Introduction
Stickler syndrome [Online Mendelian Inheritance in
Man (OMIM) nos. 108300, 609508, 604841,184840, 614134 and 614284],
first reported in 1965 by Stickler et al (1), is a group of inherited connective
tissue disorders, with an incidence of 1 in 10,000 (2,3).
Stickler syndrome is frequently misdiagnosed due to its widely
varied clinical manifestations, which may resemble other diseases
(4,5). It commonly involves distinctive
ocular and facial abnormalities, hearing loss and joint problems
(3,6-8).
Patients with Stickler syndrome typically present with shallow
supraorbital ridges, hypoplastic short nose with anteverted nares,
buphthalmic eyes, a flat hypoplastic midface with a depressed nasal
bridge, long philtrum and micrognathia (9).
Stickler syndrome is caused by mutations in collagen
genes during fetal development, and can be divided into various
subtypes based on the clinical manifestations and underlying
genetic mutations (10). The most
common form, Type 1 Stickler syndrome, is caused by a collagen type
II α1 chain (COL2A1) mutation (OMIM no. 120140), and is
characterized by membranous vitreous anomaly and megalophthalmos
(11,12). Type 2 Stickler syndrome with an
underlying collagen type XI α1 chain (COL11A1) mutation
(OMIM no. 120280) accounts for a minority of patients and presents
with a typical beaded vitreous phenotype (13). Type 3 or non-ocular Stickler
syndrome, caused by collagen type XI α2 chain (COL11A2)
mutation (OMIM no. 120290), often manifests as systemic
malformations, including midface hypoplasia and osteoarthritis
(14,15). Type 4 Stickler syndrome, caused by
collagen type IX α1 chain (COL9A1) or collagen type IX α2
chain (COL9A2) mutation (OMIM no. 120210), is associated
with sensorineural deafness, myopia, vitreoretinopathy and
epiphyseal dysplasia (16).
Stickler syndrome can lead to a variety of ocular
abnormalities, including vitreoretinal degeneration, retinal
detachment, cataract, ocular hypertension and high myopia (17). The development of Stickler
syndrome is progressive and can ultimately lead to blindness
(3). The molecular mechanism of
Stickler syndrome is not fully characterized. However, type 1
Stickler syndrome arises from aberrant type II collagen, which is
the major collagen type synthesized in the adult human vitreous
(18). Under physiological
conditions, the strongly adherent collagen fibrils (typically types
II, XI and IX) are interspersed in the extracellular matrix, which
is predominantly composed of water and glycosaminoglycans. The
interaction between collagen and hyaluronan, the most prevalent
glycosaminoglycan in the vitreous, provides swelling pressure
required to maintain the ocular structure (19). Mutation in the COL2A1 gene
can result in an abnormal fibrillar lamellar structure of the
vitreous gel (20), disrupt
collagen helices, alter fibrillogenesis and reduce collagen
secretion (20,21).
Characterizing the Stickler syndrome phenotypes and
identifying the underlying genetic mutations are initial steps to
understand the disease pathogenesis and will be useful for future
genetic counseling. The current study aimed to characterize the
clinical presentation of two young patients with Stickler syndrome
and bilateral retinal detachment, and to identify the genetic
changes in these patients using targeted next-generation sequencing
(NGS).
Materials and methods
Study subjects and clinical
examinations
Two patients from two different families presenting
with bilateral retinal detachment and peripheral retinal
degeneration were recruited in the present study. All experimental
protocols were performed according to the guidelines approved by
the Ethics Committee of Zhongshan Ophthalmic Center (Guangzhou,
China), and in accordance with the Declaration of Helsinki.
Informed consent was obtained from all subjects.
Complete ophthalmic examinations were performed at
the Zhongshan Ophthalmic Center. The best-corrected visual acuity
(BCVA) was measured using the ETDRS chart (Precision Vision,
Woodstock, IL, USA). Anterior segment images were obtained using a
BX 900 Slit Lamp (Haag-Streit, Bern, Switzerland). Anterior segment
measurements were performed using Pentacam HR version 70700
(Oculus, Wetzlar, Germany). Fundus photography was performed using
Heidelberg Retina Angiograph (Heidelberg Engineering, Heidelberg,
Germany) or ultra-wide-field 200Tx Optos system (Optos plc,
Dunfermline, UK). Optical coherence tomography (OCT) was performed
by Cirrus HD OCT (Zeiss GmbH, Jena, Germany). Physical examinations
were performed to exclude systemic diseases. Venous blood samples
from the patients, their unaffected family members and 200
unrelated control subjects from the same population were
collected.
Target capture, NGS and mutation
validation
NGS was used to identify the potential variants. The
parameters used for whole exome sequencing have been described in
our previous studies (22,23).
Identified mutations were validated using conventional polymerase
chain reaction (PCR)-based sequencing methods (24-29,23). Briefly, exons 21-22 and 33-34 of
the COL2A1 gene were amplified by PCR with respective
primers (Table I). PCRs were
conducted in 50 µl total reaction volume using an ABI2720
system (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
cycling conditions included one cycle at 94°C for 5 min, followed
by 40 cycles at 94°C for 45 sec, 59-60°C for 45 sec, 72°C for 45
sec, and one cycle at 72°C for 10 min. The PCR products were
sequenced in both directions using an ABI3730 Automated Sequencer
(PE Biosystems, Foster City, CA, USA). The sequencing results were
analyzed using Seqman (version 2.3; Technelysium Pty Ltd.,
Brisbane, Australia), and compared with the reference sequences in
National Center for Biotechnology Information (NCBI) databases
(26-28,30).
 | Table IPrimers used for the amplification of
COL2A1 in the current study. |
Table I
Primers used for the amplification of
COL2A1 in the current study.
| Gene | Exon | Forward
(5′-3′) | Reverse
(5′-3′) | Product size
(bp) | Annealing
temperature (°C) |
|---|
| COL2A1 | 21-22 |
GCCAAAGGATCTGCTGTGAG |
GCCCTGTTAAGTCTCCTCCA | 599 | 60 |
| COL2A1 | 33-34 |
CCTGGGTCCTATGCTCCTG |
AGCTTTGGTGAGAGGCTGTA | 581 | 59 |
Interpretation of the genetic
variants
To predict the effect of missense variants,
polymorphism phenotyping (PolyPhen) and sorting intolerant from
tolerant (SIFT) were used to predict the potential impact of an
amino acid substitution on the protein structure and function,
using physical and comparative considerations (23,31,32). Variants were predicted to be
pathogenic when at least one of the two programs predicted
deleterious effect of the amino acid substitution on the protein
structure and function. The Human Gene Mutation Database
(hgmd.cf.ac.uk/ac/index.php) was used
to screen mutations reported in previously published studies.
