Introduction
Rheumatoid arthritis (RA) is the most common chronic
inflammatory joint disease, with a worldwide incidence of 0.5-1%.
It is characterized by synovial inflammation of the joints.
Repeated synovitis can cause joint pain, joint compulsion/stiffness
and swelling, leading to the destruction of articular cartilage and
bone. Furthermore, it may gradually cause joint deformity and
dysfunction (1). Fibroblast-like
synoviocytes (FLS) are an important component of the synovium and
serve a major role in the pathological process of RA. One of the
major pathological features of RA is the abnormal proliferation of
FLS, manifesting as hyperplasia of synovial tissue (2). This is similar to the proliferation
of tumor cells and is termed neoplastic. Reports showed that
insufficient apoptosis and abnormal proliferation of FLS are
observed in RA, and an excess of FLS lead to the various
pathological processes of RA (3).
Thus, promoting apoptosis of FLS and inhibiting proliferation is
the primary focus of current studies surrounding RA treatment.
Aspirin is a nonsteroidal anti-inflammatory drug
(NSAID), and is also known as acetylsalicylic acid (ASA). It
functions as a cyclooxygenase (COX) inhibitor that has been widely
used in anti-oxidation, anti-microbial and anti-inflammatory
treatments, especially for RA. Aspirin has also been utilized for
its function of anti-platelet aggregation and anti-inflammatory
analgesia by inhibiting COX, which ultimately results in the
decreased production of prostaglandins (4). In recent years, numerous studies
have found that aspirin can also inhibit the proliferation of tumor
cells and promote their apoptosis in a concentration- and
time-dependent manner (5,6). These findings will be helpful in
developing the wider use of aspirin and may explain, from a new
perspective, the mechanism of aspirin as pertains to the treatment
of RA.
STAT3 is an important transcription factor that is
stimulated by exogenous signals such as IFN, IL-6 and other growth
factors. The persistent activation of STAT3 can be detected in RA
and the majority of tumor (7,8).
Downstream targets of STAT3 are involved in cell proliferation,
differentiation and apoptosis. It has also been shown that aberrant
activation of STAT3 increases proliferation and tumorigenesis. The
IL6/JAK/STAT3 pathway is also important in the pathogenesis of RA.
NF-κB is an intracellular transcription factor that controls and
participates in cell growth, survival, apoptosis, inflammatory
responses and oncogenesis. It not only serves an important role in
the whole pathological process of RA, but also in the excessive
proliferation and decreased apoptosis of RA-FLS (9).
So far, and to the best of our knowledge, there have
been no reports on the direct effect of aspirin on RA-FLS and its
potential mechanism. In our study, it was found that one of the
ways in which aspirin functions is via promoting apoptosis as well
as inhibiting proliferation of RA-FLS, in a dose-dependent manner.
Thus, a possible mechanism has been proposed herein, by which
aspirin functions to inhibit JAK/STAT3 and NF-κB signaling pathways
in RA-FLS.
Materials and methods
Drugs and reagents
Aspirin was purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany; A2093). It was dissolved in dimethyl
sulfoxide (DMSO); the pH was adjusted to 7.0 by 1 mol/l NaOH and
kept at −20°C for later use. Antibodies against Cyclin D1, P21,
STAT3, p-STAT3, P50 were purchased from Abcam (Cambridge, UK).
Secondary antibodies against GAPDH, Bax, PARP1 were from
Proteintech Group (Wuhan, China). Bcl-2 was from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). P65, p-P65 were from Cell
Signaling Technology, Inc. (Danvers, MA, USA). p-P50 was from
Affinity Company (Changzhou, China).
Cell culture
Primary human RA-FLS were obtained from the BeNa
Culture Collection Company (BNCC340230; Beijing, China). Dulbecco’s
modified Eagle’s medium (DMEM), penicillin/streptomycin and fetal
bovine serum (FBS) were purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cells were cultured in
DMEM supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin (penicillin: 10,000 U/ml, streptomycin:
10,000 µg/ml), at 37°C in a humidified atmosphere of 95% air
and 5% CO2. In this experiment RA-FLS cells were treated
with different concentrations of aspirin (0, DMSO, 1, 2, 5, 10 mM)
in vitro.
Cell proliferation assay
The cells were harvested and then seeded in 96-well
plates (1x104 cells/well) with a total volume of 200
µl culture medium. Cells were divided into 6 groups: Control
group, DMSO-treated group and aspirin-treated groups (1, 2, 5 and
10 mM). Then, cells were incubated for 12, 24 and 48 h at 37°C.
After the cells were treated for indicated times, a Cell Counting
Kit-8 (CCK-8; MedChem Express, China) was used (20 µl per
well) and cells were incubated for 3 h at 37°C. Subsequently, the
plate absorbance was read using an automated microplate
spectrophotometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
at 450 nm. Each experiment was repeated at least three times.
Cell apoptosis flow cytometry
analysis
Aspirin-induced apoptosis was detected using an
Annexin V-FITC Apoptosis Assay Kit (Hanbio, Shanghai, China).
Treated cells (0, DMSO; 1, 2, 5 and 10 mM aspirin for 24 h) were
harvested. Annexin V-FITC and propidium iodide (PI) were used based
on the manufacturer’s instructions. Annexin V specifically binds to
the phosphatidyl serine (PS) residues on the cell membrane, and
FITC is a marker of Annexin V; while PI binds to DNA once the cell
membrane becomes permeable. The cells were stained, and the data
were analyzed using BD Accuri C6 Plus (Becton-Dickinson, San Jose,
CA, USA) software. Each experiment was repeated at least three
times.
Cell cycle analysis
Cells were plated in parallel in 35-mm2
culture plates at a concentration of 1x106 cells/plate.
After 24 h of serum starvation, cells were exposed to aspirin for
various durations (0, DMSO; 1, 2, 5 and 10 mM) and then were
harvested by trypsinization, washed twice in cool PBS and placed in
75% ethanol overnight at 4°C. Following this, cells were incubated
in solution with the DNA-binding dye propidium iodide (PI) and
RNase A (KeyGEN Biotech, Nanjing, China) for 30 min at 37°C in the
dark. Finally, red fluorescence from 488 mm laser-excited PI in
every cell was analyzed using a flow cytometer (Becton-Dickinson)
using a peak fluorescence gate to discriminate aggregates. The
percentage of cells in the G0/G1, S and
G2/M phases was determined from DNA content histograms
created with BD Accuri C6 Plus software. Each experiment was
repeated at least three times.
Western blot analysis
The 6 groups of cells (2x106) were washed
in PBS and then lysed with RIPA lysis buffer (Beyotime Institute of
Biotechnology, Beijing, China). Cell debris was removed by
centrifugation (Thermo Fisher Scientific, Inc.) at 10,000 x g for
15 min at 4°C, followed by protein concentration measurement using
a BCA assay kit (Beyotime Institute of Biotechnology). Proteins
were denatured by boiling for 5 min prior to electrophoresis, so
that complete depolymerization of proteins could be achieved and a
negative charge added; then, equivalent amounts of protein (30-40
µg) samples were separated via 10-15% SDS-PAGE and
transferred to immobilon polyvinylidene difluoride membranes. The
membranes were blocked with 5% BSA in TBS-T for 1 h, and then
incubated with rabbit anti-Bax (1:2,000 dilution, Proteintech
Group; #S0599-2-lg), anti-Bcl-2 (1:1,000 dilution, Santa Cruz
Biotechnology, Inc.; #sc-7382), anti-PARP1 (1:2,000 dilution,
Proteintech Group; #13371-1-AP), anti-P21 (1:1,000 dilution,
#EPR3993), anti-cyclin D1 (1:10,000 dilution, #EPR2241; both from
Abcam), anti-P65 (1:2,000 dilution; #8242), anti-p-P65 (1:2,000
dilution; #3033; both from Cell Signaling Technology, Inc.),
anti-P50/P105 (1:1,000 dilution, Abcam: #E381), anti-p-P50/105
(1:1,000 dilution, Affinity Company: #AF3219), anti-STAT3 (1:2,000
dilution, #EPR787Y) and anti-p-STAT3 (1:200,000 dilution, #EP2147Y;
both from Abcam) antibodies for 2 h at room temperature. The
membranes were then washed three times with TBST for 10 min each
time, and incubated with horseradish peroxidase-conjugated goat
anti-rabbit secondary antibodies at a dilution of 1:5,000 for 1 h.
After three washes with TBST, the immunoreactive bands were
visualized using an ECL detection system (SmartChemi 420, Beijing,
China). GAPDH served as the loading control. Each experiment was
repeated at least three times.
Drug-target and direct protein targets
(DPT)-associated genes search
The DrugBank 5.0.11 (https://www.DrugBank.ca/) is a database that collects
detailed information about various drugs and associated materials
(10). In the present study, 11
drug targets for aspirin were found using the DrugBank database.
Information was collected on these 11 targets and the gene names
entered into the ‘Multiple Proteins by Names/Identifiers’ search
box in the STRING database (https://STRING-db.org/cgi/input.pl) (11) for primary DPT-associated genes.
Parameters were set in minimum required interaction score: Highest
confidence 0.900, then 300 secondary DPT-interacting proteins.
Finally, 311 genes were collected, and data down-loaded for the
next analysis.
