiol; TGFβ1, transforming
growth factor-β1; CD, cluster of differentiation; IHC,
immunohistochemistry. |
EGF and 17β-estradiol are known stimulators of cell
proliferation (3–9). The present study observed that
prolonged exposure resulted in faster proliferation of MCF7 cells,
and this higher rate of proliferation was maintained even upon
removal of EGF and 17β-estradiol from the medium (Fig. 1). EGF and 17β-estradiol have been
reported to promote tumor formation in mice (34,35). The present study observed that
exposure to EGF and 17β-estradiol for 40–42 days did not affect the
take or volume of the formed tumors when the treatments were
withdrawn, and the cells injected in the mice were no longer under
treatment (Fig. 2). However,
immunohistochemistry revealed that exposure to 17β-estradiol
resulted in lower proliferation rate and vascularization. This
finding is in agreement with the reported roles of estrogen in the
development of breast tissues and breast tumors (36,37). The data obtained in the present
study suggested that long-term exposure to drugs did not alter the
cellular physiology sufficiently to ensure more marked tumor growth
upon withdrawal of treatment.
Cancer stem cells are considered to be responsible
for the development of tumors (38). Human breast cancer stem cells can
be identified by the expression profile of markers, including CD44
and CD24. The CD44+/CD24− phenotype is
characteristic of breast cancer stem cells (38). The present study observed that the
fractions of CD44+/CD24− cells followed the
pattern of cell proliferation rate and formation of mammospheres
(Figs. 1A, 3A and B). This suggested that cell
proliferation, the expression of CD44+/CD24−,
and mammosphere formation may be early sustainable features induced
by carcinogenic exposure to EGF and 17β-estradiol.
Changes in cellular physiology may have an effect on
cellular responsiveness to drugs. The present study evaluated
whether the formation of mammospheres was affected upon treatment
of the cells with EGF, 17β-estradiol, TGFβ1, and drugs that inhibit
the corresponding signaling pathways, Iressa, tamoxifen and
SB431542 (Fig. 4). As expected,
EGF and 17β-estradiol induced mammosphere formation in parental and
control cells, however, this effect was negligible in the EGF-
and/or 17β-estradiol-exposed cells, and even inhibitory in the
EGF-exposed cells treated with 17β-estradiol. This response may
indicate refractoriness of the EGF and 17β-estradiol signaling
pathways. Cells exposed to 17β-estradiol were less susceptible to
the effects of EGF, TGFβ1, 17β-estradiol, Iressa, and SB431542.
This result was expected, as treatment with the ligand
17β-estradiol has been shown to reduce the expression of human
epidermal growth factor receptor 2 (HER2) in MCF7 wild-type cells
(39). The potent inhibitory
effect of tamoxifen on all cells suggested that the MCF7 cells
preserved their ERα-positive status, thus tamoxifen remained
effective independently of the exposure of cells to EGF and
17β-estradiol (Fig. 4). The cells
exposed to EGF were particularly sensitive to TGFβ-induced
proliferation, a clear indicator of crosstalk of these two
pathways, for which there is accumulating evidence (40–42). In HER2-transformed cells, TGFβ
further stimulated HER2 signaling to promote malignancy and induced
resistance to anti-HER2 therapy. The observations in the present
study showed changes in cellular responsiveness, and confirmed the
previously reported promotion of transformation by EGF and TGFβ1,
the stimulatory effect of 17β-estradiol, and the inhibitory effects
of Iressa and tamoxifen (3–8,43,44). The observations also confirmed
that long-term exposure may contribute to the development of
resistance to Iressa in 17β-estradiol-exposed cells, and to
SB431542 in EGF- and/or 17β-estradiol-exposed cells.
The response of tumor cells to external stimuli upon
short-term treatment induces a number of regulatory processes.