HomoloGene (ncbi.nlm.nih.gov/homologene) was used to assess the
conservation of the altered amino acid residues across different
species (22,33).
Results
Clinical presentations of the
patients
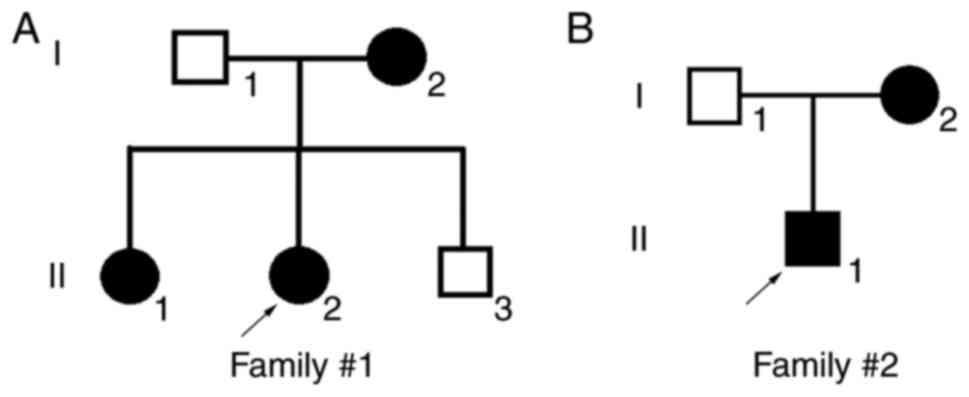
The patients reported in the present study were from
the southern area of China (the family pedigrees are illustrated in
Fig. 1). The clinical
manifestations of Patient 1 in Family 1 (II:2 in Fig. 1A) are summarized in Table II. The patient was a 24-year-old
female without a known familial history of ocular disease. The BCVA
