Introduction
The development of liver fibrosis is known to
ultimately led to liver cirrhosis (LC). It is characterized by the
excessive accumulation of extracellular matrix (ECM) and activated
hepatic stellate cells (HSCs) undergo myofibroblast transition,
which is identified by α-smooth muscle actin (α-SMA) expression
(1). Currently there are no
reliable, cost-effective and noninvasive measures available to
treat LC (2). To date, the
exploratory therapy used to treat hepatic fibrosis is the removal
or reduction of scar accumulation in experimental models of chronic
liver injury. Clinical trials evaluating the safety and efficacy of
some of these treatments are anticipated in the coming years.
HSCs are the primary targets of fibrogenic stimuli
in the damaged liver, and are known to be involved in the
pathogenesis of many types of liver disease, particularly in
fibrosis and ECM remodeling for which its key role is well
established. When the liver becomes inflamed or injured by
mechanical stimulation, HSCs rich in retinol are activated and
migrate to the liver injury site (3,4).
On the one hand, the number of activated HSCs increase in the
injured site through paracrine and autocrine effects; however, on
the other hand, these activated HSCs, which assume a
myofibroblast-like phenotype, also transform into muscle fibrotic
cells through morphological and functional changes, which then
migrate and proliferate at the sites of liver injury, perpetuating
hepatic inflammation and increasing the formation of stress fibers
and the accumulation of α-SMA (3,5-7). A
large amount of ECM is then synthesized, secreted and not degraded,
ultimately depositing in the liver. Therefore, activated HSCs are
considered to be major cellular targets in the prevention of the
progression of liver fibrosis. In fact, the majority antifibrotic
treatments that are currently under evaluation have aimed to
inhibit the activation, proliferation or synthetic products of HSCs
(3). The identification of key
cytokines involved in the accumulation process of activated HSCs in
chronic liver diseases and the elucidation of the molecular
mechanisms responsible for increased ECM levels have led to the
development of pathophysiologically based strategies to treat liver
fibrosis (7). Therefore, the
search and development of reliable antifibrotic strategies
targeting HSCs that can prevent, inhibit or reverse hepatic
fibrosis by influencing the expression of pro-fibrogenic factors is
urgently required.
Transforming growth factor-β1 (TGF-β1), a key
inflammatory and fibrogenic peptide growth factor, has complex
biological effects on cells and is a key mediator in the
development of liver fibrosis. TGF-β1 has been shown to be involved
in the activation of HSCs and/or has high expression during liver
fibrogenesis (8). TGF-β1 can
activate fibroblast cells and HSCs, and promote the cell synthesis
of matrix proteins, including fibronectin, and type I, III and IV
collagen; it also prevents the degradation of ECM, leading to a
large number of extracellular ECM deposits, which eventually form
hepatic fibrosis (1,9). In addition, it is also a
multifunctional cytokine that serves various roles in wound
healing, chemotaxis, mito-genesis, apoptosis, migration,
differentiation, ECM synthesis and immunomodulation (9). In experimental disease models, the
inhibition of the TGF-β signaling pathway, which is activated in
response to liver injury, is closely associated with the subsequent
onset of fibrogenesis (10,11).
In our previous study, weak cation-exchange magnetic
bead purification and matrix-assisted laser desorption/ionization
time-of-flight mass spectrometry (MALDI-TOF MS) was used to
identify potential serum markers for LC in protein expression
profiles. A 4,210 Da protein, coded for by the G1 to S
phase transition 2 (GSPT2) gene, was expressed differently between
the LC, hepatocellular carcinoma (HCC) and chronic hepatitis B
groups, but showed no significant difference between the HCC and LC
groups (12). It was later
identified as a eukaryotic peptide chain release factor guanosine
triphosphate-binding subunit known as eukaryotic release factor
(eRF), a part of the eRF3b lysate, including 37 amino acids
(eRF3b-37). eRF3b-37 may be involved in the progression of liver
fibrotic diseases, such as LC, which forms as a result of liver
fibrosis.
In order to clarify the role of eRF3b-37 in the
occurrence and development of liver fibrotic disease, the present
study investigated its effect on the activation state of HSCs,
which was determined by the expression of pro-fibrogenic factors
induced by TGF-β1 in LX-2 cells and its mechanism concerning HSC
proliferation, DNA synthesis, cell apoptosis and migration by
polypeptide synthesis technology in vitro.
Materials and methods
Chemicals
Recombinant human TGF-β1 protein (PeproTech, Inc.,
Rocky Hill, NJ, USA) was used to stimulate LX-2 cells. The eRF3b-37
was synthesized by purification with high performance liquid
chromatography (HPLC) and identification via mass spectrometry,
which was performed by Sangon Biotech Co., Ltd. (Shanghai, China).
The primary antibodies used were as follows: TGF-β1 (cat. no.
gtx110630), collagen I (ColI; cat. no. gtx20292), connective tissue
growth factor (CTGF; cat. no. gtx124232), α-smooth muscle actin
(SMA; cat. no. gtx100034) and glyceraldehyde-3-phosphate
dehy-drogenase (GAPDH; cat. no. gtx100118) were purchased from
GeneTex, Inc. (Irvine, CA, USA). The anti-eRF3b antibody against
human eRF3b was purchased from ProteinTech Group, Inc. (Chicago,
IL, USA; cat. no. 12989-1-AP). Primers for Col1A1, CTGF, α-SMA,
Cyclin D1, Cyclin-dependent kinase 4 (CDK4), P21, B-cell lymphoma 2
(Bcl-2), Bcl-2-associated X (Bax) and Fas mRNA were synthesized by
Beijing Huada Genomics Research Center Co., Ltd. (Beijing,
China).
Cell culture
The LX-2 cell line was a gift from Professor Sun
Dianxing (Bethune International Peace Hospital, Shijiazhuang,
China). LX-2 cells were grown in Dulbecco's modified Eagle's medium
(DMEM; cat. no. 11965-092; Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with 10% (v/v) fetal bovine serum (FBS;
cat. no. 10099141; Invitrogen: Thermo Fisher Scientific, Inc.)
supplemented with 1% penicillin and streptomycin, and incubated at
37˚C and 5% CO2. The cells were sub-cultured upon
reaching 70% confluence every 3 days.
Establishment of the TGF-β1 cell model
and experimental grouping
LX-2 cells during the exponential growth period
following 0.25% trypsin digestion at 37˚C for 1 min were seeded
into 96-well plates at a cell density of 5.0x104/ml, 100
µl/well, for 12 h at 37˚C and were made quiescent by
incubating in medium containing 0.5% FBS (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37˚C for 12 h. Cells were then treated with
TGF-β1 or eRF3b-37 at different concentrations (0.1, 1, 10, 50, 100
or 500 ng/ml) at 37˚C for 24 and 48 h, respectively. A
3-(4,5-dimethyl-2-thiazolyl)-2,5-di-phenyltetrazolium bromide (MTT)
assay was employed to obtain the optimum concentration of
TGF-β1/eRF3b-37. The experiments were repeated six times per
concentration. Following this, a TGF-β1-induced LX-2 cell model was
established using the optimum concentration of TGF-β1 to observe
the inhibitory effect of eRF3b-37 on hepatic pro-fibrogenic
factors. LX-2 cells were divided into three groups with triplicate
dishes per group: Control, TGF and TGF+eRF3b. In the TGF group, the
cells were treated with 10 ng/ml TGF-β1 following starvation with
serum-free DMEM. In the TGF+eRF3b group, the cells were treated
with 10 ng/ml TGF-β1 fat 37˚C or 1 h following starvation, and then
with 100 ng/ml eRF3b-37 at 37˚C for 24 and 48 h. Cells incubated
under the same culture conditions but without stimulation served as
the controls.
