Introduction
Angiogenesis, a multistep process by which new blood
vessels are formed from pre-existing vessels, is important in
different physiological and pathophysiological situations,
including wound healing, cancer, and ischemic diseases (1). During angiogenesis, endothelial
cells proliferate and migrate as 'tube-like sprouts' from
pre-existing vessels (2).
Cell proliferation is a key step in angiogenesis.
Gap junctions have been demonstrated to be important not only in
the regulation of cell proliferation and growth but also in the
differentiation and development of blood vessels (3-5).
The gap junctions serve as critical gatekeepers in cell
proliferation and several other cellular processes by regulating
the exchange of ions, amino acids, secondary messengers, including
ATP, and different growth regulators (6-9).
The gap junctions are composed of connexins, which are a family of
transmembrane proteins that form channels connecting the cytoplasm
of adjacent cells (10-13). The hexameric arrangement of
connexins form single-membrane channels, hemichannels or connoxons,
which align their counterparts in adjacent cell membranes to
constitute a complete intercellular channel (14,15). Several connexins, including
connexin (Cx)37, Cx40, Cx43 and Cx45, are expressed in different
cell types (16,17); however, in endo-thelial cells,
Cx43 is predominantly expressed (18).
In addition to being an important component of the
gap junction, Cx43 has been shown to regulate different cellular
processes, including cell proliferation and migration (19-22). Notably, a reduction in the
expression of Cx43 in endothelial cells by nicotine treatment
impaired angiogenesis (23). In
addition, mitogen-activated protein kinases (MAPKs) may regulate
the expression of Cx43 in endothelial cells (24). These findings suggest that Cx43 is
involved in different steps of angiogenesis.
The expression pattern of Cx43 has been shown to be
altered in cell proliferation and cell migration during ischemic
injury (25,26), cancer (27,28), atherosclerosis (29,30) or wound healing (31,32). Similarly, the overexpression or
loss of caveolin-1 (Cav-1) alters different cellular functions,
including cell proliferation and permeability under different
conditions (33,34).
Several studies have shown that Cx43 co-localizes
with other proteins, including fibroblast growth factor receptors
(22), cytoplasmic proteins
(35) and cytoskeleton proteins
(36). In addition, published
literature has demonstrated that Cx43 interacts with caveolin
proteins in different cell types (37,38). However, its interaction with Cav-1
protein appears to be cell type-specific and its functional
importance remains to be fully elucidated. Cav-1, the structural
protein of caveolae, is involved in intracellular communication in
gap junctions and also in cell cycle control (33). It is also well documented that the
overexpression of Cav-1 inhibits mitogenic signaling in a number of
cell lines (39,40).
The aim of the present study was to investigate the
mutual effect of Cx43 and Cav-1 proteins in different steps of
angiogen-esis, including cell proliferation, migration and tube
formation, in endothelial cells. Whether Cx43 counter-regulates
Cav-1 in controlling angiogenesis is of interest as the two
proteins work differently under different pathophysiological
conditions.
Materials and methods
Materials
Cell culture dishes and serum reduced Matrigel™ were
purchased from BD Biosciences (San Jose, CA, USA). Anti-Cx43 (cat.
no. 610062) and anti-Cav-1 (cat. no. 610057) antibodies were from
BD Biosciences. Cav-1 small interference (si)RNA (cat. no.
sc-29241) was purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Endothelial cell basal medium plus supplement
pack was from PromoCell (Heidelberg, Germany). Different
pharmacological inhibitors of Cx43, including 18 α-glycyrrihizinic
acid (α-GA) and niflumic acid (NA) and flufenamic acid (FA) were
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Connexin
mimetic peptides, Gap26 (G-26; cat. no. CX2605-P-1) was from Alpha
Diagnostic International, Inc. (San Antonio, TX, USA), and Gap27
(G-27; cat. no.G794) were from Sigma; Merck KGaA (Darmstadt,
Germany). Fetal calf serum (FCS) and penicillin-streptomycin were
from Gibco®; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Hank's balanced salt solution (HBSS), collagenase and
trypsin-EDTA solutions were from PAA Laboratories (Pasching,
Austria). The jetSI™-ENDO transfection reagent was purchased from
(Polyplus-transfection SA, Illkirch, France). The negative siRNA
(cat. no. SI03650318) was from Qiagen GmbH (Hilden, Germany), and
mitogen-activated protein kinase kinase (MEK)1 inhibitor, PD-98059,
was from Calbiochem; Merck KGaA. Anti-extracellular
signal-regulated kinase (ERK)1/2 (cat. no. 9102) and
anti-phosphorylated (p)ERK1/2 (cat. no. 9101) were purchased from
Cell Signalling Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase (HRP)-conjugated anti-rabbit IgG (cat no. NA9340),
HRP-conjugated anti-mouse (cat. no. NA931) antibodies and
anti-vinculin antibody (cat. no. V9131) were purchased from
Sigma-Aldrich; Merck KGaA. Pierce™ ECL solution was from Thermo
Fisher Scientific Inc. All other chemicals were of the highest
available quality.
Cell culture
Human umbilical veins were maintained at 4°C in a
sterile container and obtained once a week from the Department of
Gynaecology, University Hospital Giessen and Marburg (Giessen,
Germany) between August 2009 and August 2013. Human umbilical vein
endothelial cells (HUVECs) were isolated and cultured as previously
described (41). Briefly,
untraumatized human umbilical veins were cannulated and perfused
with HBSS to remove traces of blood. Following this, the lumen of
the vein was filled with 0.025% collagenase solution (w/v) and
incubated for 30 min at 37°C. Collagenase solution containing
endothelial cells was subsequently removed via perfusion of the
vein with 30 ml of HBSS containing 3% (v/v) FCS to terminate the
reaction. The effluent was then collected in a 50 ml falcon tube
and centrifuged for 5 min at 250 x g at room temperature. The
supernatant was then removed and the cell pellet was resuspended in
endothelial cell culture medium containing 0.1% (v/v) gentamycin.
Following this, cells were seeded to 3-4 primary culture dishes.
Following a total of 2 h of incubation at 37°C in 5%
CO2, cells were extensively washed with HBSS to remove
non-endothelial cells and cell debris. Adherent cells were then
incubated in 15-20 ml of endothelial cell culture medium containing
2% (v/v) penicillin/streptomycin at 37°C and 5% CO2.
Following 24 h, cell culture medium was replaced with fresh
endothelial cell culture medium. For experiments, HUVECs were
cultured in endothelial cell culture medium supplemented with 10%
(v/v) FCS, 0.4% (v/v) endothe-lial growth supplement with heparin,
0.1 ng/ml human epithelial growth factor, 0.1 µg/ml hydrocortisone,
1 ng/ml human basic fibroblast growth factor, 100 IU/ml penicillin
G and 100 µg/ml streptomycin. Cells were seeded either on 12-well
plates or 96-well plates. The experiments were performed when cells
in the monolayers reached 100% confluence. For the present study,
informed consent was obtained from patients, and ethical approval
was obtained from the Ethics Committee of Justus-Liebig University
of Giessen (Giessen, Germany; no. AZ132/09). The study confirms the
principles outlined in the 'Declaration of Helsinki' (42).