was 0.0 in the right eye and 0.2 in the left eye. Anterior segment
photography demonstrated transparent lenses in both eyes. When
Patient 1 was 21 years old, she exhibited a decreased vision in the
left eye and retinal detachment of the left eye was diagnosed.
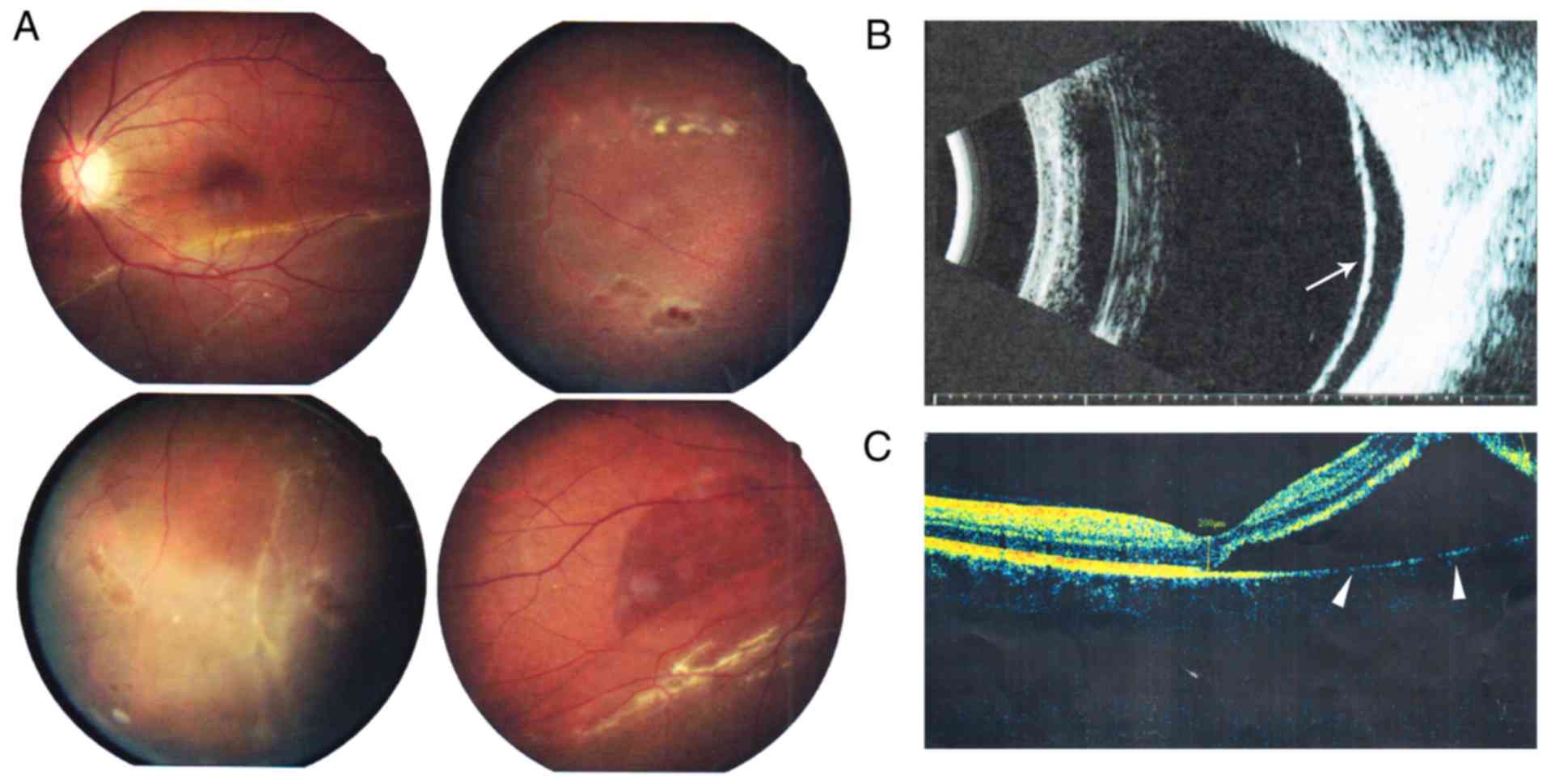
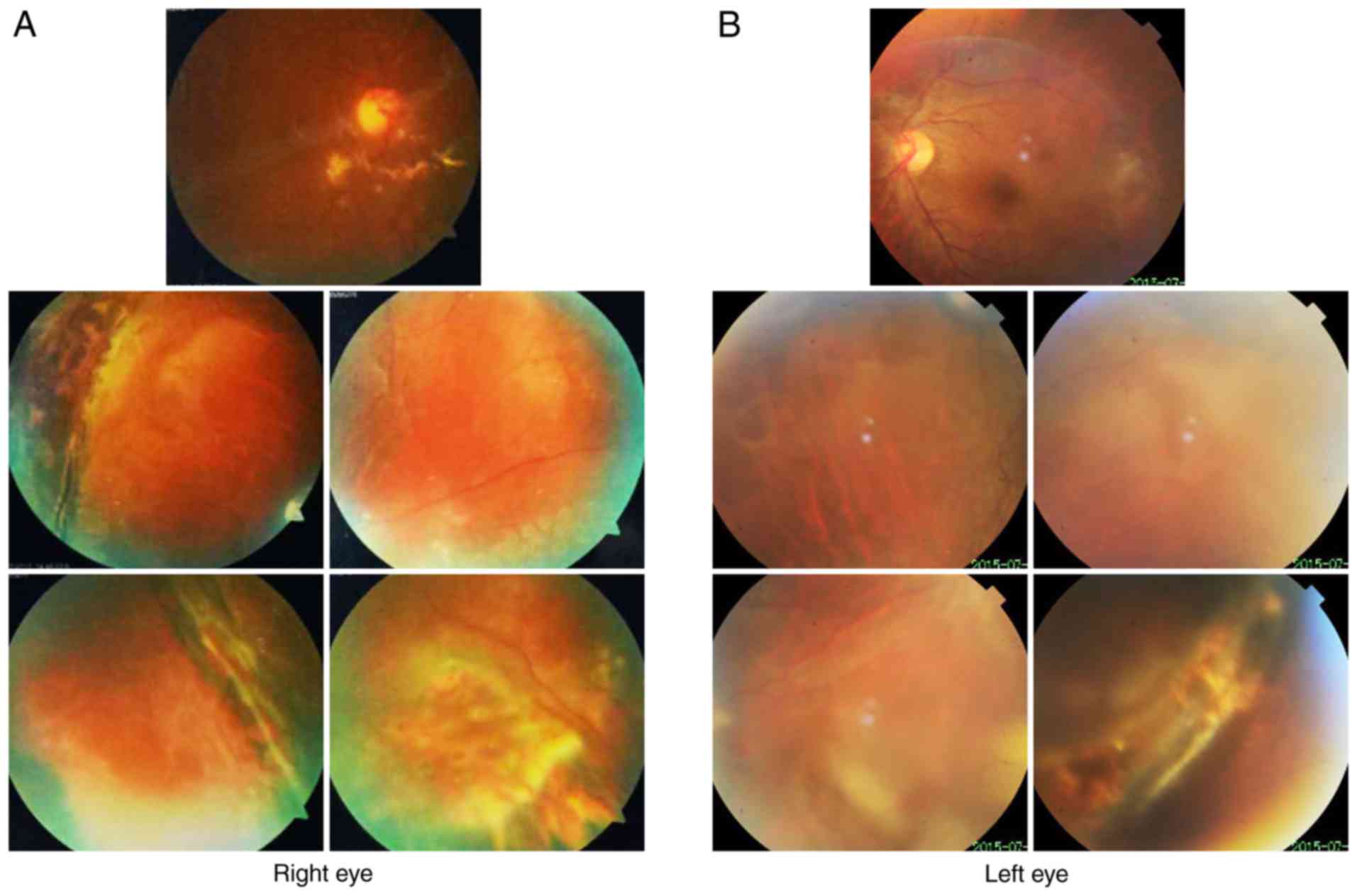
Fundus photography revealed inferior retinal detachment and
peripheral retinal degeneration (Fig.
2A). B-scan indicated localized retinal detachment (white
arrow; Fig. 2B). OCT revealed a
partially damaged macular area (Fig.
