Introduction
The cardiovascular complications of diabetes
mellitus (DM), including heart failure and arrhythmia, lead to high
rates of disability and mortality (1–3).
The molecular mechanism of myocardial injury in DM is complicated,
and further investigation of this pathogenic mechanism is
beneficial to further improving the prognosis of DM.
The posttranslational modification of O-linked
β-N-acetylglucosamine (O-GlcNAc) regulates proliferation,
differentiation, apoptosis, autophagy and other physiological and
pathological processes in a variety of cells, including
cardiomyocytes (4–9). Two genes encode the enzymes for
O-GlcNAcylation; O-GlcNAc transferase (OGT) catalyzes the addition
of UDP-GlcNAc to target proteins, whereas O-GlcNAcase (OGA) removes
the GlcNAc moiety from proteins (10). Accordingly, the selective
O-GlcNAcase inhibitor, thiamet G (TG), effectively increases the
level of O-GlcNAc modification. The concerted actions of the two
enzymes allow the O-GlcNAc modification to be a dynamic
post-translational modification; 6-diazo-5-oxo-L-norleucine (Don)
is an inhibitor of glucosamine fructose-6-phosphate
aminotransferase isomerizing 1, which is an enzyme that controls
the flux of glucose into the hexosamine pathway and catalyzes the
formation of glucosamine 6-phosphate. In 2013, a study published in
the Journal Nature (11) first
applied Don as a specific O-GlcNAc antagonist, which showed that 5
µM of Don effectively inhibited the level of O-GlcNAc in
vitro. Several studies have subsequently used Don as an
inhibitor of O-GlcNAc. Based on these previous publications
(12,13), Don was selected as a selective
O-GlcNAc inhibitor in the present study to clarify the role of
O-GlcNAc modified SNAP29 in cardiomyopathy in type I diabetic rats.
Previous studies (11,14) have found that the acute inhibition
of O-GlcNAc inhibits premature ventricular complexes activated by
diabetic hyperglycemia through O-GlcNAc- and
Ca2+/calmodulin-dependent protein kinase II-dependent
pathways. Additionally, the dysregulation of O-GlcNAcylation is
likely to be important in the rat cardiac mitochondrial dysfunction
associated with diabetes, which was reported by Banerjee et
al (15). Reportedly
(16), an excessive level of
O-GlcNAc modification in a DM animal model led to the worsening of
cardiac function, whereas a reduction in O-GlcNAc levels improved
cardiac function. However, the mechanism between the O-GlcNAc
modification and the deterioration of cardiac function remains to
be fully elucidated.
Autophagy is a dynamic process, which is regulated
by multiple genes and molecular signals. The process of
autophagosome formation, transport of the autophagic substrate to
lysosomes, and autophagosomal degradation in lysosomes is termed
autophagic flux. Autophagy is an important metabolic pathway for
maintaining eukaryotic cell homeostasis in response to starvation,
inflammation, and hypoxia/reoxygenation injury. Autolysosomes
mainly eliminate aging organelles and misfolded proteins to provide
energy for cells. In the normal myocardium, autophagy is maintained
at a low level (17). However,
energy metabolism of the myocardium is disturbed in DM, and
autophagy can be overactivated or inhibited (18), although how autophagic flux causes
myocardial injury in DM remains to be elucidated. Specifically, no
reports have determined whether myocardial injury is influenced by
interactions between the O-GlcNAc modification and autophagic flux
in type I DM.
Synaptosomal-associated protein 29 (SNAP29) is
mainly involved in the localization and fusion of organelle
membranes in the cytoplasm (19).
Several studies (20–22) have reported that SNAP29 is
involved in mediating membrane fusion between autophagosomes and
lysosomes, and forms a complex with syntaxin-17 (STX17) and
vesicle-associated membrane protein 8 (VAMP8) to participate in the
autophagy process. A previous study (23) showed that SNAP29 is modified as a
protein substrate by O-GlcNAc in Nematodes. However, whether the
O-GlcNAc modification of SNAP29 regulates autophagic flux in a
mammalian disease model remains to be elucidated.
Materials and methods
Reagents
The primary antibody mouse anti-O-GlcNAc (RL2; cat.
no. RB229146; 1:1,000) was purchased from Life Company (New York,
USA). The anti-OGT (cat. no. ab177941; 1:1,000), anti-LAMP2 (cat.
no. ab125068 1:1,000), anti-SNAP29 (cat. no. ab181151; 1:1,000),
anti-VAMP8 (cat. no. ab76021; 1:1,000), anti-STX17 (cat. no.
ab116113; 1:1,000) and anti-OGA (cat. no. ab105217; 1:1,000)
antibodies were obtained from Abcam (Cambridge, USA). The
anti-Beclin1 (cat. no. D40C5; 1:1,000) antibody was obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). The anti-P62
(cat. no. 18420-1-AP; 1:1,000) and anti-Tubulin (cat. no.
66240-1-Ig; 1:1,000) antibodies were obtained from ProteinTech
Group, Inc. (Chicago, IL, USA). Protein A/G beads were obtained
from Abmart, Inc. (Shanghai, China). Anti-LC3B antibody (cat. no.
L7543; 1:1,000), STZ (cat. no. SD130), TG (cat. no. SML0244), and
Don (cat. no. D2141) were purchased from Merck KGaA (Darmstadt,
Germany). The RNAi-LAMP2 sequences were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The three sequence pairs of
RNAi-LAMP2 were as follows: Sequence 1 forward, 5′-GAA GUU CUU AUA
UGU GCA ATT-3′ and reverse, 5′-UUG CAC AUA UAA GAA CUU CTT-3′;
sequence 2 forward, 5′-GGC AGG AGU ACU UAU UCU ATT-3′ and reverse,
5′-UAG AAU AAG UAC UCC UGC CTT-3′; sequence 3 forward, 5′-CUG CAA
UCU GAU UGA UUA UU-3′ and reverse, 5′-TAA ACA CTG CTT GAC CAC C-3′.
The most effective sequence was selected according to the
interference efficiency detected using western blot analysis.
Animal experiments
All animal experiments were approved by the Animal
Ethics and Experimentation Committee of Nanchang University
(Nanchang, China) and were performed in accordance with the ʻGuide
for the Care and Use of Laboratory Animalsʼ (revised 1996). The
experimental protocol was approved by the Second Affiliated
Hospital of Nanchang University. All experimental animals were
purchased from Hunan Slack Jingda Experimental Animal Co., Ltd.
(Hunan, China). A total of 50 adult male Sprague Dawley (SD) rats
were purchased at 8-weeks old (weight range, 180–200 g), and kept
in a specific pathogen-free conditions at room temperature 20–24°C
and humidity 50–60%. The experiment began after 1 week of
acclimation. Rats were provided with adequate food and drinking
water and maintained on a 12/12-h light-dark cycle to mimic the
normal biorhythm of the rats.
Animal model establishment and
treatment
All rats were intraperitoneally (i.p) injected with
streptozotocin (STZ) at a dose of 55 mg/kg (24). Glucose concentrations were
monitored using a commercial blood glucose-monitoring kit
(Accusoft; Roche Diagnostics, Laval, QC, Canada) using blood
samples obtained from the tail vein of nonfasting animals prior to
and following the STZ injection, and immediately prior to the start
of the experiment. Animals with tail vein blood glucose levels
>16.7 mmol/l were considered to have DM (11,25). Control animals (vehicle) received
the same volume of sodium citrate (pH 4.4, i.p.).
The rats were randomly divided into five groups (n≥6
in each group). The Vehicle group comprised the solvent control
group injected with sodium citrate (pH 4.4; i.p). The STZ group, as
the diabetic group, comprised rats injected with STZ i.p. at a dose
of 55 mg/kg. At 4 weeks post-STZ injection, rats were randomly
divided into three groups: The STZ+Don group comprised STZ rats
injected with the O-glycosylation inhibitor, Don (Sigma-Aldrich;
Merck KGaA) at a dose of 5 mg/kg, i.p. The STZ+TG group comprised
STZ rats injected with O-glycosylation agonist TG (Sigma-Aldrich;
Merck KGaA) at a dose of 25 mg/kg, i.p. The STZ+NaCl group
comprised STZ rats injected with 0.9% NaCl, i.p. Don, TG, and NaCl
injected on a 2-day interval. All the rats were sacrificed as
outlined in the experimental protocols.
Doppler echocardiography
Transthoracic echocardiography was performed prior
to sacrifice at the indicated time points, as previously described
(26,27). Briefly, the rats were anesthetized
with 2.0% isoflurane via inhalation. The chest was shaved, and the
rats were placed in a supine position. Echocardiographic images
were obtained by placing the transducer against the chest. M-mode
echocardiograms of the left ventricle (LV) were recorded at the
level of the papillary muscle using a commercially available
echocardiographic system, Vevo2100, equipped with a 17-MHz
transducer (Visualsonics, Toronto, ON, Canada). The passive LV
filling peak velocity (E, mm/sec) and atrial contraction flow peak
velocity (A, mm/sec) were acquired from the mitral valve Doppler
flow images in the apical four-chamber view.
Transmission electron microscopy (TEM),
hematoxylin and eosin (H&E) staining and Masson staining
All the cardiac tissues were fixed with 10% buffered
formalin, and a region of the ventricle was fixed with 2.5%
glutaraldehyde for analysis by TEM. The TEM, H&E staining and
Masson staining were performed by Google Biological Technology
(Wuhan, China). The quantitative results were analyzed with Image
Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD,
USA).
Cell culture and reagents
Isolation and treatment of neonatal
rat cardiac myocytes (NRCMs)
The NRCMs were derived from newborn rats within 3
days according to a conventional protocol described previously
(28,29). Newborn SD rats were purchased from
Hunan Slack Jingda Experimental Animal Co., Ltd. The mice were kept
with their mothers for 3 days after birth, and after 1 day of
stabilization in specific pathogen-free conditions at room
temperature 20–24°C and humidity 50–60%, they were subjected to
cardiomyocyte isolation. The cells were cultured in DMEM
supplemented with 15% fetal bovine serum (FBS) (both from Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1%
penicillin-streptomycin at 37°C with 5% carbon dioxide
(CO2). The NRCMs were exposed to DMEM containing 25 mM
glucose to mimic hyperglycemia injury, and the normal-glucose group
was exposed to DMEM containing 5 mM glucose.
