Introduction
Skin exposure to ultraviolet radiation and
environmental toxic chemicals, such as air pollutants, heavy metals
or ozone, can result in oxidative stress, which in turn accelerates
the skin aging process and can directly cause disease (1). In addition, skin exposure to
oxidizing chemicals present in cosmetic and pharmaceutical products
can cause oxidative stress, leading to skin tissue and cellular
damage. Oxidative stress is essentially the excess production of
reactive oxygen species (ROS) beyond the control of the antioxidant
defense system. ROS generation by cells under physiological
conditions is important for the maintenance of cellular functional
integrity. In a normal context, ROS are continuously generated
through cellular respiration and other metabolic processes
(1,2). These highly reactive molecules have
a wide range of physiological functions, acting as microbicidal
agents and second messengers involved in cell proliferation and
differentiation, and regulating numerous cellular signaling
pathways (3). However, the
overproduction of ROS can be induced in skin cells by exogenous
irritants, such as ultraviolet radiation or chemical agents,
causing direct oxidative damage to proteins, lipids and DNA
(2). A variety of endogenous
defense mechanisms exist in cells to constrain such damage,
including the induction of the antioxidative proteins, glutathione
peroxidase (GPx), catalase (CAT) and superoxide dismutase (SOD),
which have enzymatic activity. Cells can further synthesize
antioxidants that lack enzymatic activity, including glutathione
(GSH), vitamins C and E, and ubiquinol (4). These endogenous antioxidants protect
cells from free radicals by reducing and neutralizing them.
Normally there is balance between the antioxidant system and any
ROS generation, but this balance is dynamic and tenuous. Any redox
disturbances resulting in ROS overproduction or in faulty
antioxidant mechanisms in skin cells may induce or aggravate skin
diseases.
Tert-Butyl hydroperoxide (tBHP) is an organic
peroxide that can be metabolized in cells by cytochrome P450 to
produce peroxyl and alkoxyl radicals or detoxified to tert-butanol,
resulting in the rapid oxidation and depletion of cellular GSH.
These pathways lead to oxidative injury of cells. Thus, tBHP is
commonly employed as an exogenous oxidative stressor in cells and
tissues in laboratory research (5-7).
Previous work has shown that tBHP can cause oxidative damage to
keratinocytes both in vitro and in vivo (8,9). Thus,
use of an exogenous inducer of oxidative stress, such as tBHP, may
simulate situation of augmented oxidative stress in skin cells.
In mammalian cells, nuclear factor-erythroid
2-related factor 2 (Nrf2) regulates antioxidant protein expression
and is itself a redox-sensitive transcription factor (10). In the absence of oxidative stress,
Nrf2 remains in the cytoplasm and is constantly degraded via
binding to Kelch-ECH-associated protein 1 (KEAP1), an adapter for
the Cul3/Rbx1 E3 ubiquitin ligase that mediates Nrf2 ubiquitination
and degradation. Keap1 is able to respond to changes in cellular
redox conditions because it contains multiple reactive cysteine
residues, the oxidation of which causes conformational changes that
disrupt Nrf2 proteasomal degradation by altering the Keap1-Nrf2
complex. Other regulatory mechanisms, such as phosphorylation of
particular residues within Nrf2, can also stabilize the protein,
reducing its subsequent destruction (10). Kinases such as mitogen-activated
protein kinases (MAPKs), phosphatidylionositol-3-kinase/Akt,
protein kinase C and adenosine monophosphate-activated protein
kinase (AMPK) have all been proposed as upstream kinases mediating
Nrf2 phosphorylation (11). Nrf2
stabilization leads to its accumulation in the nucleus, allowing it
to drive the transcription of target genes, including antioxidant
genes, that contain the antioxidant response element (ARE).
Extensive research has demonstrated that Nrf2 can be regulated by a
range of pathways that modulate its binding to Keap1 and its
subsequent stabilization. The appropriate regulation and
cytoprotection cells may require the simultaneous activation of two
or more such pathways, which together regulate Nrf2 activity in a
cell type- and stimulant-dependent manner (12).
