Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide, accounting for 158,040
mortalities in 2015, and non-small cell lung cancer (NSCLC) is a
common histological subtype of lung carcinoma (1-3).
Epidermal growth factor receptor (EGFR) mutations commonly occur in
East Asian patients with NSCLC. Approximately 70% of NSCLC patients
with EGFR mutations exhibit a favorable response to tyrosine kinase
inhibitors (TKIs), such as gefitinib, when compared with that in
patients with wild-type EGFR (4-7).
However, drug resistance invariably emerges, and the disease
eventually progresses, which is referred to as acquired resistance.
The mechanisms underlying acquired resistance to EGFR-TKIs include
the T790M gatekeeper mutation, amplification of
mesenchymal-epithelial transition and human EGFR 2 (HER2)
amplification (8). However, ~30%
of patients with EGFR mutations do not exhibit sufficient responses
to EGFR-TKIs, which is known as primary resistance (9). Although possible mechanisms have
been investigated in several studies, the molecular background of
primary resistance remains unknown.
The Bim gene (also known as BCL2L11), which encodes
a BH3-only protein, is a pro-apoptotic member of the B-cell
lymphoma 2 (Bcl-2) family (10).
Overexpression of Bim triggers apoptosis by inducing cytochrome
c release (11-13). It has previously been demonstrated
that Bim is essential for EGFR-TKI-induced killing of NSCLC cells
(14). A recent study further
demonstrated that a common Bim deletion polymorphism contributes to
poor responses among NSCLC patients with EGFR-mutations treated
with EGFR-TKIs (15). Patients
with this polymorphism were shown to have a reduced median
progression-free survival (mPFS; 6.6 vs. 11.9 months, respectively;
P=0.003) (16). However, the
association between the Bim polymorphism and the response to
EGFR-TKIs has not been completely elucidated. By contrast, certain
studies have suggested that the presence of this polymorphism is
not associated with the efficacy of EGFR-TKIs (17). Faber et al (18) demonstrated that low Bim mRNA
expression in cancer specimens accurately predicts the apoptotic
response to targeted therapies, and confirmed that Bim levels can
serve as a correlative marker of EGFR-TKI treatment efficacy.
Human antigen R (HuR) is a widely studied
RNA-binding protein that is involved in the regulation of major
pathways required for proliferation, apoptosis, differentiation and
therapeutic resistance (19). In
the cellular context, HuR binds to the 3′-untranslated regions of
mRNAs, regulating their stability and translation (20). It has been increasingly reported
that HuR is closely associated with the development of numerous
types of malignant tumors, including breast (21), colon (22), ovarian (23) and pancreatic cancer (24,25). Furthermore, HuR has been
demonstrated to participate in the promotion of chemoresistance by
increasing Bcl-2 mRNA stability (19), and HuR displays a good potential
for diagnosis and prognosis in cancer.
Therefore, the aim of the present study was to
determine whether HuR expression affects the clinical outcomes of
NSCLC patients treated with EGFR-TKIs through the modulation of Bim
expression.
Materials and methods
Cell lines
H1650, PC-9 and HCC827 lung cancer cell lines were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). These cell lines were cultured in
RPMI containing 10% fetal bovine serum. All cells were cultured at
37°C in an incubator with 5% CO2.
Tissue samples
Paraffin-embedded tissue specimens were collected at
the General Hospital, Jinan Command of the People's Liberation Army
(Jinan, China) between October 2008 and May 2016 (Table I). All the patients involved were
diagnosed with adenocarcinoma via biopsy or surgery. Tumor
histology and subtypes were classified according to the World
Health Organization criteria (26). Eastern Cooperative Oncology Group
performance status (ECOG-PS) was used to assess the functional
status of patients (27). In
addition, the majority of the selected patients were treated with
first-line EGFR-TKI treatment. Primary drug resistance was defined
as progression within 3 months after the use of EGFR-TKIs, and the
sensitive group had a PFS duration of >6 months. Informed
consent was obtained from all patients, and the study was approved
by the Ethics Committee of the General Hospital (Jinan Command of
the People's Liberation Army, Jinan, China).
 | Table IPatient characteristics and
comparison between patients in the EGFR tyrosine kinase inhibitor
resistant and sensitive groups. |
Table I
Patient characteristics and
comparison between patients in the EGFR tyrosine kinase inhibitor
resistant and sensitive groups.
| Resistant group
(n=27) | Sensitive group
(n=54) | P-value |
|---|
| Sex, n | | | 0.01 |
| Male | 22 | 23 | |
| Female | 5 | 31 | |
| Age (years), n | | | 0.343 |
| ≥60 | 13 | 32 | |
| <60 | 14 | 22 | |
| ECOG PS, n | | | 0.717 |
| Score 0-2 | 26 | 51 | |
| Score 3 | 1 | 3 | |
| EGFR mutation,
n | | | 0.501 |
| Exon 19
deletion | 17 | 38 | |
| L858R
mutation | 10 | 16 | |
| Tumor stage, n | | | 0.069 |
| IIIa | 1 | 6 | |
| IIIb | 1 | 7 | |
| IV | 25 | 41 | |
| Surgical history,
n | | | 0.392 |
| Yes | 3 | 10 | |
| No | 24 | 44 | |
| Chemoradiotherapy
history, n | | | 0.270 |
| Yes | 17 | 27 | |
| No | 10 | 27 | |
| Brain metastases,
n | | | 0.089 |
| Yes | 3 | 15 | |
| No | 24 | 39 | |
Chemicals and antibodies
Gefitinib was purchased from LC Laboratories (cat.
no. G-4408; New Boston, MA, USA), dissolved in dimethyl sulfoxide
to a final concentration of 10 mM and stored at −20°C. Anti-Bim
antibodies used for western blotting were purchased from Cell
Signaling Technology, Inc. (cat. no. 2819; Danvers, MA, USA).
Anti-HuR and anti-β-actin antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). All other chemicals were
acquired from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
RNA interference
Small interfering RNA (siRNA) against HuR was used
for RNA interference. The siRNA target sequence for HuR was:
5′-UUUGUCAUGGUCACAAAGCTT-3′. Briefly, cells were plated at a
density of 5×105 cells/well in 12-well plates. Using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), cells were transfected with 80 nmol/l siRNA
duplex mixture (GeneChem Co., Ltd., Shanghai, China) for 48 h.
Construction, transduction and expression
of HuR lentiviral vectors
The HuR gene sequence was amplified by polymerase
chain reaction (PCR) using a PrimeScript™ One Step RT-PCR Kit
(Takara Bio, Inc., Otsu, Japan) and then subcloned into a
lentiviral expression vector (GV365; GeneChem Co., Ltd.). The PCR
primers used for HuR amplification were as follows: HuR forward,
5′-GAGGATCCCCGGGTACCGGTCGCCACCATGTCTAATGGTTATGAAGAC-3′ and reverse,
5′-TCCTTGTAGTCCATACCTTTGTGGGACTTGTTGGTTTTG-3′. The thermocycling
conditions used for PCR were as follows: Initial denaturation for 5
min at 98°C; followed by 30 cycles of denaturation for 10 sec at
98°C, annealing for 10 sec at 55°C and extension for 90 sec at
72°C; followed by a final extension for 8 min at 72°C. Lentiviral
packaging plasmids (pHelper 1.0; GeneChem Co., Ltd.) were used for
the packaging and production of lentiviral particles. GV365 has a
green fluorescent protein (GFP) marker for positive selection.
Briefly, H1650 cells were transduced with lentiviral particles as
previously described (28), and
H1650 cells that stably expressed high levels of HuR were obtained
and subsequently named GV365-H1650 cells. Subsequently, the
expression of GFP was observed using fluorescence microscopy, while
the expression of HuR was detected using reverse
transcription-quantitative PCR (RT-qPCR) and western blotting.
Cell viability assay
As aforementioned, HCC827 cells were transfected
with siRNA targeting HuR for 48 h, and the resultant cells were
subsequently names named siHCC827 cells. H1650, GV365-H1650,
siHCC827 and HCC827 cells were cultured in 96-well flat-bottomed
microliter plates and were allowed to adhere at 37°C overnight in
5% CO2 (~1×104 cells/well). After cellular
adhesion, gefitinib was added at a final concentration of 0-40
µM. After 48 h of incubation, the cytotoxic effects were
assessed using the Cell Counting Kit-8 (CCK-8; Beyotime Institute
of Biotechnology, Shanghai, China), according to the manufacturer's
protocol. Each sample was plated in triplicate.
Apoptosis assays and Annexin V
staining
Apoptosis was examined by performing Annexin V and
dye exclusion assays. Briefly, cells were simultaneously stained
with Annexin V-allophycocyanin (APC; blue staining) and propidium
iodide (PI; red staining). This assay was conducted to
differentiate between intact (APC−/PI−),
early apoptotic (APC+/PI−) and late apoptotic
(APC+/PI+) cells. Comparative experiments
were performed by bivariate flow cytometry using a FACScan (BD
Biosciences, San Jose, CA, USA). Cell Quest software (version 3.3;
BD Biosciences, Franklin Lakes, NJ, USA) was used to analyze the
obtained data of the cell populations, for which debris was gated
out.