Network generation/visualization,
founding hub gene and functional annotation
Associations between the 311 genes downloaded from
the STRING database were identified using Cytoscape. APP-Cytohubba
in Cytoscape was used to find the top 10 hub genes of the network
by degree. The Database for Annotation Visualization and Integrated
Discovery (DAVID) online tool (https://david.ncifcrf.gov/) was used to conduct
functional and pathway enrichment analyses in the present study
(12,13). Additional GO and KEGG pathway
enrichment analyses were performed to detect the potential
biological functions and pathways of the 311 genes.
Statistical analysis
All experiments were performed at least three times,
and the data (bar graphs) are presented as the means ± standard
deviations (SD). SPSS v.23.0 software (IBM Corp., Armonk, NY, USA)
was used to analyze the data. One-way ANOVA was performed for
multiple group comparisons, and the mean values of different groups
were compared using the Student-Newman-Keuls (SNK) test. P<0.05
was considered to indicate statistically significant
differences.
Results
Visualization of aspirin-linkage networks
by Cytoscape
Aspirin was input to DrugBank, which output
DB00945-aspirin with 11 targets. Aspirin can inhibit PTGS1, PTGS2
and AKR1C1, activate PRKAA1, acetylate TP53, bind to HSPA5, and
antagonize NFKB2; the actions of EDNRA, IKBKB, RPS6KA3 and NFKBIA
were not mentioned but were closely associated with aspirin.
Table I presents detailed
information on the 11 primary DPTs of aspirin. Using the STRING
database, a total of 300 proteins were identified that were
associated with aspirin and its 11 primary DPTs, and a network of
aspirin target-protein interactions was created (Fig. 1A). The 11 primary DPTs and their
secondary DPT-associated proteins are presented in Fig. 1A. Next, Cytohubba in Cytoscape was
used to analyze the top 10 hub genes in this network by degree, and
these are as follows: TP53, UBC, EP300, RELA, AKT1, NFKB1, CDK2,
MYC, CREBBP and NFKBIA (Fig.
1B).
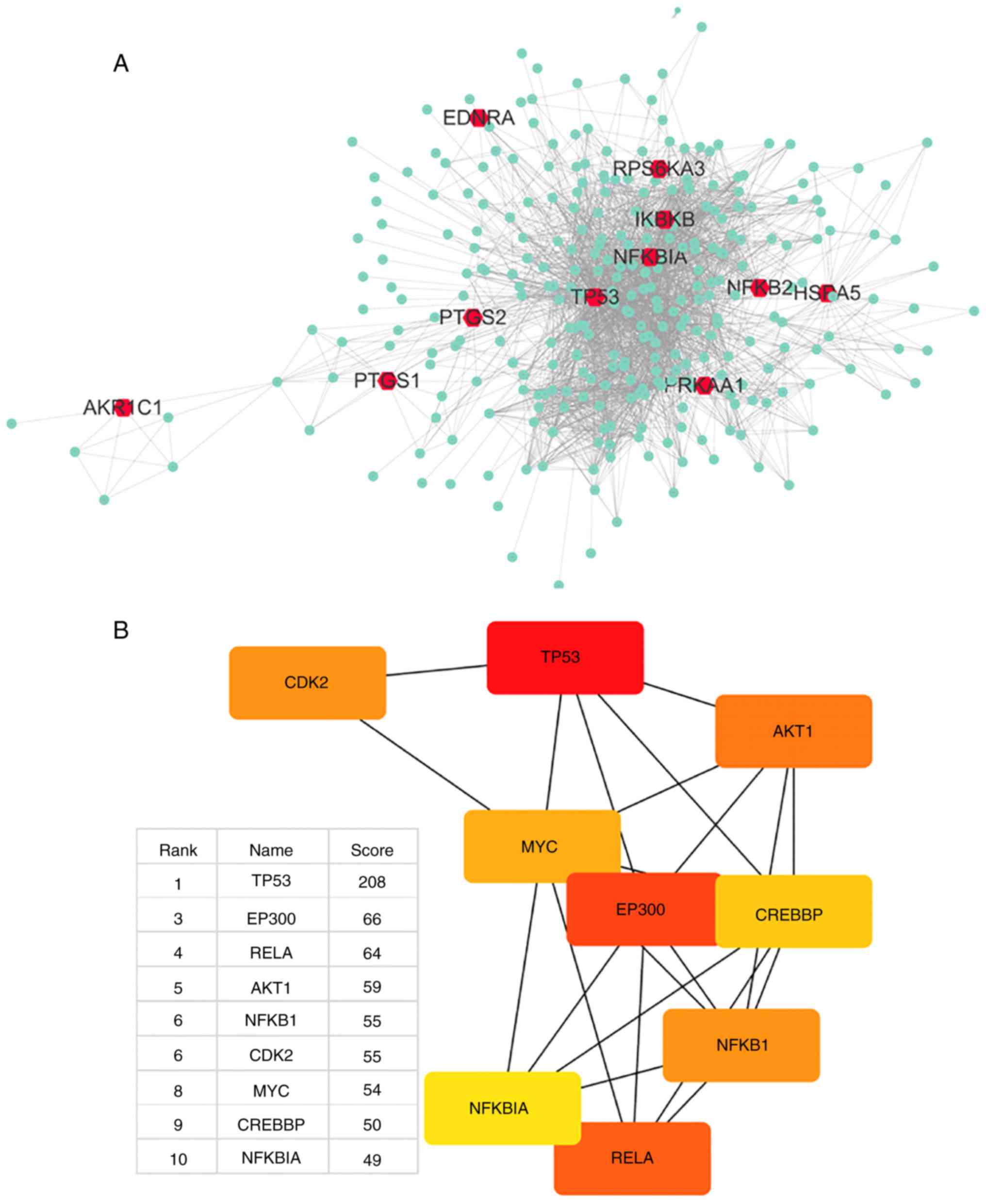 | Figure 1Network of drug-target genes and
DPT-associated genes and their enrichment analysis. (A) DrugBank
was used to find 11 primary and DPTs of aspirin (red); STRING was
used to find 300 secondary DPT-interacting proteins (green);
Cytoscape was used to create a PPI network of all 311 genes. (B)
Cytohubba was used to find network hub genes by degree, and the top
10 hub genes are displayed (red, high score; yellow, low score).
The second hub gene-UBC was not suitable as a hub gene, thus was
removed. DPT, direct protein target; STRING, Search Tool for the
Retrieval of Interacting Genes/Proteins; PPI, protein-protein
interaction. Network of drug-target genes and DPT-associated genes
and their enrichment analysis. (C) DAVID was used to analyze GO
annotations (biological process) of all 311 genes. The top 12 terms
were selected according to P-value. (D) The top 12 KEGG pathways
enriched for all 311 genes were identified using the aforementioned
method. DAVID, Database for Annotation, Visualization and
Integrated Discovery; KEGG, Kyoto Encyclopedia of Genes and
Genomes; GO, gene ontology. |
 | Table IIdentification of direct targets of
aspirin using DrugBank. |
Table I
Identification of direct targets of
aspirin using DrugBank.
| Target | Full name | Uniprot ID | Actions | Related papers
(PMID) |
|---|
| PTGS1 |
Prostaglandin-endoperoxide synthase 1 | P23219 | Inhibitor | 17030227 |
| | | | 17078596 |
| | | | 17131625 |
| | | | 17259075 |
| | | | 17319904 |
| | | | 11752352 |
| PTGS2 |
Prostaglandin-endoperoxide synthase 2 | P35354 | Inhibitor | 17033106 |
| | | | 17037745 |
| | | | 17181859 |
| | | | 17258197 |
| | | | 17301265 |
| | | | 11752352 |
| AKR1C1 | Aldoketo reductase
family 1 member C1 | Q04828 | Inhibitor | 18045204 |
| PRKAA1 | Protein kinase
AMP-activated catalytic subunit alpha 1 | Q13131 | Activator | 22406476 |
| | | | 22517326 |
| EDNRA | Endothelin receptor
type A | P25101 | – | 10727528 |
| IKBKB | Inhibitor of
nuclear factor κB kinase subunit beta | O14920 | – | 9817203 |
| TP53 | Tumor protein
p53 | P04637 | Acetylation | 21475861 |
| HSPA5 | Heat shock protein
family A (Hsp70) member 5 | P11021 | Binding | 11689471 |
| RPS6KA3 | Ribosomal protein
S6 kinase A3 | P51812 | – | 10553090 |
| NFκBIA | NF-κB inhibitor
alpha | P25963 | – | 10553090 |
| NFκB2 | Nuclear factor κB
subunit 2 | Q00653 | Antagonist | 8052854 |
The gene UBC encodes a ubiquitin precursor protein
often found in the interactome database, as almost all proteins are
ubiquitinated and degraded via the proteasome. Thus, it was
considered that UBC is not a specific interaction hub molecule and
was removed from the list of top 10 hub genes. The central position
of the TP53 gene suggests that it may largely be involved in the
biological mechanism of aspirin.
Enrichment analysis of GO functions and
pathways using DAVID
To assess the functional features of
aspirin-mediated gene sets, GO annotations of all 311 genes were
performed. The top 12 terms were selected according to P-value. Top
12 KEGG pathways enrichment was also performed (Fig. 1C and D). A few interesting terms
were selected interested us from the 12 KEGG terms and 12 GO (BP)
terms (Table II). Functional
analysis suggests that aspirin-associated genes are mainly linked
to apoptosis and proliferation (cell cycle). Cytohubba results
showed that TP53 serves the most important role in the network.