2C). Retinal detachment surgery was performed, and her vision
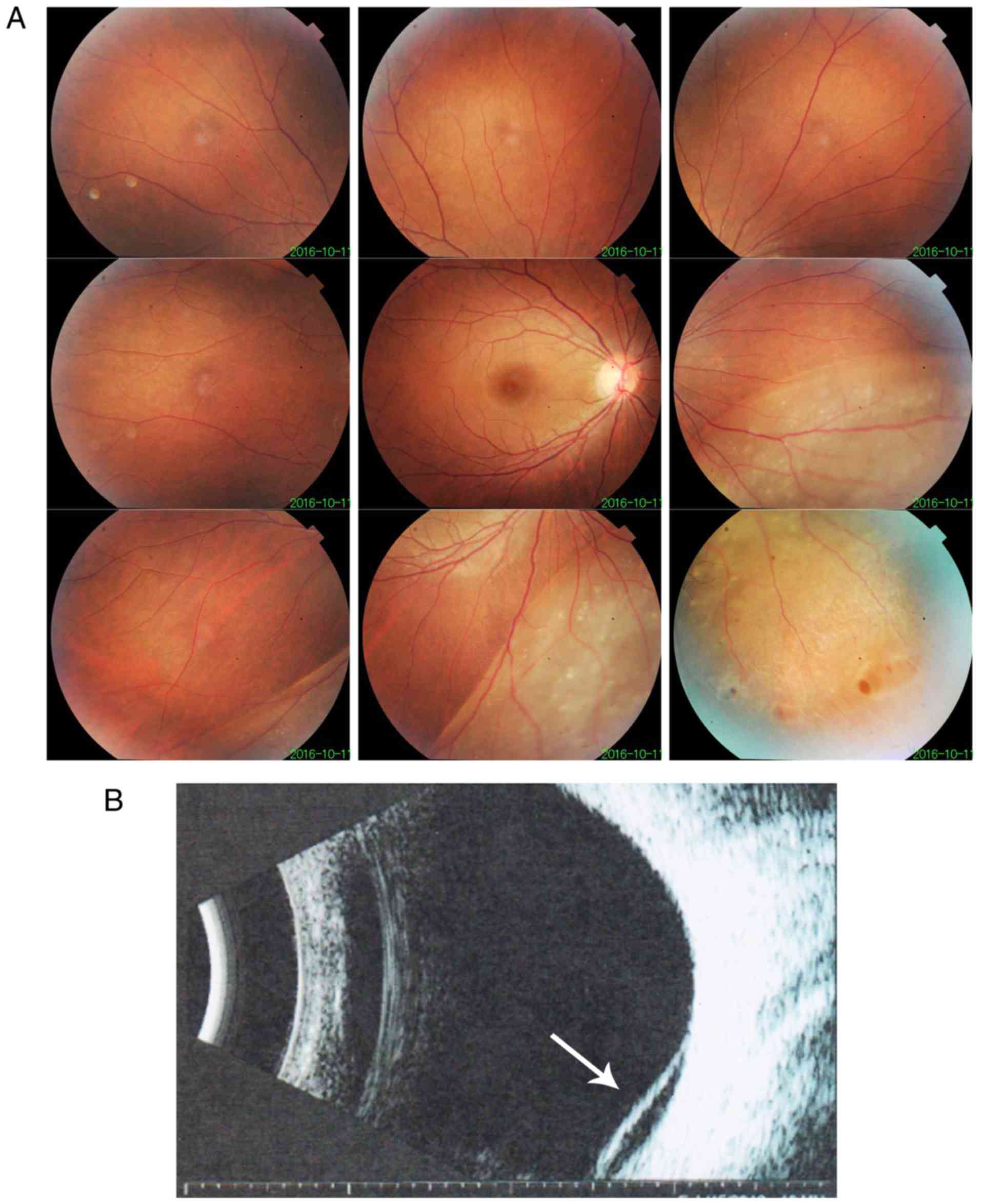
improved. After one year, vision was decreased in the right eye and
retinal detachment of the right eye without macular involvement was
diagnosed. Fundus imaging revealed inferior retinal detachment and
peripheral retinal degeneration (Fig.
3A). B-scan indicated localized retinal detachment (white
arrow; Fig. 3B). The elder sister
of this patient (II:1) also exhibited myopia and peripheral retinal
degeneration (Fig. 4).
| Table IISummary of clinical manifestations
and mutations in Family 1. |
Table II
Summary of clinical manifestations
and mutations in Family 1.
| Patient | Sex | Age | Clinical
manifestation
| Mutation |
|---|
| BCVA | Optometry | IOP | Lens/Cornea | Fundus | B-scan | OCT |
|---|
| I:1 | M | 48 | OD:0.0(0.0);
OS:0.0(0.0) | N/A | Normal | Normal | Normal | N/A | N/A | – |
| I:2 | F | 45 | OD:0.0(0.0);
OS:0.0(0.0) | N/A | Normal | Normal | Normal | N/A | N/A | c.1310G>C
(p.R437P) |
| II:1 | F | 24 | OD:0.7(0.0);
OS:0.7(0.0) |
OD:-4.25DS&-0.50DC; OS:-3.75DS
&-1.00DC | Normal | Normal | Peripheral retinal
degeneration | N/A | N/A | c.1310G>C
(p.R437P) |
| II:2 | F | 20 | OD:2.0(0.0);
OS:2.0(0.2) |
OD:-3.00DS&-1.50DC;
OS:-1.00DS&-1.00DC | Normal | Normal | Retinal detachment
of the right eye | Retinal
detachment | Retinal
detachment | C.1310G>C
(p.R437P) |
| II:3 | M | 19 |
OD:0.0(0.0);OS:0.0(0.0) | N/A | Normal | Normal | Normal | N/A | N/A | – |
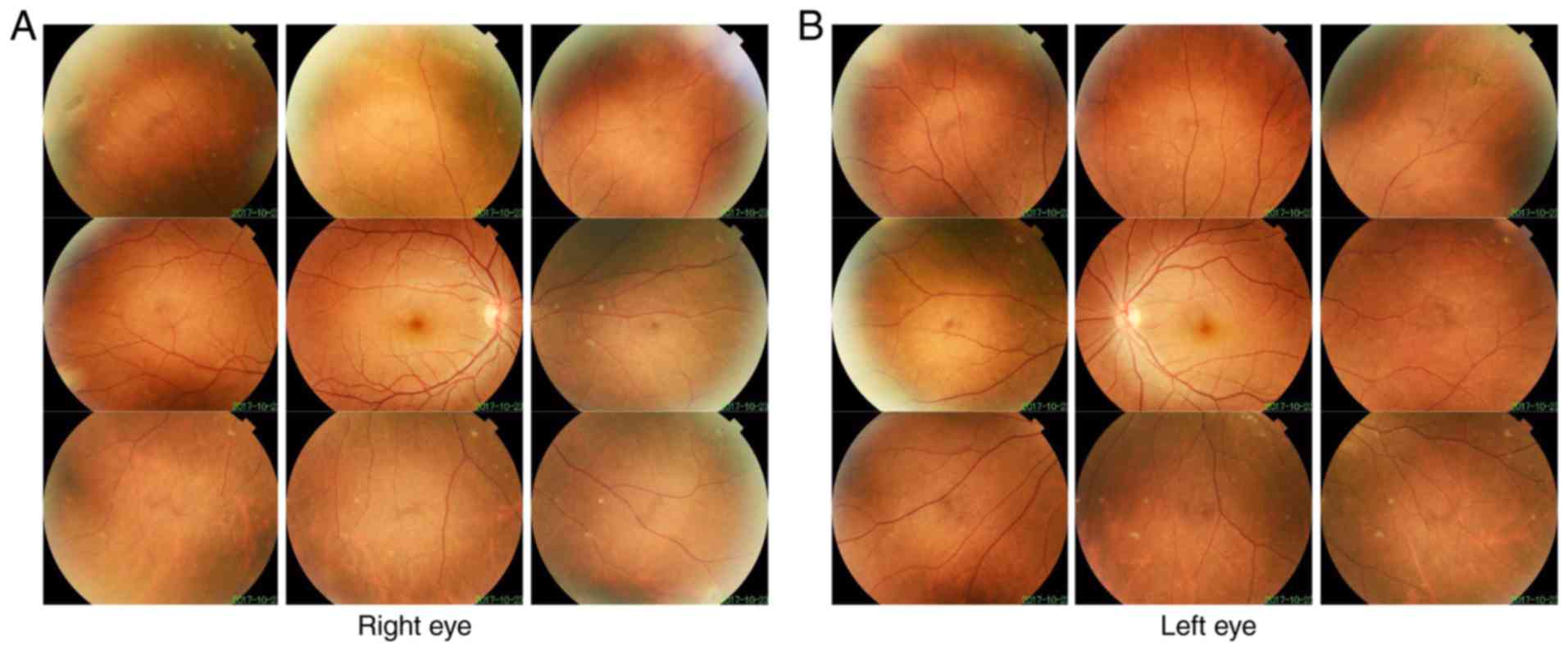
Patient 2 in Family 2 (II:1 in Fig. 1B) was a 17-year-old male. Retinal
detachment of the left eye and the right eye of Patient 2 was
diagnosed at 14 and 17 years old, respectively. Following surgery,
the BCVA was 0.7 in the right eye and 0.3 in the left eye. Fundus
imaging revealed peripheral retinal scars (Fig. 5). The mother of patient 2 also had
bilateral retinal detachment. The left eye of the mother was blind
at birth and exhibited severe atrophy. Right retinal detachment was