RNA isolation and RT-qPCR analyses
Total RNA was isolated from H1650, GV365-H1650,
siHCC827 and HCC827 cells using the TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) in accordance
with the manufacturer's instructions. Isolated RNA was
reverse-transcribed into cDNA using PrimeScript™ RT reagent kit
with gDNA Erase (Takara Bio, Inc.). To investigate the expression
of target genes, qPCR was performed in triplicate using an ABI Step
One Plus Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using SYBR® Premix Ex Taq™ (Takara
Bio, Inc.) in a 25 µl reaction with 900 nM primers. qPCR
thermocycling conditions were as follows: Initial denaturation for
30 sec at 95°C; followed by 40 cycles at 95°C for 5 sec and 60°C
for 30 sec. The PCR primers for HuR, Bim or β-actin were as
follows: HuR forward, 5′-ACATGACCCAGGATGAGTTACGAAG-3′, and reverse,
5′-TCGCGGTCACGTAGTTCACAA-3′; Bim forward,
5′-TGATTCTTGCAGCCACCCTG-3′, and reverse,
5′-GGGGAACAAGGGCCAAGAAA-3′; β-actin forward,
5′-TGGCACCCAGCACAATGAA-3′, and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′. The relative quantification of HuR
and Bim expression was calculated using the 2−ΔΔCq
method (29), and normalized to
β-actin expression.
Western blotting
Cells were lysed with lysis buffer (Beyotime
Institute of Biotechnology), and the precipitated cell debris was
discarded by centrifugation at 4°C for 15 min (1,200 x g). The
protein concentration was then determined using a Bradford protein
assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein
samples (30-50 µg/lane) were separated on a 2.5% SDS-PAGE
gel and then transferred to polyvinylidene difluoride membranes.
Following this, membranes were blocked with 5% (w/v) non-fat dry
milk in 1X Tris buffered saline with 0.1% Tween 20 for 3 h at 37°C
and then incubated overnight at 4°C with primary antibodies against
the following proteins: HuR (cat. no. sc-5261; 1:1,000) and GAPDH
(cat. no. sc-20358; 1:1,000) were purchased from Santa Cruz
Biotechnology, Inc.; and antibodies against Bim (cat. no. 2819;
1:2,000) were procured from Cell Signaling Technology, Inc.
Following this, the membranes were washed and incubated with the
following secondary antibodies for 1 h at 25°C: HRP-tagged rabbit
anti-mouse IgG (cat. no. sc-358920; 1:2,000; Santa Cruz
Biotechnology, Inc.), HRP-conjugated donkey anti-goat IgG-HRP (cat.
no. sc-2020; 1:5,000; Santa Cruz Biotechnology, Inc.) and
HRP-conjugated mouse anti-rabbit IgG (cat. no. A01827-200; 1:5,000;
Genscript, Piscataway, NJ, USA). The blots were visualized using an
enhanced chemiluminescence kit (Beyotime Institute of
Biotechnology, Shanghai, China). Images were captured using Scion
image software (version 4.0.3.2; Scion Corporation, Frederick, MD,
USA).
Immunohistochemical (IHC) analysis
Formalin-fixed, paraffin-embedded tissues collected
from patients with NSCLC (n=81) were sectioned at 5 mm. Briefly,
tissue sections were incubated at 60°C for 2 h, deparaffinized,
rehydrated in a descending alcohol series and subsequently blocked
with 3% hydrogen peroxide for 30 min at room temperature. Following
incubation with sodium citrate buffer (0.01 M) at room temperature
for 30 min, the sections were then preincubated in 10% normal goat
serum (Beyotime Institute of Biotechnology) at room temperature for
30 min to prevent non-specific staining. Subsequently, the sections
were incubated overnight at 4°C with antibodies against HuR (cat.
no. sc-5261; 1:1,000; Santa Cruz Biotechnology, Inc.) and Bim (cat.
no. 2819; 1:1,000; Cell Signaling Technology, Inc.). Following
this, sections were then incubated with corresponding horseradish
peroxidase (HRP)-tagged secondary antibodies (cat. no. sc-358920,
1:1,000; and cat. no. sc-2020, 1:2,000; Santa Cruz Biotechnology,
Inc.) for 30 min at room temperature. Two pathologists individually
examined the levels of IHC signaling under a light microscope
(magnification, x200). The algorithm output returns a number of
quantitative measurements, namely the intensity and percentage of
positive staining present (30).
Subsequently, the staining intensity and percentage of positive
staining were categorized into 4 and 5 classes, respectively,
following the determination of cut-off values, according to a
previously published protocol (30). The intensity of staining was
categorized as 0 (no staining), 1+ (weak staining), 2+ (moderate
staining) and 3+ (strong staining). In each sample, five high-power
fields were selected, and the specimens were categorized into five
semi-quantitative classes based on percentage of positive staining
as follows: 0 (<5% stained cells), 1 (6-25% stained cells), 2
(26-50% stained cells), 3 (51-75% stained cells) and 4 (>76%
stained cells). The products of percent positive and intensity
scores yielded final IHC scores of <2 (negative) and >2
(positive).
In vivo animal model experiments
A total of 14 healthy female BALB/c nude mice (aged
4 weeks old, weighing 14.6-21.8 g) were purchased from Beijing
Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China)
and subsequently housed at 21°C and 50-55% humidity with a 14/10 h
light/dark cycle under germ-free (GF) conditions at the Laboratory
Animal Research Center in General Hospital, Jinan Command of the
People's Liberation Army. Mice were permitted to acclimatize for 1
week post-arrival. Mice received a standard laboratory chow and
water ad libitum. For experiments assessing the effect of
drug treatment on tumor growth, 10 female BALB/c nude mice were
selected. The mice were randomized into two groups, including the
transfection and control groups. In the transfection and control
groups, GV365-H1650 cells or non-transfected H1650 cells were
respectively injected into the back of BALB/c nude mice. The
xenograft size was measured every 3 days, and the tumor volume was
determined as follows: (length x width2)/2. A total of 2
weeks post-tumor implantation (all tumors were ≥500 mm3
in volume), gefitinib (5 mg/kg) was administrated once a day via
intragastric administration for a total of 15 days. Animal welfare
and relevant experiments were conducted in compliance with the
Guide for the Care and Use of Laboratory Animals (Animal Scientific
Procedures, SAC/TC281), and were approved by the Ethics Committee
of the General Hospital, Jinan Command of the People's Liberation
Army.
Statistical analysis
The correlation of HuR and Bim staining with the
different clinicopathological features of patients was evaluated
using the χ2 test. Univariate survival analysis was
calculated using the Kaplan-Meier Method. Significance of the data
was analyzed by the log-rank test. A P-value of <0.05 was
considered to indicate a difference that was statistically
significant, and all P-values were two-sided. All statistical
calculations were performed using the SPSS statistical software
(version 18.0; SPSS, Inc., Chicago, IL, USA).
Results
Negative expression of HuR and Bim
correlates with lower EGFR-TKI sensitivity in tumor tissues
obtained from EGFR-mutant NSCLC patients
The associations between HuR and Bim expression
levels and clinicopathological characteristics of patients with
NSCLC were determined using χ2 tests. There was an
evident difference in terms of the sex between the two groups of
patients. Female patients accounted for 18.5% of cases in the
resistant group, whereas they comprised 57.4% of the sensitive
group cases, and this different was statistically significant
(P=0.01; Table I). However, there
were no significant differences between the EGFR-TKI-sensitive
group and EGFR-TKI-resistant group with respect to the age, ECOG-PS
(27), mutation type, tumor
stage, surgery or chemoradiotherapy history, and brain metastases
(P>0.05; Table I) (31).
For HuR staining, 81.5% (22 out of 27)
cytoplasm-negative and 18.5% (5 out of 27) cytoplasm-positive
specimens were identified in the EGFR-TKI-resistant group, whereas
20.4% (11 out of 54) specimens were cytoplasm-negative and 79.6%
(43 out of 54) were cytoplasm-positive in the EGFR-TKI-sensitive
group. These results indicated that IHC staining for HuR was higher
in the EGFR-TKI-sensitive group as compared with that in the
EGFR-TKI-resistant group (P<0.001; Table II). For Bim staining, 70.4% (19
out of 27) cytoplasm-negative and 29.6% (8 out of 27)
cytoplasm-positive specimens were detected in the
EGFR-TKI-resistant group, whereas 7.4% (4 out of 54) cases were
cytoplasm-negative and 92.6% (50 out of 54) were cytoplasm-positive
in the EGFR-TKI-sensitive group. These findings indicated that IHC
staining for Bim was also higher in the EGFR-TKI-sensitive group as
compared with that in the EGFR-TKI-resistant group (P<0.001;
Table II). Therefore, the
altered expression of HuR and Bim may modulate the therapeutic
effect of EGFR-TKIs in NSCLC and serve as an important index for
the treatment of this disease.
| Table IIExpression levels of HuR and Bim in
epidermal growth factor receptor-tyrosine kinase inhibitor
resistant and sensitive groups. |
Table II
Expression levels of HuR and Bim in
epidermal growth factor receptor-tyrosine kinase inhibitor
resistant and sensitive groups.