Determination of cell proliferation
The endothelial cells were seeded in endothelial
cell basal medium at a density of 2.5×103 in a 96-well
plates cultured in a humidified atmosphere at 37°C and 5%
CO2. After 24 h, the endothelial cells were treated at
37°C in a 5% CO2 atmosphere with Cx43 pharmacological
inhibitors, including NA (50 µM), FA (50 µM), and α-GA (25 µM) or
specific mimetic peptides G-26 and G-27 (0.25 µg/ml) and MEK1
inhibitor, PD-98059 (20 µM). Subsequently, the endothelial cells
were incubated for 48 h. Analysis of cell proliferation was
determined by crystal violet staining. At the end of the
experiments, the cells were briefly fixed for 30 min with 5.5%
gluteraldehyde at room temperature. Following three washes with
water, the cells were dried for 1 h. The cell nuclei were then
stained with 1% crystal violet solution (pH 5.4) for 20 min.
Following another washing step, the cells were dried and treated
with 10% acetic acid, and the absorbance of the crystal violet
staining was measured at 595 nm using a microplate reader
(Sunrise™; Tecan Group, Inc., Männedorf, Switzerland). Migration
assays. For migration assays, a self-made section of silicone
rubber (length, 3 mm) was fixed in the center of a 12-well plate,
and endothelial cells were seeded at a density of 1×105
per well. The removal of the silicone rubber from the culture
plates was considered as the initial time point (0 h) and the
endothelial cells were treated with Cx43 inhibitors or PD-98059.
The cells were incubated for 48 h, which was considered the
endpoint for the migration assay. At the end of experiments, images
of the migrated cells were captured with an inverted microscope
(magnification, x2) using a CCD camera, and the migration area was
quantified using Cell D software version 5.1 (Olympus Corporation,
Tokyo, Japan).
Tube formation assay
Matrigel (200 µl) was deposited into wells in a
24-well plate and allowed to solidify for 30 min at 37°C. The
endothelial cells (4×104 cells) were added to each well.
Following of incubation at 37°C for 5 h, the cells were treated
with Cx43 inhibitors or with PD-98059. The cells were incubated on
Matrigel for a further 24 h at 37°C and images were captured by
phase contrast microscopy (Olympus). Random fields of view/well
were examined for image capture. The total number of tubes from the
nodes of endothelial cells was quantified using Cell D software
(Olympus).
Transfection of endothelial cells
The endothelial cells were seeded according to the
experiments being performed. Subconfluent monolayers of endothelial
cells were transfected for 48 h with a specific siRNA against Cav-1
(50 nM) or with a negative siRNA (50 nM) using jetSI™-ENDO
transfection reagent according to the manufacturer's protocol.
SDS-PAGE and western blot analysis
For immunoblot analyses, the experimental incubation
of cultures was terminated by rapid removal of medium and the
addition of 2X SDS sample buffer [250 mM Tris/HCl (pH 6.8), 20%
glycerol, 2% (w/v) SDS, 0.001% (w/v) bromophenol blue, 10% (v/v)
2-mercaptopropandiol, 1 mM DTT (added fresh prior to use) and 10 µl
Benzonase (250 U) in 200 mM MgSO4]. Protein
concentration was determined by Pierce™ BCA protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.; cat. no. 23227), and equal
amounts of protein (30 µg) in lysis buffer were resolved by 12%
SDS-PAGE and transferred onto nitrocellulose membranes.
Subsequently, the membranes were blocked for 1 h at room
temperature under constant shaking in either 1X TBST [5% (w/v)
non-fat milk powder] or TBST containing 3% bovine serum albumin
(BSA; w/v; Sigma-Aldrich; Merck KGaA) when phospho-specific
antibodies were applied. The primary antibodies were diluted in
TBST. The membranes were incubated with the appropriate primary
antibodies (anti-Connexin43, anti Caveolin-1, anti-ERK1/2,
anti-pERK1/2 and anti-Vinculin; all used at a 1:1,000 dilution in
3% BSA) with gentle shaking overnight at 4°C. The membranes were
washed three times with TBST and incubated with the appropriate
secondary antibody (HRP-conjugated anti-rabbit IgG or
HRP-conjugated anti-mouse IgG; both used at a dilution of 1:2,000
in 3% BSA) for 1 h at room temperature. Following washing, the
protein bands were visualized by enhanced chemiluminescence and
detected with a CCD camera using the Bio-Rad ChemiDoc system
(Bio-Rad Laboratories, Inc. Hercules, CA, USA) according to the
manufacturer's protocol.
Statistical analysis
Statistical analyses were performed using Graphpad
Prism software version 5 (GraphPad Software, Inc., La Jolla, CA,
USA). Data are shown as the mean ± standard error of the mean of
3-6 experiments using independent cell preparations. The comparison
of means between groups was performed by one-way analysis of
variance followed by a Bonferroni post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibition of Cx43 impairs cell
proliferation, migration and tube formation of endothelial
cells
The pharmacological inhibition of Cx43 using NA (50
µM), FA (50 µM), and α-GA (25 µM) resulted in significant
reductions in cell proliferation and migration of endothelial
cells. Similarly, the specific inhibition of Cx43 using mimetic
peptides, Gap-26 or Gap-27, also suppressed the proliferation and
migration of endothelial cells compared with respective control
cells (Fig. 1A-C).
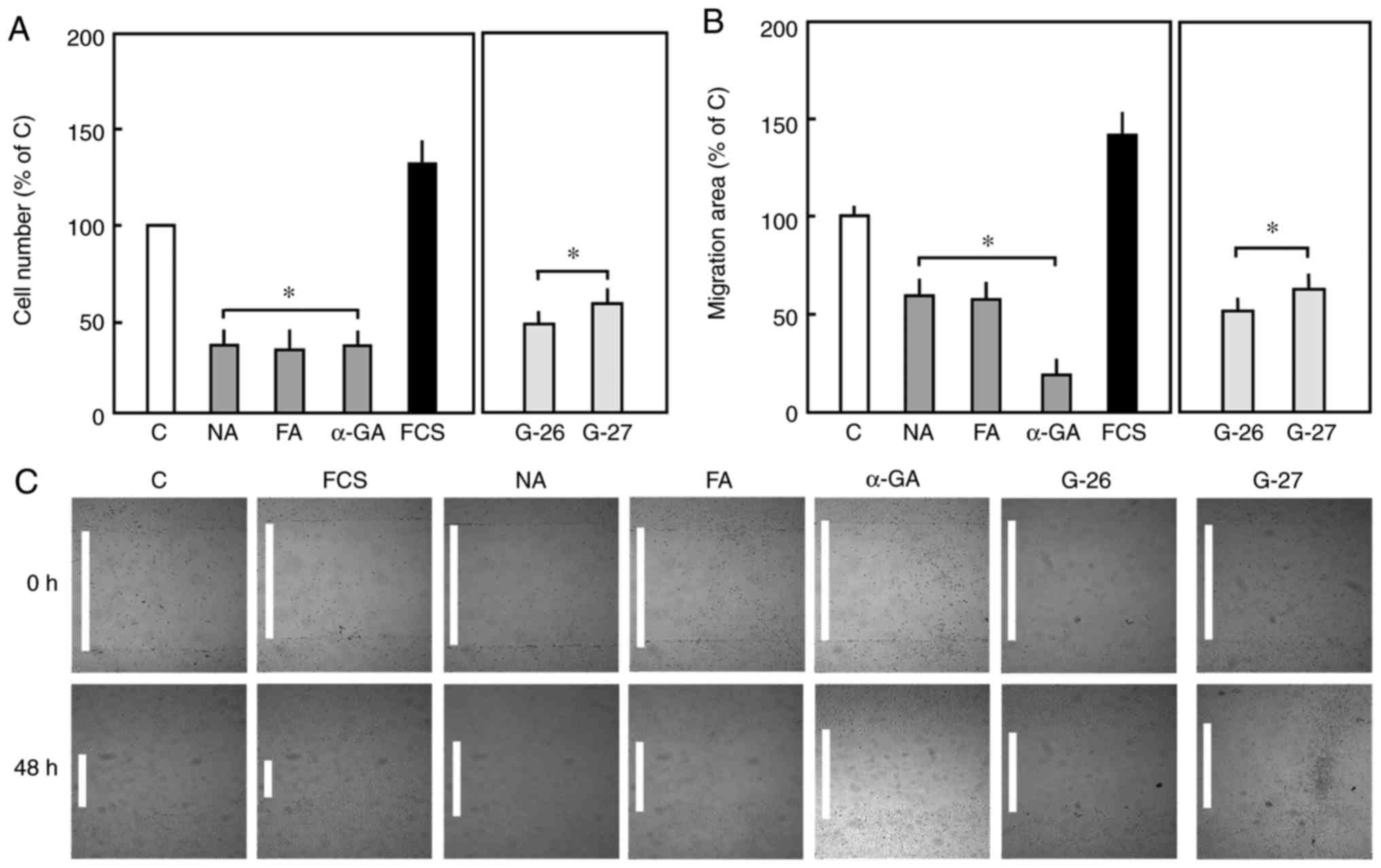 | Figure 1(A) Effect of Cx43 inhibition on the
proliferation of endothelial cells. Endothelial cells were treated
with Cx43 inhibitors (NA, 50 µM), (FA, 50 µM) or (α-GA, 25 µM), or
specific mimetic peptides (G-26 and G-27, 0.25 µg/ml) for 48 h. For
the positive control, the cells were incubated in 20% serum.