Given the importance of Nrf2 for cytoprotection from
oxidative stress, and the deleterious contributions of ROS in the
context of human physiology, developing compounds that modulate the
Nrf2/Keap1/ARE pathway is of great research interest (13). It has been shown that many natural
compounds with known antioxidant activity primarily elicit such
activity by activating Nrf2 (11). Nrf2 itself has also been
implicated in the regulation of mitochondrial homeostasis,
proteasome activity, autophagy and stem cell renewal, all of which
can in turn contribute to improved skin health and rejuvenation
(14,15). Thus, there is substantial
opportunity for groups to develop naturally-derived compounds that
can elicit Nrf2-mediated protection of the skin.
Schisandrin B (Sch B) is derived from the fruit of
the traditional Chinese herb, Schisandra chinensis, which is
traditionally used to treat hepatitis and cardiovascular disease
(16). Studies have determined
that Schisandra chinensis has numerous benefits, including
the ability to protect against numerous stressors, including heat
shock, burns, frostbite, swimming in a low oxygen environment,
aseptic inflammation, irradiation and heavy metal toxicity
(17). As one of the major active
ingredients in Schisandra chinensis, Sch B has been
extensively studied for its diverse pharmacological effects
(18). Previous studies have
demonstrated that Sch B has potent antioxidant and cytoprotective
effects against different types of oxidative stress-induced cell
injury in hepatocytes (19-21), cardiomyocytes (22,23), neurcyte (24,25), kidney tubule cells (26) and trophoblasts (27). In most of these cell types, the
antioxidant effects of Sch B derive from Nrf2 activation and
subsequent upregulation of antioxidant enzyme expression. Several
in vivo studies have shown that Sch B treatment enhances
antioxidant activity in liver, cardiac tissue, brain, and kidneys
in various oxidative stress animal models (18,28-35). More recently, the antioxidant
capacity of Sch B was tested in human skin cells, showing that Sch
B could protect BJ human fibroblasts against solar
irradiation-induced oxidative injury (36), and protect human keratinocyte-
derived HaCaT cells against UVB-induced oxidative damage (37,38). However, whether Sch B enhances
antioxidant defenses in human skin cells via Nrf2 activation
remains to be elucidated.
In the present study, activity of Sch B in
tBHP-stimulated HaCaT cells was investigated, and the underlying
mechanisms were explored. Primary areas of focus included its
effects on apoptosis, intracellular ROS generation, mitochondrial
dysfunction, oxidation biomarkers, antioxidant enzymes expression
and the requirement for Nrf2 activation for cyto-protection against
tBHP-induced oxidative damage.
Materials and methods
Materials
Sch B was obtained from Nature Standard
Biotechnology Co., Ltd. (Shanghai, China). DMEM was purchased from
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Fetal bovine
serum (FBS) and penicillin/streptomycin were from Gibco (Thermo
Fisher Scientific, Inc). Tert-Butyl hydroperoxide was purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Protein
extraction kits for total protein were obtained from Beyotime
Institute of Biochecnology (Jiangsu, China). The BCA kit was from
Thermo Fisher Scientific, Inc.
Antibodies against Nrf2 (cat. no. ab62352), p-Nrf2
(cat. no. ab76026), Keap1 (cat. no. ab218815), JNK (cat. no.
ab179461), p-JNK (cat. no. ab124956) and GAPDH (cat. no. ab181602)
were purchased from Abcam (Cambridge, UK). Antibodies against p38
mitogen-activated protein kinase (p38 MAPK; cat. no. 8690),
phospho-p38 MAPK (cat. no. 4511), Akt (cat. no. 4685), p-Akt (cat.
no. 4060), Erk1/2 (cat. no. 4695), p-Erk1/2 (cat. no. 4370), AMPK
(cat. no. 5832) and phospho-AMPK (cat. no. 2535) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA), and
anti-Lamin B (cat. no. sc-6217) was obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Goat anti-Mouse IgG H&L
(cat. no. ab216772) and Goat anti-Rabbit IgG H&L (cat. no.
ab216773) secondary antibodies were purchased from Abcam
(Cambridge, UK).
Cell culture
HaCaT cells were purchased from Shanghai Zhong Qiao
Xin Zhou Biotechnology Co., Ltd. (Shanghai, China; cat. no.,
ZQ0044) and cultured in DMEM containing 10% FBS and
penicillin/streptomycin. Cell culture took place at 37°C in 5%
CO2. Experiments were performed at 70% confluency.
Cell viability assay
Cell Counting kit-8 (CCK-8; Dojindo Laboratories,
Kumamoto, Japan) was used for viability assessment, according to
the manufactuerer's protocol. Viability is expressed as % of
control optical density (OD).