| Cytoplasmic
expression | Resistant group
(n=27) (%) | Sensitive group
(n=54) (%) | χ2
value | P-value |
|---|
| HuR, n (%) | | | 84 | <0.001 |
| Negative | 22 (81.5) | 11 (20.4) | | |
| Positive | 5 (18.5) | 43 (79.6) | | |
| Bim, n (%) | | | 10 | <0.001 |
| Negative | 19 (70.4) | 4 (7.4) | | |
| Positive | 8 (29.6) | 50 (92.6) | | |
Association among HuR expression, Bim
expression and clinicopathological variables
The correlation of Bim expression with HuR
expression in NSCLC patients was analyzed by performing
χ2 tests. In patients with a positive expression of HuR,
the positive rate of Bim was significantly higher (95.8 vs. 4.2%,
P<0.01). In addition, the Bim negative expression rate was
significantly higher in patients with negative HuR expression (63.6
vs. 36.4%, P<0.05), proving the existence of correlation between
HuR and Bim expression (Table
III). Notably, among the 27 cases with interpretable HuR and
Bim staining, negative Bim expression was associated with negative
cytoplasmic HuR expression (P=0.01; Table IV). However, Bim status was not
correlated with the sex, age or mutation type (P>0.05; Table IV).
| Table IIICorrelation between the expression
levels of HuR and Bim in all patients. |
Table III
Correlation between the expression
levels of HuR and Bim in all patients.
| HuR expression | Bim expression
| Total | Positive rate
(%) | P-value |
|---|
| Positive (%) | Negative (%) |
|---|
| Positive (%) | 46 (95.8) | 2 (4.2) | 48 | 95.8 | P<0.01 |
| | | | | P<0.05 |
| Negative (%) | 12 (36.4) | 21 (63.6) | 33 | 36.4 | |
| Total | 58 | 23 | 81 | 71.6 | |
| Table IVCorrelation of cytosolic Bim staining
with the clinicopathological features and cytoplasmic HuR
expression in the EGFR-tyrosine kinase inhibitor resistant
group. |
Table IV
Correlation of cytosolic Bim staining
with the clinicopathological features and cytoplasmic HuR
expression in the EGFR-tyrosine kinase inhibitor resistant
group.
| Clinicopathological
features | Total (n=27) | Negative Bim
taining | χ2
value | P-value |
|---|
| Sex, n | | | 1.877 | 0.221 |
| Male | 22 | 19 | | |
| Female | 5 | 3 | | |
| Age (years), n | | | 0.345 | 0.648 |
| ≥60 | 13 | 10 | | |
| <60 | 14 | 12 | | |
| EGFR mutation,
n | | | 3.610 | 0.124 |
| Exon 19
deletion | 17 | 12 | | |
| L858R
mutation | 10 | 10 | | |
| HuR staining | | | 372 | 0.010 |
| Positive | 5 | 1 | | |
| Negative | 22 | 21 | | |
Negative HuR and Bim expression is
associated with reduced PFS in patients in the sensitive group
undergoing EGFR-TKI treatment
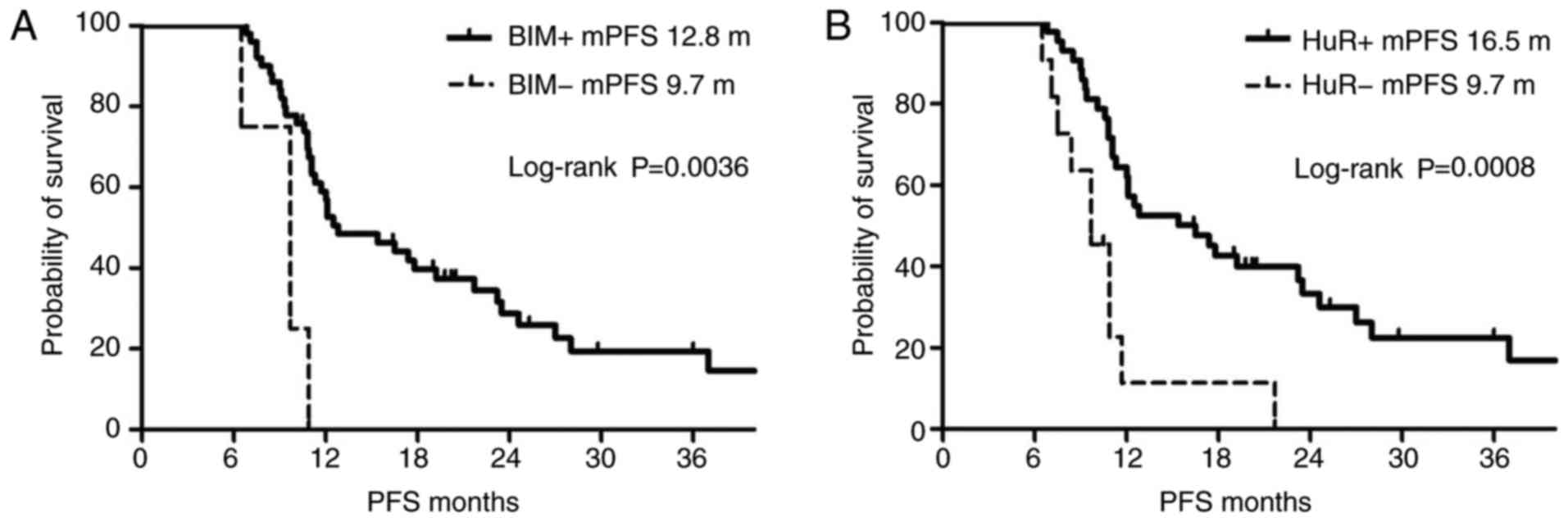
In the EGFR-TKI-sensitive group, the mPFS was 12.8
months for patients with positive Bim expression and 9.7 months for
patients with negative Bim expression. The mPFS of patients with
positive Bim expression was significantly higher in comparison with
that of patients with negative Bim expression, based on
Kaplan-Meier analysis (P=0.0036; Fig.
1). By contrast, the mPFS was 16.5 months for patients with
positive HuR expression and 9.7 months for patients with negative
HuR expression in the EGFR-TKI-sensitive group; thus, patients with
positive HuR expression exhibited a significantly longer mPFS based
on Kaplan-Meier analysis (P=0.0008; Fig. 1).
Gefitinib-resistant cells display altered
expression of HuR and Bim
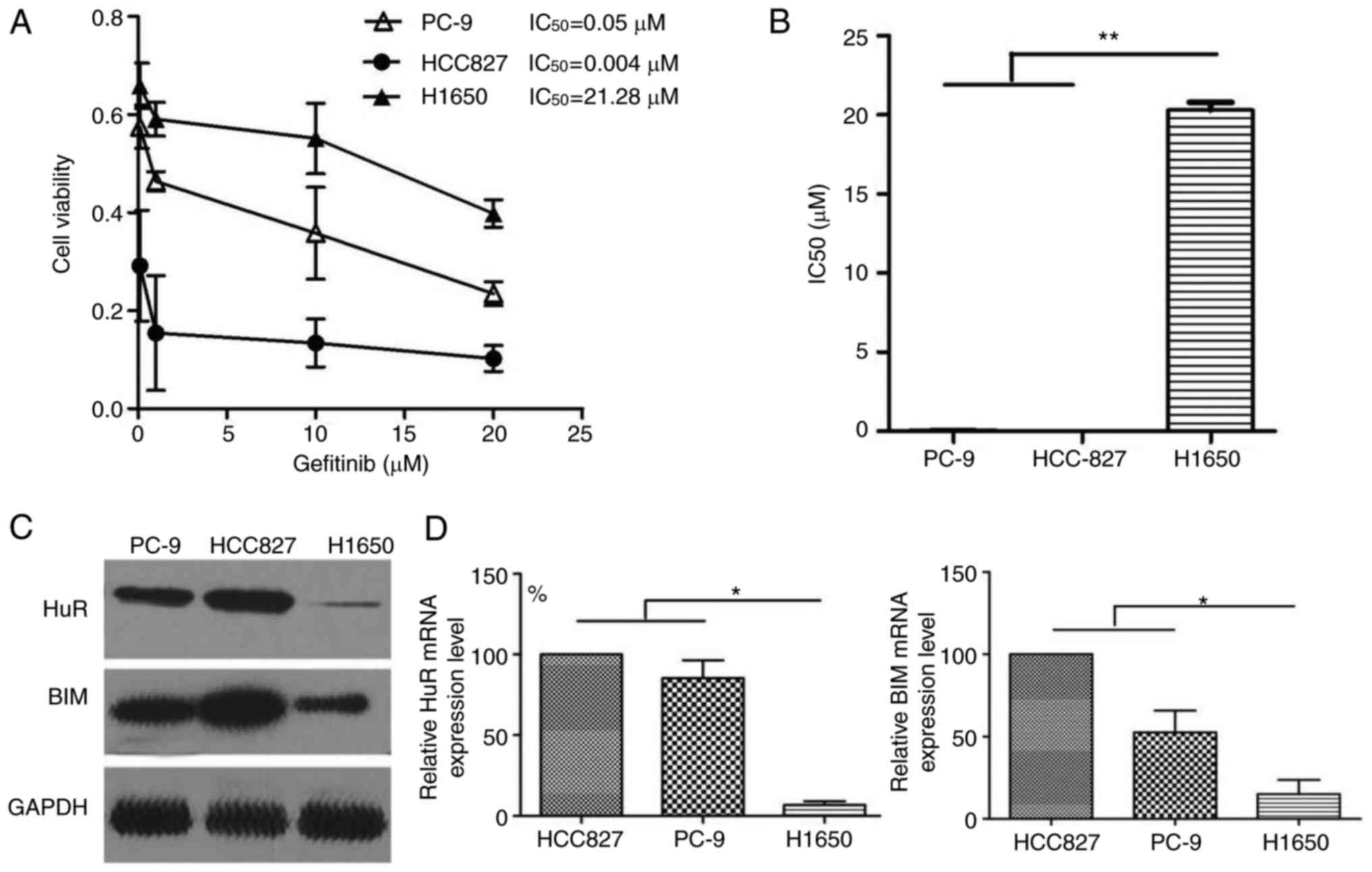
To determine whether HuR and Bim expression were
implicated in gefitinib resistance, three EGFR-mutant lung cancer
cell lines, namely PC9, HCC827 and H1650, were used in the present
study. CCK-8 assays were performed to determine the half maximal
inhibitor concentration (IC50) for gefitinib. The
IC50 value for gefitinib in the H1650 cells was 21.28
µM. By contrast, HCC827 and PC-9 cells exhibited
significantly lower IC50 values for gefitinib (0.05 and
0.004 µM, respectively; Fig.