However, the majority of these induced responses reverse to the
initial state in cells upon withdrawal of the stimulus. The present
study hypothesized the existence of stimuli that are long-term
effective and/or durable enough to irreversibly alter cellular
responsiveness, as cells are robust regulatory systems (32,33). The roles of EGF and estrogen in
breast cancer are well documented, and are associated with high
levels of signaling as a response to constantly elevated levels of
EGF and estrogen as ligands or due to mutations rendering their
signaling levels elevated and independent from ligands (3,4,34–37,42–44). The results of the present study
suggested that other stimuli may be required in addition to EGF and
17β-estradiol to promote tumor growth, or that EGF and
17β-estradiol stimulation may be constantly required during tumor
growth. The results demonstrated that exposure to EGF and/or
17β-estradiol for 40 days was sufficient to alter cell
proliferation rate; this transformation was evident in the ability
of cells to form mammospheres and express stemness markers, but was
not sufficient to affect the rate of tumor growth in mice.
Acknowledgments
The authors would like to thank Dr Luis Furuya
Kanamori for help with statistical analysis. The authors would also
like to thank Oves Minnesfond for their support and
encouragement.
Funding
This study was supported in part by grants from the
Swedish Institute, the EU program RTN 'EpiPlastCarcinoma' and
Erasmus KI-UWM (grant nos. QUST-SPR-2017-12, QUST-SPR-2017-11,
NPRP9-453-3-089, HMC-MRC-RP16354 and HMC-MRC-RP-iTRI to SS).
Availability of data and materials
Data and materials are available upon request. There
is no data to be deposited in repositories.
Authors' contributions
All authors contributed equally. SS designed and
managed the project, SIC and MJ performed experiments. All authors
were involved in writing the manuscript.
Ethics approval and consent to
participate
The animal experiments were performed according to
the Swedish national and international guidelines (ethical permit
no. C123/6, approved by the Uppsala Animal Tests Committee of the
Uppsala Court).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
PDQ Breast Cancer Treatment.
PDQ® Adult Treatment Editorial Board. Bethesda, MD:
National Cancer Institute. Updated 10/13/2017. https://www.cancer.gov/types/breast/hp/breast-treatment-pdq.
Accessed November 21, 2017. PubMed/NCBI2017
|
|
2
|
Patani N, Martin LA and Dowsett M:
Biomarkers for the clinical management of breast cancer:
International perspective. Int J Cancer. 133:1–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Masuda H, Zhang D, Bartholomeusz C,
Doihara H, Hortobagyi GN and Ueno NT: Role of epidermal growth
factor receptor in breast cancer. Breast Cancer Res Treat.
136:331–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yue W, Yager JD, Wang JP, Jupe ER and
Santen RJ: Estrogen receptor-dependent and independent mechanisms
of breast cancer carcinogenesis. Steroids. 78:161–170. 2013.
View Article : Google Scholar
|
|
5
|
Voudouri K, Berdiaki A, Tzardi M,
Tzanakakis GN and Nikitovic D: Insulin-like growth factor and
epidermal growth factor signaling in breast cancer cell growth:
Focus on endocrine resistant disease. Anal Cell Pathol (Amst).
2015:9754952015.
|
|
6
|
Fouad YA and Aanei C: Revisiting the
hallmarks of cancer. Am J Cancer Res. 7:1016–1036. 2017.PubMed/NCBI
|
|
7
|
Margan MM, Jitariu AA, Cimpean AM, Nica C
and Raica M: Molecular portrait of the normal human breast tissue
and its influence on breast carcinogenesis. J Breast Cancer.
19:99–111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hardy KM, Booth BW, Hendrix MJ, Salomon DS
and Strizzi L: ErbB/EGF signaling and EMT in mammary development
and breast cancer. J Mammary Gland Biol Neoplasia. 15:191–199.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Katzenellenbogen BS and Katzenellenbogen
JA: Estrogen receptor transcription and transactivation: Estrogen
receptor alpha and estrogen receptor beta: Regulation by selective
estrogen receptor modulators and importance in breast cancer.