Subsequently, cell proliferation was determined by crystal violet
staining. The number of cells in the control group was set to 100%.
(B) For the migration assay, the cells were seeded, and removal of
a rubber section was considered the initial time point (0 h). The
cells were treated with the same concentration of Cx43 inhibitors
or mimetic peptides against Cx43, incubated for 48 h (considered
the endpoint), and the area of migration was measured. The area of
migration for control cells was considered as 100%. (C) Images of
migration (magnification, x2). Data are shown as the mean ±
standard error of the mean of 3-5 experiments of independent cell
preparations *P<0.05, vs. C. Cx43, connexin 43; NA,
niflumic acid; FA, flufenamic acid; α-GA, 18 α-glycyrrihizinic
acid; C, control; FCS, fetal calf serum. |
To investigate the effect of Cx43 inhibition on
angiogen-esis, endothelial cells were seeded on Matrigel to perform
a tube formation assay. The treatment of endothelial cells with
Cx43 inhibitors, NA, FA and α-GA, for 24 h markedly reduced tube
formation (Fig. 2A and B).
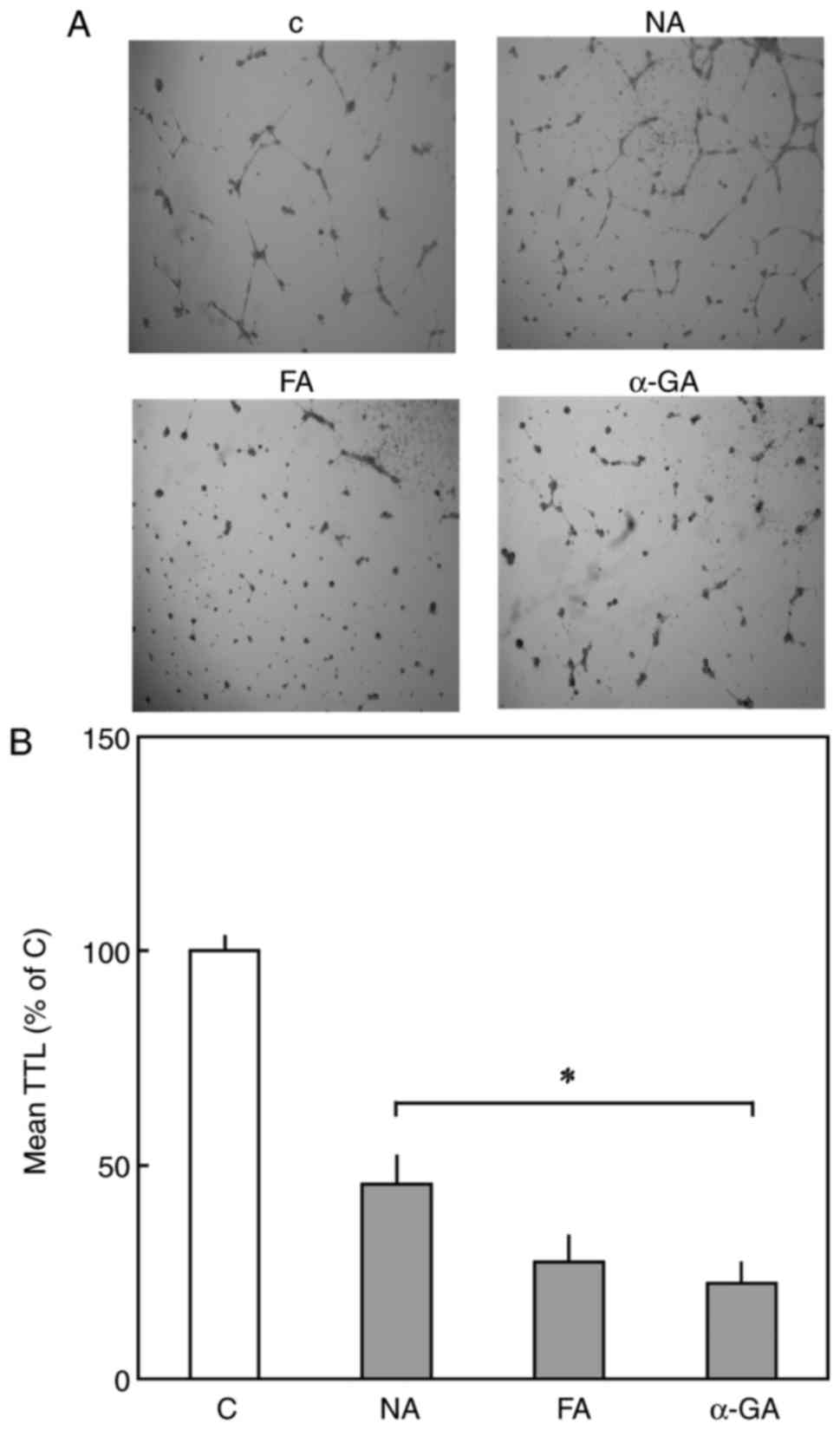 | Figure 2For the tube formation assay,
4.2×104 cells were seeded onto Matrigel coated-24-well
plates. After 3 h, cells were treated with Cx43 inhibitors and
incubated for 24 h. (A) Images of cells (magnification, x2) were
captured and (B) TTL was determined. Data are shown as the mean ±
standard error of the mean of 3-5 experiments of independent cell
preparations *P<0.05, vs. C. Cx43, connexin 43; NA,
niflumic acid; FA, flufenamic acid; α-GA, 18 α-glycyrrihizinic
acid; C, control; TTL, total tube length. |
Effect of Cx43 inhibition on the
expression of Cav-1 and activation of ERK1/2
The pharmacological inhibition of Cx43 resulted in a
significant decrease in the expression of Cx43, as shown in
Fig. 3A. However, the long-term
treatment of endothelial cells with Cx43 inhibitors enhanced the
expression of Cav-1 compared with that in the control cells
(Fig. 3B). Furthermore, Cx43
inhibitors significantly reduced the phosphorylation of ERK1/2
compared with untreated cells (Fig.