2A and B). Western blot analysis revealed an evident
downregulation of HuR and Bim levels in gefitinib-resistant H1650
cells (Fig. 2C). The
determination of mRNA levels for these genes led to similar
conclusions. Indeed, H1650 cells exhibited reduced HuR levels by
10-fold and reduced Bim levels by 5-fold, as compared with those in
parental HCC827 cells (Fig. 2D).
According to the differential expression levels of HuR, in
subsequent experiments, expression levels of HuR were downregulated
in HCC827 cells via siRNA interference. Meanwhile, expression
levels of HuR were upregulated in H1650 cells transfected with HuR
lentiviral vectors.
Knockdown of HuR confers primary EGFR-TKI
resistance and a reduction in gefitinib-induced apoptosis in HCC827
cells by decreasing Bim expression
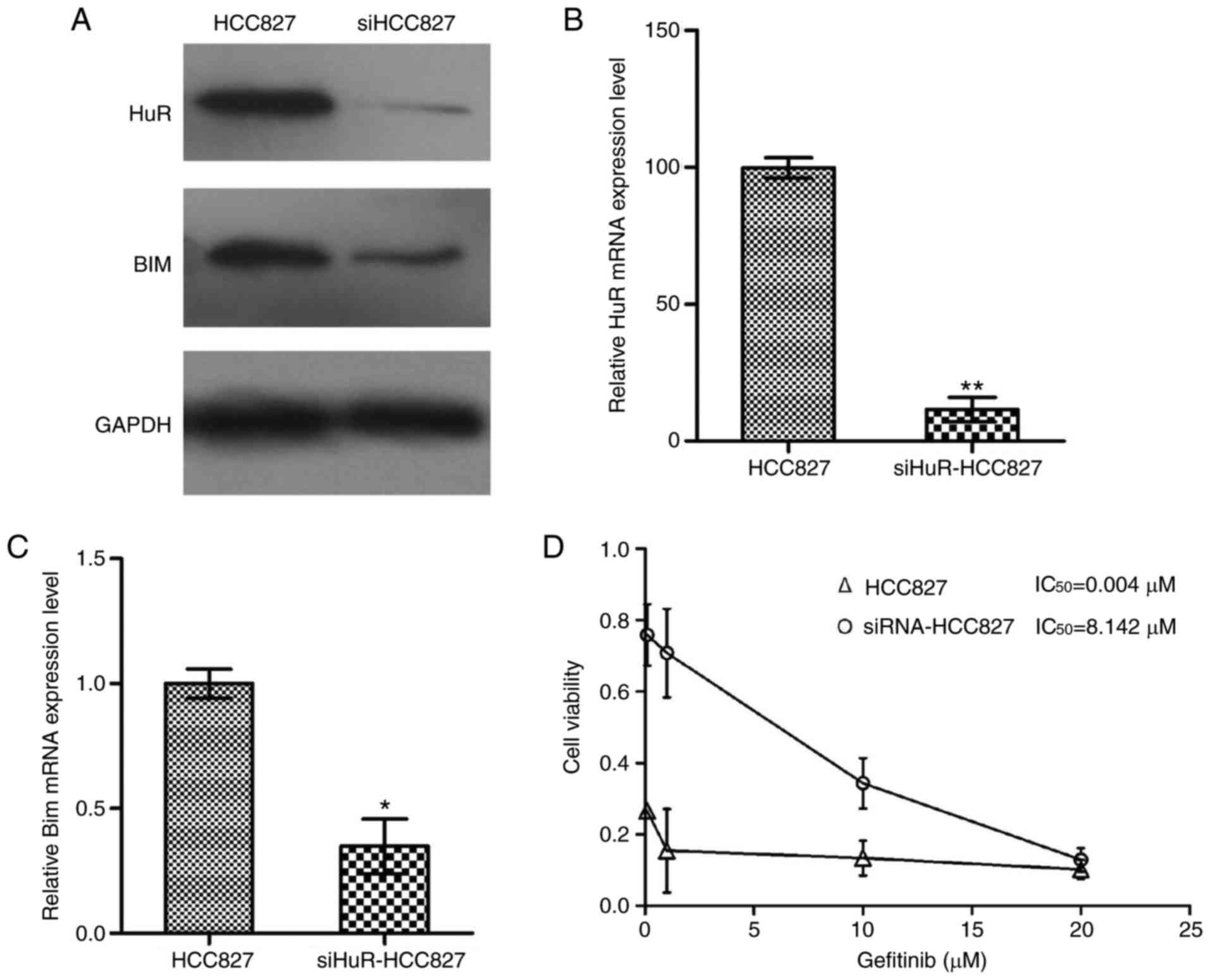
The results of the current study revealed that HuR
and Bim were upregulated in HCC827 cells compared with their
expression levels in H1650 cells. Since HCC827 cells had the lowest
IC50 value for gefitinib, it is hypothesized that
altered expression of HuR/Bim serves an important role in
modulating gefitinib sensitivity in EGFR-mutant lung cancer cells.
The study next examined HCC827 cells to determine whether lower
expression of HuR promotes gefitinib resistance. In HCC827 cells,
HuR expression was down-regulated via siRNA interference. As
expected, RT-qPCR and western blotting data indicated that HuR
expression decreased significantly following HuR knockdown as
compared with the levels in control cells (Fig. 3A-C). Furthermore, a CCK-8 assay
kit was used to detect gefitinib sensitivity at 48 h after
transfection, revealing that the cell viability increased in
HuR-knockdown HCC827 cells compared with that in control HCC827
cells. The results revealed that the cell viability of siHuR-HCC827
cells was markedly increased compared with control HCC827 cells
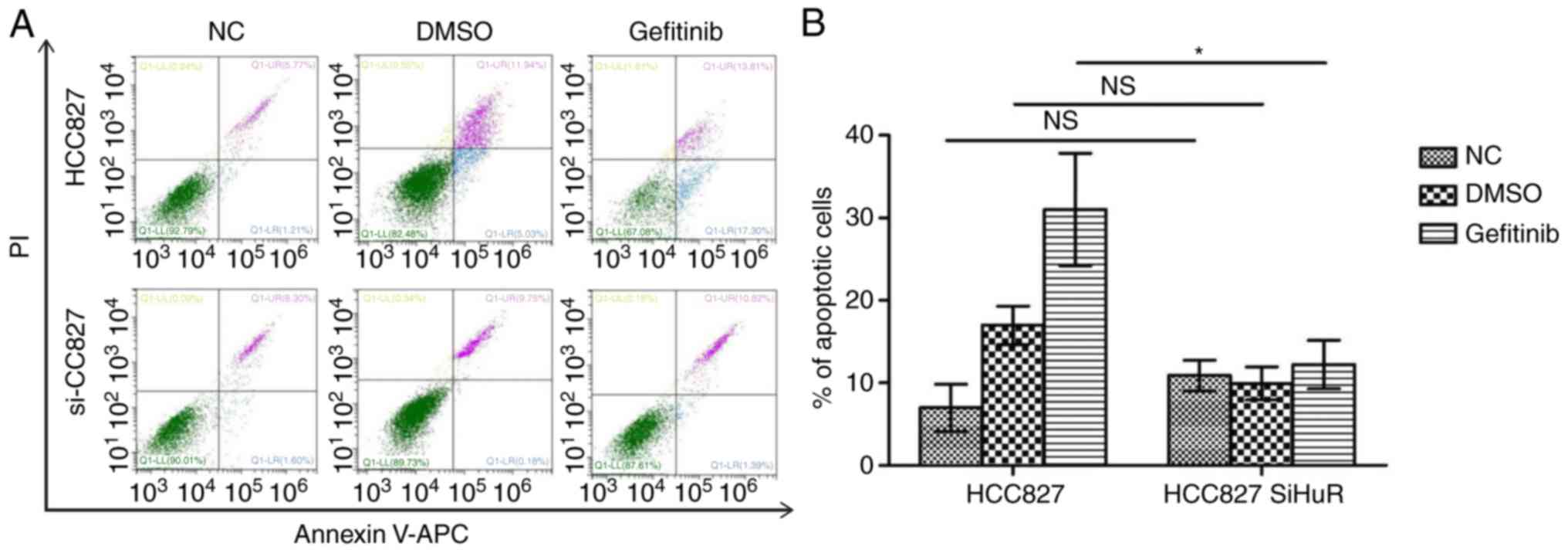
(Fig. 3D). Annexin V/PI staining
also demonstrated a marked decrease in gefitinib-induced apoptosis
in the HuR-knockdown HCC827 cells compared with that in control
cells (Fig. 4).