Additionally, genes involved in the NF-κB signaling pathway were
revealed to be among the top 10 hub genes (Fig. 1B). The aforementioned evidence
demonstrates a direct association of the biological effects of
aspirin with the p53 axis and NF-κB signaling pathway. Furthermore,
aspirin can directly affect multiple key genes in the NF-κB
signaling pathway, including IKBKB, NFKBIA and NFKB2.
| Table IINotable pathway/biological process
enrichment terms from the 12 significant KEGG/GO terms. |
Table II
Notable pathway/biological process
enrichment terms from the 12 significant KEGG/GO terms.
| KEGG term | P-value | Count | Genes |
|---|
| hsa04115:p53
signaling pathway | 2.99E-36 | 38 | CHEK1, CHEK2, SFN,
PMAIP1, RRM2B, CCNG1, SESN2, PTEN, SESN1, CCNE2, CCNE1, CASP3,
TP53I3, CDKN2A, CASP8, SERPINE1, FAS, RCHY1, TP53AIP1, TP53, ATR,
ATM, CDK2, CCNB1, CCND1, CDKN1A, SERPINB5, BBC3, CD82, BAX, TSC2,
DDB2, MDM2, APAF1, MDM4, PERP, GADD45A, IGFBP3 |
| hsa04068:FoxO
signaling pathway | 1.51E-28 | 43 | PRKAG3, USP7, HRAS,
STK11, PRKAG1, PRKAG2, FOXO1, FOXO3, PTEN, AKT1, PRMT1, PDPK1,
PIK3CA, BCL6, PRKAA1, PRKAA2, CHUK, EGFR, CREBBP, PRKAB2, SKP2,
PRKAB1, SIRT1, IRS1, ATM, CDK2, CCNB1, MAPK1, CCND1, CDKN1A, PLK3,
EP300, CDKN1B, PLK2, CSNK1E, MAPK14, MAPK3, MDM2, SETD7, MAPK9,
MAPK8, IKBKB, GADD45A |
| hsa04064:NF-kappa B
signaling pathway | 6.09E-22 | 31 | TRAF2, TNF, PTGS2,
NFKBIA, NFKB1, TLR4, NFKB2, BCL2L1, MAP3K7, CSNK2A2, TNFRSF1A,
MYD88, CSNK2A1, BCL2, TRAF6, CHUK, IRAK1, BCL10, RELA, RELB, UBE2I,
MALT1, BIRC3, TAB1, TAB2, BIRC2, ATM, LCK, IKBKG, ERC1, IKBKB |
|
hsa04210:Apoptosis | 1.06E-21 | 27 | TRAF2, TNF, NFKBIA,
NFKB1, BCL2L1, AKT1, TNFRSF1A, CASP6, CASP3, BCL2, CASP8, PIK3CA,
FAS, CHUK, RELA, TP53, BIRC3, BIRC2, ATM, TNFRSF10A, TNFRSF10C,
TNFRSF10B, TNFRSF10D, BAX, IKBKG, APAF1, IKBKB |
|
| GO term (BP) | P-value | Count | Genes |
|
|
GO:0042981~regulation of apoptotic
process | 6.81E-74 | 45 | TRAF2, MCL1,
BCL2L1, PMAIP1, CALR, GLS2, CASP6, TNFRSF1A, TRIAP1, TP53I3, CASP8,
BCL3, BCL6, NDRG1, FAS, CASP1, TP53AIP1, BCL10, TP53BP2, CREBBP,
TP53, SKP2, MALT1, BIRC5, WRN, BIRC3, CDK5, BIRC2, BRCA1, ATM,
TNFRSF10A, TNFRSF10C, TNFRSF10B, DUSP1, TNFRSF10D, BBC3, BAX,
BNIP3L, JAK2, PPP1R13B, APAF1, GDF15, PERP, IGFBP3, TP53INP1 |
| GO:0006977~DNA
damage response, signal transduction by p53 class mediator
resulting in cell cycle arrest | 1.02E-32 | 29 | E2F4, PML, AURKA,
CHEK2, SFN, TRIAP1, PRMT1, PCBP4, NPM1, TP53, ARID3A, CDC25C, ATM,
CDK2, CCNB1, CDKN1A, CDKN1B, EP300, PLK3, BTG2, PLK2, BAX, RGCC,
PCNA, UBC, MDM2, MDM4, CARM1, GADD45A |
| GO:0043066~negative
regulation of apoptotic process | 1.21E-31 | 58 | FOXO1, NFKB1,
AURKA, PTEN, AKT1, TRIAP1, CASP3, MYD88, SIN3A, FAS, MYC, EGFR,
IRAK1, RELA, TP53, HMGA2, TNFRSF10D, UBC, MDM2, MAPK8, MDM4, YWHAZ,
MCL1, FHL2, NFKBIA, PRKDC, BCL2L1, SRC, BCL2, NPM1, DNAJA1, BCL3,
TAF9, PRKAA1, HSPA5, PRKAA2, TRAF6, MALT1, BIRC5, RPS6, BIRC3,
SIRT1, BIRC2, RPS6KA3, CDKN1A, HDAC3, HSP90B1, PLK3, CDKN1B, HDAC2,
DUSP1, HDAC1, PLK2, PSMD10, GSK3B, BNIP3L, IKBKB, BARD1 |
| GO:0007050~cell
cycle arrest | 3.12E-26 | 33 | PRKAG3, HRAS,
STK11, PRKAG1, PRKAG2, PML, CALR, CDKN2A, PCBP4, PRKAA1, CAB39,
PRKAA2, MYC, CUL1, KAT2B, MSH2, PRKAB2, TP53, PRKAB1, RB1, RPTOR,
ATM, JMY, CDKN1A, CDKN1B, TSC1, TSC2, ERN1, RHEB, MTOR, GADD45A,
TP53INP1, BARD1 |
| GO:0006974~cellular
response to DNA damage stimulus | 6.03E-24 | 36 | BLM, STK11, FOXO1,
CHEK1, PMAIP1, CHEK2, AKT1, BCL2, BCL3, BCL6, TAF9, DYRK2, TRAF6,
MYC, TAF1, TP53BP1, YY1, TP53, TOPBP1, ATR, WRN, SIRT1, BRCA1, ATM,
RAD50, MAPK1, CCND1, CDKN1A, PLK3, BTG2, OTUB1, BBC3, IKBKG, MAPK3,
SETD7, BARD1 |
Aspirin inhibits RA-FLS cell
proliferation
The effect of aspirin on cell proliferation was
examined by using a CCK-8 to test cell viability. After
administering aspirin, proliferation in each tested group was
significantly and dose-dependently inhibited when compared with the
control. Fig. 2A-C shows the
effects of different concentrations of aspirin on cell viability at
12, 24 and 48 h. It was observed that cell viability gradually
decreased with increasing concentrations of aspirin, and that DMSO
had a slight effect on cell viability, but the effect is not
statistically significant. Fig.
2D shows the percentage reduction in cell proliferation at each
of the three time periods. These results indicate that aspirin can
reduce cell viability and inhibit proliferation in a
concentration-dependent manner.