The NRCMs were treated with drugs and randomly
divided into eight groups: Normal glucose (Vehicle group), normal
glucose+Don (Don group), normal glucose+TG (Sigma-Aldrich; Merck
KGaA), normal glucose+3-methyladenine (3-MA group), high glucose
(Glu group), high glucose+Don (Don+Glu group), high glucose+TG
(TG+Glu group) and high glucose+3-MA (3-MA+Glu). In the present
study, the dose and time schedule of drug application were as
follows: In vitro, NRCMs were treated with DMEM containing
25 mM glucose for 24 h to mimic hyperglycemic injury, Don (40
µM) for 24 h, TG (5 µM) for 24 h, or 3-MA (5 mM) for
24 h.
Plasmid and adenovirus
transfection
All cells were seeded in 6-well plates 1 day prior
to transfection at a density of 1×105 cells/well. The
cells were infected with either the OGT knockdown adenovirus
(sh-OGT), OGT-overexpression adenovirus (ad-OGT) or
OGA-overexpression adenovirus (ad-OGA) at an MOI of 50, and
re-incubated with DMEM supplemented with 10% FBS 6 h following
transfection. After 48 h, the transfection efficiency was detected
via fluorescence microscopy and western blot analysis.
Small interfering (si)RNA oligonucleotides specific
for LAMP2A were synthesized by Santa Cruz Biotechnology, Inc.
All-star negative siRNA was used as a negative control. The siRNA
sequences targeting the indicated proteins were as follows:
si-LAMP2, forward 5′-CUG CAA UCU GAU UGA UUA UU-3′ and reverse
5′-TAA ACA CTG CTT GAC CAC C-3′. The NRCMs were seeded in 6-well
plates (1×105 cells/well). After 24 h, the cells were
transfected with 50 pmol of siRNA/well using Lipofectamine 3,000
(cat. no. 1781682, Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer’s protocol.
Western blot analysis and
coimmunoprecipitation (co-IP)
Proteins were extracted with a mixed lysis buffer
containing a proteasome inhibitor (cat. no. RL2274422, Thermo
Fisher Scientific, Inc.) and phenylmethanesulfonyl fluoride (cat.
no. ST506; Beyotime Institute of Biotechnology, Shanghai, China).
Western blot analysis was performed as previously described
(29). Briefly, following protein
extraction, the protein concentration was detected using a
bicinchoninic acid assay. A total of 100 µg protein/lane
lysates were separated by 5-12% SDS-PAGE and subsequently
transferred onto polyvinylidene difluoride membranes. Following
blocking with Blocking reagent (cat.no. P0023B; Beyotime Institute
of Biotechnology) at room temperature for 1 h, the membranes were
incubated overnight at 4°C with the aforementioned primary
antibodies and the corresponding goat anti-mouse (1:5,000; cat. no.
SA00001-1; ProteinTech Group, Inc.) or goat anti-rabbit (1:5,000;
cat. no. SA00001-15; ProteinTech Group, Inc.) horseradish
peroxidase-conjugated IgG (H+L) secondary antibodies at room
temperature for 1 h. The protein bands were visualized using ECL
western blotting reagent (cat. no. 32209; Thermo Fisher Scientific,
Inc.). The band intensities were quantified using Image Lab 4.0.1
software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
NRCMs, treated as mentioned above, were harvested, and co-IP was
performed as previously described (29). Briefly, 400 µg of total
cell lysate was incubated with 4 µg of primary antibody for
the co-IP. The remaining cell lysates were incubated with rabbit
IgG at 4°C overnight as a negative control. Following incubation of
the cells with the primary antibodies, the lysates were mixed with
protein A/G beads overnight. The co-IP samples were separated by
SDS-PAGE and analyzed using western blot analysis as
aforementioned.
Statistical analysis
The results are expressed as the mean ± standard
error of the mean of at least three independent experiments, and
the differences between two groups were analyzed using Student’s
t-test. When more than two groups were compared, one-way analysis
of variance followed by Tukey’s post hoc test was used. Statistical
analyses were performed using Graph Pad Prism 5 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Myocardial injury is accompanied by an
increase in O-GlcNAc modification and inhibited autophagic flux in
rats at 8 weeks following STZ induction
Compared with the vehicle group, the STZ-induced
rats manifested as hyper-glycemic (30.49±0.6211, vs.
5.881±0.2036 mmol/l, P<0.01), a loss of body weight, and an
increased heart/body weight ratio beginning at 4 weeks post-STZ
administration (data not shown). Diabetic myocardial injury is
mainly manifested as early diastolic dysfunction characterized by
an abnormal E/A ratio. The echocardiographic results showed that
the E/A ratio decreased significantly in rats at 8 weeks post-STZ
induction compared with the vehicle rats (1.30±0.04, vs.
1.68±0.05, P<0.01; Fig. 1A);
no significant difference in cardiac systolic function was
observed, however, the cardiac structure was expanded (LVEF%:
81.47±2.92, vs. 72.92±4.45, n≥6, P>0.05). Compared with the
vehicle group, the STZ group had higher levels of cardiomyocyte
disorganization and fat accumulation as detected by H&E
staining. Additionally, the diabetic rats showed increased
myocardial fibrosis on Masson staining (Fig. 1B and C).
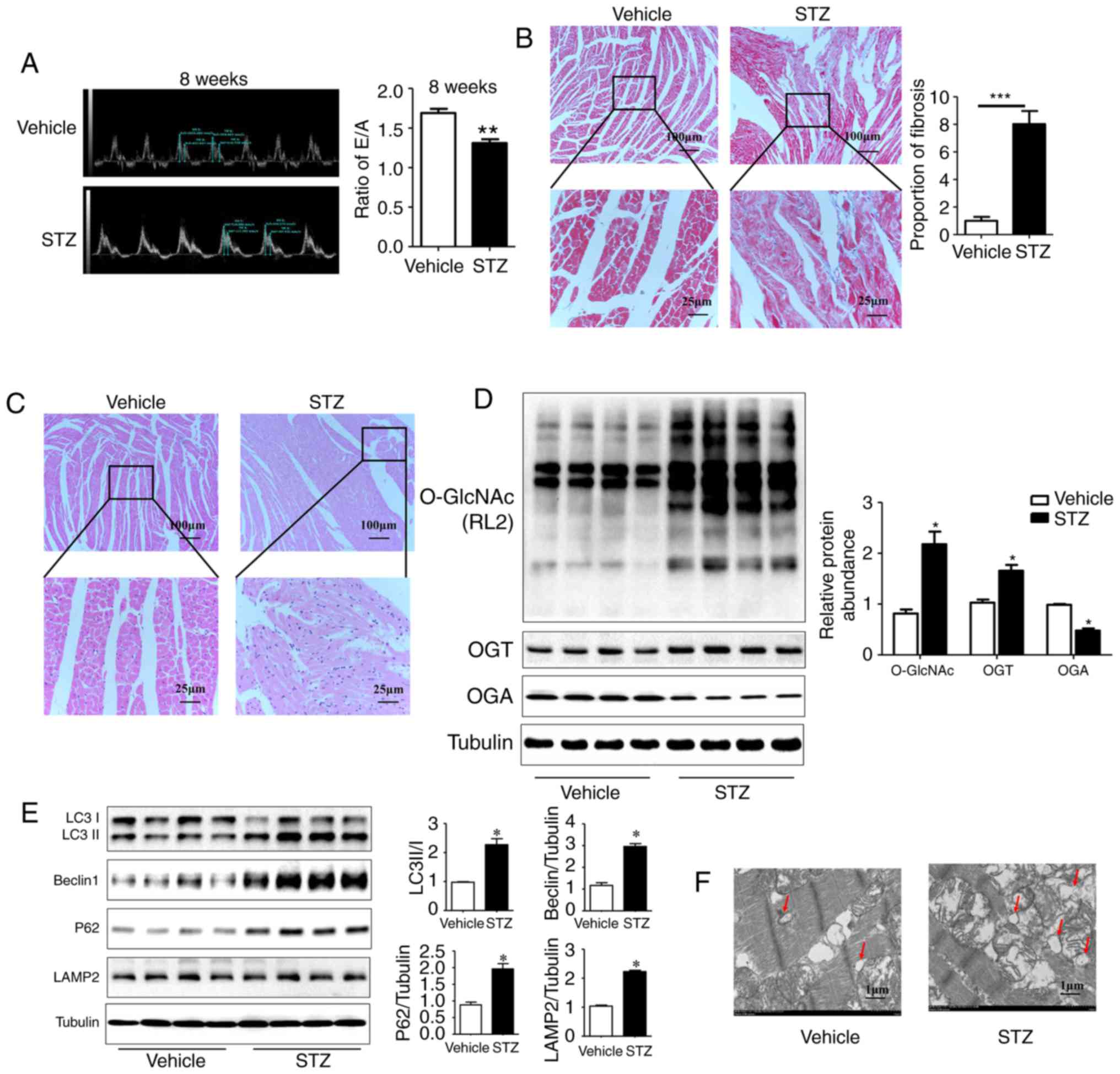 | Figure 1Myocardial injury is accompanied by
an increase in O-GlcNAc modification and inhibited autophagic flux
in rats at 8 weeks post-STZ induction. (A) M-mode echocardiography
shows the left ventricular diastolic function as the ratio of E/A
in rats at 8 weeks post-STZ induction. Morphological changes in the
myocardium were assessed by (B) Masson staining and (C) hematoxylin
and eosin staining (scale bar=100 and 25 µm). (D) Protein
was extracted from heart tissue, and the expression levels of
O-GlcNAc (RL2), OGT and OGA were detected by western blot analysis;
(E) Expression levels of LC3II/I, LAMP2, Beclin-1 and P62 in each
group were detected by western blot analysis in the vehicle group
and the 8-week STZ-induced group. (F) Transmission electron
microscopy shows autophagosomes, characterized by a double-layer
membrane structure, as indicated with the red arrows (scale bar=1
µm). Data are expressed as the mean ± standard error of the
mean (n≥6). *P<0.05 and **P<0.01, vs.