The effect of HuR knockdown in HCC827 cells on Bim
expression was also examined. Western blotting and RT-qPCR were
used to detect the expression of Bim protein and mRNA in HCC827
cells, respectively. The expression levels of Bim protein and mRNA
were significantly decreased in the HuR-knockdown group in
comparison with that in the control group (P<0.01; Fig. 3).
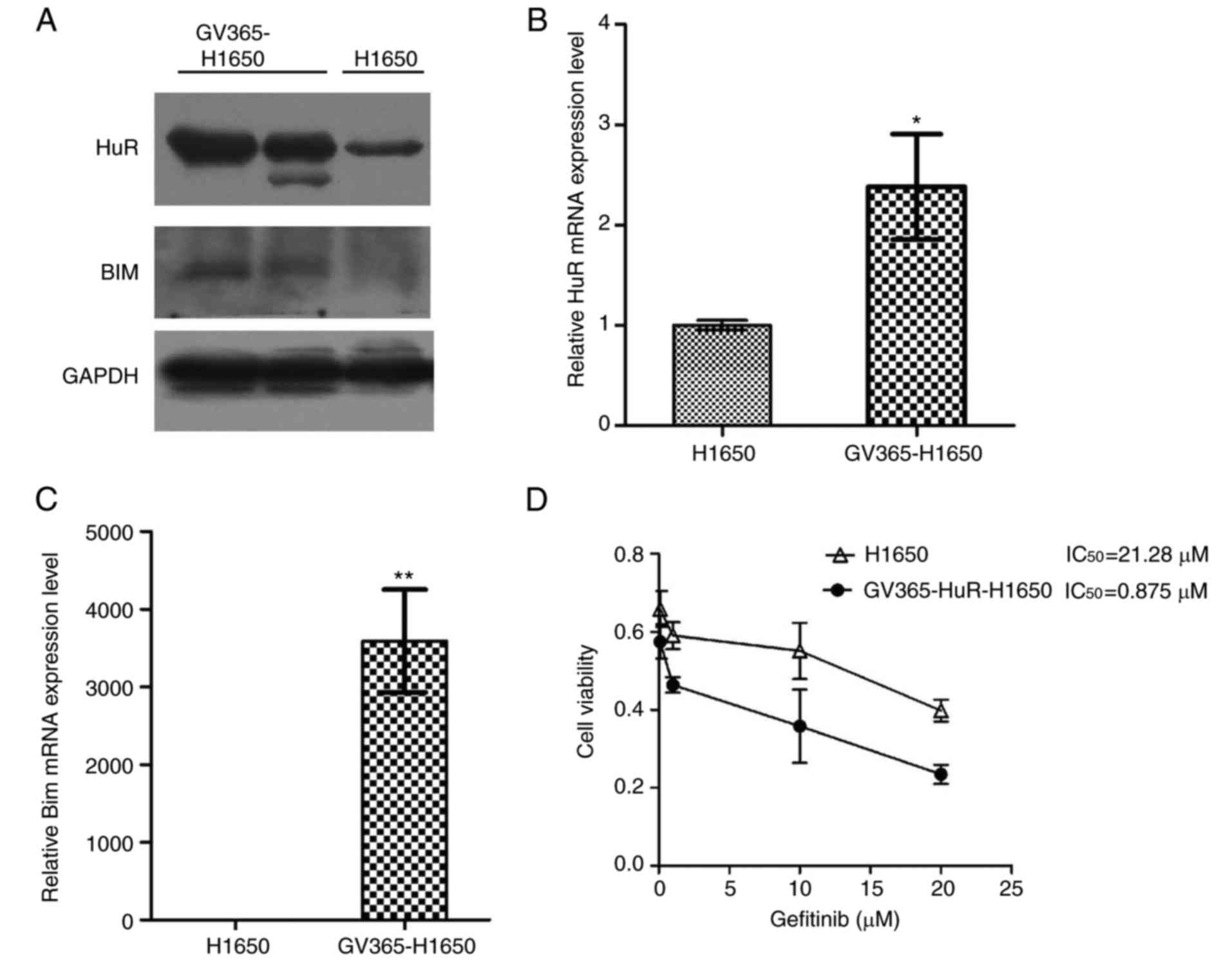
Elevated expression of HuR restores
gefitinib sensitivity and enhances gefitinib-induced apoptosis in
H1650 cells by increasing Bim expression
As mentioned earlier, the expression of HuR was
lower in H1650 cells. Next, these cells were used to determine
whether a higher expression of HuR was able to restore gefitinib
sensitivity by manipulating Bim expression using a HuR lentiviral
expression vector. HuR and Bim expression levels in infected and
control cells were then examined using western blotting and
RT-qPCR. Western blotting indicated that the protein expression of
HuR and Bim was increased with lentiviral infection (Fig. 5A). HuR and Bim mRNA levels were
also significantly higher in the H1650 infected group as compared
with those in the control group (P<0.01; Fig. 5B and C). The results revealed that
the cell viability of GV365-H1650 cells was markedly decreased
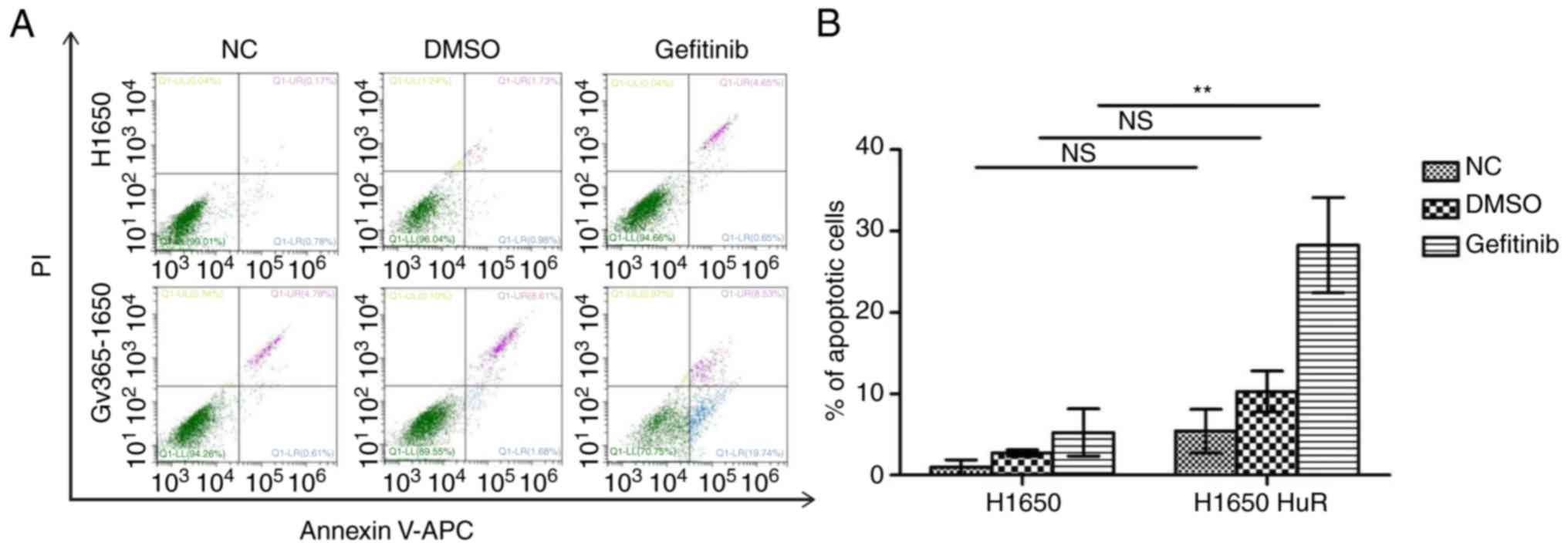
compared with the control H1650 cells (Fig. 5D). Furthermore, Annexin V/PI
staining indicated that HuR overexpression increased the apoptosis
rates when compared with those in control cells (Fig. 6).