diagnosed at 30 years old. Patient 2 and the mother had cleft
palate.
Mutation screening and bioinformatics
analysis of the mutations
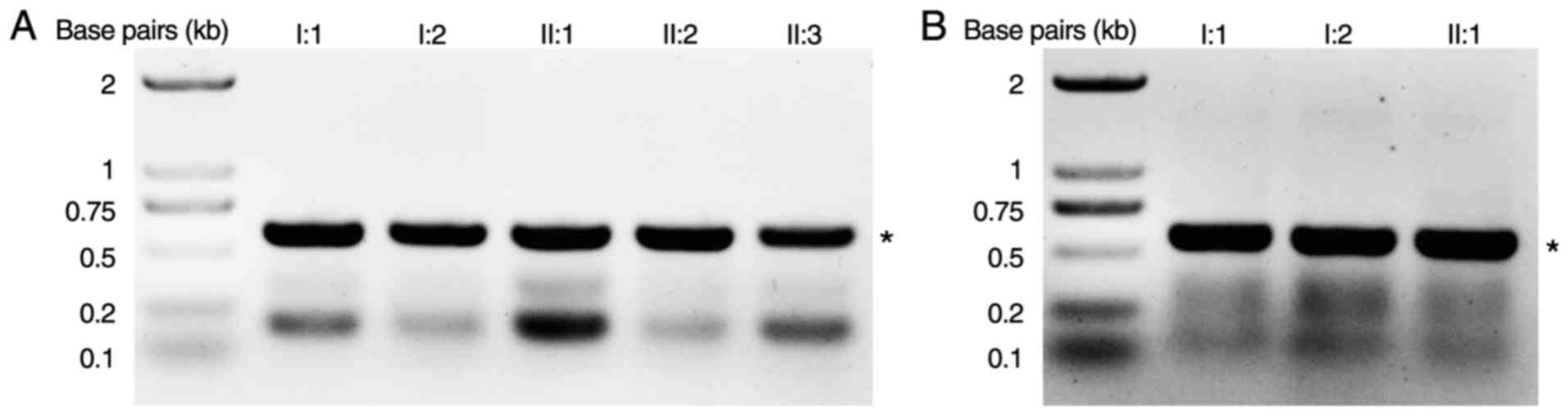
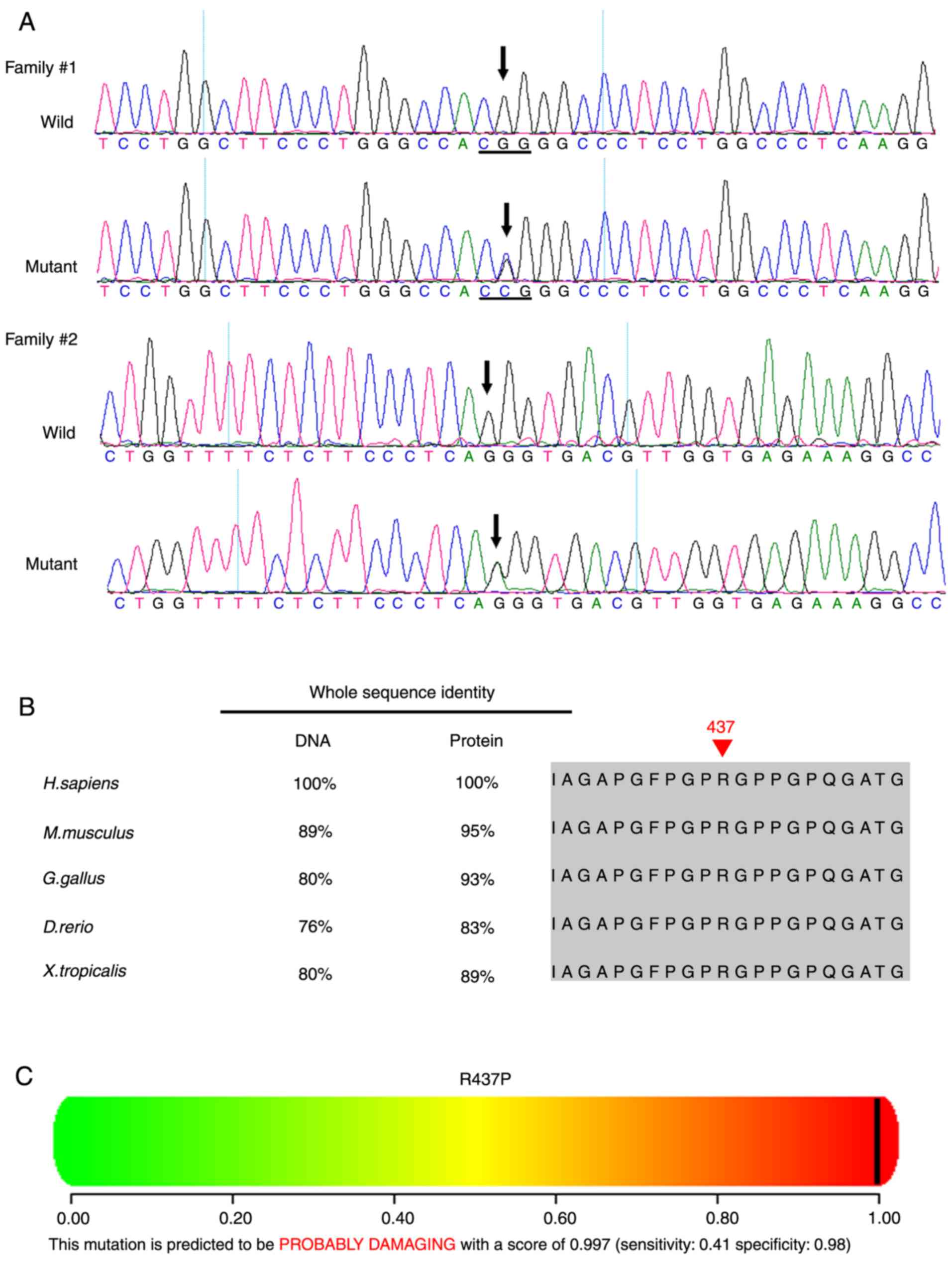
A novel heterozygous COL2A1 mutation
c.1310G>C (p.R437P) in exon 21 was identified in Family 1 (I:2,
II:1, II:2; Table II, Figs. 6 and 7A). Multiple sequence alignment
indicated that the arginine residue at position 437 of collagen
type II α1 chain is highly conserved (Fig. 7B). PolyPhen and SIFT predicted
that this mutation is damaging (Fig.
7C). A novel heterozygous COL2A1 mutation c.2302-1G>A
in intron 34 was identified in Family 2 (I:2, II:1; Figs. 6 and 7A). This mutation is likely to result in
a splicing defect as it occurs at the exon-intron border. These two
mutations were not present in the unaffected family members and the
other unrelated control subjects from the same population.
Discussion
The clinical manifestation of Stickler syndrome is
heterogeneous (3-5). Retinal detachment is the most severe
consequence of Stickler syndrome (34), and there is a high incidence of
blindness. Approximately 55-73% of Caucasian patients with a
clinical diagnosis of Stickler syndrome exhibit retinal detachment
(35,36). Thus, Stickler syndrome should be
considered and excluded if a patient presents with multiple
peripheral degeneration spots in both eyes (37-40). In the current report, both
patients exhibited sequential bilateral retinal detachment and
multiple peripheral retinal degeneration at a young age. In Patient
1, the localized retinal detachment of the right eye did not extend
to the macular area; thus, the visual impairment was less
severe.
The diagnostic criteria for Stickler syndrome have
not been well-established (5,10).
Adult patients diagnosed with Stickler syndrome typically do not
present with typical facial anomalies as children (41). In Family 2, the patient and his
mother had cleft palate, which is an important clinical indicator
of Stickler syndrome (6,37,42,43). Other typical extraocular