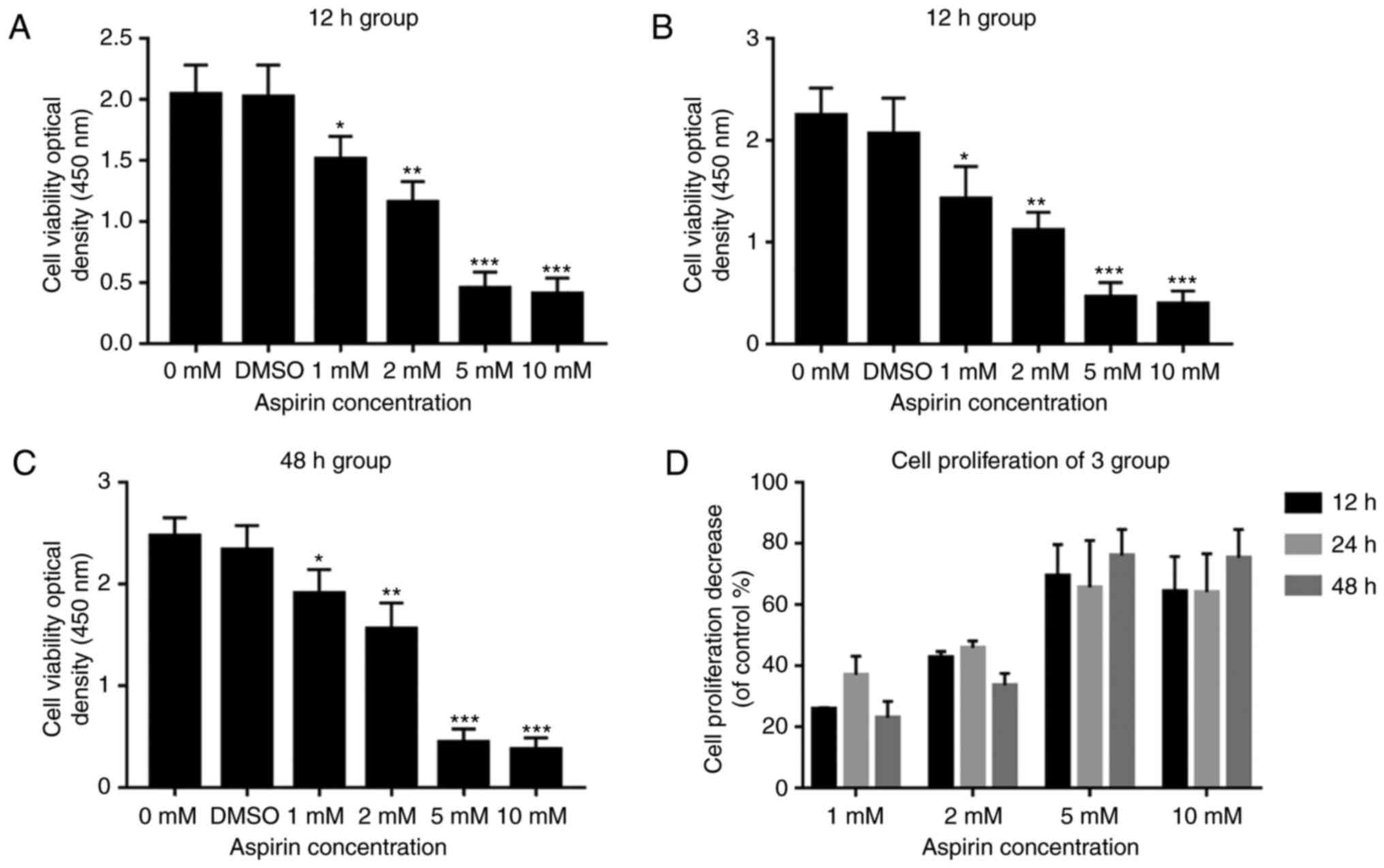 | Figure 2The CCK-8 assay results are presented
as bar graphs for the antiproliferative effects of aspirin on
RA-FLS. (A-C) RA-FLS were treated with (0, DMSO, 1, 2, 5 and 10 mM)
aspirin for 12, 24 and 48 h. At each time interval, cell viability
was determined by CCK-8 analysis. Data are presented as the means ±
SD (error bars) from three independent experiments. Aspirin
inhibited the growth of RA-FLS in a dose-dependent manner. (D) Cell
proliferation decreased at 12, 24 and 48 h. Data are presented as
the means ± SD (error bars) from three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 vs. DMSO group. CCK-8: Cell-Counting
Kit-8; RA-FLS, rheumatoid arthritis-fibroblast-like synoviocytes;
DMSO, dimethyl sulfoxide; SD, standard deviation. |
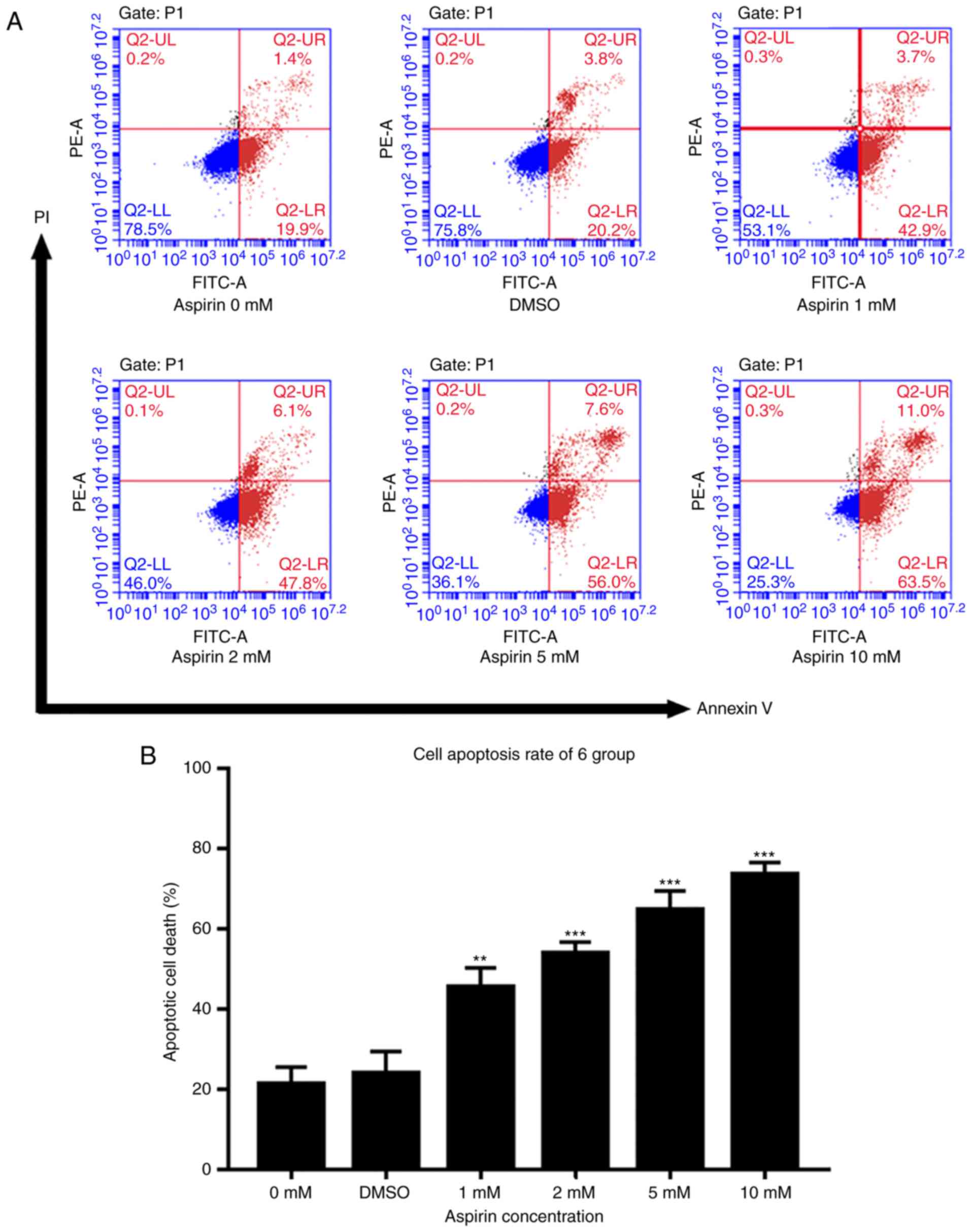
Aspirin triggers RA-FLS cell
apoptosis
An Annexin V- FITC/PI assay and flow cytometry were
used to test the effects of aspirin on cell apoptosis. As described
in Fig. 3A, the percentage of
apoptotic cells in treated groups increased gradually with the drug
concentration. DMSO groups showed a slightly higher percentage of
apoptosis than the control group, but this was not statistically
significant. Fig. 3B shows a
histogram of the apoptotic proportions in three independent
experiments. These results suggest that aspirin can induce
apoptosis in a concentration-dependent manner.
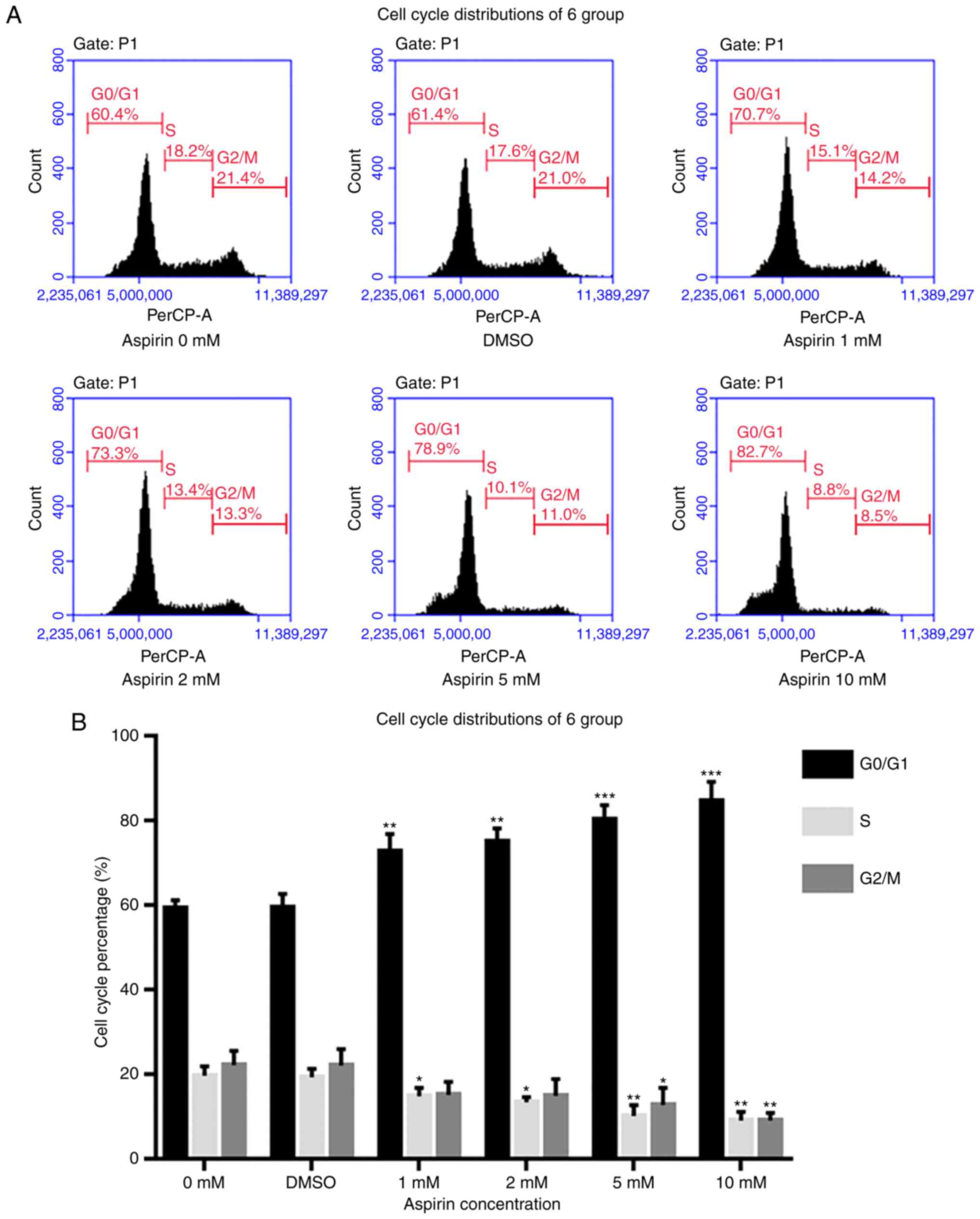
Aspirin induces cell cycle arrest at the
G0/G1 phase and decreases the S phase
fraction in RA-FLS
To further under-stand the role of aspirin in
inhibiting proliferation and to explore its mechanism, flow
cytometry was used to detect changes in cell cycle distribution. As
presented in Fig. 4A, after
aspirin administration, the S phase fraction decreased, while cells
in the G0/G1 phase increased in a
dose-dependent manner. Therefore, it is hypothesized that aspirin
may arrest cells in the G0/G1 phase,
preventing them from entering the S phase and thus decreasing
proliferation.