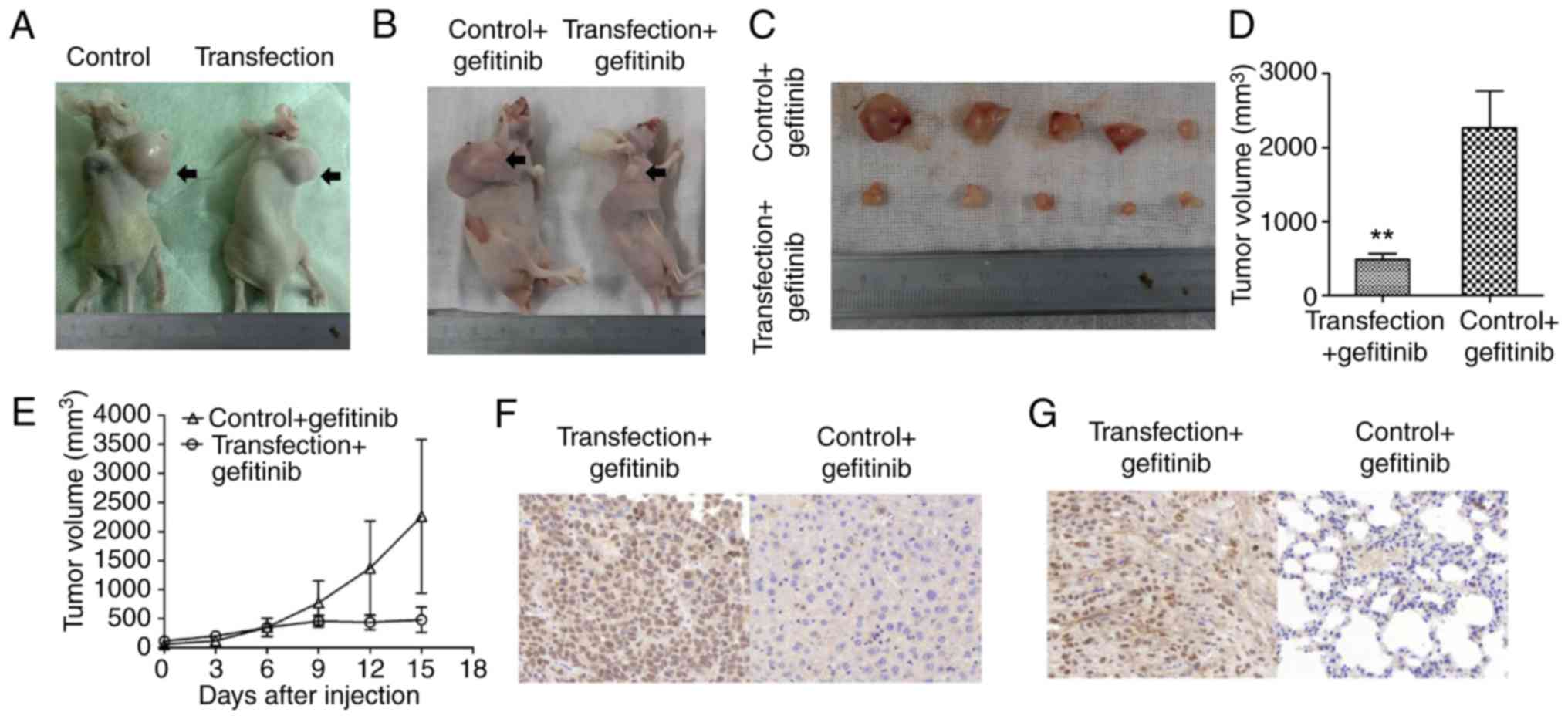
H1650 xenograft tumor burden is nearly
suppressed by gefitinib treatment with HuR overexpression
Prior to the administration of gefitinib treatment,
xenograft tumors originating from the HuR-overexpressing H1650
stable clone and control clone exhibited continued and rapid
growth. As shown in Fig. 7A, the
size of tumors in the two groups 15 days post-treatment grew
rapidly, with no significant difference between the two groups
without gefitinib. By contrast, when gefitinib treatment was
administered for 15 days in mice with xenograft tumors, the tumor
burden induced by HuR-overexpressing H1650 cells was evidently
suppressed by gefitinib in comparison with that in the control
group (Fig. 7B). In addition,
suppression of the tumor burden in the transfection + gefitinib
group continued for 15 days. However, the tumor burden induced by
the control + gefitinib clone was not evidently changed following
gefitinib treatment. Tumor volumes in the transfection + gefitinib
group were significantly smaller in comparison with those in the
control + gefitinib group (Fig. 7C
and D). The growth trend of xenograft tumors in each group
exhibited an upward trend; however, the growth increase in the
transfection + gefitinib group was significantly slower compared
with that of the control + gefitinib group (Fig. 7E; Table V). IHC analysis further revealed
that HuR expression was significantly increased and that Bim
expression was simultaneously elevated in transfection + gefitinib
group following gefitinib treatment (Fig. 7F and G).
| Table VVolume of xenograft tumors in the two
groups subsequent to gefitinib treatment, examined at 3-day
intervals (mean ± standard error of the mean). |
Table V
Volume of xenograft tumors in the two
groups subsequent to gefitinib treatment, examined at 3-day
intervals (mean ± standard error of the mean).
| Day | Volume
(mm3)
| P-value |
|---|
| Control +
gefitinib | Transfection +
gefitinib |
|---|
| 0 | 00±26.49 | 112.57±72.35 | 0.093 |
| 3 | 118.64±47.90 | 200.29±62.10 | 0.017 |
| 6 | 349.79±153.93 | 347.36±68.54 | 0.970 |
| 9 | 771.71±375.42 | 455.07±98.86 | 0.041 |
| 12 |
1,369.43±811.87 | 437.43±126.81 | 0.012 |
| 15 |
2,260.28±1,322.97 | 479.14±216.38 | 0.004 |
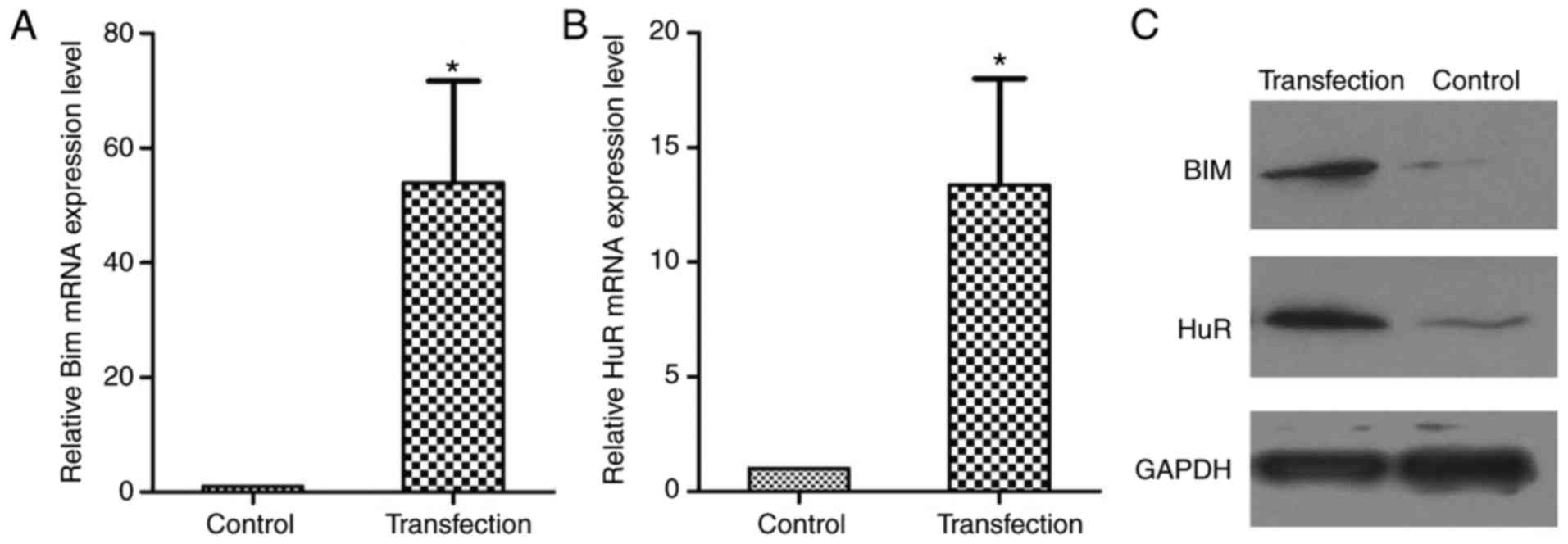
HuR and Bim expression levels were next measured by
western blotting and RT-qPCR in the transplanted tumors. The levels
of HuR and Bim were significantly higher in the transduction +
gefitinib group as compared with the control + gefitinib group
following gefitinib treatment (Fig.
8). These results strongly support the mechanism implied by the
cell model, and suggest that HuR-mediated Bim overexpression
confers EGFR-TKI resistance.
Discussion
The present study demonstrated that different HuR
expression levels may explain the contradictory findings regarding
the EGFR-TKI clinical outcomes; specifically, low-HuR expression
was associated with an unfavorable response to EGFR-TKIs.
Mechanistically, HuR affected the clinical outcomes of EGFR-TKI
treatment of NSCLC by influencing Bim expression. The current study
also found that lower HuR expression resulted in resistance to
EGFR-TKIs by inhibiting apoptosis. In addition, xenograft tumor
growth in nude mice induced by H1650 cells overexpressing HuR was
completely suppressed by an EGFR inhibitor (gefitinib); however,
complete tumor inhibition was not observed in the control (low HuR
expression) group. These findings strongly supported the
observations in the cell model and revealed that HuR-mediated Bim
expression confers EGFR-TKI sensitivity.