collagenopathies include achondrogenesis, hypochondrogenesis and
early onset osteoarthritis (44).
However, diagnosing Stickler syndrome only based on clinical
manifestations is often challenging. Genetic analysis, therefore,
is an important tool for the diagnosis of Stickler syndrome,
particularly in patients with myopia and peripheral retinal
degeneration (12,39,45-47). Early diagnosis and close-follow up
will help to decrease the incidence of the retinal detachment
(3,38). Currently, the Cambridge
prophylactic cryotherapy protocol has been demonstrated to be a
safe intervention and can markedly reduce the risk of retinal
detachment in patients with Stickler syndrome (48).
Although the affected patients in the present study
had different genetic mutations, they exhibited similar clinical
presentations of retinal detachment and degeneration. Previous
studies have also reported that different mutations in
COL2A1 can lead to similar phenotypes, with various degrees
of expressivity (42,49). The majority of COL2A1
mutations identified in Stickler syndrome are loss-of-function
mutations, as they are predicted to result in nonsense-mediated
decay of transcripts (42).
Splice site mutations, as identified in Family 2 in the current
study, are commonly identified in Stickler syndrome, and are likely
to cause unusual RNA isoforms with premature stop codons (42). Additionally, silent mutations in
COL2A1 can also result in splicing defects and reading frame
shifts (50).
In summary, the present study characterized the
clinical presentation of two Chinese families with Stickler
syndrome, and identified two novel mutations in the COL2A1
gene in the affected family members. These findings expand the
known mutation spectrums of COL2A1, and may facilitate
genetic counseling and development of therapeutic strategies for
patients with Stickler syndrome.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81500709, 81570862 and
81670872), Guangzhou Science and Technology Project (grant no.