Cell proliferation assay
Cell proliferation was determined using the MTT
method. Following cell starvation, as aforementioned, LX-2 cells
were treated with TGF-β1 or TGF-β-eRF3b as aforementioned. At the
designated time (24 or 48 h), 20 µl MTT (5 mg/ml) was added
to each well. The medium was removed 4 h later, then 150 µl
DMSO was added to dissolve the dye for 10 min. The absorbance of
each well was read at 490 nm by a Biotek Microplate Reader (BioTek
Instruments, Inc., Winooski, VT, USA). The experiments were
performed six times per concentration, and the following were
applied:
Proliferation vitality (PV; %)= (A490experimental group/A490control group−1)×100
Inhibition rate (IR; %)=(1−A490experimental group/A490control group)×100
Detection of the cell cycle and
apoptosis
The cell cycle and apoptosis were detected by flow
cytometry (FCM). LX-2 cells in the exponential growth period were
seeded into 6-well culture plates at a density of
5x104/ml and made quiescent as aforementioned. Following
24 or 48 h treatment with TGF-β1 or TGF-β1 combined with eRF3b-37,
respectively, the cells were digested with 0.25% trypsin at 37˚C
for 1 min and collected by centrifugation at 110 x g and 37˚C for 5
min. Cells were washed twice with cooled, sterile phosphate buffer
saline (PBS) and fixed with 500 µl ice-cold 70% ethanol per
105 cells at 4˚C for 24 h. Cells were resuspended in a
staining solution containing DNase-free RNase (100 µg/ml)
and prop-idium iodide (50 µg/ml) was added at a final
concentration of 0.05 mg/ml at 4˚C for 30 min in the dark and
subjected to cell cycle and apoptosis analysis using an Epics XL
flow cytometer (Beckman Coulter, Inc., Brea, CA, USA) with
MultiCycle AV version 3.0 software (Innovative Cell Technologies,
Inc., San Diego, CA, USA) for cell cycle analysis and EXPO™ 32 ADC
version 1.1C software (Beckman Coulter, Inc.) for apoptosis
analysis. The cell proliferating index (PI) was calculated
according to the cell cycles as follows:
PI=[(S+G2/M)/(G0/G1+S+G2/M)]
x100%.
Cell migration viability assay
Cell migrating viability was observed and migrating
distances were measured under a fluorescent microscope. LX-2 cells
were seeded and treated as aforementioned. The migrating distances
of cells (D) in the scratched band were obtained by scratching the
bottom of the dishes with a sterile tip. The cells were then
measured under a fluorescent microscope at 0, 24 and 48 h following
the interventions with TGF-β1 or TGF-β1 combined with eRF3b-37. The
experiments were performed in triplicate in each group. The formula
used for calculating the relative migrating distance was as
follows: Relative Migrating Distance=[(D0-D24/48)/D0] x100%.
Whole cell extracts and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to detect the gene expressions
of pro-fibrogenic factors in cell proliferation, migration,
apoptosis and the cell cycle. LX-2 cells were seeded and the three
groups were treated as aforementioned. Subsequently, total RNA was
extracted at 24 h post-treatment using a total RNA isolation kit
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China).
First strand cDNA was synthesized with the Thermo
Scientific RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.) and the 2720 Thermal Cycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). RT-PCR was performed in a
20 µl reaction mixture containing 1 µl Random Primer,
1 µl RevertAid M-MuLV Reverse Transcriptase (200
U/µl), 2 µl 10 nM dNTP Mix and 1 µl RiboLock
RNase Inhibitor (20 U/µl). RT-PCR was performed with the
following temperature protocol: 25˚C for 5 min, 42˚C for 60 min and
70˚C for 5 min. RT-qPCR quantification was performed using the
SYBR-Green Real-Time kit (Beijing Transgen Biotech Co., Ltd.,
Beijing, China) and the SYBR-Green and Real-Time Rotor-Gene™ 6000
cycler (Corbett Life Science; Qiagen, Inc., Valencia, CA, USA). The
primers used are listed in Table
I. The 20 µl reaction mix contained: 1 µl cDNA,
0.5 µl of each primer and 10 µl 2X SYBR Master Mix.
RT-qPCR was performed with the following thermal cycling
conditions: 94˚C for 30 sec followed by 40 cycles of 94˚C for 5
sec, 58-60˚C for 15 sec and 72˚C for 10 sec. Experiments for each
gene were repeated three times. Relative gene quantities were
obtained using the 2-∆∆Cq method (13) with GAPDH as an internal
reference.
 | Table IPrimer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer sequences
(5'-3') | Fragment length
(bp) |
|---|
| GAPDH | Forward:
TGGTATCGTGGAAGGACTCA | 325 |
| Reverse:
CCAGTAGAGGCAGGGATGAT | |
| GSPT2 | Forward:
GGAGATCCGCTCACACACAA | 230 |
| Reverse:
TCCATGGTCTCGGAACTTGC | |
| TGF-β1 | Forward:
ACAGCAACAATTCCTGGCGATAC | 140 |
| Reverse:
GCTAAGGCGAAAGCCCTCAAT | |
| Col1A1 | Forward:
CCCAGCCACAAAGAGTCTACAT | 183 |
| Reverse:
TCATGGTACCTGAGGCCGTT | |
| CTGF | Forward:
CAACTATGATGCGAGCCAACTG | 286 |
| Reverse:
CACACCCCACAGAACTTAGCC | |
| α-SMA | Forward:
ATCAAGGCCCAAGAAAAGCAAG | 241 |
| Reverse:
CCTCTTCTTCACACATAGCTGG | |
| Cyclin D1 | Forward:
CCCGATGCCAACCTCCTCAA | 184 |
| Reverse:
GGAAGACCTCCTCCTCGCAC | |
| CDK4 | Forward:
TCTGGTGACAAGTGGTGGAAC | 228 |
| Reverse:
TGGTCGGCTTCAGAGTTTCC | |
| P21 | Forward:
CAGGAGACCTCTAAAGACCCCA | 179 |
| Reverse:
GAATACTCCCCACATAGCCCG | |
| Bcl-2 | Forward:
GAACTGGGGGAGGATTGTGG | 183 |
| Reverse:
CCGTACAGTTCCACAAAGGC | |
| Bax | Forward:
CGGGTTGTCGCCCTTTTCTA | 105 |
| Reverse:
GAAGTCCAATGTCCAGCCCA | |
| Fas | Forward:
GCATCTGGACCCTCCTACCT | 186 |
| Reverse:
GAGGACAGGGCTTATGGCAG | |
Protein expression determination
Western blotting was performed to detect
pro-fibrogenic factor expression. Following treatment for 48 h,
LX-2 cells were lysed in Radioimmunoprecipitation assay buffer, and
protein concentration was determined using BCA protein assay kits
(both from Beijing Solarbio Science & Technology Co., Ltd.).
The protein solution was denatured with an equal volume of 2X SDS
loading buffer for 5 min, then an equal quantity of protein (20
µg/lane) was separated by 12% SDS-PAGE and
electro-transferred onto polyvinylidene difluoride membranes.
Following blocking with 5% skimmed milk in PBS at 4˚C overnight,
the blots were incubated with the following specific primary
antibodies: TGF-β1 (1:1,000), ColI (1:1,000), CTGF (1:5,000), α-SMA
(1:1,000), GAPDH (1:10,000) and GSPT2 (1:2,000) overnight at 4˚C,
followed by incubation with a DyLight™ 800-conjugated goat
anti-rabbit immunoglobulin G (H&L) secondary antibody (Rockland
Immunochemicals Inc., Pottstown, PA, USA) diluted in 1% bovine
serum albumin-PBS (cat. no. A604481; Sangon Biotech Co. Ltd.) at
room temperature for 2 h in the dark. The band intensity on the
membrane was measured using an Odyssey Infrared Laser Imaging
System) with Odyssey version 3.0 software (LI-COR Biosciences,
Lincoln, NE, USA), and GAPDH was used as an internal control.
Statistical analysis
Values are expressed as the mean ± standard
deviation from duplicate samples. The differences among means of
>3 groups were tested by one-way analysis of variance followed
by the least significant difference post hoc test. The associations
between two indices were evaluated by correlation analysis. SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA) was used for
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
Validation of the eRF3b-37 sequence and
determination of the optimum dosage of TGF-β1 and eRF3b-37 for LX-2
cells
The amino acid sequence of eRF3b-37 in the NCBI
BLAST protein database (blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins)
is KEQSD FCPWY TGLPF IPYLD NLPNF NRSID GPIRL P (Locus no.
IPI00642097 GSPT2; Fig. 1A).