Aspirin treatment regulates the
expression of Bcl-2, Bax, PARP1, Cyclin D1 and P21 in RA-FLS
As indicated in Fig.
5, Bcl-2 levels decreased after the addition of aspirin, in a
dose-dependent manner (Fig. 5A).
Simultaneously, Bax levels increased with the addition of aspirin
(Fig. 5A). Intracellular PARP1
content also decreased with increasing aspirin concentration
(Fig. 5A). Cyclin D1, which
promotes progression in the cell cycle, exhibited decreased
intracellular levels after the addition of aspirin, in a
concentration-dependent manner (Fig.
5A). P21 was also decreased after the addition of aspirin,
contrary to expectation (Fig.
5A). This suggests that P21 may not serve a role in this
process. The results showed no significant differences between the
DMSO and control groups. Fig. 5B
depicts a bar graph with the gray value analysis for Bcl-2, Bax,
PARP1, Cyclin D1 and P21.
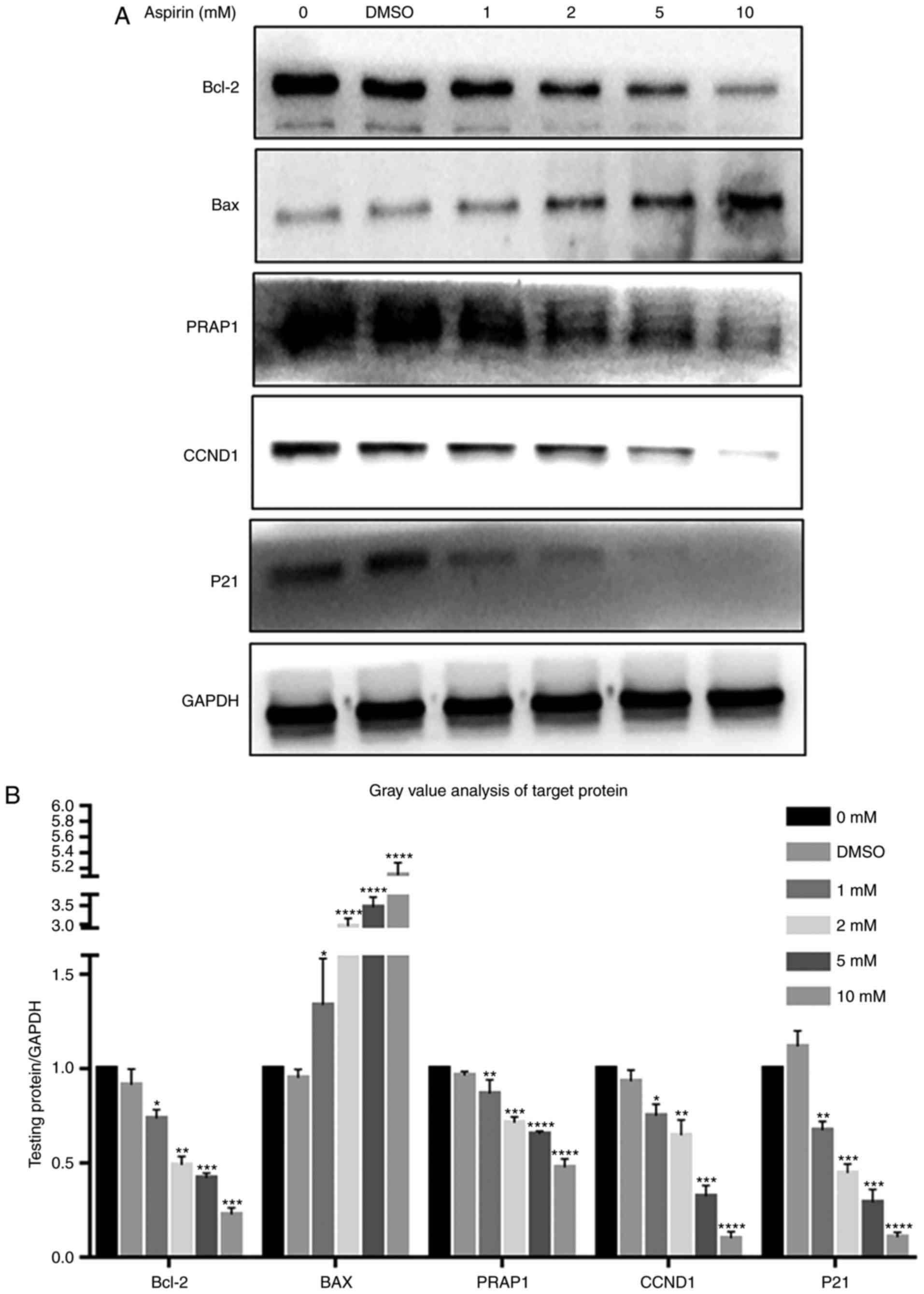 | Figure 5Effects of aspirin on Bcl-2, BAX,
PARP1, Cyclin D1 and P21 expression in RA-FLS. (A) After RA-FLS
were exposed to various concentration of aspirin for 24 h, western
blotting was used to determine Bcl-2, BAX, PARP1, Cyclin D1 and P21
protein levels. The corresponding internal control was GADPH.
Changes in Bcl-2, BAX, PARP1, Cyclin D1 and P21 protein expression
in RA-FLS treated with aspirin are shown: Bcl-2, PARP1, Cyclin D1
and P21 were decreased while BAX was increased, each with
increasing aspirin concentration. (B) Bar graph shows the gray
value analysis of Bcl-2, BAX, PARP1, Cyclin D1 and P21. Data are
presented as the means ± SD (error bars) from three independent
experiments. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001 vs. DMSO
group. We set the value as target protein vs. GAPDH, and set the
value of control group as 1. CCND1, Cyclin D1; RA-FLS, rheumatoid
arthritis-fibroblast-like synoviocytes; Bcl-2, B-cell lymphoma-2;
BAX, Bcl-2-associated X protein; PARP1, poly (ADP-ribose)
polymerase 1. |
Aspirin downregulates JAK/STAT3 and NF-κB
signaling by inhibiting STAT3 and P65, P50 phosphorylation in
RA-FLS
Protein levels of STAT3, p-STAT3, P65, p-P65, P50,
p-P50, P105 and p-P105 were investigated via western blotting. It
was observed that the JAK/STAT3 and NF-κB signaling pathways were
each inhibited after the addition of aspirin. p-P65, p-P50 and
p-STAT3 levels decreased gradually with an increase in aspirin
concentration, while the P65, P50, P105 and p-P105 levels were not
affected (Fig. 6A). Fig. 6B depicts a bar graph representing
protein expression. STAT3 was not affected, while p-STAT3 levels
decreased (Fig. 7A); Fig. 7B shows the protein expression in a
bar plot. This indicates that aspirin inhibits the phosphorylation
of P65, P50 and STAT3, thereby inhibiting JAK/STAT3 and NF-κB
signal transduction; this inhibitory effect of phosphorylation was
concentration-dependent. The DMSO group was similar to the control,
and the differences between them were not statistically
significant.