Vehicle group. Tubulin was the loading control. O-GlcNAc, O-linked
β-N-acetylglucosamine; STZ, streptozotocin; OGT, O-GlcNAc
transferase; OGA, O-GlcNAcase; LAMP2, lysosome-associated membrane
protein 2; LC3, microtubule-associated protein 1 light chain 3α;
E/A, left ventricular filling peak velocity/atrial contraction flow
peak velocity. |
Additionally, the results of the western blot
analysis showed a significant increase in the O-GlcNAc modification
and the expression of OGT, and a reduction in the expression of OGA
in the myocardium of rats at 8 weeks post-STZ induction compared
with the vehicle rats (Fig. 1D;
P<0.05). The expression levels of the autophagy markers Beclin1
(30) and LC3II/I were increased
significantly in the STZ group. The expression levels of SQSTM1/P62
and lysosome-associated membrane protein LAMP2 (31) were also significantly increased.
These results indicated that the autophagy-mediated degradation
process and autophagic flux were inhibited in rats at 8 weeks
post-STZ induction (Fig. 1E;
P<0.05). As the gold standard for the identification of
autophagy, TEM showed a marked increase in autophagosomes with a
double-membrane structure in the STZ group (Fig. 1F). These results suggested that
the significant increase in myocardial O-GlcNAc modification was
simultaneously accompanied by autophagic flux inhibition in the DM
rat hearts.
Increased O-GlcNAc modification in vivo
aggravates myocardial injury and inhibits autophagic flux in type I
DM rats
To further clarify the effect of O-GlcNAc
modification on myocardial injury in type I DM rats, the selective
O-GlcNAcase inhibitor TG and the glutamine antagonist Don were used
to investigate the effects of O-GlcNAc modification on cardiac
function and myocardial structure in type I DM rats. The
preliminary results (data not shown) demonstrated that, at 4 weeks
post-STZ induction, no significant difference was observed in the
level of O-GlcNAc modification, myocardial structure or the levels
of autophagy, compared with those in the vehicle rats, however, the
E/A ratio was marginally decreased (P<0.05). Therefore, drug
intervention in the STZ-induced rats was started at 4 weeks. The
results showed that TG significantly enhanced the heart/body weight
ratio of the rats at 8 weeks post-STZ induction. By contrast, Don
significantly mitigated the above phenotype in type I DM rats. The
E/A ratio in the STZ+TG group was significantly higher than that in
the Vehicle group (2.28±0.14, vs. 1.67±0.067, P<0.05) and
the STZ+NaCl group (2.28±0.14, vs. 1.29±0.081, P<0.01),
which suggested that the LV pressure was increased and the
diastolic function was deteriorated further. In addition, the E/A
ratio was increased in the STZ+Don group compared to that in the
STZ+NaCl group (1.62±0.13, vs. 1.29±0.081, P<0.05), which
suggested that the cardiac diastolic function was improved in the
DM rats following treatment with the O-GlcNAc inhibitor (Fig. 2A). No significant difference in
cardiac systolic function was observed among the STZ-induced
groups. Additionally, the agonist TG aggravated myocardial
disorganization and fat deposition, as detected by H&E
staining, and intercellular fibrosis, as assessed by Masson
staining, in the heart tissues of STZ rats. Furthermore, Don
significantly rescued the abnormal myocardial structure in type I
DM rats (Fig. 2B and C). The
above results suggested that the increased level of O-GlcNAc
modification aggravated myocardial injury in the type I DM
rats.
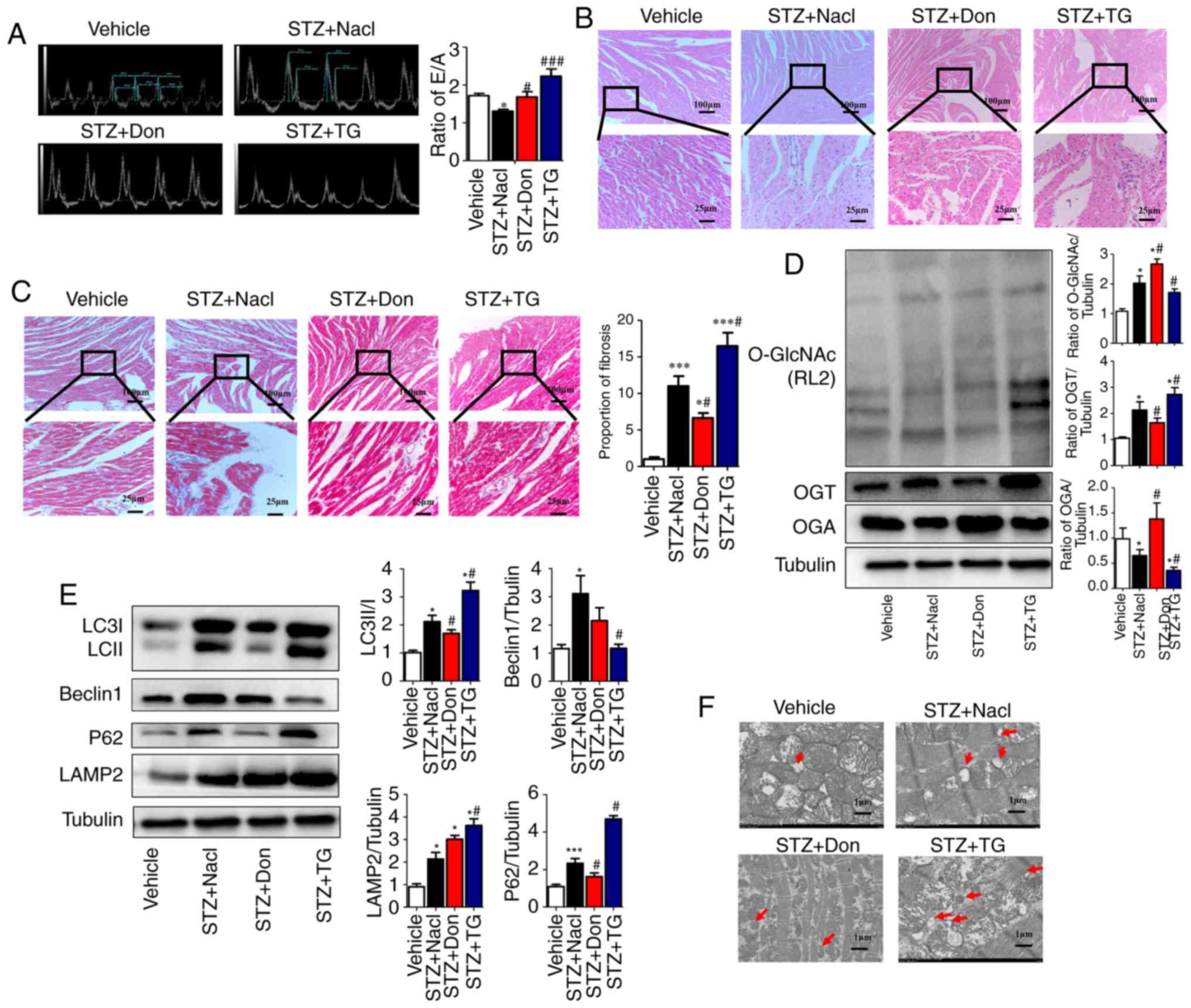 | Figure 2Increased O-GlcNAc modification in
vivo inhibits autophagic flux and aggravates myocardial
function in type I DM rats. Rats were randomly divided into five
groups (n≥6 in each group) for treatment: Vehicle, STZ, STZ+NaCl,
STZ+Don, and STZ+TG. (A) M-mode echocardiography showed the left
ventricular diastolic function as the ratio of E/A. Morphological
changes in the myocardium were assessed by (B) hematoxylin and
eosin staining and (C) Masson staining (scale bar=100 and 25
µm); (D) Expression levels of O-GlcNAc (RL2), OGA and OGT in
each group were detected by western blot analysis. (E) Western blot
analysis was used to detect the expression of autophagy markers
LC3II/I, Beclin1, P62, and LAMP2. Data are expressed as the mean ±
standard error of the mean (n≥6). *P<0.05 and
***P<0.001, vs. Vehicle group; #P<0.05
and ###P<0.001, vs. STZ+NaCl group. Tubulin was the
loading control. (F) Transmission electron microscopy showed the
autophagosomes, which are characterized by a double-layer membrane
structure, as indicated by red arrows (scale bar=1 µm).
O-GlcNAc, O-linked β-N-acetylglucosamine; STZ, streptozotocin; Don,
6-diazo-5-oxo-L-norleucine; TG, thiamet G; OGT, O-GlcNAc
transferase; OGA, O-GlcNAcase; LAMP2, lysosome-associated membrane
protein 2; LC3, microtubule-associated protein 1 light chain 3α;
E/A, left ventricular filling peak velocity/atrial contraction flow
peak velocity. |
To investigate the correlation between myocardial
O-GlcNAc modification and autophagy in type I DM rats, Don and TG
were administered in vivo. Western blot analysis was used to
detect the levels of myocardial O-GlcNAc modification and autophagy
in each group. Following drug administration, the level of O-GlcNAc
modification and the expression of OGT were significantly increased
in the STZ+TG group compared with those in the STZ+NaCl group
(P<0.05), whereas the expression of OGA was notably decreased
(P<0.05). Compared with that in the STZ+NaCl group, the
expression of OGA was significantly increased in the STZ+Don group
(P<0.05), and the level of O-GlcNAc modification and expression
of OGT were significantly decreased (Fig. 2D, P<0.05). Compared with the
STZ+NaCl group, the expression levels of LC3II/I, P62 and LAMP2
were markedly increased in the STZ+TG group, and the expression of
Beclin1 was significantly decreased (P<0.05), which suggested
that the O-GlcNAc modification level was increased and that
autophagy-mediated degradation was inhibited. Following treatment
with Don, the expression levels of LC3II/I and P62 were decreased,
and the expression of LAMP2 was significantly increased (Fig. 2E, P<0.05). In addition, the TEM
results showed that autophagosomes were increased in the STZ+TG and
STZ+NaCl groups, compared with those in the vehicle and STZ+Don
groups, respectively (Fig. 2F).
The above results indicated that myocardial O-GlcNAc modification
was increased in the heart of type I DM rats and that the
degradation of autophagosomes was significantly increased when
O-GlcNAc modification was inhibited, thereby ensuring the
progression of autophagy.