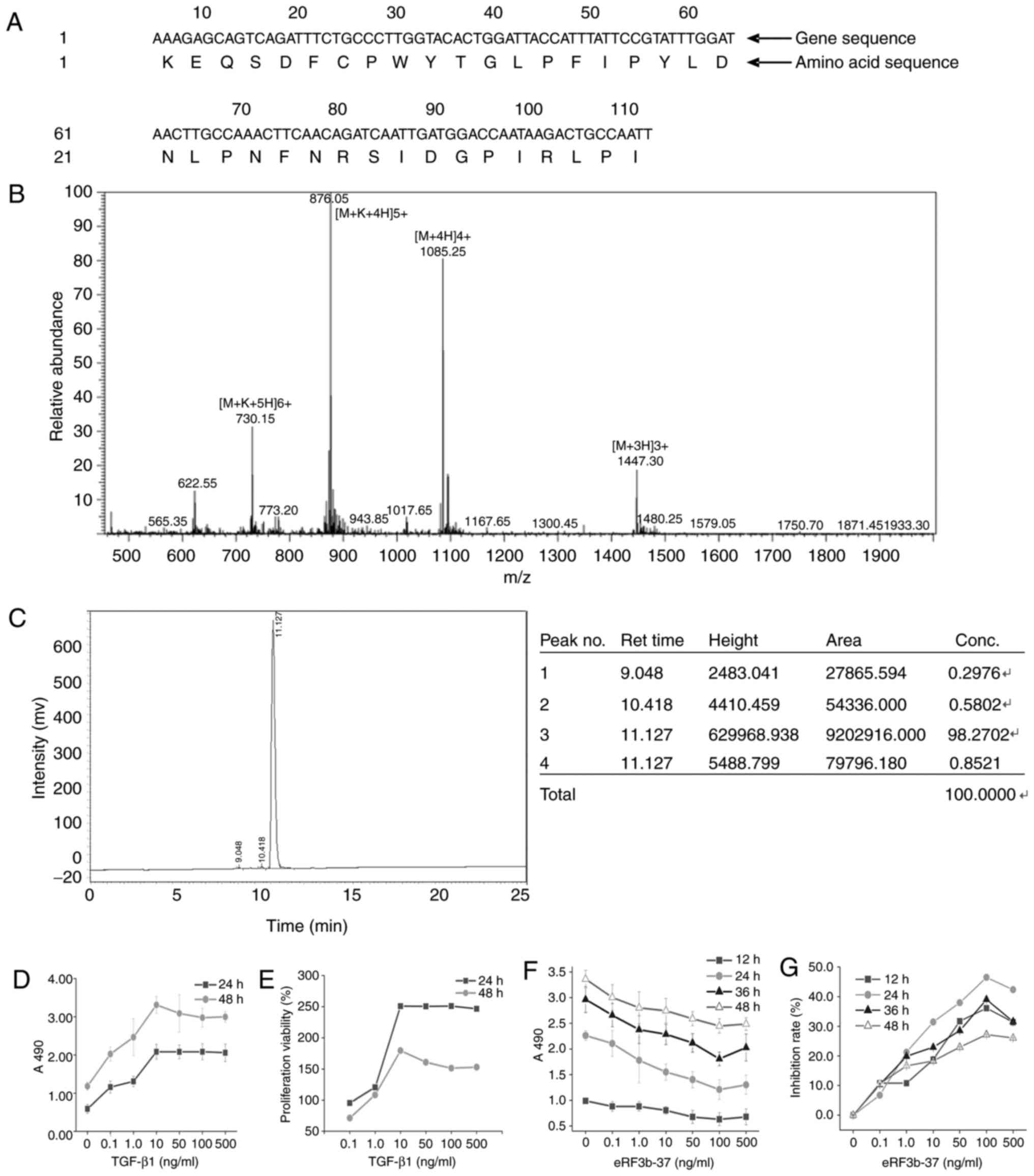 | Figure 1Validation of eRF3b-37 sequences and
determination of the optimum dosage of TGF-β1 and eRF3b-37 for
LX-2. (A) Nucleotide sequences and the corresponding amino acid
alignments of the eRF3b-37 in Homo species were obtained
from the NCBI BLAST protein database. eRF3b-37 identified by (B)
mass-spectrogram and (C) liquid chromatogram satisfied the
requirements of the experiment. (D) The A490 and (E) proliferation
viability of LX-2 cells were determined by MTT following
stimulation with 0, 0.1, 1, 10, 100 or 500 ng/ml TGF-β1: It reached
the plating point at 10 ng/ml at 24 and 48 h. LX-2 cells were
stimulated with 0, 0.1, 1, 10, 100 or 500 ng/ml eRF3b-37, and the
(F) cell proliferation A490 and (G) inhibition rate reached the
optimal level at 100 ng/ml at 12, 24, 36 and 48 h, as determined by
MTT. Data are presented as the mean ± standard deviation of six
independent repeated experiments per group. TGF, transforming
growth factor; eRF3b, eukaryotic peptide chain releasing factor 3b
polypeptide; MTT,
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide. |
The eRF3b-37 mass spectrogram is presented in
Fig. 1B. The charge-mass ratio
was 876.05 at the highest peak in the mass spectrogram and 1,085.25
at the second highest peak. According to the formula: Charge-mass
ratio=(Molecular weight+Charge quality)/Charge number, the
molecular weight was calculated as 4,337.25 and 4,337.00 at the two
peaks, respectively. Their errors were 0.006 and 0.000%,
respectively, which were within the 1% normal range in the
experimental study when compared with the real molecular weight
4,337. The peak figure of eRF3b-37 was specific and obtained by
HPLC. The peak area was 9,202,916.00 and the eRF3b-37 concentration
was 98.27% (Fig. 1C).
In order to identify the optimum dosage of TGF-β1
and eRF3b-37, the present study determined the cell proliferation
activity with and without TGF-β1 and eRF3b-37 treatment.
Proliferation measured at A490 and the proliferation viability of
LX-2 cells treated with TGF-β1 at 0, 0.1, 1, 10, 50, 100 or 500
ng/ml at 24 and 48 h reached the highest point at 10 ng/ml
(Fig. 1D and E). Proliferation
measured at A490 and the inhibition rate of LX-2 cells by eRF3b-37
at 0, 0.1, 1, 10, 50, 100 or 500 ng/ml at 12, 24, 36 and 48 h were
inhibited with a minimum of 100 ng/ml (Fig. 1F and G). The dose of eRF3b-37 was
positively correlated with the cell inhibition rate in the
concentration range of 0.1-100 ng/ml at all 4 time-points; the
absolute values of Pearson correlation coefficient were 0.926,
0.992, 0.946 and 0.968, respectively. Within the range of 0.1-100
ng/ml, the inhibition of LX-2 cells by eRF3b-37 was dose-dependent.
The results indicated that eRF3b-37 was of high purity, satisfying
the requirement of experimental research. Therefore, TGF-β1 and
eRF3b-37 acted on LX-2 with optimum concentrations of 10 and 100
ng/ml, respectively.