2014Y2-00064), and the State Scholarship Fund from the China
Scholarship Council.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
XH, YL, TL, CJ, XL and LL analyzed and interpreted
the patient data. HG, BL, CL, YH, QW and HL examined the patients
and performed PCR and gene sequence analysis. YL, CC and YZ
interpreted the sequencing data, drafted the manuscript and revised
it critically. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
ethics committee of Zhongshan Ophthalmic Center (Guangzhou, China).
Informed consent was obtained from all subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stickler GB, Belau PG, Farrell FJ, Joes
JD, Pugh DG, Steinberg AG and Ward LE: Hereditary progressive
arthro-ophthalmopathy. Mayo Clin Proc. 40:433–455. 1965.PubMed/NCBI
|
|
2
|
Robin NH, Moran RT and Ala-Kokko L:
Stickler syndrome. GeneReviews®. Adam MP, Ardinger HH,
Pagon RA, et al: Seattle, WA: 1993
|
|
3
|
Rishi P, Maheshwari A and Rishi E:
Stickler syndrome. Indian J Ophthalmol. 63:614–615. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bennett JT and McMurray SW: Stickler
syndrome. J Pediatr Orthop. 10:760–763. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aylward B, daCruz L, Ezra E, Sullivan P,
MacLaren RE, Charteris D, Gregor Z, Bainbridge J and Minihan M:
Stickler syndrome. Ophthalmology. 115:1636–1637. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Snead MP, McNinch AM, Poulson AV,
Bearcroft P, Silverman B, Gomersall P, Parfect V and Richards AJ:
Stickler syndrome, ocular-only variants and a key diagnostic role
for the ophthalmologist. Eye (Lond). 25:1389–1400. 2011. View Article : Google Scholar
|
|
7
|
Savasta S, Salpietro V, Spartà MV,
Foiadelli T, Laino D, Lobefalo L, Marseglia GL and Verrotti A:
Stickler syndrome associated with epilepsy: Report of three cases.
Eur J Pediatr. 174:697–701. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goyal M, Kapoor S, Ikegawa S and Nishimura
G: Stickler syndrome type 1 with short stature and atypical ocular
manifestations. Case Rep Pediatr. 2016:31985972016.PubMed/NCBI
|
|
9
|
Faber J, Winterpacht A, Zabel B, Gnoinski
W, Schinzel A, Steinmann B and Superti-Furga A: Clinical
variability of Stickler syndrome with a COL2A1 haploinsufficiency
mutation: Implications for genetic counselling. J Med Genet.
37:318–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ang A, Ung T, Puvanachandra N, Wilson L,
Howard F, Ryalls M, Richards A, Meredith S, Laidlaw M, Poulson A,
et al: Vitreous phenotype: A key diagnostic sign in Stickler
syndrome types 1 and 2 complicated by double heterozygosity. Am J
Med Genet A. 143A:604–607. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alexander P, Poulson A, McNinch A,
Richards A and Snead M: Type I membranous anomaly in Stickler
syndrome. Ophthalmic Genet. 39:1472018. View Article : Google Scholar
|
|
12
|
Leung L, Hyland JC, Young A, Goldberg MF
and Handa JT: A novel mutation in intron 11 of the COL2A1 gene in a
patient with type 1 Stickler syndrome. Retina. 26:106–109. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richards AJ, Yates JR, Williams R, Payne
SJ, Pope FM, Scott JD and Snead MP: A family with Stickler syndrome
type 2 has a mutation in the COL11A1 gene resulting in the
substitution of glycine 97 by valine in a1 (XI) collagen. Hum Mol
Genet. 5:1339–1343. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brunner HG, van Beersum SE, Warman ML,
Olsen BR, Ropers HH and Mariman EC: A Stickler syndrome gene is
linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet.
3:1561–1564. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giedion A, Brandner M, Lecannellier J,
Muhar U, Prader A, Sulzer J and Zweymüller E:
Oto-spondylo-megaepiphyseal dysplasia (OSMED). Helv Paediatr Acta.
37:361–380. 1982.PubMed/NCBI
|
|
16
|
Van Camp G, Snoeckx RL, Hilgert N, van den
Ende J, Fukuoka H, Wagatsuma M, Suzuki H, Smets RM, Vanhoenacker F,
Declau F, et al: A new autosomal recessive form of Stickler
syndrome is caused by a mutation in the COL9A1 gene. Am J Hum
Genet. 79:449–457. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rose PS, Levy HP, Liberfarb RM, Davis J,
Szymko-Bennett Y, Rubin BI, Tsilou E, Griffith AJ and Francomano
CA: Stickler syndrome: Clinical characteristics and diagnostic
criteria. Am J Med Genet A. 138A:199–207. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barat-Houari M, Sarrabay G, Gatinois V,
Fabre A, Dumont B, Genevieve D and Touitou I: Mutation update for
COL2A1 gene variants associated with type II collagenopathies. Hum
Mutat. 37:7–15. 2016. View Article : Google Scholar
|
|
19
|
Scott JE: The chemical morphology of the
vitreous. Eye (Lond). 6:553–555. 1992. View Article : Google Scholar
|
|
20
|
Richards AJ, Baguley DM, Yates JR, Lane C,
Nicol M, Harper PS, Scott JD and Snead MP: Variation in the
vitreous phenotype of Stickler syndrome can be caused by different
amino acid substitutions in the X position of the type II collagen
Gly-X-Y triple helix. Am J Hum Genet. 67:1083–1094. 2000.PubMed/NCBI
|
|
21
|
Prockop DJ and Kivirikko KI: Collagens:
Molecular biology, diseases, and potentials for therapy. Annu Rev
Biochem. 64:403–434. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin Y, Gao H, Chen C, Zhu Y, Li T, Liu B,
Ma C, Jiang H, Li Y, Huang Y, et al: Clinical and next-generation
sequencing findings in a Chinese family exhibiting severe familial
exudative vitreoretinopathy. Int J Mol Med. 41:773–782. 2018.
|
|
23
|
Li T, Lin Y, Gao H, Chen C, Zhu Y, Liu B,
Lian Y, Li Y, Zhou W, Jiang H, et al: Two heterozygous mutations
identified in one Chinese patient with bilateral macular coloboma.