Increased O-GlcNAc modification inhibits
autophagic flux in NRCMs under high-glucose conditions
The NRCMs were subjected to high-glucose stimulation
(25 mM) to further verify the impact of the changes in the level of
O-GlcNAc modification on autophagy. The administration of TG
increased the level of O-GlcNAc modification in the normal- and
high-glucose environments, whereas the inhibitor Don exerted a
potent inhibitory effect on O-GlcNAc modification compared with
that in the control group (Fig.
3A). Consistent with the results in the animal experiments,
autophagic flux was significantly inhibited in the NRCMs under
high-glucose conditions, as demonstrated by the increased
expression levels of LC3II/I, Beclin1, P62, and LAMP2. In addition,
the O-GlcNAc modification agonist TG inhibited autophagic flux but
increased the level of O-GlcNAc modification (Fig. 3B). By contrast, the O-GlcNAc
modification inhibitor Don significantly reduced O-GlcNAc levels
and promoted autophagic flux.
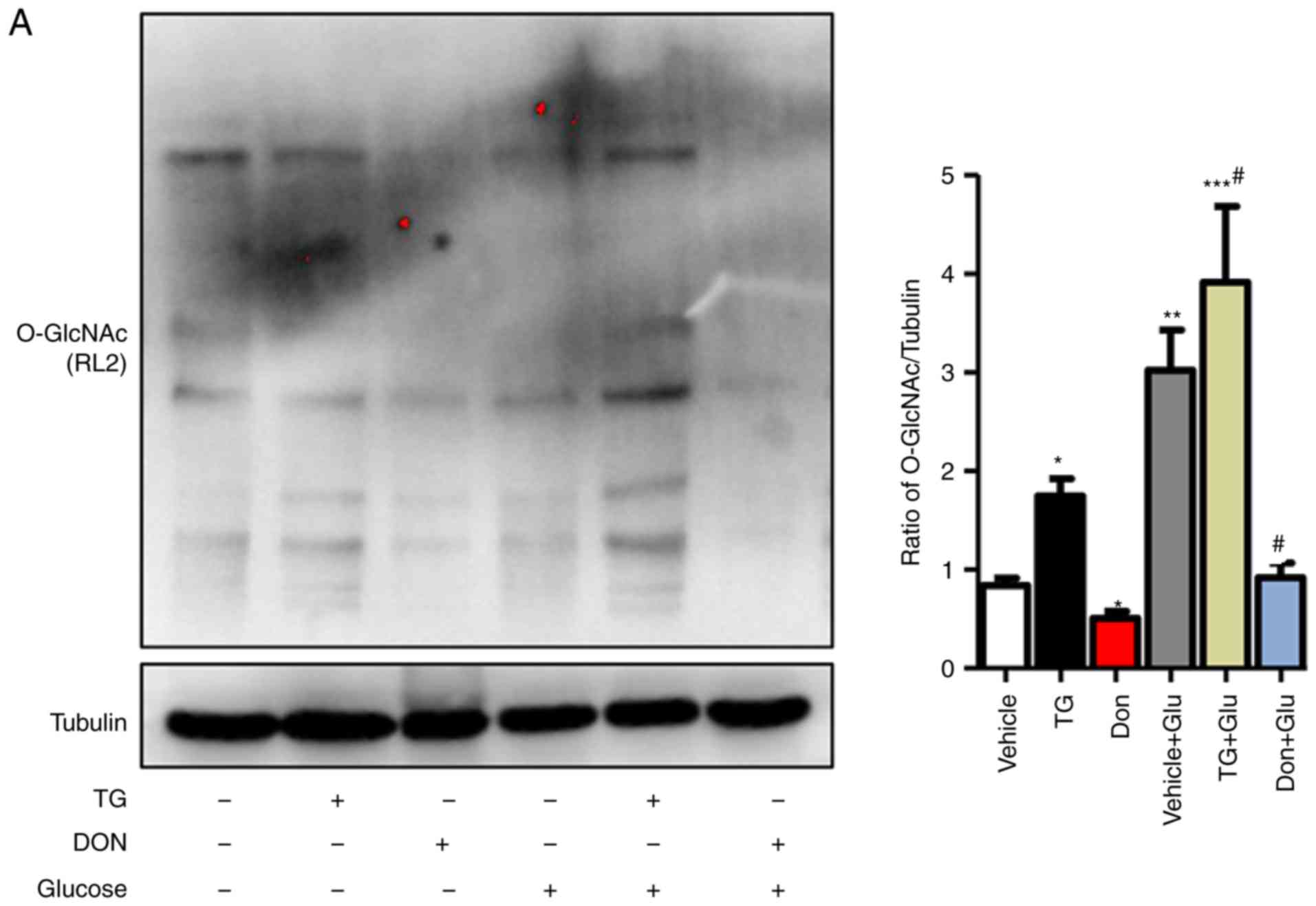 | Figure 3Increased O-GlcNAc modification
inhibits autophagic flux in NRCMs under high-glucose conditions.
NRCMs were exposed to high glucose (25 mM) and were treated with TG
(5 µM) or Don (40 µM) for 24 h. (A) Supernatants were
extracted, and the expression of O-GlcNAc (RL2) was analyzed by
western blot analysis. (B) Expression levels of LAMP2, Beclin-1,
P62, and LC3II/I in each group were detected by western blot
analysis. NRCMs were transfected with sh-OGT or ad-OGA for 48 h and
were exposed to high glucose (25 mM, Glu group) for 24 h. (C)
Supernatants were extracted, and the expression levels of O-GlcNAc
(RL2), OGT, and OGA were analyzed by western blot analysis. (D)
Expression levels of LAMP2, Beclin-1, P62, and LC3II/I in each
group were detected by western blot analysis. Data are expressed as
the mean ± standard error of the mean (n≥6). *P<0.05,
**P<0.01 and ***P<0.001, vs. Vehicle
group; #P<0.05 and ##P<0.01, vs. Glu
group. Tubulin was the loading control. NRCMs, neonatal rat
cardiomyocytes; O-GlcNAc, O-linked β-N-acetylglucosamine; STZ,
streptozotocin; sh-OGT, OGT-knockdown adenovirus; ad-OGA,
OGA-overexpression adenovirus; Ctrl, control; Glu, glucose; Don,
6-diazo-5-oxo-L-norleucine; TG, thiamet G; OGT, O-GlcNAc
transferase; OGA, O-GlcNAcase; LAMP2, lysosome-associated membrane
protein 2; LC3, microtubule-associated protein 1 light chain
3α. |
Aside from changing the level of O-GlcNAc
modification by Don and TG administration, NRCMs were separately
trans-fected with adenoviruses that either suppressed the
expression of OGT (sh-OGT) or overexpressed OGA (ad-OGA) to reduce
the level of O-GlcNAc modification. Western blot analysis was used
to detect the levels of autophagy under normal- and high-glucose
conditions. However, regardless of the glucose level, sh-OGT and
ad-OGA significantly reduced the levels of O-GlcNAc modification
(Fig. 3C, P<0.05). Measurement
of autophagy-related markers showed that, under high-glucose
stimulation, the expression levels of LC3II/I, Beclin1, P62, and
LAMP 2 were significantly increased (P<0.001), which indicated
that autophagy was significantly increased (Fig. 3D). OGT interference or OGA
overexpression resulted in a significant decrease in autophagy and
autophagic flux degradation in NRCMs under normal- and high-glucose
conditions. Therefore, consistent with the results of the
aforementioned drug intervention, interference with the O-GlcNAc
modification promoted autophagic flux in the NRCMs under normal-
and high-glucose conditions.
High glucose levels inhibit the
degradation stage of autophagy
To clarify the specific stage of autophagy that is
affected by high glucose in NRCMs, the autophagy inhibitor 3-MA and
LAMP2-interference sequences were used. The optimal interfering
sequence of LAMP2 was selected according to the efficiency of
transfection, which was verified by western blot analysis (data not
shown). The results showed that, under the normal-glucose
condition, disrupted expression of LAMP2 in NRCMs was associated
with increased levels of Beclin1, LC3II/I, and P62 (P<0.01).
Under the high-glucose condition, LAMP2 interference did not
increase the levels of Beclin1 or LC3II/I, which indicated that
high glucose levels affected the degradation process of autophagic
flux (Fig. 4A). To further
validate this hypothesis, the autophagy inhibitor 3-MA was used to
treat NRCMs at different glucose levels. In normal glucose levels,
the expression levels of LC3II/I and P62 decreased following 3-MA
treatment, which was in line with the function of 3-MA as an
inhibitor of class III phosphoinositide 3-kinase in the initial
stage of autophagy. In the high glucose-induced NRCMs, the
expression levels of LC3II/I and P62 decreased with 3-MA treatment
compared with those in NRCMs induced with high glucose alone, which
indicated that high glucose levels did not affect the initial stage
of autophagy (Fig. 4B). The above
results showed that high glucose levels inhibited
autophagy-mediated degradation rather than the initial stage of
autophagy.