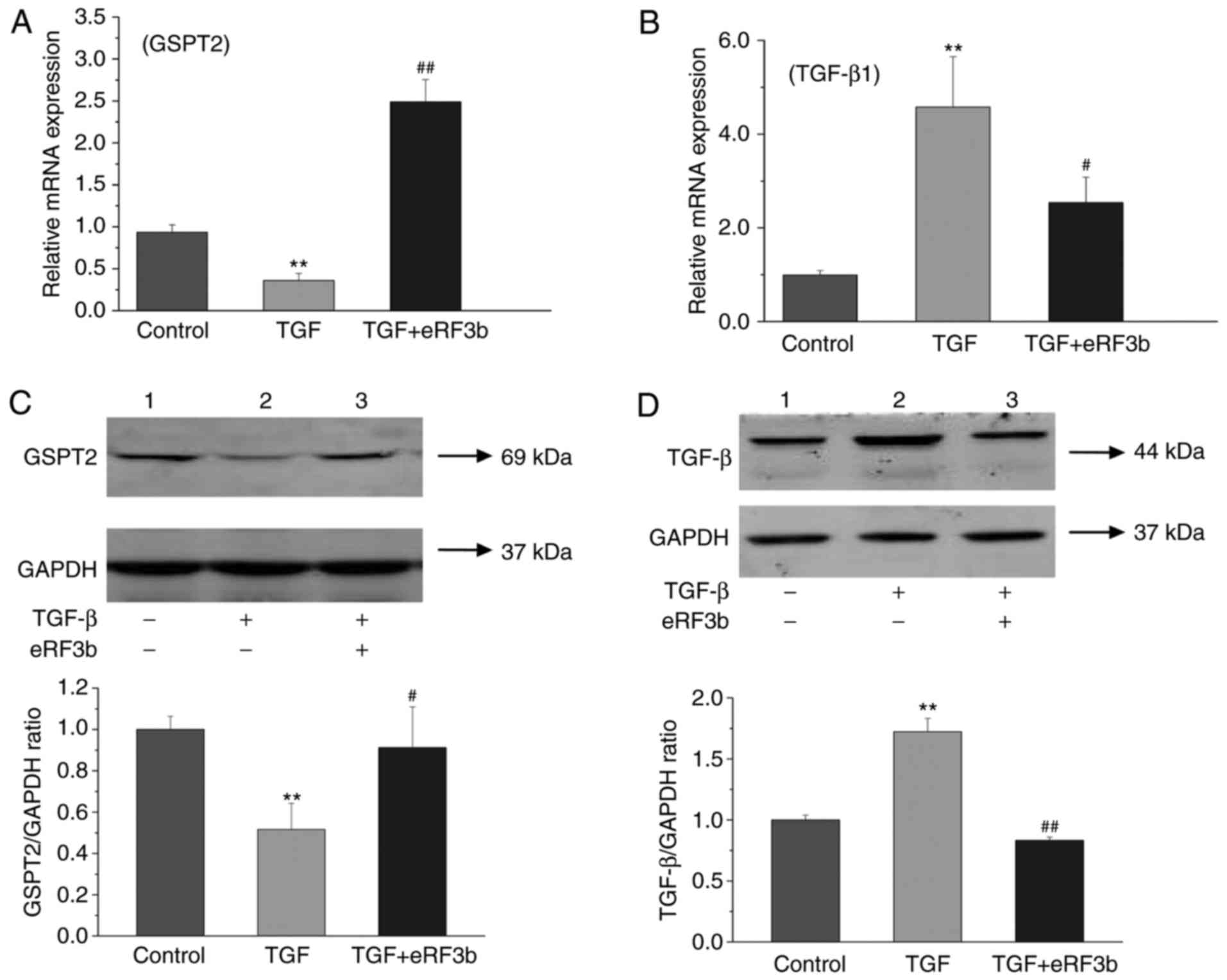
eRF3b-37 promotes GSPT2 and inhibits the
TGF-β1 expression induced by TGF-β1 in LX-2 cells
In order to determine whether eRF3b-37 influences
the expression of GSPT2 and TGF-β1, the cells were divided into
three groups: Control, TGF (10 ng/ml) and TGF (10 ng/ml)+eRF3b (100
ng/ml). In regard to GSPT2, the mRNA and protein expressions of the
TGF group were lower than those observed in the control or
TGF+eRF3b groups at 24 and 48 h following treatment (Fig. 2A and C). With respect to TGF-β1,
the mRNA and protein expressions in the TGF group were markedly
higher when compared with the control and TGF+eRF3b groups
(Fig. 2B and D). The results
revealed that eRF3b-37 treatment increased GSPT2 expression and
inhibited TGF-β1 expression. It is thought that eRF3b-37 may exert
its effects on LX-2 by inhibiting TGF-β signaling. Thus, TGF-β1 was
successfully used to activate HSCs and construct a fibrogenic
expression model of HSCs.
eRF3b-37 decreases the mRNA and protein
expression of pro-fibrogenic factors induced by TGF-β1 in LX-2
cells
To evaluate the effects of eRF3b-37 on HSC
activation, the mRNA and protein expression levels of
pro-fibrogenic factors induced by TGF-β1 in LX-2 cells were
determined. The mRNA and protein expressions of ColI, CTGF and
α-SMA in the TGF group were greater than those observed in the
control or TGF+eRF3b groups (Fig.
3A-F). In addition, the protein expression of GSPT2 increased,
while that of α-SMA decreased with the increased dosage of eRF3b-37
(Fig. 3G). A negative correlation
was also observed between GSPT2 and α-SMA expression, with a
Pearson's correlation coefficient of -0.991 (Fig. 3H). The results revealed that HSC
activation occurred as the TGF-β1-induced increased expression of
ColI, CTGF and α-SMA by was inhibited by eRF3b-37 application;
eRF3b-37 increased GSPT2 expression and inhibited α-SMA expression
dose-dependently in LX-2 cells.
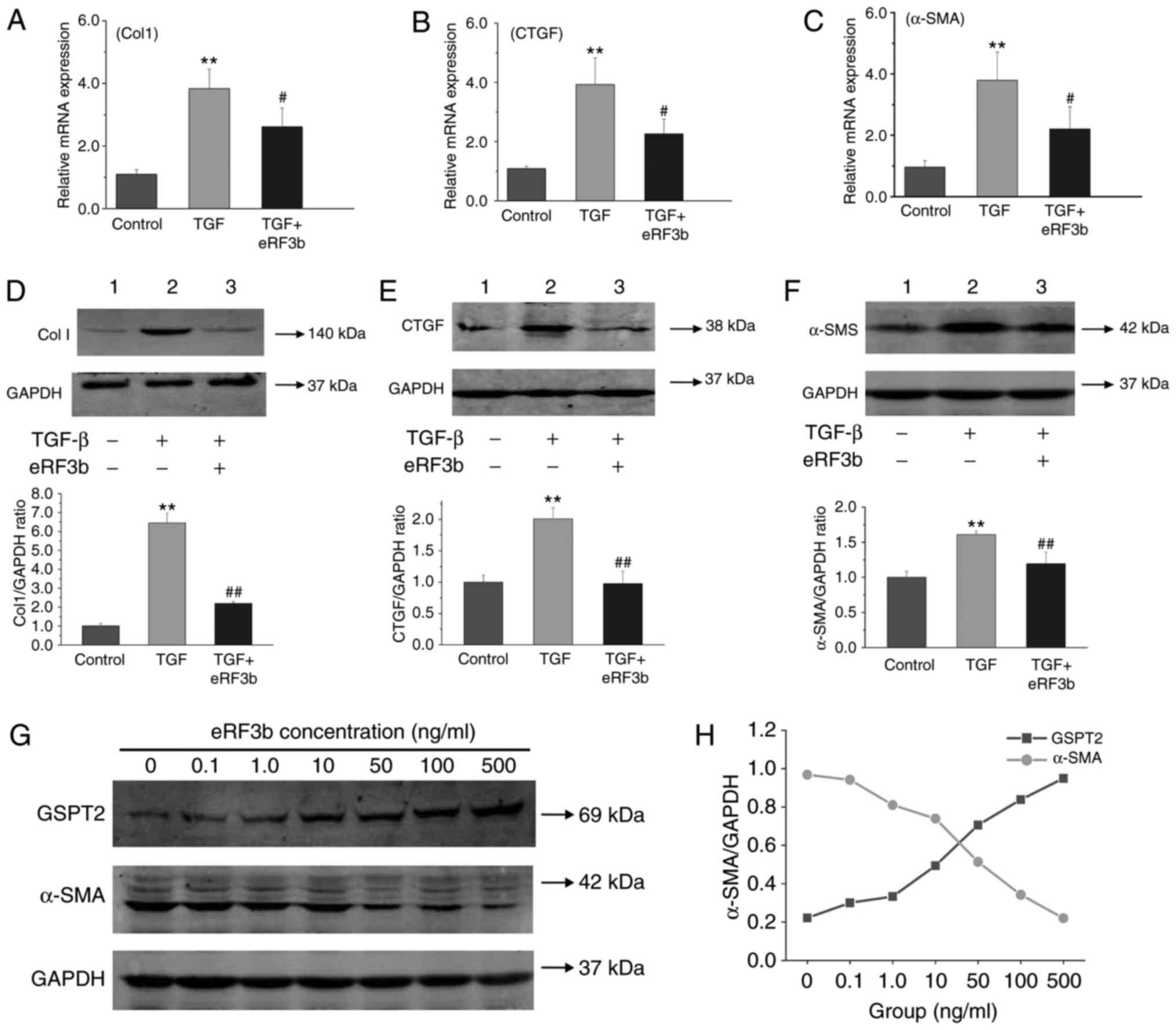 | Figure 3eRF3b-37 decreases the mRNA and
protein levels of the pro-fibrotic factors ColI, CTGF and α-SMA
TGF-β1-stimulated in LX-2 cells. eRF3b-37 inhibited the mRNA
expression of (A) ColI, (B) CTGF and (C) α-SMA, as determined by
reverse transcription-quantitative polymerase chain reaction at 24
h. eRF3b-37 inhibited the protein expression of (D) Col I, (E) CTGF
and (F) α-SMA as determined by western blotting at 48 h. (G) Doses
of 0.1, 1, 10, 100 or 500 ng/ml of eRF3b-37 increased GSPT2
expression and inhibited the expression of α-SMA dose-dependently
at 48 h. (H) Statistical analysis indicated that there was a
negative correlation between GSPT2 and α-SMA protein expression.