The Bcl-2 family consists of anti-apoptotic and
pro-apoptotic proteins regulating apoptosis (32-35). As a tumor suppressor, Bim is one
of the most powerful BH3-only proteins and is able to engage all
pro-survival proteins. Deregulating Bim expression at the genetic,
transcriptional and post-translational levels may confer resistance
to apoptosis (10,36). In addition, Bim upregulation is
associated with gefitinib-induced apoptosis in EGFR-mutant lung
cancer cells (37). Therefore,
Bim expression may correlate with the sensitivity to EGFR-TKIs
(38). In addition to the
contribution of Bim to the sensitivity to EGFR-TKIs, HuR expression
may also confer such sensitivity in EGFR-mutant lung cancer
cells.
HuR is a RNA-binding protein in the placenta lethal
abnormal visual (embryonic lethal abnormal vision) family, and is
located on chromosome 19p13.2 (27,39). HuR has been reported to be
associated with numerous human carcinomas, including breast
(40), liver (41), pancreatic (42), ovarian (43,44) and colorectal cancer, as well as
NSCLC (45,46). Recently published studies
indicated that elevated HuR expression is associated with
aggressiveness and poor prognosis in patients with invasive breast
carcinoma and lung carcinoma (47,48). Furthermore, HuR also stabilizes
mRNAs encoding crucial regulators of cellular proliferation,
differentiation and stress response, such as cyclins (49), cyclooxygenase-2 (50,51), p21 (52), HER2 (53) and Bcl-2 (54,55). However, the role of HuR in
EGFR-TKI resistance in NSCLC and the associated mechanism have not
been reported.
In the present study, specimens were collected from
81 NSCLC patients who received EGFR-TKI therapy to examine whether
HuR and Bim expression levels were associated with the response to
EGFR-TKIs and the patients' PFS. The study indicated that the HuR
protein expression in tumor tissues was positively correlated with
EGFR-TKI response in the patients with NSCLC. Furthermore, the
expression of cytoplasmic Bim was closely correlated with HuR
expression (P<0.01), but was not correlated with the sex, age
and mutation type (P>0.05). In the EGFR-TKI-sensitive group,
patients with high HuR tumor expression exhibited longer mPFS
following EGFR-TKI therapy in comparison with those with low HuR
expression (16.5 months vs. 9.7 months, respectively; P<0.01).
Similarly, the mPFS of patients with positive Bim expression was
significantly higher than that of patients with negative Bim
expression (12.8 months vs. 9.7 months, respectively; P<0.01).
Kaplan-Meier analysis also revealed that patients with low HuR and
Bim expression levels exhibited a shorter PFS as compared with
those with high HuR and Bim expression levels. In three lung cancer
cell lines that harbor an EGFR mutation, RT-qPCR and western
blotting demonstrated that HuR and Bim expression was significantly
lower in the EGFR-TKI-resistant cell line H1650 when compared with
the EGFR-TKI-sensitive cell lines HCC827 and PC9. Therefore, the
present study suggests that HuR and Bim levels may be predictive of
the treatment response and outcome of EGFR-TKI therapy in NSCLC
patients harboring EGFR mutations. However, the collected cases all
involved patients with lung adenocarcinoma, with the majority of
patients presenting IV stage disease, which is a limitation of the
current study. Therefore, further expansion of the corresponding
cases and further subgroup analysis should be performed in future
studies.
Knockdown of HuR expression was also conducted in
the current study, which resulted in resistance to EGFR-TKIs in
HCC827 cells by decreasing apoptosis. In addition, the expression
levels of HuR and Bim in HuR-knockdown HCC827 cells was lower
compared with that in the control group. By contrast,
overexpression of HuR restored the sensitivity to EGFR-TKIs in
H1650 cells by increasing apoptosis. Accordingly, HuR and Bim
expression levels in the HuR-transduced H1650 group were higher
than those in the control group. High expression of HuR was
associated with EGFR-TKI sensitivity through upregulation of Bim
expression. These findings indicated that HuR serves functionally
important roles in the sensitivity of cancer cells to EGFR-TKIs.
Furthermore, resistance to EGFR-TKIs was observed when HuR was
suppressed in cancer cells. To further validate the role of HuR in
regulating EGFR-TKI resistance, this protein was overexpressed in a
xenograft mouse model. In agreement with the in vitro data,
HuR overexpression restored gefitinib sensitivity in the mouse
model.
However, a limitation of the present study is that
the results did not indicate the direct regulation of HuR on Bim.
It was observed that low expression of HuR can lead to low
expression of Bim and the change of cell apoptosis. Therefore, HuR
may be able to affect apoptosis by regulating Bim expression In the
follow-up experiments, further investigation of the correlation
between HuR and other associated factors that can cause apoptosis
will be performed. Further investigation is also required to
identify the underlying molecular mechanisms. In addition, the
correlation between HuR and Bim was only confirmed in one cell
line; thus, subsequent studies should confirm this correlation in
more cell lines, and further prove these results.
In conclusion, the present study suggested that
decreased Bim expression, resulting from HuR downregulation, may
confer primary resistance to EGFR-TKIs in EGFR-mutant lung cancer
cells. Therefore, it is suggested that decreasing HuR expression in
patients with NSCLC may predict primary resistance to EGFR-TKI
treatment. The data also indicated that altered HuR/Bim expression
constitutes a novel mechanism of primary EGFR-TKI resistance in
NSCLC, and provided a rationale for therapeutic strategies to
reverse gefitinib resistance in NSCLC.
Funding
This study was supported by the National Nature
Science Foundation of China (grant nos. 81572875 and 81272619).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY, HC, JW and BW made substantial contributions to
the concept and design of the present study. YY and BW drafted the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the General Hospital (Jinan Command of the People's
Liberation Army, Jinan, China). Written informed consent was
obtained from all patients at the time of enrollment.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Naidoo J, Sima CS, Rodriguez K, Busby N,
Nafa K, Ladanyi M, Riely GJ, Kris MG, Arcila ME and Yu HA:
Epidermal growth factor receptor exon 20 insertions in advanced
lung adenocarcinomas: Clinical outcomes and response to erlotinib.
Cancer. 121:3212–3220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ettinger DS, Akerley W, Bepler G, Blum MG,
Chang A, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Ganti AK, et
al: Non-small cell lung cancer. J Natl Compr Canc Netw. 8:740–801.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Harman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takeuchi S and Yano S: Clinical
significance of epidermal growth factor receptor tyrosine kinase
inhibitors: Sensitivity and resistance. Respir Investig.
52:348–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Jänne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar
|
|
10
|
O'Connor L, Strasser A, O'Reilly LA,
Hausmann G, Adams JM, Cory S and Huang DC: Bim: A novel member of
the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harada H and Grant S: Targeting the
regulatory machinery of BIM for cancer therapy. Crit Rev Eukaryot.
Gene Expr. 22:117–129. 2012.
|
|
12
|
Bouillet P, Metcalf D, Huang DC, Tarlinton
DM, Kay TW, Köntgen F, Adams JM and Strasser A: Proapoptotic Bcl-2
relative Bim required for certain apoptotic responses, leukocyte
homeostasis, and to preclude autoimmunity. Science. 286:1735–1738.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bouillet P, Purton JF, Godfrey DI, Zhang
LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM and
Strasser A: BH3-only Bcl-2 family member Bim is required for
apoptosis of autoreactive thymocytes. Nature. 415:922–926. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong YX, Somwar R, Politi K, Balak M,
Chmielecki J, Jiang X and Pao W: Induction of BIM is essential for
apoptosis triggered by EGFR kinase inhibitors in mutant
EGFR-dependent lung adenocarcinomas. PLoS Med. 4:e2942007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cardona AF, Rojas L, Wills B, Arrieta O,
Carranza H, Vargas C, Otero J, Corrales-Rodrigues L, Martin C,
Reguart N, et al: BIM deletion polymorphisms in Hispanic patients
with non-small cell lung cancer carriers of EGFR mutations.
Oncotarget. 7:68933–68942. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garofalo M, Romano G, Di Leva G, Nuovo G,
Jeon YJ, Ngankeu A, Sun J, Lovat F, Alder H, Condorelli G, et al:
EGFR and MET receptor tyrosine kinase-altered microRNA expression
induces tumorigenesis and gefitinib resistance in lung cancers. Nat
Med. 18:74–82. 2012. View
Article : Google Scholar :
|
|
17
|
Lee JY, Ku BM, Lim SH, Lee MY, Kim H, Kim
M, Kim S, Jung HA, Sun JM, Ahn JS, et al: The BIM deletion
polymorphism and its clinical implication in patients with
EGFR-mutant non-small-cell lung cancer treated with EGFR tyrosine
kinase inhibitors. J Thorac Oncol. 10:903–909. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Faber AC, Corcoran RB, Ebi H, Sequist LV,
Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et
al: BIM expression in treatment-naive cancers predicts
responsiveness to kinase inhibitors. Cancer Discov. 1:352–365.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Filippova N, Yang X, Wang Y, Gillespie GY,
Langford C, King PH, Wheeler C and Nabors LB: The RNA-binding
protein HuR promotes glioma growth and treatment resistance. Mol
Cancer Res. 9:648–659. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brennan CM and Steitz JA: HuR and mRNA
stability. Cell Mol Life Sci. 58:266–277. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heinonen M, Fagerholm R, Aaltonen K,
Kilpivaara O, Aittomäki K, Blomqvist C, Heikkilä P, Haglund C,
Nevanlinna H and Ristimäki A: Prognostic role of HuR in hereditary
breast cancer. Clin Cancer Res. 13:6959–6963. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giammanco A, Blanc V, Montenegro G, Klos
C, Xie Y, Kennedy S, Luo J, Chang SH, Hla T, Nalbantoglu I, et al:
Intestinal epithelial HuR modulates distinct pathways of
proliferation and apoptosis and attenuates small intestinal and
colonic tumor development. Cancer Res. 74:5322–5335. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Erkinheimo TL, Lassus H, Sivula A,
Sengupta S, Furneaux H, Hla T, Haglund C, Butzow R and Ristimäki A:
Cytoplasmic HuR expression correlates with poor outcome and with
cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer
Res. 63:7591–7594. 2003.PubMed/NCBI
|
|
24
|
Denkert C, Koch I, von Keyserlingk N,
Noske A, Niesporek S, Dietel M and Weichert W: Expression of the
ELAV-like protein HuR in human colon cancer: Association with tumor
stage and cyclooxygenase-2. Mod Path. 19:1261–1269. 2006.