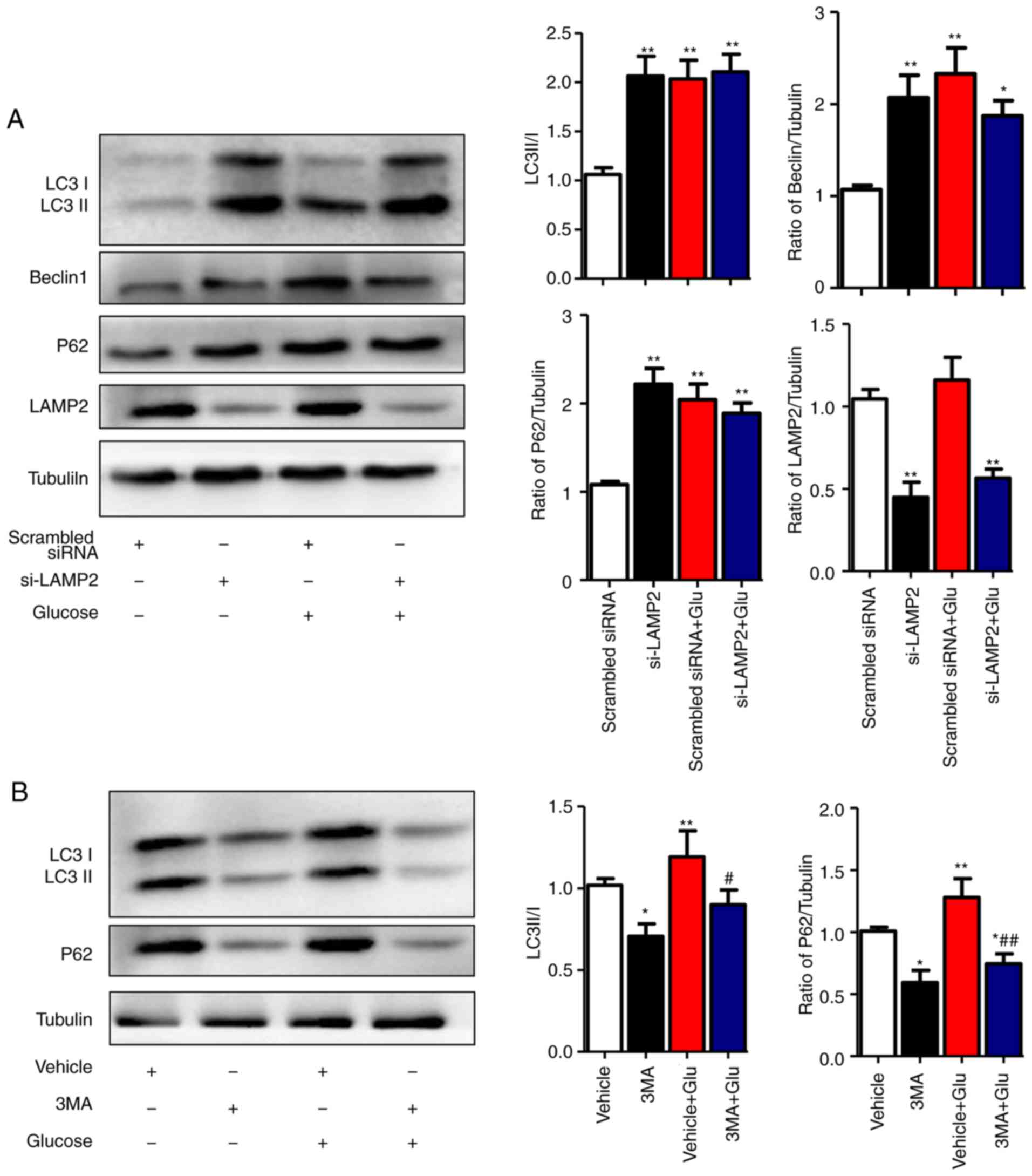 | Figure 4High glucose inhibits the degradation
stage of autophagy. (A) NRCMs were transfected with LAMP2 siRNA for
48 h and were exposed to high glucose for another 24 h. The
expression of LAMP2, Beclin-1, P62, and LC3II/I in each group was
detected by western blot analysis. (B) NRCMs were pretreated with
3-MA (5 mM) for 24 h and with high glucose for another 24 h; the
expression levels of P62 and LC3II/I in each group were detected by
western blot analysis. Data are expressed as the mean ± standard
error of the mean (n≥6). *P<0.05 and
**P<0.01, vs. Vehicle group; #P<0.05
and ##P<0.01, vs. Glu group. Tubulin was the loading
control. NRCMs, neonatal rat cardiomyocytes; O-GlcNAc, O-linked
β-N-acetylglucosamine; Glu, glucose; siRNA, small interfering RNA;
LAMP2, lysosome-associated membrane protein 2; LC3,
microtubule-associated protein 1 light chain 3α; 3-MA,
3-methyladenine. |
O-GlcNAc modification of SNAP29 inhibits
SNAP29-STX17-VAMP8 complex formation and inhibits
autophagy-mediated degradation
To reveal the specific role of SNAP29 in the
regulation of autophagic flux, co-IP was used to detect
O-GlcNAc-modified SNAP29 and observe differences in the formation
of the SNAP29-STX17-VAMP8 complex. In the rat heart tissues,
SNAP29, but not VAMP8 or STX17, was modified by O-GlcNAc (Fig. 5A). In addition, the level of
O-GlcNAc modification in SNAP29 was markedly increased in the STZ
group, whereas binding of the SNAP29-STX17-VAMP8 complex was
decreased significantly (Fig.
5B-D). High-glucose stimulation did not alter the relevant
expression levels of SNAP29, VAMP8 or STX17 in the NRCMs. These
results suggested that only O-GlcNAc-modified SNAP29 was involved
in the regulation of autophagy. Under continuous high-glucose
stimulation in the presence of the agonist TG, O-GlcNAc
modification of SNAP29 was significantly increased, and the
interactions among SNAP29, STX17 and VAMP8 were significantly
decreased in the NRCMs (Fig.
6A-C). Following incubation with the inhibitor Don, sh-OGT and
ad-OGA, the level of O-GlcNAc-modified SNAP29 was decreased, but
the formation of the SNAP29-VAMP8-STX17 complex was increased
(Fig. 6D-F). The above results
suggested that O-GlcNAc modification of SNAP29 affected the
degradation of autoph-agic flux via negative regulation of
SANP29-VAMP8-STX17 complex formation.
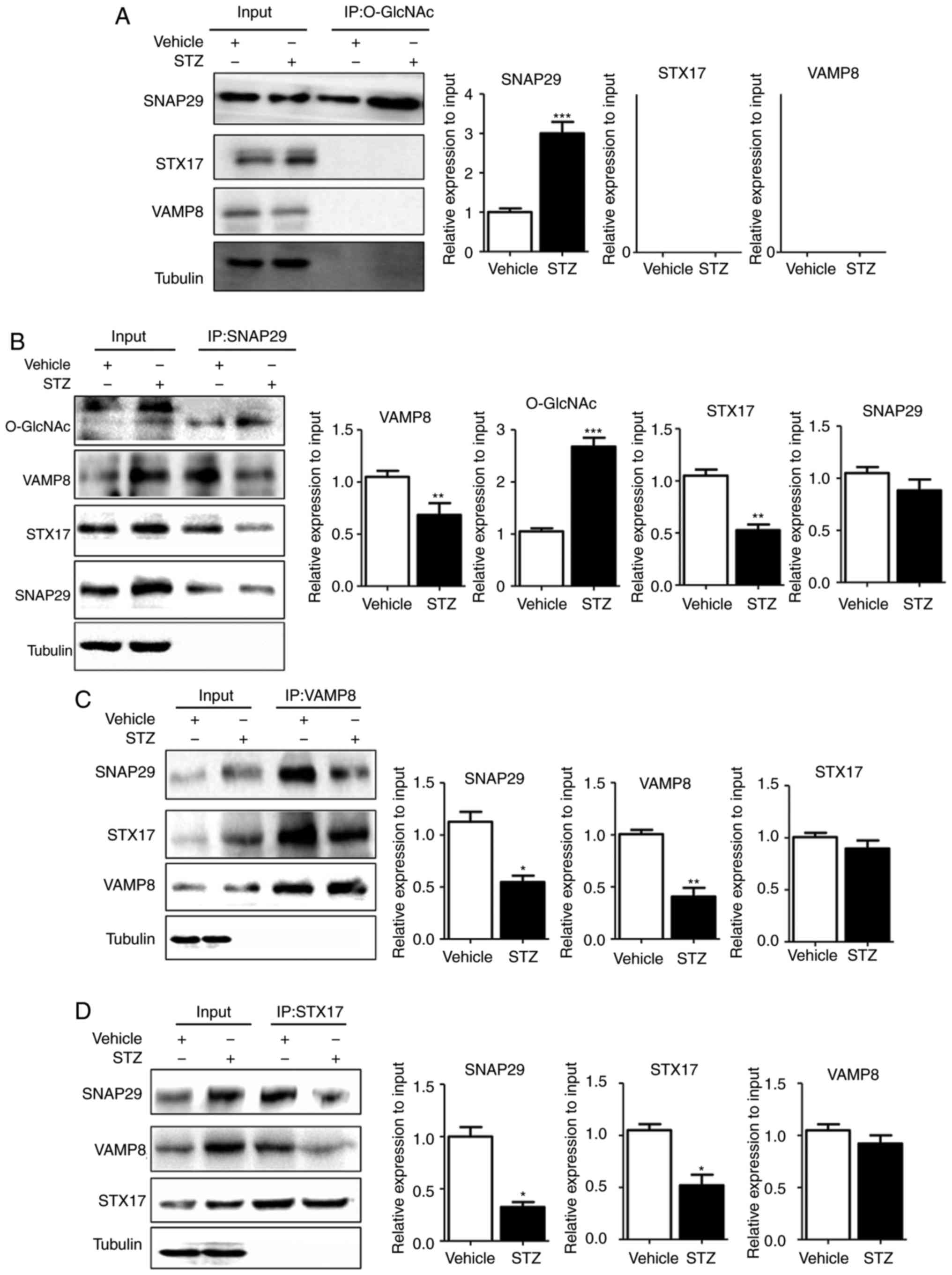 | Figure 5SNAP29 is modified by O-GlcNAc and
form a complex with STX17 and VAMP8. In heart tissues from vehicle
and streptozotocin-induced rats, the interaction between the
O-GlcNAc modification and SNAP29-VAMP8-STX17 was evaluated by
co-IP. (A) O-GlcNAc antibody was used as the known target to pull
down the complex proteins SNAP29, VAMP8 and STX17, and Tubulin was
used as the loading control. (B) Similarly, SNAP29 antibody was
used as pull down the O-GlcNAc, VAMP8 and STX17, SNAP29 and Tubulin
were used as the loading control; (C) VAMP8 antibody was applied to
pull down the complex SNAP29 and STX17, VAMP8 and Tubulin were used
as the loading control. (D) STX17 antibody was used to pull down
the complex SANP29 and VAMP8, both STX17 and Tubulin acted as the
loading control. (*P<0.05, **P<0.01,
***P<0.001 vs. vehicle group). O-GlcNAc, O-linked
β-N-acetylglucosamine; SNAP29, synaptosomal-associated protein 29;