Mol Med Rep. 16:2505–2510. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
25
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: PAX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.PubMed/NCBI
|
|
26
|
Lin Y, Gao H, Ai S, Eswarakumar JVP, Chen
C, Zhu Y, Li T, Liu B, Liu X, Luo L, et al: C278F mutation in FGFR2
gene causes two different types of syndromic craniosynostosis in
two Chinese patients. Mol Med Rep. 16:5333–5337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin Y, Gao H, Ai S, Eswarakumar JVP, Zhu
Y, Chen C, Li T, Liu B, Jiang H, Liu Y, et al: FGFR2 mutations and
associated clinical observations in two Chinese patients with
Crouzon syndrome. Mol Med Rep. 16:5841–5846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin Y, Li T, Gao H, Lian Y, Chen C, Zhu Y,
Li Y, Liu B, Zhou W, Jiang H, et al: Bestrophin 1 gene analysis and
associated clinical findings in a Chinese patient with Best
vitelliform macular dystrophy. Mol Med Rep. 16:4751–4755. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.PubMed/NCBI
|
|
30
|
Burland TG: DNASTAR's Lasergene sequence
analysis software. Methods Mol Biol. 132:71–91. 2000.
|
|
31
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin Y, Li T, Ma C, Gao H, Chen C, Zhu Y,
Liu B, Lian Y, Huang Y, Li H, et al: Genetic variations in
Bestrophin 1 and associated clinical findings in two Chinese
patients with juvenile onset and adult onset best vitelliform
macular dystrophy. Mol Med Rep. 17:225–233. 2018.
|
|
34
|
Ang A, Poulson AV, Goodburn SF, Richards
AJ, Scott JD and Snead MP: Retinal detachment and prophylaxis in
type 1 Stickler syndrome. Ophthalmology. 115:164–168. 2008.
View Article : Google Scholar
|
|
35
|
Wang X, Jia X, Xiao X, Li S, Li J, Li Y,
Wei Y, Liang X and Guo X: Mutation survey and genotype-phenotype
analysis of COL2A1 and COL11A1 genes in 16 Chinese patients with
Stickler syndrome. Mol Vis. 22:697–704. 2016.PubMed/NCBI
|
|
36
|
Vilaplana F, Muiños SJ, Nadal J, Elizalde
J and Mojal S: Stickler syndrome. Epidemiology of retinal
detachment. Arch Soc Esp Oftalmol. 90:264–268. 2015.In English,
Spanish. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Antunes RB, Alonso N and Paula RG:
Importance of early diagnosis of Stickler syndrome in newborns. J
Plast Reconstr Aesthet Surg. 65:1029–1034. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wilson MC, McDonald-McGinn DM, Quinn GE,
Markowitz GD, LaRossa D, Pacuraru AD, Zhu X and Zackai EH:
Long-term follow-up of ocular findings in children with Stickler's
syndrome. Am J Ophthalmol. 122:727–728. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Watanabe Y, Ueda M and Adachi-Usami E:
Retinal detachment in identical twins with Stickler syndrome type
1. Br J Ophthalmol. 80:976–981. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bowling EL, Brown MD and Trundle TV: The
Stickler syndrome: Case reports and literature review. Optometry.
71:177–182. 2000.PubMed/NCBI
|
|
41
|
De Keyzer TH, De Veuster I and Smets RM:
Stickler syndrome: An underdiagnosed disease. Report of a family.
Bull Soc Belge Ophtalmol. 45–49. 2011.PubMed/NCBI
|
|
42
|
Hoornaert KP, Vereecke I, Dewinter C,
Rosenberg T, Beemer FA, Leroy JG, Bendix L, Björck E, Bonduelle M,
Boute O, et al: Stickler syndrome caused by COL2A1 mutations:
Genotype-phenotype correlation in a series of 100 patients. Eur J
Hum Genet. 18:872–880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee KH and Hayward P: Retrospective review
of Stickler syndrome patients with cleft palate 1997–2004. ANZ J
Surg. 78:764–766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Parke DW: Stickler syndrome: Clinical care
and molecular genetics. Am J Ophthalmol. 134:746–748. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brown DM, Vandenburgh K, Kimura AE,
Weingeist TA, Sheffield VC and Stone EM: Novel frameshift mutations
in the procollagen 2 gene (COL2A1) associated with Stickler
syndrome (hereditary arthro-ophthalmopathy). Hum Mol Genet.
4:141–142. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yoshida S, Yamaji Y, Kuwahara R, Yoshida
A, Hisatomi T, Ueno A and Ishibashi T: Novel mutation in exon 2 of
COL2A1 gene in Japanese family with Stickler Syndrome type I. Eye
(Lond). 20:743–745. 2006. View Article : Google Scholar
|
|
47
|
Higuchi Y, Hasegawa K, Yamashita M, Tanaka
H and Tsukahara H: A novel mutation in the COL2A1 gene in a patient
with Stickler syndrome type 1: A case report and review of the
literature. J Med Case Rep. 11:2372017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fincham GS, Pasea L, Carroll C, McNinch
AM, Poulson AV, Richards AJ, Scott JD and Snead MP: Prevention of
retinal detachment in Stickler syndrome: The Cambridge prophylactic
cryotherapy protocol. Ophthalmology. 121:1588–1597. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kondo H, Matsushita I, Nagata T, Hayashi
T, Kakinoki M, Uchio E, Kondo M, Ohji M and Kusaka S: Novel
mutations in the COL2A1 gene in Japanese patients with Stickler
syndrome. Hum Genome Var. 3:160182016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Richards AJ, Laidlaw M, Meredith SP,
Shankar P, Poulson AV, Scott JD and Snead MP: Missense and silent
mutations in COL2A1 result in Stickler syndrome but via different
molecular mechanisms. Hum Mutat. 28:6392007. View Article : Google Scholar : PubMed/NCBI
|