Data are presented as the mean ± standard deviation of three
independent repeated experiments per group. **P<0.01
vs. Control group; #P<0.05 and ##P<0.01
vs. TGF group. eRF3b, eukaryotic peptide chain releasing factor 3b
polypeptide; CTGF, connective tissue growth factor; SMA, smooth
muscle actin; TGF, transforming growth factor; Col, collagen;
GSPT2, G1 to S phase transition 2. |
eRF3b-37 inhibits proliferation, promotes
the G0/G1 arrest of the cell cycle and
affects proliferative factor expression in LX-2 cells stimulated
with TGF-β1
To determine the effects of eRF3b-37 on cell
proliferation and the cell cycle when induced by TGF-β1, HSCs were
divided into five experimental groups: Control, TGF, TGF+1.0 ng/ml
eRF3b-37, TGF+10 ng/ml eRF3b-37 and TGF+100 ng/ml eRF3b-37.
eRF3b-37 inhibited LX-2 cell proliferation when stimulated with
TGF-β1, with inhibition rates of 34.80 and 19.90% at 24 and 48 h,
respectively, for the TGF+100 ng/ml eRF3b-37 group (Fig. 4A). Subsequently, the cells were
divided into three groups as aforementioned. In the TGF group,
there were fewer G0/G1 phase cells, while the
number of S cells increased when compared with the control group.
In the TGF+eRF3b group, the number of G0/G1
phase cells increased, while the number of S phase cells decreased
in the TGF group; the number of G2/M phase cells did not
change significantly at 48 h (Fig.
4B-D). The PI of the TGF group was greater than that observed
in control or TGF+eRF3b groups at 48 h (Fig. 4E). Cyclin D1, CDK4 and P21 are
factors that regulate proliferation and cell cycle (14). The mRNA expressions of Cyclin D1
and CDK4 in the TGF group increased when compared with the control
and TGF+eRF3b groups. However, the mRNA expression of P21 in the
TGF group was reduced when compared the control or TGF+eRF3b groups
(Fig. 4F-H). These results
revealed that eRF3b-37 may inhibit cell proliferation viability,
promote G0/G1 arrest and block DNA synthesis
by downregulating the expression of Cyclin D1 and CDK4 and
upregulating the level of P21 in LX-2 cells stimulated with
TGF-β1.
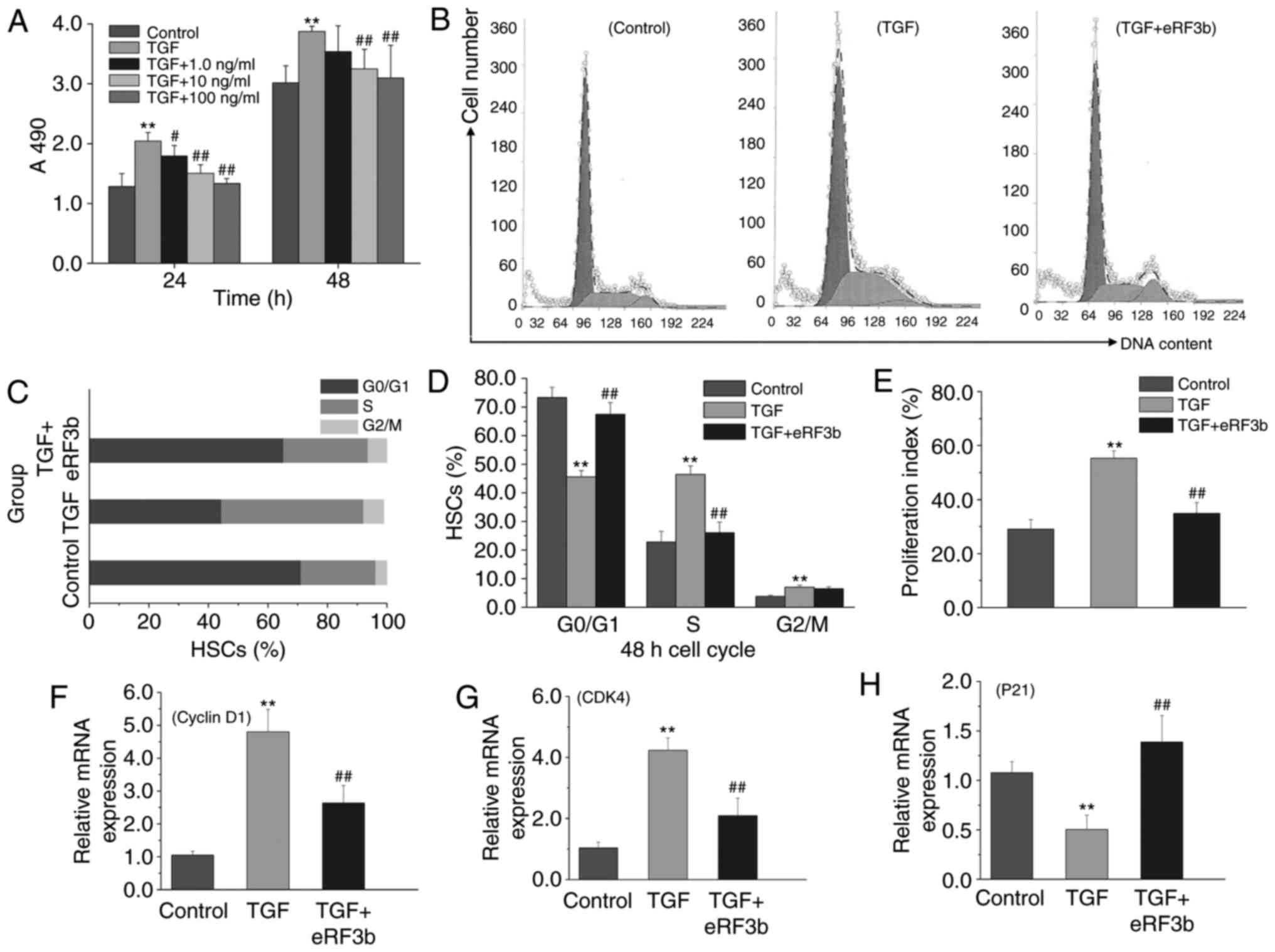 | Figure 4eRF3b-37 inhibits cell proliferation
and promotes G0/G1 cell cycle arrest, as well
as markedly affecting the expression of proliferative factors in
LX-2 cells stimulated with TGF-β1. (A) eRF3b-37 inhibited LX-2
proliferation when stimulated with TGF-β1, as determined by
(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide assay at
24 and 48 h. (B) eRF3b-37 increased the number of
G0/G1 phase cells and downregulated the
number of S phase cells, as determined by FCM. (C) A percentage bar
chart and (D) statistical analysis results presenting the effects
of eRF3b-37 on the cell cycle. (E) eRF3b-37 inhibited the
proliferation index (%) of HSCs stimulated with TGF-β1, as
determined by FCM. eRF3b-37 regulated the mRNA expression of (F)
Cyclin D1, (G) CDK4 and (H) P21, as determined by reverse
transcription-quantitative polymerase chain reaction. Data are
presented as the mean ± standard deviation of three independent
repeated experiments per group. **P<0.01 vs. Control
group; #P<0.05 and ##P<0.01 vs. TGF
group. eRF3b, eukaryotic peptide chain releasing factor 3b
polypeptide; TGF, transforming growth factor; CDK, Cyclin-dependent
kinase; FCM, flow cytometry; HSCs, hepatic stellate cells. |
eRF3b-37 reverses cell apoptosis and
decreases cell migration viability by regulating the expression of
related factors in LX-2 cells stimulated with TGF-β1
At 48 h, the apoptotic rate of HSCs was lower in the
TGF group than in the control group; however, no statistical
difference was observed as the apoptotic rate in the control group
was quite low. The rate of apoptosis was significantly greater in
the eRF3b group when compared with the TGF group (Fig. 5A and B). Bcl-2, Bax, and Fas are
factors that regulate cell apoptosis (15,16). The mRNA expression of Bcl-2 in the
TGF group was higher than that of the control and TGF+eRF3b groups.
By contrast, the expressions of Bax and Fas in the TGF group were
lower than those observed in the control or TGF+eRF3b groups
(Fig. 5C-E).