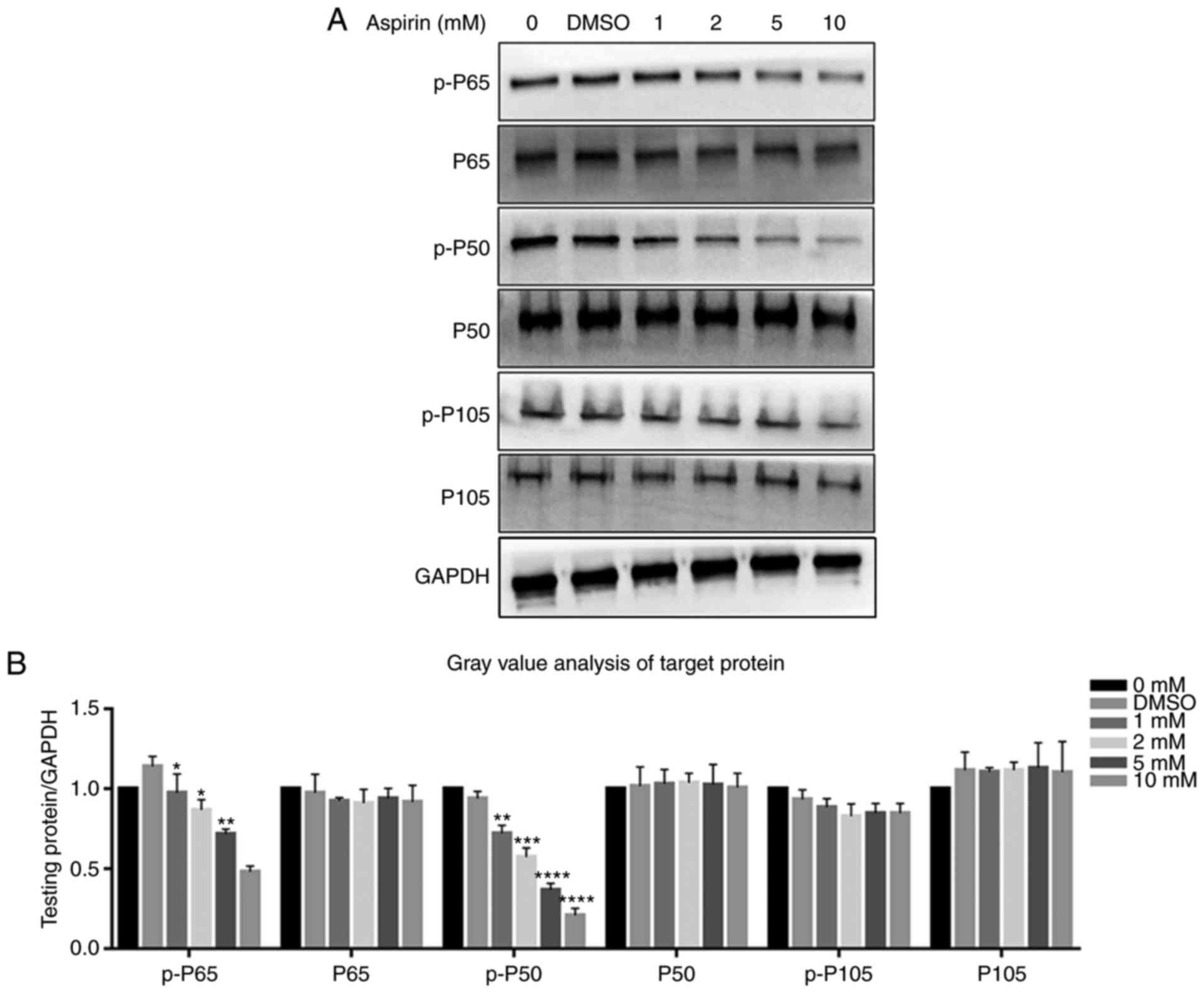 | Figure 6Effects of aspirin on the NF-κB
signaling pathway. It was observed that aspirin significantly
affects the phosphorylation levels of P65 and P50. (A) Cells were
treated with various concentrations of aspirin for 24 h, and then
whole cell lysates were obtained and subjected to western blotting
to detect p-P65, P65, p-P50, P50, p-P105 and P105. GAPDH served as
the loading control. The levels of p-P65 and p-P50 decreased,
whilst P65, P50, p-P105 and P105 remained the same. Phosphorylation
of P65 and P50 was inhibited to varying degrees by aspirin. (B) Bar
graph shows p-P65, P65, p-P50, P50, p-P105 and P105 protein
expression, which was analyzed relative to GAPDH expression by
densitometry. Data are presented as the means ± SD (error bars)
from three independent experiments. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001 vs. the DMSO group. We set the value as
target protein vs. GAPDH, and set the value of control group as 1.
RA-FLS, rheumatoid arthritis-fibroblast-like synoviocytes; NF-κB,
nuclear transcription factor-κB; DMSO, dimethyl sulfoxide; p-,
phosphorylated. |
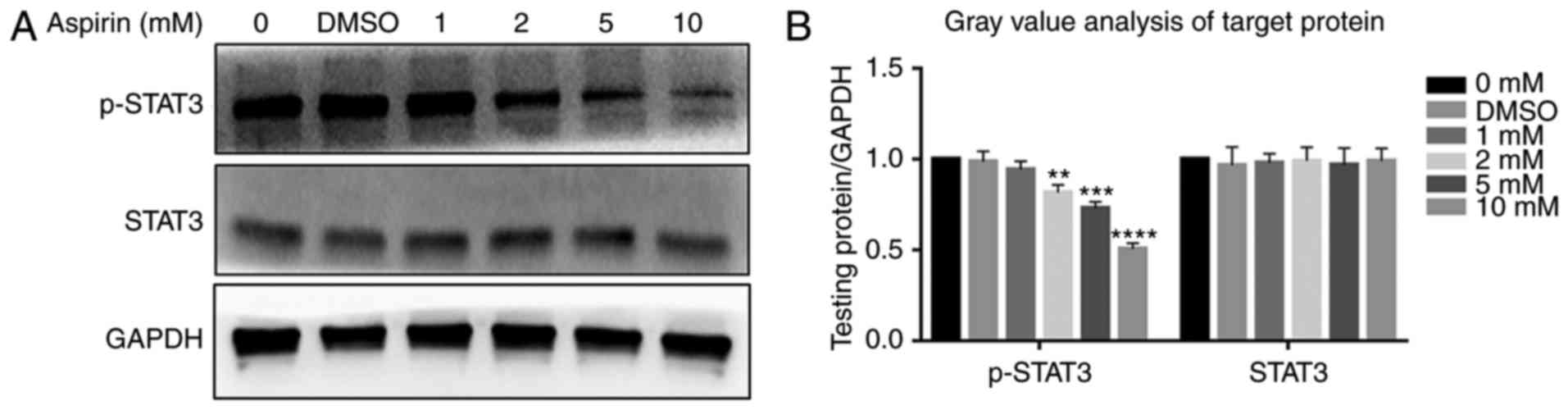 | Figure 7Effects of aspirin on the JAK/STAT3
signaling pathway. Aspirin significantly affects the
phosphorylation level of STAT3. (A) Cells were treated with various
concentrations of aspirin for 24 h, after which whole cell lysates
were obtained and subjected to western blotting to detect p-STAT3
and STAT3. GAPDH served as the loading control. Results showed a
decrease in p-STAT3, whilst STAT3 remained unchanged.
Phosphorylation of STAT3 was inhibited to varying degrees by
aspirin. (B) Bar graph represents the expression of p-PSTAT3 and
STAT3, which were analyzed relative to GAPDH expression by
densitometry. Data are presented as the means ± SD (error bars)
from three independent experiments. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001 vs. DMSO group. We set the value as
target protein vs. GAPDH and set the value of control group as 1.
RA-FLS, rheumatoid arthritis-fibroblast-like synoviocytes; STAT3,
signal transducer and activator of transcription 3; DMSO, dimethyl
sulfoxide; p-, phosphorylated. |
Discussion
Aspirin and other NSAIDs are commonly used as anti-
inflammatory and analgesic drugs (14). As research progresses, other
functions of aspirin are being discovered, such as its potential in
cancer prevention (15,16). In vivo, low doses (75-150
mg/day) of aspirin can prevent and treat thrombosis, though higher
doses of aspirin (3,000-6,000 mg/day) are required for treating RA.
High doses of aspirin can cause serious side effects, while low
doses may be less effective; reports have shown that for long-term
use, 50-160 mg/day is optimal (17). In vitro experiments
demonstrated that the maximum effective dose of aspirin varies
between different cells, but generally remains in the 5-10 mM
range; it may even reach 20 mM in cervical cancer cells (18). Therefore, four doses, 1, 2, 5 and
10 mM, were selected using the literature and preliminary
experiments. The majority of in vivo experiments regarding
aspirin in RA have been about its anti-inflammatory effects on FLS
and immunocytes, resulting in a reduction in inflammatory factors
(19). In RA, abnormal excessive
inflammatory factors lead to abnormal FLS proliferation; aspirin
has anti-inflammation effects that reduce these inflammation
factors and block the associated regulation of FLS, resulting in
decreased abnormal proliferation. However, the direct effects of
aspirin on FLS have not yet been studied, thus the current study
aimed to investigate the antitumor effects of aspirin, particularly
the mechanisms by which aspirin can inhibit proliferation and
promote the apoptosis of multiple tumor cell types. Bioinformatics
analyses were used to investigate biological processes and
signaling pathways in which aspirin is involved.
Functional/activity network (FAN) analysis of gene-phenotype
connectivity liaised by aspirin (20) was used, producing 10 genes: TP53,
EP300, RELA, AKT1, NFKB1, CDK2, MYC, CREBBP and NFKBIA. Many of
these genes are associated with cell proliferation and apoptosis.
TP53 is an important gene involved in proliferation (21), indicating that aspirin affects
cell proliferation (22).