3C). Taken together, these data suggested that the expression
of Cx43 and Cav-1 is critical in the proliferation and migration of
endothelial cells.
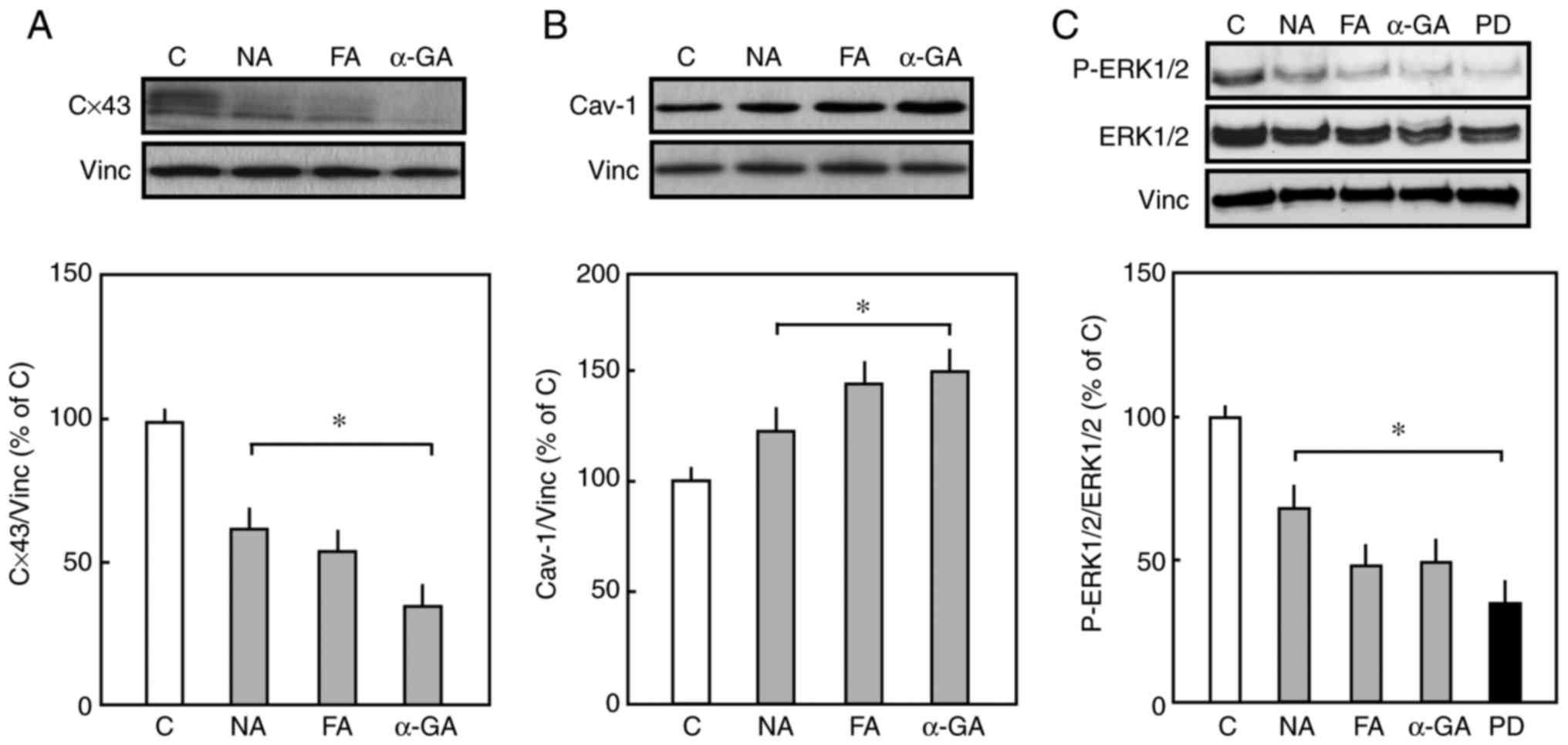 | Figure 3(A) Effect of Cx43 inhibitors on
expression levels of Cx43 and Cav-1. Representative western blots
of total Cx43 and Vinc as an internal control. The endothelial
cells were treated with Cx43 inhibitors (NA, FA, or α-GA) for 48 h.
Densitometric analysis of the expression of Cx43 is shown. The mean
total expression of Cx43 in control cells in the absence of
inhibitors was set to 100%. (B) Representative western blots of
total Cav-1 and Vinc as an internal control. Densitometric analysis
of total expression of Cav-1 is shown. The mean of total expression
of cav-1 in the control cells in the absence of inhibitors was set
to 100%. (C) Representative western blots of p-ERK1/2, total
ERK1/2, and Vinc as endogenous control. Densitometric analysis of
p-ERK1/2 is shown. The mean expression of p-ERK1/2 in the control
was set to 100%. Data are presented as the mean ± standard error of
the mean of three separate experiments of independent cell
preparations, *P<0.05, vs. C. Cx43, connexin 43;
Cav-1, caveolin 1; Vinc, vinculin; ERK, extracellular
signal-regulated kinase; p-ERK, phosphorylated ERK; NA, niflumic
acid; FA, flufenamic acid; α-GA, 18 α-glycyrrihizinic acid; PD,
PD-98059; C, control. |
ERK1/2 is involved in the expression of
Cx43 and Cav-1
As ERK1/2 is known to be involved in the regulation
of Cx43 and Cav-1 protein expression in different cell types
(43,44), the present study examined whether
ERK1/2 affects the protein expression of Cx43 and Cav-1 in
endothelial cells. PD-98059 (20 µM), an MEK inhibitor, was used to
assess the expression of these proteins. The pre-treatment of
endothelial cells with PD-98059 significantly reduced the
expression of Cx43. By contrast, the inhibition of ERK1/2 by
PD-98059 enhanced the expression of Cav-1, which is a protein that
is known to be associated with reduced cell proliferation (Fig. 4A). Therefore, the effect of
PD-98059 on the proliferation and migration of endo-thelial cells
was examined. The pharmacological inhibition of ERK1/2 resulted in
a significant reduction in the proliferation and migration in
endothelial cells (Fig. 4B),
suggesting the involvement of Cx43 and Cav-1 via the MEK/ERK
pathway.