vesicle-associated membrane protein 8; STX17, syntaxin-17; Ctrl,
control; co-IP, coimmunoprecipitation. |
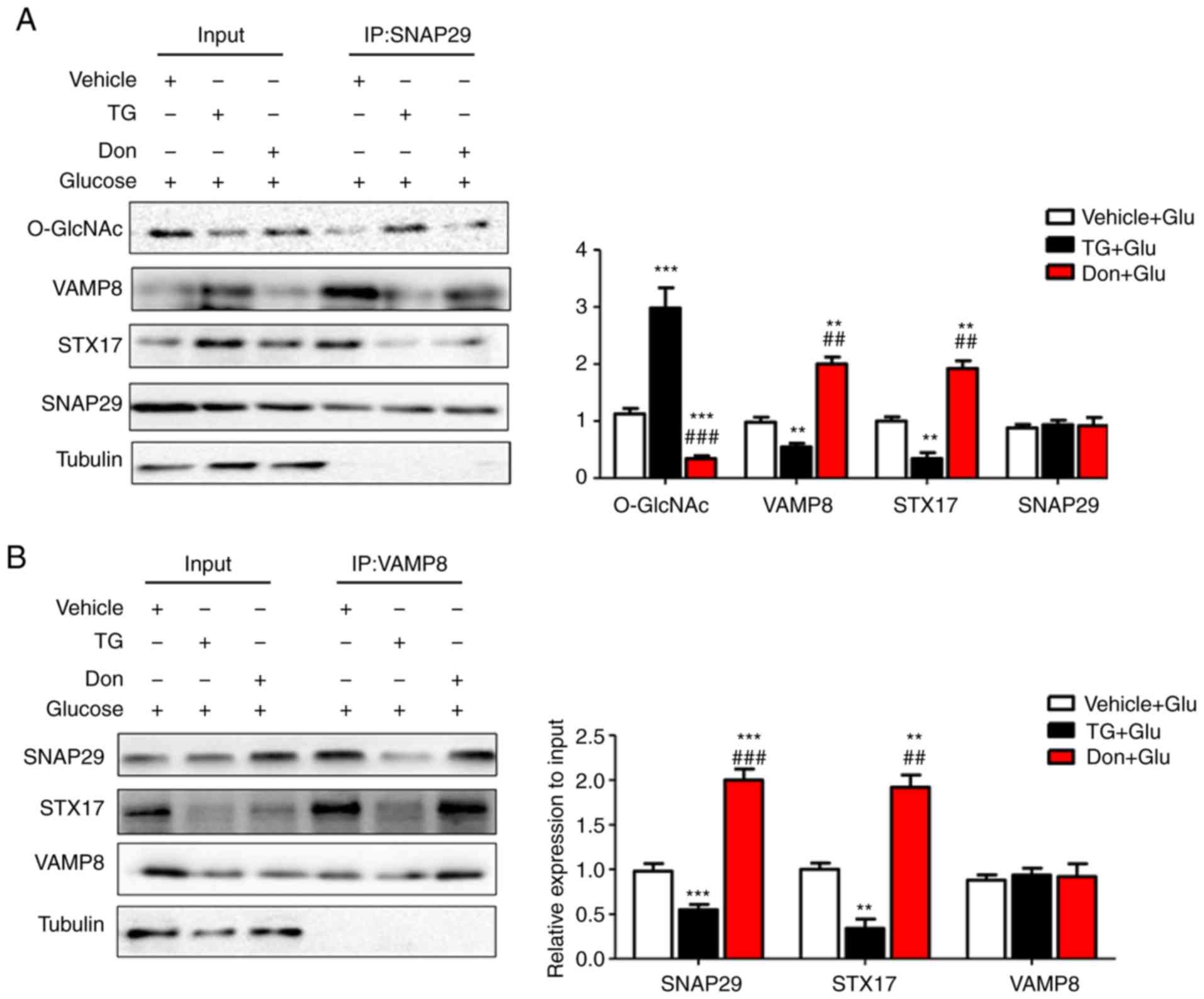 | Figure 6O-GlcNAc modification of SNAP29
inhibits SNAP29-STX17-VAMP8 complex formation and inhibits
autophagy-mediated degradation. NRCMs were exposed to high glucose
(25 mM) and were treated with TG (5 µM) or Don (40
µM) for 24 h. The combination of O-GlcNAc-modified SNAP29
into the SNAP29-STX17-VAMP8 complex was observed by co-IP. (A)
SNAP29 antibody was used as pull down the VAMP8 and STX17, SNAP29
and Tubulin were used as the loading control; (B) VAMP8 antibody
was applied to pull down the complex SNAP29 and STX17, VAMP8 and
Tubulin were used as the loading control. (C) STX17 antibody was
used to pull down the complex SANP29 and VAMP8, STX17 and Tubulin
acted as the loading control. (**P<0.01,
***P<0.001 vs. Vehicle+Glu group;
##P<0.01, ###P<0.001 vs. TG+Glu group);
NRCMs were transfected with sh-OGT or ad-OGA for 48 h and to high
glucose for another 24 h, following which the formation of the
SNAP29-STX17-VAMP8 complex was observed by co-IP. (D) SNAP29
antibody was used as pull down the VAMP8 and STX17, SNAP29 and
Tubulin were used as the loading control; (E) VAMP8 antibody was
applied to pull down the complex SNAP29 and STX17, VAMP8 and
Tubulin were used as the loading control. (F) STX17 antibody was
used to pull down the complex SANP29 and VAMP8, STX17 and Tubulin
acted as the loading control. (*P<0.05,
**P<0.01, ***P<0.001 vs. sh-Ctrl+Glu
group; ##P<0.01, ###P<0.001 vs.
sh-OGT+Glu group); NRCMs, neonatal rat cardiomyocytes; O-GlcNAc,
O-linked β-N-acetylglucosamine; SNAP29, synaptosomal-associated
protein 29; vesicle-associated membrane protein 8; STX17,
syntaxin-17; Don, 6-diazo-5-oxo-L-norleucine; TG, thiamet G; OGT,
O-GlcNAc transferase; sh-OGT, OGT-knockdown adenovirus; ad-OGA,
OGA-overexpression adeno-virus; Ctrl, control; co-IP,
coimmunoprecipitation. |
Discussion
To the best of our knowledge, the present study is
the first to examine the regulatory mechanism between
O-GlcNAc-modified SNAP29 and autophagy in the progression of
diabetic myocardial injury in an animal model. As shown in Fig. 7, under continuous high-glucose
stimulation, the increased O-GlcNAc modification of SNAP29
effectively inhibited the formation of the SANP29-STX17-VAMP8
complex, which acts as the mediator of autophagosome and lysosome
fusion and inhibits autophagy-mediated degradation, resulting in
myocardial injury in the type I DM heart.
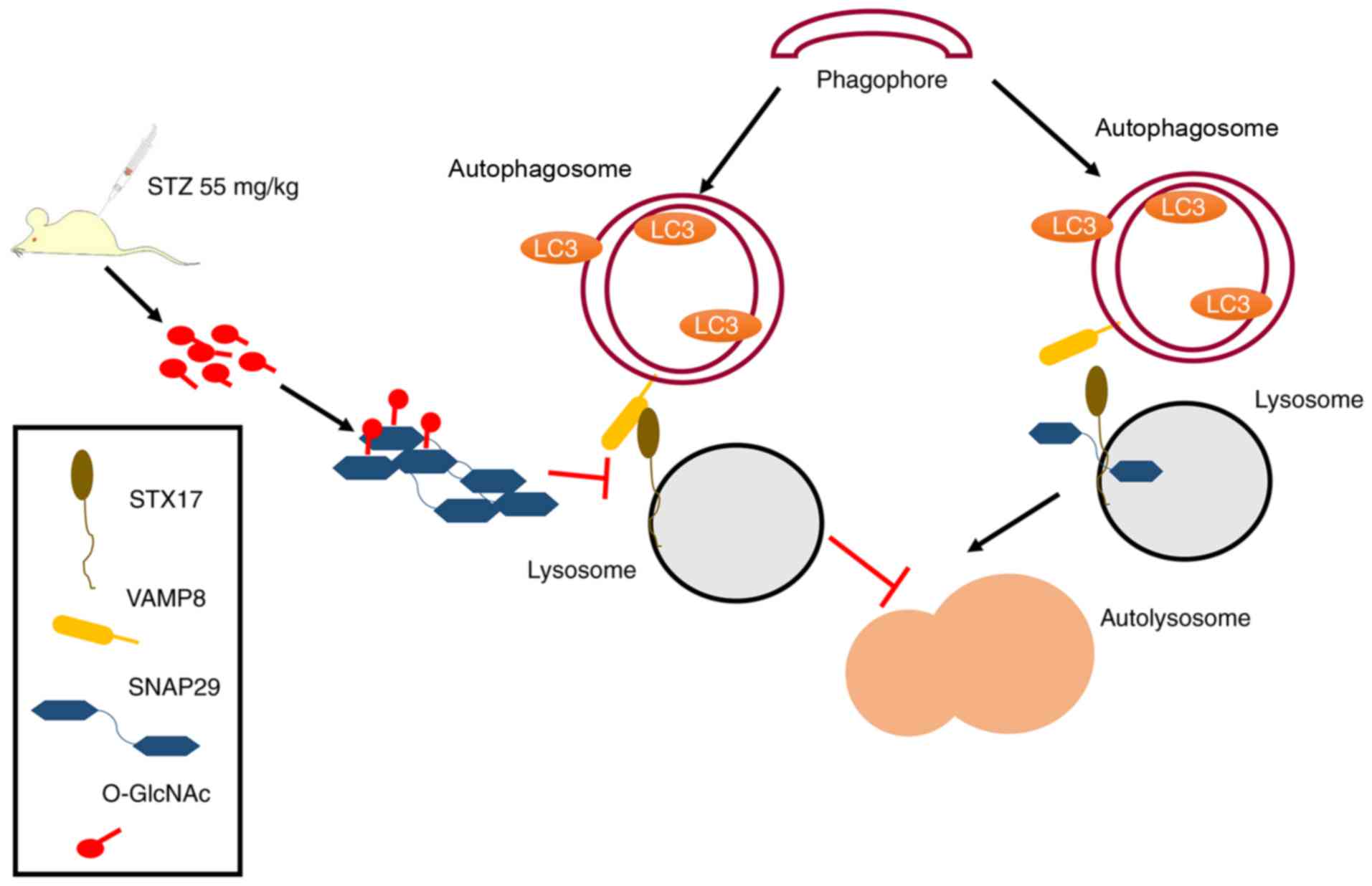 | Figure 7Schematic of the present study,
including a description of the mechanism. Under normal
circumstances, SNAP29 combines with SXT17 and VAMP8, forming a
complex that is involved in the process of autophagosome and
lysosome formation. In STZ-induced type I DM, the increase in
O-GlcNAc-modified SNAP29 inhibits the formation of the
SNAP29-STX17-VAMP8 complex, subsequently affecting autophagosome
and lysosomal membrane fusion, which triggers myocardial injury in
type I DM. O-GlcNAc, O-linked β-N-acetylglucosamine; SNAP29,
synaptosomal-associated protein 29; vesicle-associated membrane
protein 8; STX17, syntaxin-17; LC3, microtubule-associated protein
1 light chain 3α; DM, diabetes mellitus. |
The O-GlcNAc modification is affected by multiple
factors, including intracellular glucose and lipid metabolism.