Furthermore, RELA, NFKB1 and NFKBIA are important molecules in the
NF-κB signaling pathway. Therefore, we hypothesized that the NF-κB
signaling pathway is significantly regulated by aspirin, which has
been confirmed in multiple myeloma cells using in vivo and
in vitro experiments (23). Enrichment analysis results
revealed that many biological processes and pathways affected by
aspirin are associated with cell proliferation (cell cycle) and
apoptosis (Table II). These
results indicate that aspirin has a significant regulatory effect
on cell proliferation and apoptosis. In both GO (BP) and KEGG,
signals associated with the NF-κB cascade were evident, suggesting
that aspirin may regulate cells through this pathway (24). According to these bioinformatics
results, it was initially concluded that aspirin is able to
regulate cell proliferation and apoptosis, mainly through the p53
and NF-κB signaling pathways (25).
FLS are similar to cancer cells without the
restriction of excessive proliferation (26,27). Apoptotic defects are another
important cause of synovial hyperproliferation (28). Aspirin is a first-line treatment
drug in RA, working to prevent the conversion of arachidonic acid
to prostaglandin by inhibiting COX (4). In other words, it serves the role of
an anti-inflammatory analgesic drug. Among the emerging
biotherapeutic approaches to RA is cell-based therapy, which
targets synovial cells. Therefore, it is essential to understand
whether and how aspirin is able to directly affect FLS. Data from
the present study indicate that the activity of RA-FLS decreases
significantly after the addition of aspirin. Decreased RA-FLS was
also noted to persist as the concentration of aspirin increased,
indicating it was concentration-dependent. This result is similar
to the effect of aspirin on glandular tumors (29). Flow cytometry revealed that
apoptosis occurred irrespective of aspirin use; however, after
adding aspirin the degree of apoptosis increased. With increased
aspirin concentration, the level of apoptosis increased relatively,
regardless of whether cells were in the early or late apoptotic
stage. It was speculated that this effect is
concentration-dependent. In colon cancer, aspirin can induce tumor
cell apoptosis (30). There are
two known pathways of apoptosis: The death receptor pathway and the
intrinsic mitochondrial pathway. The intrinsic mitochondrial
pathway is considered to be the more critical pathway of the two in
terms of the mechanism of apoptosis (31-34). Mitochondrial pathway-mediated
apoptosis is largely regulated by the Bcl-2 protein family. Bax
exerts pro-apoptotic effects while Bcl-2 exerts inhibitory effects
on apoptosis (33). In this
study, the expression of Bax protein was significantly increased in
FLS treated with aspirin, while the expression of Bcl-2 protein was
significantly decreased. This resulted in an increased Bax/Bcl-2
protein ratio, which is observed in many other cell types during
external stimulus-induced apoptosis (23). That aspirin treatment
significantly increases the expression of PARP1 protein, relative
to the control, suggests that aspirin-induced FLS apoptosis may be
mediated by the caspase-mitochondrial apoptotic pathway.
We examined the distribution of the cell cycle by
flow cytometry and the expression of cell cycle-associated proteins
was measured via western blotting. According to the results, as
aspirin concentration increased, the G0/G1
phase fraction increased, indicating that aspirin can prevent cells
entering the S phase thus arresting cells in the
G0/G1 phase and affecting proliferation
(35). The molecular mechanisms
of the cell cycle regulation involve cyclin, cyclin-dependent
kinase (CDK) and CDK inhibitor (CKI) interactions (34,36). After adding aspirin, Cyclin D1
decreased in a concentration-dependent manner, demonstrating that
aspirin causes cell cycle arrest in the G0/G1
phase, potentially via the regulation of Cyclin D1 affecting the
normal cell cycle, thus inhibiting proliferation. This is similar
to results obtained with numerous other tumor cell types (36). The P21 gene is a cyclin-dependent
kinase inhibitor able to arrest cells in the G1 phase
(37-40). The results showing the gradual
decrease of P21 following the addition of aspirin are inconsistent
with our expectations, and contrary to those obtained from other
experiments on inhibiting the cell cycle (29). Therefore, there are significant
differences between cell types. We hypothesized that aspirin might
inhibit the cell cycle in RA-FLS not through P21, but via other
molecules such as P27, though further experiments are required for
specificity and for verification.
The IL-6/JAK/STAT3 pathway serves a key role in RA
(41); there are many genes
regulating the cell cycle and apoptosis downstream of the JAK/STAT3
pathway. Therefore, further study of JAK/STAT3 signaling pathway
may reveal the mechanism of aspirin on FLS (41). In the present study, the content
of STAT3 and p-STAT3 was compared using western blotting before and
after the addition of aspirin, revealing that the content of STAT3
was not affected by the addition of aspirin though p-STAT3 levels
decreased with increasing aspirin concentration. These results
suggest that aspirin likely affects the phosphorylation of STAT3,
leading to JAK/STAT3 signaling pathway inhibition. These results
are concordant with those in other tumor cells, in which aspirin
decreased STAT3 phosphorylation by blocking the formation of STAT3
phosphodiesterase (42). Aspirin
inhibits the proliferation of RA-FLS and promotes apoptosis. This
may be associated with blockade of the JAK/STAT3 signaling pathway,
leading to decreased downstream anti-apoptotic and cell cycle
regulatory gene expression.
The NF-κB signaling pathway also serves a key role
in the pathogenesis of RA. NF-κB is an important nuclear
transcription factor in the pathogenesis of RA, which has two
principal mechanisms: Firstly, NF-κB activation can increase the
transcription of inflammatory mediators, and these inflammatory
mediators can, in turn, activate NF-κB expression; both constitute
a positive feedback mechanism leading to the inflammatory response
of RA (43). Secondly, it is able
to block synovial cell apoptosis, resulting in synovial cell
hyperplasia (44). The
transcription factor NF-κB regulates the expression of
anti-apoptosis proteins and antagonizes the TNF-induced apoptosis
of FLS (45). Studies have shown
that aspirin can inhibit NF-κB activity and prevent its transfer to
the nucleus (46). According to
previous bioinformatics results, aspirin may affect the NF-κB
signaling pathway by affecting P65 and P50. Our results showed that
the content of P65, P50, P105 and p-P105 did not change after the
addition of aspirin, while p-P65 and p-P50 levels gradually
decreased with increased aspirin concentration. This indicates that
aspirin affects P65 and P50 phosphorylation, resulting in NF-κB
pathway inhibition. There are anti-apoptotic genes and cell
cycle-promoting genes downstream of the NF-κB pathway (47); therefore, it may be considered
that aspirin can inhibit the phosphorylation of P65 and P50 to
inhibit NF-κB, thus affecting apoptosis and proliferation. This is
consistent with results obtained in osteoclast cells (48).
In summary, our results indicate that aspirin is
capable of promoting apoptosis in RA-FLS, and of inhibiting
proliferation by blocking the cell cycle, in a
concentration-dependent manner. Aspirin was found to reduce STAT3,
P65 and P50 phosphorylation, thereby inhibiting the JAK/STAT3 and
NF-κB signaling pathways. Aspirin promoting apoptosis and
inhibiting proliferation may be achieved by blocking both pathways.
This direct effect of aspirin on RA-FLS, which has not been
previously reported to the best of our knowledge, may provide a
novel perspective for revealing the underlying molecular mechanisms
and exploring new therapeutic targets for RA. However, many
problems remain unsolved that require further study, including the
in vivo effects of aspirin on FLS for which we intend to use
an RA rat model to investigate.
Funding
This study was supported by the National Nature
Science Foundation of China (grant nos. 81470719 and
81611140133).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article, and are available from the
corresponding author on reasonable request.
Authors’ contributions
ML, TH and NA conceived and designed the study; XZ
and HF analyzed the data; JS and DL created the figures and tables;
XZ, HF, JD, JS and DL conducted the experiments; XZ wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Cooles FA and Isaacs JD: Pathophysiology
of rheumatoid arthritis. Curr Opin Rheumatol. 23:233–240. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Müller-Ladner U and Pap T: Pathogenesis of
RA: More than just immune cells. Z Rheumatol. 64:396–401. 2005.In
German. View Article : Google Scholar
|
|
3
|
Pope RM: Apoptosis as a therapeutic tool
in rheumatoid arthritis. Nat Rev Immunol. 2:527–535. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vane JR and Botting RM: The mechanism of
action of aspirin. Thromb Res. 110:255–258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao RS, Rioux N, Castonguay A and You M:
Inhibition of COX-2 and induction of apoptosis: Two determinants of
nonsteroidal anti-inflammatory drugs’ chemopreventive efficacies in
mouse lung tumorigenesis. Exp Lung Res. 26:731–742. 2000.
View Article : Google Scholar
|
|
6
|
Richter M, Weiss M, Weinberger I,
Fürstenberger G and Marian B: Growth inhibition and induction of
apoptosis in colorectal tumor cells by cyclooxygenase inhibitors.
Carcinogenesis. 22:17–25. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krause A, Scaletta N, Ji JD and Ivashkiv
LB: Rheumatoid arthritis synoviocyte survival is dependent on
Stat3. J Immunol. 169:6610–6616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Makarov SS: NF-kappa B in rheumatoid
arthritis: A pivotal regulator of inflammation, hyperplasia and
tissue destruction. Arthritis Res. 3:200–206. 2001. View Article : Google Scholar
|
|
10
|
Wishart DS, Feunang YD, Guo AC, Lo EJ,
Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al:
DrugBank 5.0: A major update to the DrugBank database for 2018.