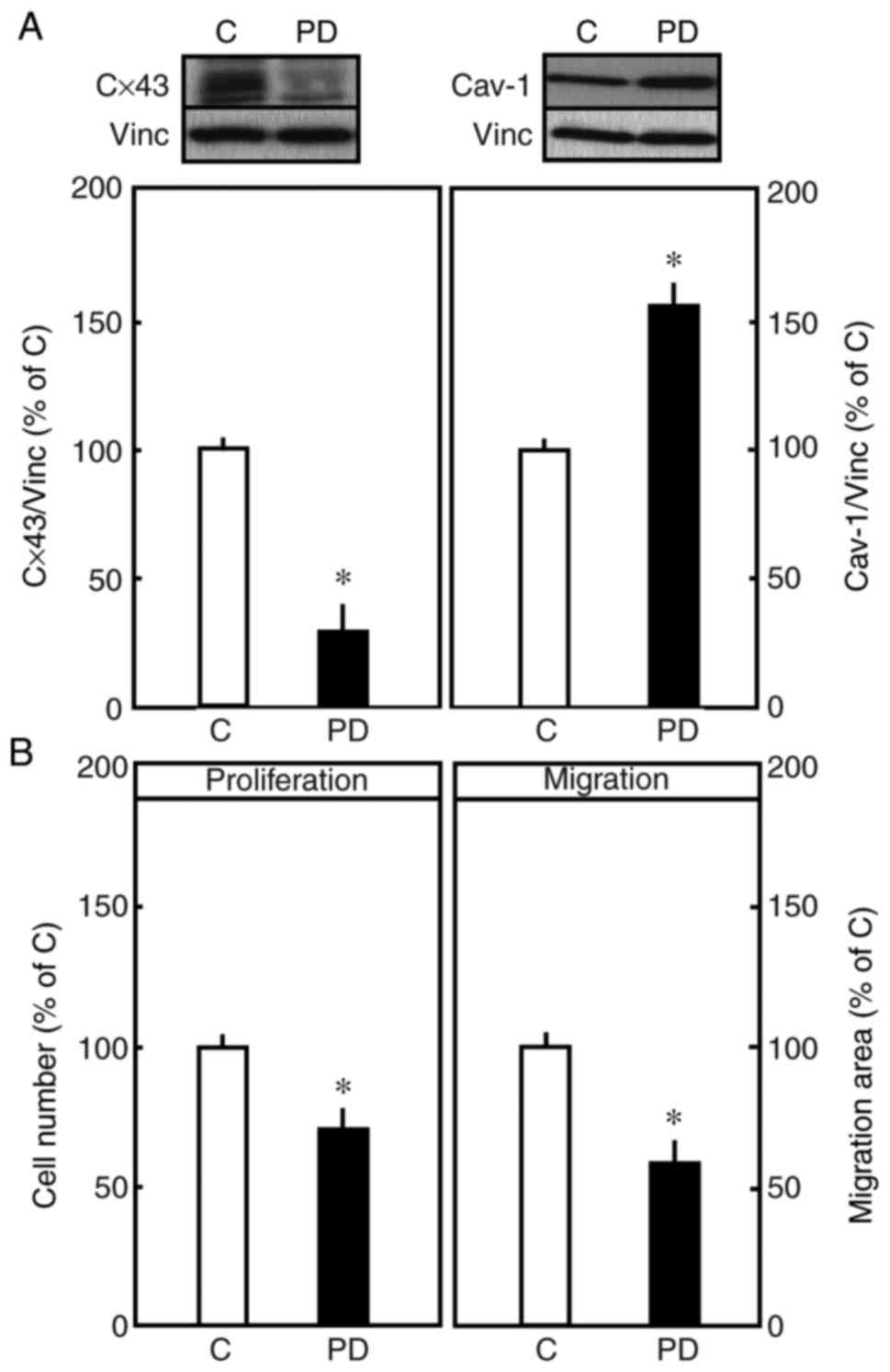 | Figure 4(A) Effect of PD-98059 (MEK1
inhibitor) on the expression of Cx43 and Cav-1. Representative
western blots of total Cav-1 and Vinc (internal control). The
endothelial cells were treated with PD-98059 (20 µM) for 48 h.
Densitometric analysis of total expression of Cx43 is shown. The
mean of total expression of Cx43 in the control cells in the
absence of inhibitors was set to 100%. Representative western blots
of total Cav-1 and Vinc (internal control). Densitometric analysis
of total expression of Cav-1 is shown. The mean total expression of
Cav-1 in the control cells in the absence of inhibitors was set to
100%. (B) Endothelial cells were treated with MEK1 inhibitor
(PD-98059, 20 µM) for 48 h. Cell proliferation was determined by
crystal violet staining. The number of cells in the control was set
to 100%. In the migration assay, the cells were seeded at a density
of 1×105 in 12-well plates. After 48 h, the cells were
treated with PD-98059 and incubated for 48 h, and the area of
migration was measured. The area of migration of control cells was
considered as 100%. Data are shown as the mean ± standard error of
the mean of three separate experiments of independent cell
preparations, *P<0.05, vs. C. Cx43, connexin 43;
Cav-1, caveolin 1; Vinc, vinculin; MEK1, mitogen-activated protein
kinase kinase 1; C, control; PD, PD-98059. |
In the above data, the inhibition of Cx43 increased
the expression of Cav-1, and the MEK/ERK pathway was found to serve
as an intermediate signalling element. Limited data in the
literature show the interaction of these two proteins (37,38), however, it may be that an
intermediate signalling element has not been identified. In the
present study, it was identified that the MEK/ERK pathway serves as
an intermediate signaling element.
siRNA mediated-downregulation of Cav-1
increases the expression of Cx43 and promotes ERK1/2
activation
A siRNA transfection strategy was used to
selectively down-regulate the protein expression of Cav-1. The
immunoblot probed for Cav-1 in endothelial cells, 48 h following
transfection with specific Cav-1 siRNA (Cav-1 siRNA) is shown in
Fig. 5A, and quantification is
shown in Fig. 5B. The expression
of Cav-1 was efficiently downregulated with Cav-1 siRNA, whereas
the expression of Cav-1 remained unchanged in the untransfected
cells or in the cells that were transfected with negative siRNA.
Furthermore, the specific downregulation of Cav-1 protein
significantly enhanced the expression of Cx43 by 25±5% compared
with that in the control. The downregulation of Cav-1 resulted in
increased ERK1/2 phosphorylation compared with that in the
control.