View Article : Google Scholar
|
|
25
|
Costantino CL, Witkiewicz AK, Kuwano Y,
Cozzitorto JA, Kennedy EP, Dasgupta A, Keen JC, Yeo CJ, Gorospe M
and Brody JR: The role of HuR in gemcitabine efficacy in pancreatic
cancer: HuR upregulates the expression of the gemcitabine
metabolizing enzyme deoxycytidine kinase. Cancer Res. 69:4567–4572.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beasley MB, Brambilla E and Travis WD: The
2004 World Health Organization classification of lung tumors. Semin
Roentgenol. 40:90–97. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
De K I, Mirhosseini M, Lau F, Thai V,
Downing M, Quan H, Lesperance M and Yang J: Conversion of Karnofsky
Performance Status (KPS) and Eastern Cooperative Oncology Group
Performance Status (ECOG) to Palliative Performance Scale (PPS),
and the interchangeability of PPS and KPS in prognostic tools. J
Palliat Care. 29:163–169. 2013.
|
|
28
|
Yu X, Zhan X, D'Costa J, Tanavde VM, Ye Z,
Peng T, Malehorn MT, Yang X, Civin CI and Cheng L: Lentiviral
vectors with two independent internal promoters transfer high-level
expression of multiple transgenes to human hematopoietic
stem-progenitor cells. Mol Ther. 7:827–838. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar
|
|
30
|
Wang J, Li D, Wang B and Wu Y: Predictive
and prognostic significance of cytoplasmic expression of ELAV-like
protein HuR in invasive breast cancer treated with neoadjuvant
chemotherapy. Breast Cancer Res Treat. 141:213–224. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maimaiti Y, Dong L, Aili A, Maimaitiaili
M, Huang T and Abudureyimu K: Bim may be a poor prognostic
biomarker in breast cancer patients especially in those with
luminal A tumors. Cancer Biomarkers. 19:411–418. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nature Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar
|
|
33
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sarosiek KA and Letai A: Directly
targeting the mitochondrial pathway of apoptosis for cancer therapy
using BH3 mimetics-recent successes, current challenges and future
promise. FEBS J. 283:3523–3533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu DW, Chen CY, Chu CL and Lee H: Paxillin
confers resistance to tyrosine kinase inhibitors in EGFR-mutant
lung cancers via modulating BIM and Mcl-1 protein stability.
Oncogene. 35:621–630. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Zhou S, Li X, Wang D, Wang Y, Zhou C
and Schmid-Bindert G: Gefitinib-resistance is related to BIM
expression in non-small cell lung cancer cell lines. Cancer Biother
Radiopharm. 28:115–123. 2013. View Article : Google Scholar :
|
|
38
|
Costa C, Molina MA, Drozdowskyj A,
Giménez-Capitán A, Bertran-Alamillo J, Karachaliou N, Gervias R,
Massuit B, Wei J, Moran T, et al: The impact of EGFR T790M
mutations and BIM mRNA expression on outcome in patients with
EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the
randomized phase III EURTAC trial. Clin Cancer Res. 20:2001–2010.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang D, Wang M, Hu C, Shuang T, Zhou Y and
Yan X: Expression of the ELAV-like protein HuR in the cytoplasm is
associated with endometrial carcinoma progression. Tumor Biol.
35:11939–11947. 2014. View Article : Google Scholar
|
|
40
|
Wang J, Guo Y, Chu H, Guan Y, Bi J and
Wang B: Multiple functions of the RNA-binding protein HuR in cancer
progression, treatment responses and prognosis. Int J Mol Sci.
14:10015–10041. 2014. View Article : Google Scholar
|
|
41
|
Zhu H, Berkova Z, Mathur R, Sehgal L,
Khashab T, Tao RH, Ao X, Feng L, Sabichi AL, Blechacz B, et al: HuR
suppresses Fas expression and correlates with patient outcome in
liver cancer. Mol Cancer Res. 13:809–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Romeo C, Weber MC, Zarei M, DeCicco D,
Chand SN, Lobo AD, Winter JM, Sawicki JA, Sachs JN, Meisner-Kober
N, et al: HuR contributes to TRAIL resistance by restricting death
receptor 4 expression in pancreatic cancer cells. Mol Cancer Res.
14:599–611. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Prislei S, Martinelli E, Mariani M,
Raspaglio G, Sieber S, Ferrandina G, Shahabi S, Scambia G and
Ferlini C: MiR-200c and HuR in ovarian cancer. BMC Cancer.
13:722013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sawicki JA, Huang YH, Brody JR, Getts RC,
Rhodes K and Gerhart J: Abstract 3542: Inhibition of HuR
effectively suppresses ovarian tumor growth in mice. Cancer Res.
75(15 Suppl): Abstract 3542. 2015. View Article : Google Scholar
|
|
45
|
Wang J, Wang B, Bi J and Zhang C:
Cytoplasmic HuR expression correlates with angiogenesis,
lymphangiogenesis, and poor outcome in lung cancer. Med Oncol.
28:S577–S585. 2011. View Article : Google Scholar
|
|
46
|
Wang J, Zhao W, Guo Y, Zhang B, Xie Q,
Xiang D, Gao J, Wang B and Chen Z: The expression of RNA-binding
protein HuR in non-small cell lung cancer correlates with vascular
endothelial growth factor-C expression and lymph node metastasis.
Oncology. 76:420–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vigouroux C, Casse JM, Battaglia-Hsu SF,
Brochin L, Luc A, Paris C, Lacomme S, Gueant JL, Vignaud JM and
Gauchotte G: Methyl(R217)HuR and MCM6 are inversely correlated and
are prognostic markers in non small cell lung carcinoma. Lung
Cancer. 89:189–196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Giaginis C, Sampani A, Kotta-Loizou I,
Giannopoulou I, Danas E, Politi E, Tsourouflis G, Kouraklis G,
Patsouris E, Keramopoulos A, et al: Elevated hu-antigen receptor
(HuR) expression is associated with tumor aggressiveness and poor
prognosis but not with COX-2 expression in invasive breast
carcinoma patients. Pathol Oncol Res. 24:631–640. 2018. View Article : Google Scholar
|
|
49
|
Wang W, Caldwell MC, Lin S, Furneaux H and
Gorospe M: HuR regulates cyclin A and cyclin B1 mRNA stability
during cell proliferation. EMBO J. 19:2340–2350. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fernau NS, Fugmann D, Leyendecker M,
Reimann K, Grether-Beck S, Galban S, Ale-Agha N, Krutmann J and
Klotz LO: Role of HuR and p38MAPK in ultraviolet
B-induced post-transcriptional regulation of COX-2 expression in
the human keratinocyte cell line HaCaT. J Biol Chem. 285:3896–3904.
2010. View Article : Google Scholar
|
|
51
|
Serini S, Fasano E, Piccioni E, Monego G,
Cittadini AR, Celleno L, Ranelletti FO and Calviello G: DHA induces
apoptosis and differentiation in human melanoma cells in vitro:
Involvement of HuR-mediated COX-2 mRNA stabilization and β-catenin
nuclear translocation. Carcinogenesis. 33:164–173. 2012. View Article : Google Scholar
|
|
52
|
Wang W, Furneaux H, Cheng H, Caldwell MC,
Hutter D, Liu Y, Holbrook N and Gorospe M: HuR regulates p21 mRNA
stabilization by UV light. Mol Cell Biol. 20:760–769. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hung CM, Huang WC, Pan HL, Chien PH, Lin
CW, Chen LC, Chien YF, Lin CC, Leow KH, Chen WS, et al: Hepatitis B
virus X upregulates HuR protein level to stabilize HER2 expression
in hepatocellular carcinoma cells. Biomed Res Int. 2014:8274152014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishimaru D, Ramalingam S, Sengupta TK,
Bandyopadhyay S, Dellis S, Tholanikunnel BG, Fernandes DJ and
Spicer EK: Regulation of Bcl-2 expression by HuR in HL60 leukemia
cells and A431 carcinoma cells. Mol Cancer Res. 7:1354–1366. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Spicer EK, Ishimaru D, Ramalingam S,
Tholanikunnel BG and Fernandes DJ: Role of HuR in the regulation of
bcl-2 mRNA stability in human HL60 leukemia cells. FASEB J. 22(1
Suppl): 786.52008.
|