Breast Cancer Res. 2:335–344. 2000. View
Article : Google Scholar
|
|
10
|
Sukocheva O, Wadham C and Xia P: Estrogen
defines the dynamics and destination of transactivated EGF receptor
in breast cancer cells: Role of S1Ps receptor and Cdc42. Exp Cell
Res. 319:455–465. 2013. View Article : Google Scholar
|
|
11
|
Lichmer RB: Estrogen/EGF receptor
interactions in breast cancer: Rationale for new therapeutic
combination strategies. Biomed Pharmacother. 57:447–451. 2003.
View Article : Google Scholar
|
|
12
|
Arpino G, Wiechmann L, Osborne CK and
Schiff R: Crosstalk between the estrogen receptor and the HER
tyrosine kinase receptor family: Molecular mechanism and clinical
implications for endocrine therapy resistance. Endocr Rev.
29:217–233. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moerkens M, Zhang Y, Wester L, van de
Water B and Meerman JHN: Epidermal growth factor receptor signaling
in human breast cancer cells operates parallel to estrogen receptor
alpha signaling and results in tamoxifen insensitive proliferation.
BMC Cancer. 14:2832006. View Article : Google Scholar
|
|
14
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-Perspectives on current status and future directions:
AACR workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Hajj M, Wicha MS, Ito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ponti D, Costa A, Zaffaroni N, Pratesi G,
Petrangolini G, Coradini D, Pilotti S, Pierotti MA and Daidone MG:
Isolation and in vitro propagation of tumorigenic breast cancer
cells with stem/progenitor cell properties. Cancer Res.
65:5506–5511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fillmore CM and Kuperwasser C: Human
breast cancer cell lines contain stem-like cells that self-renew,
give rise to phenotypically diverse progeny and survive
chemotherapy. Breast Cancer Res. 10:1–13. 2008. View Article : Google Scholar
|
|
18
|
Ischenko I, Seeliger H, Schaffer M, Jauch
KW and Bruns CJ: Cancer stem cells: How can we target them? Curr
Med Chem. 15:3171–3184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Farnie G, Willan PM, Clarke RB and Bundred
NJ: Combined inhibition of ErbB1/2 and Notch receptors effectively
targets breast ductal carcinoma in situ (DCIS) stem/progenitor cell
activity regardless of ErbB2 status. PLos One. 8:00568402013.
View Article : Google Scholar
|
|
20
|
Asselin-Labat ML, Vaillant F, Sheridan JM,
Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ
and Visvader JE: Control of mammary stem cell function by steroid
hormone signalling. Nature. 465:798–802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fillmore CM, Gupta PB, Rudnick JA,
Caballero S, Keller PJ, Lander ES and Kuperwasser C: Estrogen
expands breast cancer stem-like cells through paracrine FGF/Tbx3
signaling. Proc Natl Acad Sci USA. 107:21737–21742. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suurmeijer AJ and Boon M: Pretreatment in
a high-presure microwave processor for MIB-immunostaining of
cytological smears and paraffin tissue sections to visualize the
various phases of the mitotic cycle. J Histochem Cytochem.
47:1015–1020. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kraby MR, Opdahl S, Akslen LA and Bofin
AM: Quantifying tumour vascularity in non-luminal breast cancers. J
Clin Pathol. 70:766–774. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Von Willebra nd factor processing.
Hamostaseologie. 37:59–72. 2017.
|
|
25
|
Saadin K and White IM: Breast cancer stem
cell enrichment and isolation by mammosphere culture and its
potential diagnostic applications. Expert Rev Mol Diagn. 13:49–60.
2013. View Article : Google Scholar
|
|
26
|
Ishiguro T, Ohata H, Sato A, Yamawaki K,
Enomoto T and Okamoto K: Tumor-derived spheroids: Relevance to
cancer stem cells and clinical applications. Cancer Sci.