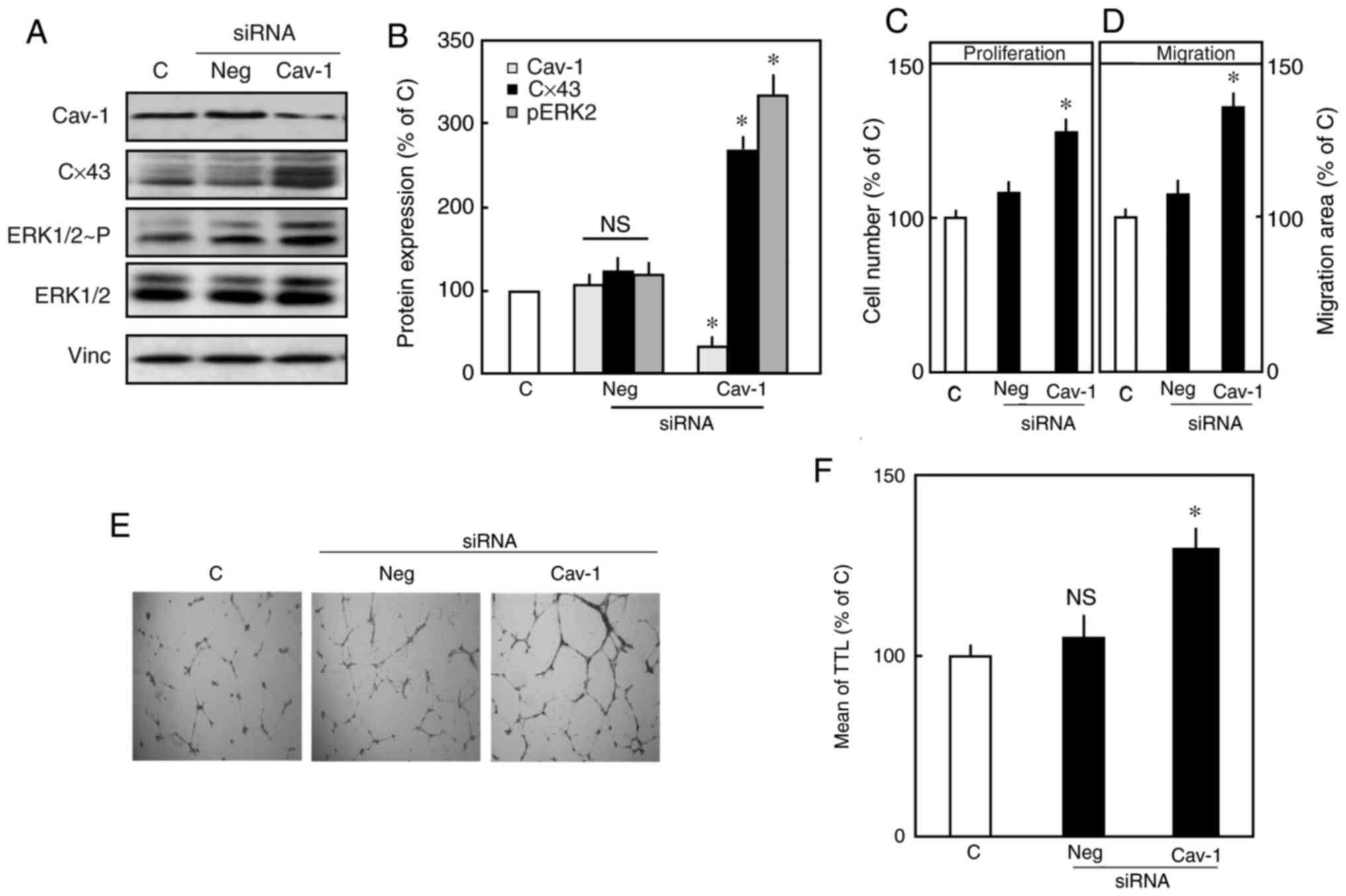 | Figure 5siRNA mediated-downregulation of
Cav-1 increases the expression of Cx43 and promotes ERK1/2
activation. Endothelial cells were transfected with either specific
siRNA against Cav-1 or negative siRNA (50 nm), or left untreated as
a control, for 48 h. Following transfection, the cells were
harvested and lysed, and proteins levels were analyzed in
immunoblots probed with Cav-1, Cx43 and p-ERK1/2 antibodies. Vinc
was probed as an internal control. (A) Representative western blots
of total Cav-1, Cx43 and P-ERK1/2, total ERK1/2, and Vinc. (B)
Densitometric analysis of total Cav-1, Cx43 and P-ERK1/2. The mean
total expression in the control was set to 100%. Data are presented
as the mean ± standard error of the mean of three separate
experiments of independent cell preparations,
*P<0.05, vs. C. (C) Effect of downregulating Cav-1 on
the proliferation of endothelial cells. Following transfection,
cell proliferation was determined, and the number of control cells
was set to 100%. (D) Results of the migration assay. Following
transfection, the area of migration was measured, with the area of
migration of the control cells set as 100%. (E) Images
(magnification, x2) and (F) quantification of the tube formation
assay. Following 48 h transfection, the cells were trypsinized and
seeded onto a Matrigel coated-24-well plate. After 24 h, the TTL
was determined. Data are shown as the mean ± standard error of the
mean of 3-5 experiments of independent cell preparations,
*P<0.05, vs. C. Cx43, connexin 43; Cav-1, caveolin 1;
Vinc, vinculin; ERK, extracellular signal-regulated kinase; P-ERK,
phosphorylated ERK; siRNA, small interference RNA; Neg, negative
control; C, control; TTL, total tube length; n.s.
non-significant. |
An increased expression of Cx43 is linked to cell
proliferation, migration and the angiogenic activity of endothelial
cells (18). The downregulation
of Cav-1 increased the expression of Cx43 and consequently
increased the proliferation and migration of endothelial cells
(Fig. 5C and D). Tube formation
was also markedly increased in the Cav-1-downregulated cells
compared with that in the control (Fig. 5E and F).
Discussion
The major findings of the present study were that
the inhibition of Cx43 impaired the proliferation, migration and
angiogenic activity of endothelial cells. In addition, the
inhibition of Cx43 led to increased expression of Cav-1. Targeted
downregulation of the Cav-1 protein increased the expression of
Cx43 and promoted the activation of ERK1/2. The MEK/ERK pathway was
found to be an intermediate signalling element.
The pharmacological inhibition of Cx43 significantly
reduced the proliferation, migration and tube formation in
endothelial cells. These findings are consistent with previous
studies, which reported that the expression of Cx43 regulated cell
proliferation (18). However, the
role of the expression of Cx43 remains to be fully elucidated.
Previous studies described that change in the expression of Cx43
affects cellular processes in a cell-type-dependent manner
(18,19,23). Inhibition of the expression of
Cx43 in keratinocytes led to increased cell proliferation at wound
sites (38), whereas its effect
was found to be the opposite in endothelial cells. Furthermore, a
reduction in the expression of Cx43 inhibited the proliferation of
endothelial cells by enhancing the expression of co-regulatory
factors, plasminogen activator inhibitor-1 and von Willebrand
factor. The c-jun N-terminal kinase pathway is reported to be
involved in the regulation of this process (18,23). Taken together, these data are in
line with previous findings that the expression of Cx43 is involved
in the angiogenic activity of endothelial cells (23).
The role of MAPKs in regulating the expression of
Cx43 remains controversial and may, to a certain extent, be
dependent on the cell type. Polontchouk et al (24) demonstrated that the
phosphorylation of ERK1/2 enhanced the expression of Cx43 in
cardiomyocytes. Polontchouk et al showed that endo-thelin-1
enhanced the expression of Cx43 via the ETA receptor by activating
ERK1/2. In the present study, it was observed that inhibition of
the phosphorylation of ERK1/2 led to decreased expression of Cx43,
however, this mechanism requires further elucidation. The present
study demonstrated that the pharmacological inhibition of Cx43
reduced the phosphorylation of ERK1/2 but not the expression of
ERK1/2.
It has been shown previously that Cx43 mediates
signalling to adjacent cells by releasing secondary messengers into
the extracellular environment. A reduction of Cx43 function in
osteoblast cells by pharmacological inhibitors markedly reduces the
activation of ERK1/2 (7). In the
literature, it has been shown that ATP is released across the Cx43
channels, and the disruption of gap junctions by pharmacological
treatment impairs the release of ATP from Cx43 channels (8,9,45,46). ATP stimulates cell proliferation
via purinergic receptor, which is dependent on the ERK1/2
activation (47). However, Cx43
dependent-ATP release and its role in paracrine inter-cellular
signaling remain to be fully elucidated. The data obtained in the
present study suggest that the expression of Cx43 and activation of
ERK1/2 are interdependent, and ATP release across Cx43 may be
involved.
Previous studies have shown that Cav-1 controls the
cell cycle (33,40). Schubert et al (37) first reported that Cx43 interacts
and co-localizes with Cav-1. Cx43 directly interacts and binds with
the scaffolding domain (residues 82-101) and C-terminal domain
(135-178) of Cav-1 protein (37).
The present study found that the inhibition of Cx43 enhanced the
expression of Cav-1, and data shows that the overexpression of
Cav-1 protein inhibits mitogenic signaling and acts as a cell cycle
suppressor protein by inhibiting ERK1/2 activity (39). From these findings, it is likely
that the suppression in ERK1/2 activity results in the reduced
expression of Cx43, which was shown in the results from the present
study.