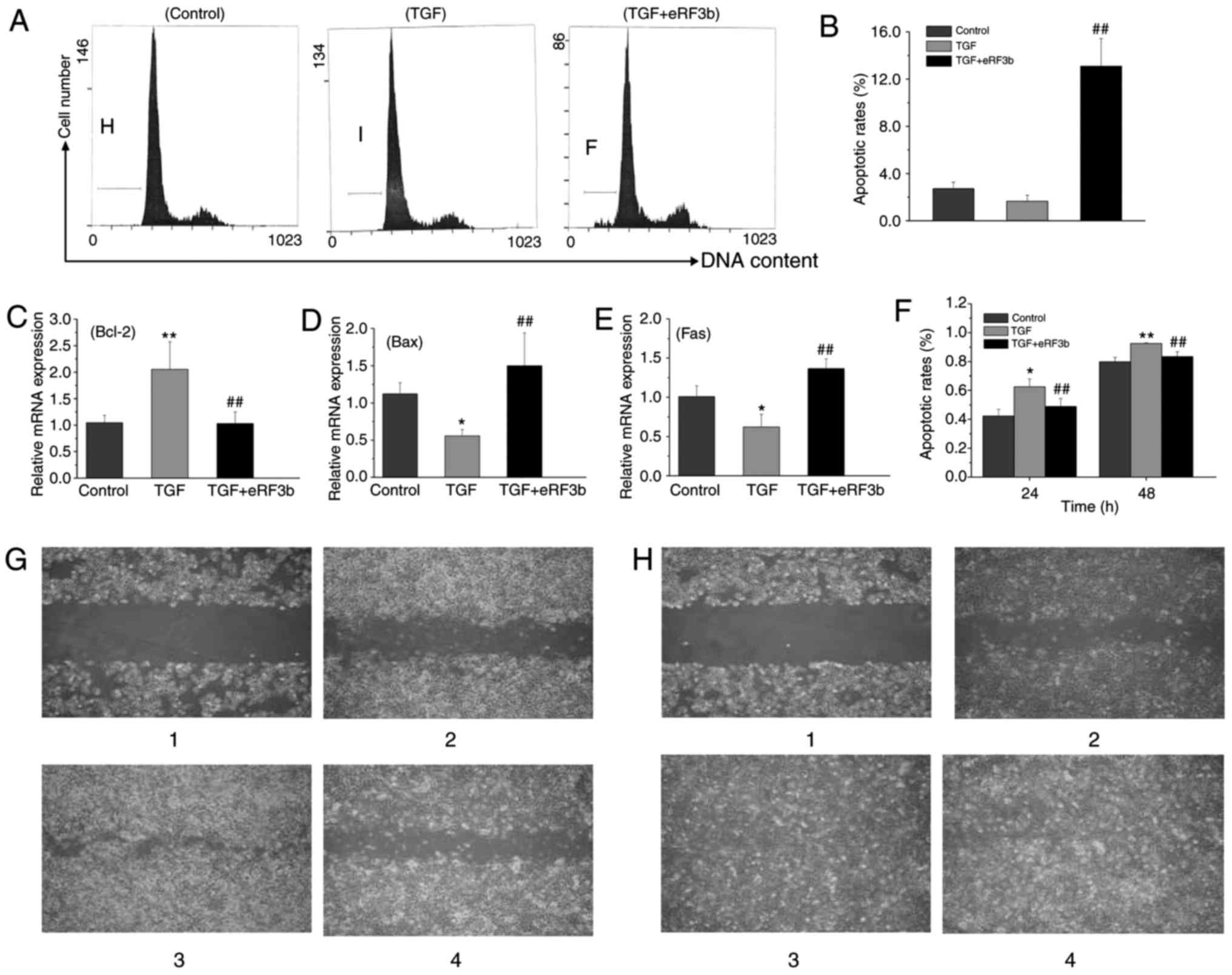 | Figure 5eRF3b-37 reverses cell apoptosis and
reduces cell migration viability by regulating associated factor
expression in LX-2 cells stimulated with TGF-β1. (A) eRF3b-37
reverses the cell apoptosis induced by TGF-β1, as determined by
flow cytometry at 48 h. (B) Statistical analysis revealed that the
apoptotic rate was higher in the TGF+eRF3b group when compared with
the TGF group. eRF3b-37 regulates the mRNA expression of (C) Bcl-2,
(D) Bax and (E) Fas, as determined by reverse
transcription-quantitative polymerase chain reaction at 24 h. (F)
Statistical analysis of the effects of eRF3b-37 on cell migration.
eRF3b-37 reduced cell migration abilities at (G) 24 and (H) 48 h
respectively (fluorescence microscope; magnification, x4.2). Image
1 is day 0; images 2, 3 and 4 are the control, TGF and TGF+eRF3b
groups, respectively. Data are presented as the mean ± standard
deviation of three independent repeated experiments per group.
*P<0.05 and **P<0.01 vs. Control group;
##P<0.01 vs. TGF group. eRF3b, eukaryotic peptide
chain releasing factor 3b polypeptide; TGF, transforming growth
factor; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein. |
In addition, the increase in HSC migrating activity
is indicative of cell activation. The migrating distances of the
three groups were observed, photographed, measured, and compared
under the microscope at the same magnification and position on the
scratch at 0, 24 and 48 h (Fig. 5G
and H). Under the microscope, the HSC morphology in the control
group was complete and orderly, and the size was uniform; however,
the rate of proliferation was slow. While in the TGF group, the
body of HSCs gradually increased, with cell protrusions expanding
into a spindle shape; the cells grew in clusters and proliferated
rapidly. The relative migrating distance was larger in the TGF
group than in the control or TGF+eRF3b groups at 24 and 48 h
(Fig. 5F).
The results indicated that eRF3b-37 may promote cell
apoptosis by downregulating Bcl-2, and upregulating the expression
of Bax and Fas. Furthermore, eRF3b-37 restricts cell migration
abilities by decreasing α-SMA expression in LX-2 cells stimulated
with TGF-β1.
Discussion
TGF-β1 is a pro-fibrotic factor involved in the
fibrotic process, which induces the formation of HSC collagen
through the classical TGF-β/mothers against decapentaplegic homolog
and P38/mitogen-activated protein kinase signaling pathways
(8,17). TGF-β1 also elevates CTGF
expression and in turn, enhances the mRNA expression of ColI and
III. HSCs are mesenchymal cells that respond to liver injury in a
stepwise manner, thereby transitioning from quiescent, vitamin
A-rich, non-proliferative cells to an activated contractile
myofibroblast phenotype characterized by an increase in DNA
synthesis, proliferation viability, α-SMA expression, and the
synthesis of various ECM components (18,19). This activation consists of an
initiation stage, a perpetuation stage and a resolution phase,
where liver injury is resolved (20). Therefore, the activation of HSC,
stimulated by TGF-β1, was employed as the starting point to explore
the mechanism underlying the effect of eRF3b-37 on HSC activation
and pro-fibrogenic-associated factor expression. TGF-β1 was used to
successfully construct a HSC fibrogenic expression model with an
optimal dose of 10 ng/ml, which is consistent with previous
literature (21).
When the liver is injured by inflammation or
mechanical stimulation, HSCs are activated, which is followed by an
increase in proliferation activity and thus, the number of cells.
In addition, their morphology and function also change into
myofibroblast-like cells, which express α-SMA and secrete a
substantial amount of collagen fibers that mainly contain type I
and III (3,22). Freshly isolated quiescent HSCs
synthesize minimal quantities of collagen, whereas activated HSCs
express high levels of the genes encoding procollagen I and III,
and secrete large amounts of collagen (6). CTGF belongs to the calponin family
of proteins and is a downstream mediator of TGF-β1 (23,24). The CTGF promoter sequence contains
a unique TGF-β response regulatory element. TGF-β1 elevated the
expression of CTGF at the mRNA and protein levels in the fibrotic
hypertrophy of the ligamentum flavum (25). Williams et al (26) reported that CTGF appears to be
directly involved in HSC biology, as it is produced as a function
of the activation or exposure of cells to TGF-β1. Accordingly, the
results of the present study demonstrated that the increased
expressions of ColI, CTGF and α-SMA reflect the pro-fibrogenic
tendency of HSCs stimulated with TGF-β1. It was deduced that TGF-β1
may directly stimulate the expression of ColI, CTGF and α-SMA in
HSCs, or stimulate ColI and α-SMA by activating its downstream
cytokine, CTGF. The inhibitory effect of eRF3b-37 on ColI, CTGF and
α-SMA expression may be attributable to the downregulation of
TGF-β1. eRF3b-37 is thought to serve a role in HSC by inhibiting
TGF-β signaling.
The cell cycle assay is a rapid and accurate method
used to study cell proliferation. The cell cycle is the basic
process of cell life that contains four phases: The
G0/G1 phase (DNA synthesis prophase), S phase
(DNA synthesis phase), G2 phase (DNA synthesis anaphase)
and M phase (DNA division phase) (14,27). The cell is prepared for DNA
replication in the G1 phase, and then chromosomes are
replicated during the S phase. A gap period, G2, allows
for preparation for mitosis prior to chromosome segregation and
cytokines are produced in the M phase (mitosis).