Nucleic Acids Res. 46:D1074–D1082. 2018. View Article : Google Scholar :
|
|
11
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar
|
|
12
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar
|
|
14
|
Kodela R, Chattopadhyay M, Goswami S, Gan
ZY, Rao PP, Nia KV, Velázquez-Martínez CA and Kashfi K: Positional
isomers of aspirin are equally potent in inhibiting colon cancer
cell growth: Differences in mode of cyclooxygenase inhibition. J
Pharmacol Exp Ther. 345:85–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsieh CC, Hernández-Ledesma B and de Lumen
B: Lunasin, a novel seed peptide, sensitizes human breast cancer
MDA-MB-231 cells to aspirin-arrested cell cycle and induced
apoptosis. Chem Biol Interact. 186:127–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Luna-Bertos E, Ramos-Torrecillas J,
García-Martínez O, Díaz-Rodríguez L and Ruiz C: Effect of aspirin
on cell growth of human MG-63 osteosarcoma line. Sci World J. 2012.
View Article : Google Scholar
|
|
17
|
Cuzick J, Thorat MA, Bosetti C, Brown PH,
Burn J, Cook NR, Ford LG, Jacobs EJ, Jankowski JA, La Vecchia C, et
al: Estimates of benefits and harms of prophylactic use of aspirin
in the general population. Ann Oncol. 26:47–57. 2015. View Article : Google Scholar :
|
|
18
|
Xiang S, Sun Z, He Q, Yan F, Wang Y and
Zhang J: Aspirin inhibits ErbB2 to induce apoptosis in cervical
cancer cells. Med Oncol. 27:379–387. 2010. View Article : Google Scholar
|
|
19
|
Fries JF, Ramey DR, Singh G, Morfeld D,
Bloch DA and Raynauld JP: A reevaluation of aspirin therapy in
rheumatoid arthritis. Arch Intern Med. 153:2465–2471. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsieh TC, Wu ST, Bennett DJ, Doonan BB, Wu
E and Wu JM: lFunctional/activity network (FAN) analysis of
gene-phenotype connectivity liaised by grape polyphenol
resveratrol. Oncotarget. 7:38670–38680. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alshatwi AA: Catechin hydrate suppresses
MCF-7 proliferation through TP53/Caspase-mediated apoptosis. J Exp
Clin Canc Res. 29:1672010. View Article : Google Scholar
|
|
22
|
Clària J, Lee MH and Serhan CN:
Aspirin-triggered lipoxins (15-epi-LX) are generated by the human
lung adenocarcinoma cell line (A549)-neutrophil interactions and
are potent inhibitors of cell proliferation. Mol Med. 2:583–596.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding JH, Yuan LY, Huang RB and Chen GA:
Aspirin inhibits proliferation and induces apoptosis of multiple
myeloma cells through regulation of Bcl-2 and Bax and suppression
of VEGF. Eur J Haematol. 93:329–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kopp E and Ghosh S: Inhibition of Nf-kappa
B by sodium-salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shao J, Fujiwara T, Kadowaki Y, Fukazawa
T, Waku T, Itoshima T, Yamatsuji T, Nishizaki M, Roth JA and Tanaka
N: Overexpression of the wild-type p53 gene inhibits NF-kappaB
activity and synergizes with aspirin to induce apoptosis in human
colon cancer cells. Oncogene. 19:726–736. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gay S, Gay RE and Koopman WJ: Molecular
and cellular mechanisms of joint destruction in
rheumatoid-arthritis: Two cellular mechanisms explain joint
destruction. Ann Rheum Dis. 52:S39–S47. 1993. View Article : Google Scholar
|
|
27
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: Key serveers in rheumatoid
arthritis. Rheumatology (Oxford). 45:669–675. 2006. View Article : Google Scholar
|
|
28
|
Pattacini L, Boiardi L, Casali B and
Salvarani C: Differential effects of anti-TNF-alpha drugs on
fibroblast-like synoviocyte apoptosis. Rheumatology (Oxford).
49:480–489. 2010. View Article : Google Scholar
|
|
29
|
Yang C, Liu J, Wang YX, Tong JJ, Wu YH and
Liu Y: Aspirin inhibits the proliferation of canine mammary gland
tumor cells in vitro and in vivo. Transl Cancer Res. 6:188–197.
2017. View Article : Google Scholar
|
|
30
|
Stark LA, Din FVN, Zwacka RM and Dunlop
MG: Aspirin-induced activation of the NF-kappaB signaling pathway:
A novel mechanism for aspirin-mediated apoptosis in colon cancer
cells. Faseb J. 15:1273–1275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J, Yuan L, Xiao HF, Xiao CX, Wang YT
and Liu XB: Momordin Ic induces HepG2 cell apoptosis through MAPK
and PI3K/Akt-mediated mitochondrial pathways. Apoptosis.
18:751–765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fulda S and Debatin KM: Extrinsic vs.
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Li X, Wang XL, Lau W, Wang Y, Xing
Y, Zhang X, Ma X and Gao F: Ginsenoside Rd attenuates myocardial
ischemia/reperfusion injury via Akt/GSK-3β signaling and
inhi-bition of the mitochondria-dependent apoptotic pathway. PLoS
One. 8:e709562013. View Article : Google Scholar
|
|
34
|
Hunter T: Braking the cycle. Cell.
75:839–841. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan WS, Li JH, Chen JF, Zhu L, Wang Y, Sun
B, Hua B, Guo C and Yan Z: Aspirin inhibits the proliferation of
synovium-derived mesenchymal stem cells by arresting the cell cycle
in the G0/G1 phase. Am J Transl Res. 9:5056–5062. 2017.PubMed/NCBI
|
|
36
|
Ou YQ, Zhu WB, Li Y, Qiu PX, Huang YJ, Xie
J, He SM, Zheng XK, Leng TD, Xu D and Yan GM: Aspirin inhibits
proliferation of gemcitabine-resistant human pancreatic cancer
cells and augments gemcitabine-induced cytotoxicity. Acta Pharmacol
Sin. 31:73–80. 2010. View Article : Google Scholar
|
|
37
|
Liu Y, Martindale JL, Gorospe M and
Holbrook NJ: Regulation of p21WAF1/CIP1 expression through
mitogen-activated protein kinase signaling pathway. Cancer Res.
56:31–35. 1996.PubMed/NCBI
|
|
38
|
Lloyd RV, Erickson LA, Jin L, Kulig E,
Qian X, Cheville JC and Scheithauer BW: p27kip1: A multifunctional
cyclin-dependent kinase inhibitor with prognostic significance in
human cancers. Am J Pathol. 154:313–323. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sherr CJ: The Pezcoller lecture: Cancer
cell cycles revisited. Cancer Res. 60:3689–3695. 2000.PubMed/NCBI
|
|
40
|
Agus DB, Cordon-Cardo C, Fox W, Drobnjak
M, Koff A, Golde DW and Scher HI: Prostate cancer cell cycle
regulators: Response to androgen withdrawal and development of
androgen independence. J Natl Cancer Inst. 91:1869–1876. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu JX, Fei D, Xing J and Du J:
MicroRNA-29a inhibits proliferation and induces apoptosis in
rheumatoid arthritis fibroblast-like synoviocytes by repressing
STAT3. Biomed Pharmacother. 96:173–181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo H, Liu J, Ben Q, Qu Y, Li M, Wang Y,
Chen W and Zhang J: The aspirin-induced long non-coding RNA OLA1P2
blocks phosphorylated STAT3 homodimer formation. Genome Biol.
17:242016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang W, Sun W and Jin L: Caffeic acid
alleviates inflammatory response in rheumatoid arthritis
fibroblast-like synoviocytes by inhibiting phosphorylation of IκB
kinase α/β and IκBα. Int Immunopharmacol. 48:61–66. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ni S, Miao K, Zhou X, Xu N, Li C, Zhu R,
Sun R and Wang Y: The involvement of follistatin-like protein 1 in
osteoarthritis by elevating NF-κB-mediated inflammatory cytokines
and enhancing fibroblast like synoviocyte proliferation. Arthritis
Res Ther. 17:912015. View Article : Google Scholar
|
|
45
|
Kim SK, Park KY, Yoon WC, Park SH, Park
KK, Yoo DH and Choe JY: Melittin enhances apoptosis through
suppression of IL-6/sIL-6R complex-induced NF-κB and STAT3
activation and Bcl-2 expression for human fibroblast-like
synoviocytes in rheumatoid arthritis. Joint Bone Spine. 78:471–477.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen JY and Stark LA: Aspirin prevention
of colorectal cancer: Focus on NF-κB signalling and the nucleolus.
Biomedicines. 5:E432017. View Article : Google Scholar
|
|
47
|
Guesmi F, Prasad S, Tyagi AK and Landoulsi
A: Antinflammatory and anticancer effects of terpenes from oily
fractions of Teucruim alopecurus, blocker of IκBα kinase, through
downregulation of NF-κB activation, potentiation of apoptosis and
suppression of NF-κB-regulated gene expression. Biomed
Pharmacother. 95:1876–1885. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang P, Wu C, Huang XH, Shen CL, Li L,
Zhang W and Yao CZ: Aspirin suppresses TNF-α-induced MMP-9
expression via NF-κB and MAPK signaling pathways in RAW264.7 cells.
Exp Ther Med. 14:5597–5604. 2017.PubMed/NCBI
|