Disruption of O-GlcNAc modification exerts an important biological
effect on diabetes, various types of cancer, neurodegeneration, and
the cardiovascular system (32).
Enhanced O-GlcNAc modification regulates a variety of molecular
signaling pathways to severely impair conduction-contractile
coupling, the uptake and release of Ca2+, and energy
metabolism of the mitochondria in cardiomyocytes. Consistent with
previous studies, the results of the present study demonstrated
that O-GlcNAc modification was gradually enhanced over time in type
I DM rat hearts. The activation of O-GlcNAc modification aggravated
the disordered cardiomyocyte arrangement, fat accumulation, and
intercellular fibrosis, and the deterioration of cardiac diastolic
function, whereas inhibition of the O-GlcNAc modification improved
cardiac function and ameliorated the abnormalities in myocardial
structure. No significant difference in cardiac systolic function
(predominantly LVEF) was observed between the Vehicle and STZ
groups at 8 weeks post-STZ injection. Due to the different
strategies of STZ injection, there are differences between the
results of the present study and those of other studies. In
general, cardiac dysfunctions are observed in STZ-induced diabetic
rats, including diastolic and systolic dysfunction, which are
dependent on the dose of STZ administration and the duration of
time following induction of STZ. Studies (33,34) have reported that a high dose of
STZ injection (65 mg/kg) resulted in a decreased systolic function
8 weeks post-STZ administration. Generally (35,36), it was not until 11-12 weeks
post-STZ injection that the cardiac systolic function was
decreased.
Low-level autophagy is an important method by which
cardiomyocyte homeostasis is maintained. The change in autophagy in
the DM rats was mainly affected by abnormal energy metabolism
caused by high glucose. The overactivation or inhibition of any
step of the process of autophagy can cause myocardial injury. Under
different conditions, diabetic cardiomyocytes exhibit different
autophagic statuses due to inconsistent detection methods. Studies
have suggested that the oxidation rates of fatty acids, glucose,
and lactate are decreased in type I DM. Furthermore, insufficient
energy due to low levels of ATP causes a cellular starvation state,
and accumulated adenosine monophosphate (AMP) induces an increase
in autophagy and myocardial injury by activating the AMP-activated
protein kinase pathway (37).
However, in a study by Kanamori et al (25), reduced autophagy was observed in
the heart tissues of type I DM mice, and the administration of
metformin prevented high glucose-induced myocardial injury by
activating myocardial autophagy. Studies (38,39) have demonstrated that STZ-induced
type I DM mice exhibit autophagosome accumulation in the heart. The
overactivation or inhibition of any step of autophagy can cause
myocardial injury. The results of the present study also showed
autophagosome accumulation in the STZ-induced type I DM rats and
the significant inhibition of autophagic flow.
The O-GlcNAc modification and autophagy are
regulated by high glucose, and the O-GlcNAc modification can
regulate a variety of pathophysiological processes, including
autophagy. The results of the present study showed that the
autophagic flux of cardiomyocytes was significantly inhibited under
high-glucose conditions, with enhancement of the O-GlcNAc
modification in vivo and in vitro. The present study
also verified that the process of autophagic flux was negatively
regulated by the O-GlcNAc modification in type I DM. The results of
the present study demonstrated for the first time, to the best of
our knowledge, that the formation of autophagosomes in
cardiomyocytes was not affected by changes in the O-GlcNAc
modification; however, the degradation stage of autophagy was
affected by enhancement of the O-GlcNAc modification.
The regulation of autophagy by the O-GlcNAc
modification is achieved by activating a substrate protein. As
shown in previous studies (22,40,41), membrane anchoring and fusion are
central to the release of neurotransmitters by vesicles, but also
in the process of membrane fusion between autophagosomes and
lysosomes and the degradation in autophagic flux. The release of
neurotransmitters is regulated mainly by SNARE protein complexes to
ensure the accurate localization and fusion of the membrane.
Therefore, SNARE complexes may be simultaneously involved in the
regulation of the fusion between autophagosomes and lysosomes. The
newly identified SNAP29 can bind to multiple syntaxin family
members (STX7, STX8, and STX17) and is subsequently distributed on
the plasma membrane and in the cytoplasm (19,20). The SNAP29-STX17-VAMP8 complex
mediates the membrane fusion process between autophagosomes and
lysosomes. Studies (40-43) on SNAP29-mediated autophagy have
mainly focused on the fruit fly, zebrafish, nematode and other
biological models. In another previous study (44), siRNA-mediated silencing of STX17
in cultured human cells was found to result in a disrupted
SNAP29-STX17-VAMP8 complex and inhibited autophagic flux.
However, no single study on SNAP29-mediated
autophagy has been reported in mammalian cardiomyocytes or in
disease models. The present study is the first, to the best of our
knowledge, to demonstrate that SNAP29 is modified by O-GlcNAc in
rat cardiomyocytes. Under high-glucose conditions,
O-GlcNAc-modified SNAP29 was increased in NRCMs, impeding the
formation of the SNAP29-STX17-VAMP8 complex and consequently
inhibiting autophagic flux. By contrast, a reduction in
O-GlcNAc-modified SNAP29 promoted the formation of the
SNAP29-STX17-VAMP8 complex and mediated degradation via autophagic
flux. Therefore, the results of the present study revealed that
O-GlcNAc-modified SNAP29 was increased and that the formation of
the SNAP29-STX17-VAMP8 complex, which is involved in autophagy, was
impaired in type I DM. The findings also demonstrated the
inhibition of autophagic degradation as the underlying mechanism of
myocardial injury in type I DM rats.
Although the present study demonstrated that
O-GlcNAc-modified SNAP29 inhibited autophagy-mediated degradation
in DM-related myocardial injury, the specific site of O-GlcNAc
modification in SNAP29 requires further investigation. Furthermore,
the incidence of type II DM is higher than that of type I DM, and
the pathogenesis and autophagic state are not the same between type
I and type II DM. The present study focused on the heart of type I
DM rats; therefore, whether the results are generalizable to type
II DM requires further investigation. The observed effect on the
SNAP29-STX17-VAMP8 complex requires validation via protein
knockdown experiments, to provide more evidence to support the
findings of the present study.
In conclusion, O-GlcNAc-modified SNAP29 inhibited
autophagic flux by inhibiting the formation of the
SNAP29-STX17-VAMP8 complex, which is involved in the process of
myocardial injury in type I DM. Elucidation of this mechanism
clarifies the regulatory mechanism between the O-GlcNAc
modification and autophagy in myocardial injury in type I DM rats.
The inhibition of O-GlcNAc-modified SNAP29 can improve autophagic
flux; therefore, O-GlcNAc-modified SNAP29 may be a potential
therapeutic target for DM-related myocardial injury.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81400188 to JZH,
81360031 to YHT, 81500257 to RW and 81860152 to LH), the Natural
Science Foundation of Jiangxi (grant nos. 20151BBB70266 and
20161BAB215242 to KH and LH, respectively) and the Innovation Fund
Project in Jiangxi Province (grant no. YC2016-B020 to PiY).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
LH and PiY analyzed the data and wrote the
manuscript; PeY, QK and ZX looked after the animals and performed
western blot and co-IP analyses. YS performed the echocardiography
and XY performed the histological examination of the hearts. JY and
RW were primarily responsible for primary neonatal rat
cardiomyocyte isolation and cell culture, and managed the drug
treatments. KH performed data analysis and contributed to image
editing. YT and JH conceived and designed the study, and gave the
final approval of the version to be published. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics and Experimentation Committee of Nanchang University and
were performed in accordance with the ʻGuide for the Care and Use
of Laboratory Animalsʼ (revised 1996). The experimental protocol
was approved by the Second Affiliated Hospital of Nanchang
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
O-GlcNAc
|
O-linked β-N-acetylglucosamine
|
|
SNAP29
|
synaptosomal-associated protein 29
|
|
STZ
|
streptozotocin
|
|
TG
|
thiamet G
|
|
Don
|
6-diazo-5-oxo-L-norleucine
|
|
NRCMs
|
neonatal rat cardiomyocytes
|
|
DM
|
diabetes mellitus
|
|
OGT
|
O-GlcNAc transferase
|
|
OGA
|
O-GlcNAcase
|
|
TEM
|
transmission electron microscopy
|
|
co-IP
|
coimmunoprecipitation
|
|
H&E
|
hematoxylin and eosin
|
|
3-MA
|
3-methyladenine
|
|
LAMP2
|
lysosome-associated membrane protein
2
|
|
LC3
|
microtubule-associated protein 1 light
chain 3α
|
|
VAMP8
|
vesicle-associated membrane protein
8
|
|
STX17
|
syntaxin-17
|
Acknowledgments
Not applicable.
References
|
1
|
Huynh K, Bernardo BC, McMullen JR and
Ritchie RH: Diabetic cardiomyopathy: Mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chavali V, Tyagi SC and Mishra PK:
Predictors and prevention of diabetic cardiomyopathy. Diabetes
Metab Syndr Obes. 6:151–160. 2013.PubMed/NCBI
|
|
3
|
Dei CA, Khan SS, Butler J, Mentz RJ, Bonow
RO, Avogaro A, Tschoepe D, Doehner W, Greene SJ, Senni M, et al:
Impact of diabetes on epidemiology, treatment, and outcomes of
patients with heart failure. JACC Heart Fail. 3:136–145. 2015.