G0/G1 is an important checkpoint; the
restriction point occurs mid-G1. Following this
checkpoint, cells become independent of growth factors and commit
to cell division. S phase cells participate in cell division,
thereby representing the number of cells involved in the division
(14,27). In fact, changes in the ratios of
G0/G1 to S+G2/M cells can reflect
cell proliferation and cell cycle status (14). In the present study, eRF3b-37
inhibited cell proliferation, increased G0/G1
arrest and blocked DNA synthesis in HSCs by inhibiting TGF-β1.
eRF3b-37 may have inhibited proliferation capabilities by promoting
G0/G1 arrest and blocking DNA synthesis.
Proliferation and cell-cycle progression are
regulated by two protein classes: Cyclins and their associated
kinase partners, CDKs. Cyclin D is the prime integrator of these
cellular signals, initiating progression through the early phase of
the G1 period (14,28). Cyclin D activity is required to
regulate the progression from G1 into the S phase partly
mediated through coordinated CDK4/6-dependent phosphorylation of
the retinoblastoma substrate (Rb). The regulation of Cyclin/CDK
activity is central to restriction-point passage (14). Cyclin D1 serves a more prominent
role in driving cell cycle progression when in comparison with
other members of the Cyclin D family. Once bound to Cyclin, CDK4
can exert its important biological role in protein kinase activity
(14,29). Cyclin D1 alone has been reported
to bind CDK4 and initiate the phosphorylation of Rb family
proteins, which in turn, dictate the events that orchestrate
mitosis; these events may regulate cell proliferation,
differentiation and the cell cycle, inducing cell cycle progression
from G1 to S (14,28,30). P21 belongs to the Cip/Kip family
of cell cycle inhibitors. The P21 family can interact with cyclin
and CDK subunits. In the G1/S transition phase of the
cell-cycle, the negative cell-cycle regulator is responsible for
suppressing Cyclin D1/CDK4 complexes (23). Qin et al (31) transfected the P21 gene into
hepatoma Hep3B cells and revealed that the increased expression of
P21 inhibited the growth of Hep3B cells. The P21 C terminal domain
can directly bind to the proliferating cell nuclear antigen (PCNA),
and inhibit the binding of DNA polymerase δ and PCNA in the process
of G1/S transformation, thereby blocking DNA replication
in the cell cycle (32).
Therefore, it could be deduced that eRF3b-37 can arrest cell
proliferation, the G0/G1 cell cycle and DNA
synthesis by inhibiting the expressions of Cyclin D1 and CDK4, and
upregulating the expression of P21 induced by TGF-β1.
Apoptosis is the major pathway for the reduction of
HSCs. TGF-β may decrease the spontaneous apoptosis of HSCs and
support activated HSC survival by regulating Fas Ligand (FasL)
synthesis and Bcl, NF-kB, and p53/P21WAF1 signaling
(15,16). The results of the present study
are consistent with these previous reports. eRF3b-37 may increase
the apoptosis of TGF-β1-induced LX-2 cells; however, the result
does not have much practical significance due to the very low
apoptotic rate of HSCs.
Many relevant factors are involved in the regulation
of this apoptotic process. Previous studies have demonstrated that
the regulation of apoptosis primarily depends on two conserved
signaling pathways: The mitochondrial pathway and the death
receptor pathway. The major regulator of the mitochondrial
apoptosis pathway, the Bcl-2 gene family, inhibits or promotes
apoptosis by blocking or stimulating, respectively, the release of
cytochrome C into the cytoplasm (33,34). The Bcl-2 gene family is divided
into two groups: Anti-apoptotic factors, such as Bcl-2; and
pro-apoptotic factors, such as Bax. The apoptosis of HSC is
believed to be due to the imbalanced expression of Bcl-2 and Bax.
Bcl-2 can decrease apoptosis mediated by TNF-α or the Fas system;
in particular, it can change human HSC/MF sensitivity to
TNF-α-induced apoptosis (35).
Bax proteins induce the apoptosis of HSC by translocating directly
from the cytoplasm to the mitochondria and the cytosolic liberation
of cytochrome C (35-37). Bcl-2 prevents Bax from
translocating from the cytosol to the mitochondria by capturing Bax
monomers prior to their dimerization, thereby preventing Bax from
forming channels in the mitochondrial outer membrane (38). The death receptor pathway is
mediated by the death factor FasL, and its corresponding death
factor receptor, Fas. Fas is located in the target cell membrane,
binding to the specific ligand FasL and then initiating the
apoptosis process. Saile et al (37) reported that the occurrence of
spontaneous apoptosis in HSCs during activation was accompanied by
an increased expression of Fas/FasL in HSCs. Therefore, eRF3b-37
was hypothesized to promote HSC apoptosis in association with the
downregulation of Bcl-2 and the upregulation of Bax and Fas genes,
when induced by TGF-β1. Consequently, eRF3b-37 may regulate cell
apoptosis through the mitochondrial and death receptor
pathways.
HSC directional migration and adhesion also serves
important roles in an injured or inflamed liver. HSCs release many
cytokines that are involved in the process of cell migration. In
particular, TGF-β1 can promote the cytoskeletal remodeling and
migration of HSCs by inducing the robust generation of stress
fibers and focal adhesions, acquisition of spindle morphology, and
increased motility (6,39). Actin is closely associated with
cell migration. One component of the HSC cytoskeleton is actin
filament, such as α-SMA, which provides structural support for cell
migration and contraction. Actin constitutes the chemical machinery
system of cell movement, using chemical energy to produce
mechanical movement (40).
Increased α-SMA expression in fibroblasts enhances the ability of
actin to generate contractile force (41). Consequently, α-SMA acts as an
indicator of HSC activation and indirectly reflects its migration
capacity in vitro. In the present study, it was assumed that
TGF-β1 induced cell migration by upregulating α-SMA expression. The
inhibitory action of eRF3b-37 on HSC migration may be partly
associated with the downregulation of α-SMA induced by TGF-β1.
In conclusion, eRF3b-37 inhibited the activation of
HSCs and the expression of pro-fibrogenic factors in LX-2 cells
stimulated with TGF-β1 by decreasing cell proliferation viability,
reducing DNA synthesis and cell apoptosis arrest, and inhibiting
cell migration, which are closely associated with changes in their
corresponding regulatory factors in HSCs. In addition, eRF3b-37 is
thought to exert its effects on HSCs by inhibiting TGF-β signaling.
Consequently, the present results may contribute to the
understanding of the roles of TGF-β1 and eRF3b-37 in the
pathogenesis of HSCs in liver fibrosis. It should be noted that the
present study only focuses on our initial work associated with the
effects of eRF3b-37 on HSC and thus, one limitation was that only
one cell line was used.
In the future, the group will further investigate
the underlying mechanism of eRF3b-37 in HSC and animal models, for
example, how it works via various cytokine signal pathways and
calcium ions. eRF3b-37 may be a novel therapeutic agent for
targeting HSCs for hepatic fibrosis, though its mechanism still
requires more in-depth study.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Major Projects of Hebei Province Natural Science Foundation of
China (grant no. H2016206576).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZX drafted the manuscript and performed the major
experiments. DL gave final approval of the version to be published
and revised it critically for important intellectual content. TL
was involved in project design, supervision and cell culture
experiments. ML analyzed and interpreted the data and performed
RT-qPCR. LY conducted western blotting and was involved in
designing the study. RX, LL and XC were involved in the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gressner AM, Yagmur E, Lahme B, Gressner O
and Stanzel S: Connective tissue growth factor in serum as a new
candidate test for assessment of hepatic fibrosis. Clin Chem.
52:1815–1817. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsukada S, Parsons CJ and Rippe RA:
Mechanisms of liver fibrosis. Clin Chim Acta. 364:33–60. 2006.
View Article : Google Scholar
|
|
4
|
Zhang CY, Yuan WG, He P, Lei JH and Wang
CX: Liver fibrosis and hepatic stellate cells: Etiology,
pathological hallmarks and therapeutic targets. World J
Gastroenterol. 22:10512–10522. 2016. View Article : Google Scholar
|
|
5
|
Peelman F, Waelput W, Iaerentant H, Lavens
D, Eyckerman S, Zabeau L and Tavenier J: Leptin: Linking adipocyte
metabolism with cardiovascular and autoimmune diseases. Prog Lipid
Res. 43:283–301. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Wang JY, Yang CQ and Jiang W: Effect
of RhoA on transforming growth factor β1-induced rat hepatic
stellate cell migration. Liver Int. 32:1093–1102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bataller R and Brenner DA: Hepatic
satellite cells as a target for the treatment of liver fibrosis.