The pharmacological inhibition of either Cx43 or
ERK1/2 enhanced the expression of Cav-1. Data from previous studies
demonstrates that Cav-1 may interact and regulate the activities of
several signaling molecules [33-35]. The targeted down-regulation
of Cav-1 has been shown to selectively activate the MEK/ERK pathway
(48-50). It is also well established that
the MEK/ERK pathway is important in cell proliferation (51). To further characterize the role of
the expression of Cx43 in angiogenesis, an siRNA transfection
strategy was used to downregulate the expression of Cav-1. The
targeted down-regulation of Cav-1 not only increased the expression
of Cx43 but also the phosphorylation of ERK1/2. Accordingly, the
cell proliferation, migration and tube formation of endothelial
cells were increased.
Therefore, based on the above findings, a mechanism
is suggested where the inhibition of Cx43 attenuated the activation
of ERK1/2 and enhanced the expression of Cav-1, leading to
suppressed cell proliferation. By contrast, the siRNA
mediated-downregulation of Cav-1 rescued the expression of Cx43 by
activating ERK1/2 and consequently increasing cell
proliferation.
In the present study, the inhibition of Cx43
increased the expression of Cav-1, but reduced cell proliferation
and angiogenesis. As Cx43 and Cav-1 are membranous proteins, this
observation led to investigation of the combined effect of Cx43 and
Cav-1 in the control of angiogenesis. It was observed that the
inhibition of one molecule affected the expression of the other
molecule and vice versa. Therefore, it may be that one protein
counter-regulates the effect of other protein. Of note, it was
found that the MEK/ERK pathway served as an intermediate signaling
element in this context.
In conclusion, the data obtained in the present
study support the hypothesis that Cx43 and Cav-1 are important in
angiogenesis. The MEK/ERK pathway was identified to be an
intermediate signaling element. The expression of Cx43 regulated
cell proliferation, migration and tube formation in endothelial
cells. Cx43 counter-regulated Cav-1, and the two proteins converged
on the MEK/ERK pathway. Therefore, the present study suggested that
pharmacological interventions may improve endothelial cell
functions in different physiological and pathophysiological
situations, including ischemic injuries, wound healing, diabetes,
atherosclerosis, or cancer therapy.
Funding
This study was supported by an Anschubsfinanzierung
and Von-Behring-Röntgen-Stiftung (2011) grant from The
Justus-Liebig University (Giessen, Germany) and (grant no. SFB547;
Projects A3 and A4) Deutsche Forschungsgemeinschaft.
Availability of data and materials
Datasets used during the current study are available
from the corresponding author on a reasonable request.
Authors' contributions
MA performed the majority of the experiments,
analysed the data and wrote the manuscript. MA and CC performed the
cell proliferation experiments. MAR contributed to the siRNA
transfection experiments, evaluated the data and proof read the
manuscript. TN and DG designed, supervised the study and approved
the final manuscript. DG provided the financial support.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Justus-Liebig University (Giessen, Germany).
Patient consent for publication
Patient consent was obtained for the current
study.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mrs H. Thomas, Mrs
D. Reiz, Mrs G. Albohn, and Mrs S. Schäfer (Department of
Cardiology and Angiology, University Hospital Giessen and Marburg,
Giessen Germany) for their technical support. Authors wish to thank
Dr M. Aslam (Department of Cardiology and Angiology, University
Hospital Giessen and Marburg) for editing the manuscript.
References
|
1
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Augustin HG: Tubes, branches, and pillars:
The many ways of forming a new vasculature. Circ Res. 89:645–647.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Larson DM and Haudenschild CC: Junctional
transfer in wounded cultures of bovine aortic endothelial cells.
Lab Invest. 59:373–379. 1988.PubMed/NCBI
|
|
4
|
Homkajorn B, Sims NR and Muyderman H:
Connexin 43 regulates astrocytic migration and proliferation in
response to injury. Neurosci Lett. 486:197–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Walker DL, Vacha SJ, Kirby ML and Lo CW:
Connexin43 deficiency causes dysregulation of coronary
vasculogenesis. Dev Biol. 284:479–498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vinken M, Decrock E, De Vuyst E, Ponsaerts
R, D'hondt C, Bultynck G, Ceelen L, Vanhaecke T, Leybaert L and
Rogiers V: Connexins: Sensors and regulators of cell cycling.
Biochim Biophys Acta. 1815:13–25. 2011.
|
|
7
|
Stains JP and Civitelli R: Gap junctions
regulate extracellular signal-regulated kinase signaling to affect
gene transcription. Mol Biol Cell. 16:64–72. 2005. View Article : Google Scholar :
|
|
8
|
Gomes P, Srinivas SP, Vereecke J and
Himpens B: ATP-dependent paracrine intercellular communication in
cultured bovine corneal endothelial cells. Invest Ophthalmol Vis
Sci. 46:104–113. 2005. View Article : Google Scholar
|
|
9
|
Faigle M, Seessle J, Zug S, El Kasmi KC
and Eltzschig HK: ATP release from vascular endothelia occurs
across Cx43 hemichannels and is attenuated during hypoxia. PloS
One. 3:e28012008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brisset AC, Isakson BE and Kwak BR:
Connexins in vascular physiology and pathology. Antioxid Redox
Signal. 11:267–282. 2009. View Article : Google Scholar
|
|
11
|
Goodenough DA, Goliger JA and Paul DL:
Connexins, connexons, and intercellular communication. Annu Rev
Biochem. 65:475–502. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Söhl G and Willecke K: Gap junctions and
the connexin protein family. Cardiovasc Res. 62:228–232. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hervé JC and Derangeon M:
Gap-junction-mediated cell-to-cell communication. Cell Tissue Res.
352:21–31. 2013. View Article : Google Scholar
|
|
14
|
Bruzzone R, White TW and Paul DL:
Connections with connexins: The molecular basis of direct
intercellular signaling. Eur J Biochem. 238:1–27. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maeda S and Tsukihara T: Structure of the
gap junction channel and its implications for its biological
functions. Cell Mol Life Sci. 68:1115–1129. 2011. View Article : Google Scholar
|
|
16
|
Haefliger JA, Nicod P and Meda P:
Contribution of connexins to the function of the vascular wall.
Cardiovasc Res. 62:345–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Van Rijen H, van Kempen MJ, Analbers LJ,
Rook MB, van Ginneken AC, Gros D and Jongsma HJ: Gap junctions in
human umbilical cord endothelial cells contain multiple connexins.
Am J Physiol. 272:C117–C130. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang HH, Kung CI, Tseng YY, Lin YC, Chen
CH, Tsai CH and Yeh HI: Activation of endothelial cells to
pathological status by down-regulation of connexin43. Cardiovasc
Res. 79:509–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dhein S, Gaertner C, Georgieff C, Salameh
A, Schlegel F and Mohr FW: Effects of isoprenaline on endothelial
connexins and angiogenesis in a human endothelial cell culture
system. Naunyn Schmiedebergs Arch Pharmacol. 388:101–108. 2015.
View Article : Google Scholar
|
|
20
|
Wang HH, Su CH, Wu YJ, Li JY, Tseng YM,
Lin YC, Hsieh CL, Tsai CH and Yeh HI: Reduction of connexin43 in
human endothelial progenitor cells impairs the angiogenic
potential. Angiogenesis. 16:553–560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu X, Francis R, Wei CJ, Linask KL and Lo
CW: Connexin 43-mediated modulation of polarized cell movement and
the directional migration of cardiac neural crest cells.