View Article : Google Scholar
|
|
4
|
Yi W, Clark PM, Mason DE, Keenan MC, Hill
C, Goddard WA III, Peters EC, Driggers EM and Hsieh-Wilson LC:
Phosphofructokinase 1 glycosylation regulates cell growth and
metabolism. Science. 337:975–980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang P and Hanover JA: Nutrient-driven
O-GlcNAc cycling influences autophagic flux and neurodegenerative
proteotoxicity. Autophagy. 9:604–606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pekkurnaz G, Trinidad JC, Wang X, Kong D
and Schwarz TL: Glucose regulates mitochondrial motility via Milton
modification by O-GlcNAc transferase. Cell. 158:54–68. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ruan HB, Dietrich MO, Liu ZW, Zimmer MR,
Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL and Yang X:
O-GlcNAc transferase enables AgRP neurons to suppress browning of
white fat. Cell. 159:306–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peng C, Zhu Y, Zhang W, Liao Q, Chen Y,
Zhao X, Guo Q, Shen P, Zhen B, Qian X, et al: Regulation of the
Hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol Cell.
68:591–604. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ngoh GA, Facundo HT, Zafir A and Jones SP:
O-GlcNAc signaling in the cardiovascular system. Circ Res.
107:171–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Z, Costa FC, Tan EP, Bushue N,
DiTacchio L, Costello CE, McComb ME, Whelan SA, Peterson KR and
Slawson C: O-Linked N-Acetylglucosamine (O-GlcNAc) Transferase and
O-GlcNAcase Interact with Mi2β Protein at the Aγ-Globin Promoter. J
Biol Chem. 291:15628–15640. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM and
Bers DM: Diabetic hyperglycaemia activates CaMKII and arrhythmias
by O-linked glycosylation. Nature. 502:372–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park MJ, Kim DI, Lim SK, Choi JH, Han HJ,
Yoon KC and Park SH: High glucose-induced O-GlcNAcylated
carbohydrate response element-binding protein (ChREBP) mediates
mesangial cell lipogenesis and fibrosis: The possible role in the
development of diabetic nephropathy. J Biol Chem. 289:13519–13530.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie S, Jin N, Gu J, Shi J, Sun J, Chu D,
Zhang L, Dai CL, Gu JH, Gong CX, et al: O-GlcNAcylation of protein
kinase A catalytic subunits enhances its activity: A mechanism
linked to learning and memory deficits in Alzheimer’s disease.
Aging Cell. 15:455–464. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma J and Hart GW: Protein O-GlcNAcylation
in diabetes and diabetic complications. Expert Rev Proteomics.
10:365–380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Banerjee PS, Ma J and Hart GW:
Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac
mitochondria. Proc Natl Acad Sci USA. 112:6050–6055. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu Y, Belke D, Suarez J, Swanson E, Clark
R, Hoshijima M and Dillmann WH: Adenovirus-mediated overexpression
of O-GlcNAcase improves contractile function in the diabetic heart.
Circ Res. 96:1006–1013. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gustafsson AB and Gottlieb RA: Recycle or
die: The role of autophagy in cardioprotection. J Mol Cell Cardiol.
44:654–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yorimitsu T and Klionsky DJ: Autophagy:
Molecular machinery for self-eating. Cell Death Differ. 12(Suppl
2): S1542–S1552. 2005. View Article : Google Scholar
|
|
19
|
Hohenstein AC and Roche PA: SNAP-29 is a
promiscuous syntaxin-binding SNARE. Biochem Biophys Res Commun.
285:167–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diao J, Liu R, Rong Y, Zhao M, Zhang J,
Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al: ATG14 promotes
membrane tethering and fusion of autophagosomes to endolysosomes.
Nature. 520:563–566. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bernard A and Klionsky DJ: Toward an
understanding of autophagosome-lysosome fusion: The unsuspected
role of ATG14. Autophagy. 11:583–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu R, Zhi X and Zhong Q: ATG14 controls
SNARE-mediated autophagosome fusion with a lysosome. Autophagy.
11:847–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P,
Ma Y, Zhao B, Kovács AL, Zhang Z, et al: O-GlcNAc-modification of
SNAP-29 regulates autophagosome maturation. Nat Cell Biol.
16:1215–1226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bell RC, Carlson JC, Storr KC, Herbert K
and Sivak J: High-fructose feeding of streptozotocin-diabetic rats
is associated with increased cataract formation and increased
oxidative stress in the kidney. Br J Nutr. 84:575–582.
2000.PubMed/NCBI
|
|
25
|
Kanamori H, Takemura G, Goto K, Tsujimoto
A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M,
et al: Autophagic adaptations in diabetic cardiomyopathy differ
between type 1 and type 2 diabetes. Autophagy. 11:1146–1160. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barefield DY, Puckelwartz MJ, Kim EY,
Wilsbacher LD, Vo AH, Waters EA, Earley JU, Hadhazy M,
Dellefave-Castillo L, Pesce LL and McNally EM: Experimental
modeling supports a role for MyBP-HL as a Novel myofilament
component in arrhythmia and dilated cardiomyopathy. Circulation.
136:1477–1491. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riha H, Papoušek F, Neckář J, Pirk J and
Ošťádal B: Effects of isoflurane concentration on basic
echocardiographic parameters of the left ventricle in rats. Physiol
Res. 61:419–423. 2012.PubMed/NCBI
|
|
28
|
Reinecke H, Zhang M, Bartosek T and Murry
CE: Survival, integration, and differentiation of cardiomyocyte
grafts: A study in normal and injured rat hearts. Circulation.
100:193–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peng X, Shao J, Shen Y, Zhou Y, Cao Q, Hu
J, He W, Yu X, Liu X, Marian AJ and Hong K: FAT10 protects cardiac
myocytes against apoptosis. J Mol Cell Cardiol. 59:1–10. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mellor KM, Bell JR, Young MJ, Ritchie RH
and Delbridge LM: Myocardial autophagy activation and suppressed
survival signaling is associated with insulin resistance in
fructose-fed mice. J Mol Cell Cardiol. 50:1035–1043. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tanida I, Wakabayashi M, Kanematsu T,
Minematsu-Ikeguchi N, Sou YS, Hirata M, Ueno T and Kominami E:
Lysosomal turnover of GABARAP-phospholipid conjugate is activated
during differentiation of C2C12 cells to myotubes without
inactivation of the mTor kinase-signaling pathway. Autophagy.
2:264–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Darley-Usmar VM, Ball LE and Chatham JC:
Protein O-linked β-N-acetylglucosamine: A novel effector of
cardiomyocyte metabolism and function. J Mol Cell Cardiol.
52:538–549. 2012. View Article : Google Scholar
|
|
33
|
Jesmin S, Zaedi S, Shimojo N, Iemitsu M,
Masuzawa K, Yamaguchi N, Mowa CN, Maeda S, Hattori Y and Miyauchi
T: Endothelin antagonism normalizes VEGF signaling and cardiac
function in STZ-induced diabetic rat hearts. Am J Physiol
Endocrinol Metab. 292:E1030–E1040. 2007. View Article : Google Scholar
|
|
34
|
Chen ZC, Cheng YZ, Chen LJ, Cheng KC, Li Y
and Cheng J: Increase of ATP-sensitive potassium (K(ATP)) channels
in the heart of type-1 diabetic rats. Cardiovasc DiabetoL.
11:82012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li HT, Wu XD, Davey AK and Wang J:
Antihyperglycemic effects of baicalin on
streptozotocin-nicotinamide induced diabetic rats. Phytother Res.
25:189–194. 2011.
|
|
36
|
Sun D, Shen M, Li J, Li W, Zhang Y, Zhao
L, Zhang Z, Yuan Y, Wang H and Cao F: Cardioprotective effects of
tanshinone IIA pretreatment via kinin B2 receptor-Akt-GSK-3β
dependent pathway in experimental diabetic cardiomyopathy.
Cardiovasc Diabetol. 10:42011. View Article : Google Scholar
|
|
37
|
Qiao L, Guo B, Zhang H, Yang R, Chang L,
Wang Y, Jin X, Liu S and Li Y: The clock gene, brain and muscle
Arnt-like 1, regulates autophagy in high glucose-induced
cardiomyocyte injury. Oncotarget. 8:80612–80624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xie Z, Lau K, Eby B, Lozano P, He C,
Pennington B, Li H, Rathi S, Dong Y, Tian R, et al: Improvement of
cardiac functions by chronic metformin treatment is associated with
enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes.
60:1770–1778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang B, Yang Q, Sun YY, Xing YF, Wang YB,
Lu XT, Bai WW, Liu XQ and Zhao YX: Resveratrol-enhanced autophagic
flux ameliorates myocardial oxidative stress injury in diabetic
mice. J Cell Mol Med. 18:1599–1611. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Q, Frank M, Akiyama M, Shimizu H, Ho
SY, Thisse C, Thisse B, Sprecher E and Uitto J: Abca12-mediated
lipid transport and Snap29-dependent trafficking of lamellar
granules are crucial for epidermal morphogenesis in a zebrafish
model of ichthyosis. Dis Model Mech. 4:777–785. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morelli E, Ginefra P, Mastrodonato V,
Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA and
Vaccari T: Multiple functions of the SNARE protein Snap29 in
autophagy, endocytic, and exocytic trafficking during epithelial
formation in Drosophila. Autophagy. 10:2251–2268. 2014. View Article : Google Scholar
|
|
42
|
Jiu Y, Hasygar K, Tang L, Liu Y, Holmberg
CI, Bürglin TR, Hietakangas V and Jäntti J: par-1, atypical pkc,
and PP2A/B55 sur-6 are implicated in the regulation of
exocyst-mediated membrane trafficking in Caenorhabditis elegans. G3
(Bethesda). 4:173–183. 2014. View Article : Google Scholar
|
|
43
|
Sato M, Saegusa K and Sato K, Hara T,
Harada A and Sato K: Caenorhabditis elegans SNAP-29 is required for
organellar integrity of the endomembrane system and general
exocytosis in intestinal epithelial cells. Mol Biol Cell.
22:2579–2587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Itakura E, Kishi-Itakura C and Mizushima
N: The hairpin-type tail-anchored SNARE syntaxin 17 targets to
autophagosomes for fusion with endosomes/lysosomes. Cell.
151:1256–1269. 2012. View Article : Google Scholar : PubMed/NCBI
|