Semin Liver Dis. 21:437–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung YJ, Lee JI, Chong S, Seok JW, Park
SJ, Jang HW, Kim SW and Chung JH: Anti-proliferative effect and
action mechanism of dexamethasone in human medullary thyroid cancer
cell line. Endocr Res. 36:149–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi W, Chen X, Poronnik P and Pollock CA:
Transforming growth factor- beta/connective tissue growth factor
axis in the kidney. Int J Biochem Cell Biol. 40:9–13. 2008.
View Article : Google Scholar
|
|
10
|
Marquez-Aguirre A, Sandoval-Rodriguez A,
Gonzalez-Cuevas J, Bueno-Topete M, Navarro-Partida J,
Arellano-Olivera I, Lucano-Landeros S and Armendariz-Borunda J:
Adenoviral delivery of dominant-negative transforming growth factor
beta type II receptor upregulates transcriptional repressor
SKI-like oncogene, decreases matrix metalloproteinase 2 in hepatic
stellate cell and prevents liver fibrosis in rats. J Gene Med.
11:207–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
George J, Roulot D, Koteliansky VE and
Bissell DM: In vivo inhibition of rat stellate cell activation by
soluble transforming growth factor beta type II receptor: A
potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA.
96:12719–12724. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li M, Wang J, Dai E and Liu D:
Identification and investigation of disease-related peptides in
sera from patients with chronic hepatitis B. Wei Sheng Yan Jiu.
40:315–319. 2011.In Chinese. PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Swanton C: Cell-cycle targeted therapies.
Lancet Oncol. 5:27–36. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saile B, Matthes N, Knittel T and Ramadori
G: Transforming growth factor beta and tumor necrosis factor alpha
inhibit both apoptosis and proliferation of activated rat hepatic
stellate cells. Hepatology. 30:196–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saile B, Matthes N, EI Armouche H,
Neubauer K and Ramadori G: The bcl, NFkappaB and p53/p21WAF1
systems are involved in spontaneous apoptosis and in the
anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic
stellate cells. Eur J Cell Biol. 80:554–561. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Breitkopf K, Godoy P, Ciuclan L, Singer MV
and Dooley S: TGF-beta/Smad signaling in the injured liver. Z
Gastroenterol. 44:57–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao R, Ball DK, Perbal B and Brigstock DR:
Connective tissue growth factor (CCN2) induces c-fos gene
activation and cell proliferation through p42/44 MAP kinase
(ERK1/2) in primary rat hepatic stellate cells. J Hepatol.
40:431–438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng ZY, Li J, Jin Y, Chen XL and Lü XW:
Effect of oxymatrine on the p38 mitogen-activated protein kinases
signalling pathway in rats with CCl4 induced hepatic fibrosis.
Chinese Med J (Engl). 122:1449–1454. 2009.
|
|
20
|
Kocabayoglu P and Friedman SL: Cellular
basis of hepatic fibrosis and its role in inflammation and cancer.
Front Biosci (Schol Ed). 5:217–230. 2013. View Article : Google Scholar
|
|
21
|
Shimada H, Staten NR and Rajagopalan LE:
TGF-β1 mediated activation of Rho kinase induces TGF-β2 and
endothelin-1 expression in human hepatic stellate cells. J Hepatol.
54:521–528. 2011. View Article : Google Scholar
|
|
22
|
Higashi T, Friedman SL and Hoshida Y:
Hepatic stellate cells as key target in liver fibrosis. Adv Drug
Deliv Rev. 121:27–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qi W, Twigg S, Chen X, Polhill TS,
Poronnik P, Gilbert RE and Pollock CA: Integrated actions of
transforming growth factor-beta1 and connective tissue growth
factor in renal fibrosis. Am J Physiol Renal Physiol.
288:F800–F809. 2005. View Article : Google Scholar
|
|
24
|
Yokoi H, Sugawara A, Mukoyama M, Mori K,
Makino H, Suganami T, Nagae T, Yahata K, Fujinaga Y, Tanaka I and
Nakao K: Role of connective tissue growth factor in profibrotic
action of transforming growth factor-beta: A potential target for
preventing renal fibrosis. Am J Kidney Dis. 38(4 Suppl 1):
S134–S138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao YL, Duan Y, Zhu LX, Zhan YN, Min SX
and Jin AM: TGF-β1, in association with the increased expression of
connective tissue growth factor, induce the hypertrophy of the
ligamentum flavum through the p38 MAPK pathway. Int J Mol Med.
38:391–398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Williams EJ, Gaça MD, Brigstock DR, Arthur
MJ and Benyon RC: Increased expression of connective tissue growth
factor in fibrotic human liver and in activated hepatic stellate
cells. J Hepatol. 32:754–761. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hopkins M, Tyson JJ and Novák B:
Cell-cycle transitions: A common role for stoichiometric
inhibitors. Mol Biol Cell. 28:3437–3446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jung KH, Kim JK, Noh JH, Eun JW, Bae HJ,
Xie HJ, Ahn YM, Park WS, Lee JY and Nam SW: Targeted disruption of
Nemo-like kinase inhibits tumor cell growth by simultaneous
suppression of cyclin D1 and CDK2 in human hepatocellular
carcinoma. J Cell Biochem. 110:686–696. 2010. View Article : Google Scholar
|
|
29
|
Meng L, Feng B, Tao H, Yang T, Meng Y, Zhu
W and Huang C: A novel antiestrogen agent (3R,6R)-bassiatin
inhibits cell proliferation and cell cycle progression by
repressing Cyclin D1 expression in 17β-estradiol-treated MCF-7
cells. Cell Biol Int. 35:599–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lange C, Huttner WB and Calegari F:
Cdk4/CyclinD1 overex-pression in neural stem cells shortens G1,
delays neurogenesis, and promotes the generation and expansion of
basal progenitors. Cell Stem Cell. 5:320–331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin LF and Ng IO: Exogenous expression of
p21 (WAF1/CIP1) exerts cell growth inhibition and enhances
sensitivity to cisplatin in hepatoma cells. Cancer Lett. 172:7–15.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cazzalini O, Perucca P, Riva F, Stivala
LA, Bianchi L, Vannini V, Ducommun B and Prosperi E: p21CDKN1A does
not interfere with loading of PCNA at DNA replication sites, but
inhibits subsequent binding of DNA polymerase delta at the G1/S
phase transition. Cell Cycle. 2:596–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Green DR: Apoptotic pathways: Paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng X, Yu Z, Liang N, Chi X, Li X, Jiang
M, Fang J, Cui H, Lai W, Zhou Y and Zhou S: The mitochondrial and
death receptor pathways involved in the thymocytes apoptosis
induced by aflatoxin B1. Oncotarget. 7:12222–12234. 2016.PubMed/NCBI
|
|
35
|
Kawada N: Human hepatic stellate cells are
resistant to apop-tosis: Implications for human fibrogenic liver
disease. Gut. 55:1073–1074. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Novo E, Marra F, Zamara E, Valfrè di Bonzo
L, Monitillo L, Cannito S, Petrai I, Mazzocca A, Bonacchi A, De
Franco RS, et al: Overexpression of Bcl-2 by activated human
hepatic stellate cells: Resistance to apoptosis as a mechanism of
progressive hepatic fibrogenesis in humans. Gut. 55:1174–1182.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Saile B, Knittel T, Matthes N, Schott P
and Ramadori G: CD95/CD95L-mediated apoptosis of the hepatic
stellate cell. A mechanism terminating uncontrolled hepatic
stellate cell proliferation during hepatic tissue repair. Am J
Pathol. 151:1265–1272. 1997.PubMed/NCBI
|
|
38
|
Tan C, Dlugosz PJ, Peng J, Zhang Z,
Lapolla SM, Plafker SM, Andrews DW and Lin J: Auto-activation of
the apoptosis protein Bax increases mitochondrial membrane
permeability and is inhibited by Bcl-2. J Biol Chem.
281:14764–14775. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhowmick NA, Ghiassi M, Bakin A, Aakre M,
Lundquist CA, Engel ME, Arteaga CL and Moses HL: Transforming
growth factor-beta1 mediates epithelial to mesenchymal
transdifferen-tiation through a RhoA-dependent mechanism. Mol Biol
Cell. 12:27–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Y, Ma J, Chen L, Xie XL and Jiang H:
Inhibition of focal adhesion kinase on hepatic stellate-cell
adhesion and migration. Am J Med Sci. 353:41–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hinz B, Celetta G, Tomasek JJ, Gabbiani G
and Chaponnier C: Alpha-smooth muscle actin expression upregulates
fibroblast contractile activity. Mol Biol Cell. 12:2730–2741. 2001.
View Article : Google Scholar : PubMed/NCBI
|