Development. 133:3629–3639. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kardami E, Stoski RM, Doble BW, Yamamoto
T, Hertzberg EL and Nagy JI: Biochemical and ultrastructural
evidence for the association of basic fibroblast growth factor with
cardiac gap junctions. J Biol Chem. 266:19551–19557.
1991.PubMed/NCBI
|
|
23
|
Gartner C, Ziegelhöffer B, Kostelka M,
Stepan H, Mohr FW and Dhein S: Knock-down of endothelial connexins
impairs angio-genesis. Pharmacol Res. 65:347–357. 2012. View Article : Google Scholar
|
|
24
|
Polontchouk L, Ebelt B, Jackels M and
Dhein S: Chronic effects of endothelin 1 and angiotensin II on gap
junctions and intercellular communication in cardiac cells. FASEB
J. 16:87–89. 2002. View Article : Google Scholar
|
|
25
|
Schulz R, Görge PM, Gorbe A, Ferdinandy P,
Lampe PD and Leybaert L: Connexin 43 is an emerging therapeutic
target in ischemia/reperfusion injury, cardioprotection and
neuropro-tection. Pharmacol Ther. 153:90–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bian B, Yu X, Wang Q, Teng T and Nie J:
Atorvastatin protects myocardium against ischemia-reperfusion
arrhythmia by increasing Connexin 43 expression: A rat model. Eur J
Pharmacol. 768:13–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Strale PO, Clarhaut J, Lamiche C, Cronier
L, Mesnil M and Defamie N: Down-regulation of Connexin43 expression
reveals the involvement of caveolin-1 containing lipid rafts in
human U251 glioblastoma cell invasion. Mol Carcinog. 51:845–860.
2012. View
Article : Google Scholar
|
|
28
|
Wang WK, Chen MC, Leong HF, Kuo YL, Kuo CY
and Lee CH: Connexin 43 suppresses tumor angiogenesis by
down-regulation of vascular endothelial growth factor via
hypoxic-induced factor-1α. Int J Mol Sci. 16:439–451. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwak BR, Mulhaupt F, Veillard N, Gros DB
and Mach F: Altered pattern of vascular connexin expression in
atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 22:225–230.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chadjichristos CE, Derouette JP and Kwak
BR: Connexins in atherosclerosis. Adv Cardiol. 42:255–267. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li H, He J, Yu H, Green CR and Chang J:
Bioglass promotes wound healing by affecting gap junction connexin
43 mediated endothelial cell behavior. Biomaterials. 84:64–75.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tarzemany R, Jiang G, Jiang JX,
Gallant-Behm C, Wiebe C, Hart DA, Larjava H and Häkkinen L:
Connexin 43 regulates the expression of wound healing-related genes
in human gingival and skin fibroblasts. Exp Cell Res. 367:150–161.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang K, Fu W, Beardsley AR, Sun X, Lisanti
MP and Liu J: Overexpression of caveolin-1 inhibits endothelial
cell proliferation by arresting the cell cycle at
G0/G1 phase. Cell Cycle. 6:199–204. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin MI, Yu J, Murata T and Sessa WC:
Caveolin-1-deficient mice have increased tumor microvascular
permeability, angiogenesis, and growth. Cancer Res. 67:2849–2856.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sotkis A, Wang XG, Yasumura T, Peracchia
LL, Persechini A, Rash JE and Peracchia C: Calmodulin colocalizes
with connexins and plays a direct role in gap junction channel
gating. Cell Commun Adhes. 8:277–281. 2001. View Article : Google Scholar
|
|
36
|
Giepmans BN, Verlaan I, Hengeveld T,
Janssen H, Calafat J, Falk MM and Moolenaar WH: Gap junction
protein connexin-43 interacts directly with microtubules. Curr
Biol. 11:1364–1368. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schubert AL, Schubert W, Spray DC and
Lisanti MP: Connexin family members target to lipid raft domains
and interact with caveolin-1. Biochemistry. 41:5754–5764. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Langlois S, Cowan KN, Shao Q, Cowan BJ and
Laird DW: Caveolin-1 and -2 interact with connexin43 and regulate
gap junctional intercellular communication in keratinocytes. Mol
Biol Cell. 19:912–928. 2008. View Article : Google Scholar :
|
|
39
|
Razani B, Schlegel A, Liu J and Lisanti
MP: Caveolin-1, a putative tumour suppressor gene. Biochem Soc
Trans. 29:494–499. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Galbiati F, Volonté D, Liu J, Capozza F,
Frank PG, Zhu L, Pestell RG and Lisanti MP: Caveolin-1 expression
negatively regulates cell cycle progression by inducing G(0)/G(1)
arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell.
12:2229–2244. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hartel FV, Holl M, Arshad M, Aslam M,
Gündüz D, Weyand M, Micoogullari M, Abdallah Y, Piper HM and Noll
T: Transient hypoxia induces ERK-dependent anti-apoptotic cell
survival in endothelial cells. Am J Physiol Cell Physiol.
298:C1501–C1509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
World Medical Association Declaration of
Helsinki: Recommendations guiding physicians in biomedical research
involving human subjects. Cardiovasc Res. 35:2–3. 1997.
|
|
43
|
Zhao Y, Yu L, Xu S, Qiu F, Fan Y and Fu G:
Down-regulation of connexin43 gap junction by serum deprivation in
human endo-thelial cells was improved by (-)-Epigallocatechin
gallate via ERK MAP kinase pathway. Biochem Biophys Res Commun.
404:217–222. 2011. View Article : Google Scholar
|
|
44
|
Li B, Jia S, Yue T, Yang L, Huang C,
Verkhratsky A and Peng L: Biphasic regulation of Caveolin-1 gene
expression by fluoxetine in astrocytes: Opposite effects of
PI3K/AKT and MAPK/ERK signaling pathways on c-fos. Front Cell
Neurosci. 11:3352017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Braet K, Vandamme W, Martin PE, Evans WH
and Leybaert L: Photoliberating inositol-1,4,5-trisphosphate
triggers ATP release that is blocked by the connexin mimetic
peptide gap 26. Cell Calcium. 33:37–48. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leybaert L, Braet K, Vandamme W, Cabooter
L, Martin PE and Evans WH: Connexin channels, connexin mimetic
peptides and ATP release. Cell Commun Adhes. 10:251–257. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nandigama R, Padmasekar M, Wartenberg M
and Sauer H: Feed forward cycle of hypotonic stress-induced ATP
release, puri-nergic receptor activation, and growth stimulation of
prostate cancer cells. J Biol Chem. 281:5686–5693. 2006. View Article : Google Scholar
|
|
48
|
Galbiati F, Volonte D, Engelman JA,
Watanabe G, Burk R, Pestell RG and Lisanti MP: Targeted
downregulation of caveolin-1 is sufficient to drive cell
transformation and hyperac-tivate the p42/44 MAP kinase cascade.
EMBO J. 17:6633–6648. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Park H, Go YM, St John PL, Maland MC,
Lisanti MP, Abrahamson DR and Jo H: Plasma membrane cholesterol is
a key molecule in shear stress-dependent activation of
extracellular signal-regulated kinase. J Biol Chem.
273:32304–32311. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Engelman JA, Chu C, Lin A, Jo H, Ikezu T,
Okamoto T, Kohtz DS and Lisanti MP: Caveolin-mediated regulation of
signaling along the p42/44 MAP kinase cascade in vivo. A role for
the caveolin-scaffolding domain. FEBS Lett. 428:205–211. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar
|