108:283–289. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saxena R and Dwivedi A: ErbB family
receptor inhibitors as therapeutic agents in breast cancer: Current
status and future clinical perspective. Med Res Rev. 32:166–215.
2012. View Article : Google Scholar
|
|
28
|
Sainsbury R: The development of endocrine
therapy for women with breast cancer. Cancer Treat Rev. 39:507–517.
2013. View Article : Google Scholar
|
|
29
|
Connolly EC, Freimuth J and Akhurst RJ:
Complexities of TGF-β targeted cancer therapy. Int J Biol Sci.
8:964–978. 2012. View Article : Google Scholar :
|
|
30
|
Mints M and Souchelnytskyi S: Impact of
combinations of EGF, TGFβ, 17β-oestradiol, and inhibitors of
corresponding pathways on proliferation of breast cancer cell
lines. Exp Oncol. 36:67–71. 2014.PubMed/NCBI
|
|
31
|
Subik K, Lee JF, Baxter L, Strzepek T,
Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG and
Tang P: The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67
and AR by immunohistochemical analysis in breast cancer cell lines.
Breast Cancer (Auckl). 4:35–41. 2010.
|
|
32
|
Tian T, Olson S, Whitacre JM and Harding
A: The origins of cancer robustness and evolvability. Integr Biol
(Camb). 3:17–30. 2011. View Article : Google Scholar
|
|
33
|
Kitano H: Cancer as a robust system:
Implications for anticancer therapy. Nat Rev Cancer. 4:227–235.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kubota T, Josui K, Fukutomi T and Kitajima
M: Growth regulation by estradiol, progesterone and recombinant
human epidermal growth factor of human breast carcinoma xenografts
grown serially in nude mice. Anticancer Res. 15:1275–1278.
1995.PubMed/NCBI
|
|
35
|
Kenney NJ, Bowman A, Korach KS, Barrett JC
and Salomon DS: Effect of exogenous epidermal-like growth factors
on mammary gland development and differentiation in the estrogen
receptor-alpha knockout (ERKO) mouse. Breast Cancer Res Treat.
79:161–173. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Arendt LM and Kuperwasser C: Form and
function: How estrogen and progesterone regulate the mammary
epithelial hierarchy. J Mammary Gland Biol Neoplasia. 20:9–25.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hilakivi-Clarke L, Cabanes A, Olivo S,
Kerr L, Bouker KB and Clarke R: Do estrogens always increase breast
cancer risk? J Steroid Biochem Mol Biol. 80:163–174. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Da Cruz Paula A and Lopes C: Implications
of different cancer stem cell phenotypes in breast cancer.
Anticancer Res. 37:2173–2183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lattrich C, Juhasz-Boess I, Ortmann O and
Treeck O: Detection of an elevated HER2 expression in MCF-7 breast
cancer cells overexpressing estrogen receptor beta1. Oncol Rep.
19:811–817. 2008.PubMed/NCBI
|
|
40
|
Chow A, Arteaga CL and Wang SE: When tumor
suppressor TGFβ meets the HER2 (ERBB2) oncogene. J Mammary Gland
Biol Neoplasia. 16:81–88. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang SE: The functional crosstalk between
HER2 tyrosine kinase and TGF-β signaling in breast cancer
malignancy. J Signal Transduct. 2011:8042362011. View Article : Google Scholar
|
|
42
|
Jia M and Souchelnytstkyi S: Comments on
the cross-talk of TGFβ and EGF in cancer. Exp Oncol. 33:170–173.
2011.PubMed/NCBI
|
|
43
|
Yamaoka T, Ohba M and Ohmori T:
molecular-targeted therapies for epidermal growth factor receptor
and its resistance mechanisms. Int J Mol Sci. 18:pii: E2420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jameera Begam A, Jubie S and Nanjan MJ:
Estrogen receptor agonists/antagonists in breast cancer therapy: A
critical review. Bioorg Chem. 71:257–274. 2017. View Article : Google Scholar : PubMed/NCBI
|