Apoptosis assay
Annexin V, FITC Apoptosis Detection kit (Dojindo
Laboratories, Kumamoto, Japan) was used to stain the cells,
according to the manufactuerer's protocol. Fluorescence was
quantified on a FACS Calibur flow cytomoeter (BD Biosciences,
Franklin Lakes, NJ, USA). Numbers of Annexin V-FITC positive cells
allowed for determination of apoptotic frequency.
Intracellular ROS measurement
Reactive Oxygen Species Assay kit (Beyotime
Institute of Biotechnology) was employed to assess ROS production,
according to the manufacturer's protocol. A flow cytometer was used
to monitor production, and the results were analysed using
CellQuest Pro software (version 5.2.1; BD Biosciences).
Mitochondrial membrane potential (MMP)
assessment
MMP loss was evaluated by Mitochondrial membrane
potential assay kit with JC-1 (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. The cells
were then assessed using a flow cytometer. The results were
analyzed using CellQuest Pro software (version 5.2.1; BD
Biosciences).
ATP level measurement
Intercellular ATP levels in HaCaT cells were
measured via bioluminescence assay kit (Beyotime, Jiangsu, China)
based on provided protocols. Levels of ATP were determined based on
luciferase luminescence, after normalizing to total protein
content.
Measurement of oxidation biomarkers
Malondialdehyde (MDA) content was assessed using
Lipid Peroxidation MDA Assay kit (Beyotime Institute of
Biotechnology), according to the manufacturer's instructions. The
amount of 8-oxo-2'-deoxyguanosine (8-oxo-dG), a DNA damage marker,
was determined with a Human 8-OhdG ELISA Kit (AMEKO, Shanghai,
China), according to the manufacturer's instructions. Protein
carbonyl levels were measured by Protein Carbonyl Colorimetric
Assay Kit (Cayman Chemical, Ann Abor, MI, USA), according to the
manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated from HaCaT cells following total
RNA extraction using RNAfast 200 (Shanghai Fastagen
Biotechnology Co., Ltd., Shanghai, China). Reverse transcription
cDNA synthesis was performed using a High Capacity cDNA Reverse
Transcription kit (Applied Biosystems, Foster City, CA, USA). Gene
expression of various antioxidant enzymes was assessed with a
SYBR-Green Master mix (Applied Biosystems). The specific primers
are listed as follows: CAT forward, 5′-ATTCTG GAG AAG TGC GGA GA-3′
and reverse, 5′-CGG CAA TGT TCT CAC ACA GA-3′; HO-1 forward, 5′-CCA
GGC AGA GAA TGC TGA GT-3′ and reverse, 5′-CTT GTT GCG CTC AAT CTC
CT-3′; SOD forward, 5′-AGG CTG TAC CAG TGC AG G TC-3′ and reverse,
5′-CAA TAG ACA CAT CGG CCA CA-3′; GPx forward, 5′-CCA AGC TCA TCA
CCT GGT CT-3′ and reverse, 5′-TCG ATG TCA ATG GTC TGG AA-3′; and
GAPDH forward, 5′-CAG GAG GCA TTG CTG ATG AT-3′ and reverse, 5′-GAA
GGC TGG GGC TCA TTT-3′. The thermocycling conditions were as
follows: Pre-denaturation at 95°C for 30 sec, amplification for 40
cycles by denaturing at 95°C for 5 sec, annealing at 60°C for 34
sec, followed by a final dissociation cycle of 95°C for 15 sec,
60°C for 1 min and 95°C for 15 sec. GAPDH served as an internal
normalization control. The relative expression of target genes were
analyzed by 2-ΔΔCt method (39).
Western blotting
Total protein and nucleoprotein was extracted from
HaCaT cells and quantified via BCA assay. A total of 40 µg
protein was loaded per lane and separated by 8% SDS-PAGE prior to
transfer into 0.45-µm nitrocellulose membranes. Membrane
blocking was conducted for 1 h at room temperature with 5% non-fat
dry milk. The membrane was probed at 4°C overnight with primary
antibodies against Nrf2 (dilution, 1:1,000), phospho-Nrf2
(dilution, 1:3,000), Keap1 (dilution, 1:500), JNK (dilution,
1:2,000), p-JNK (dilution, 1:2,000), GAPDH (dilution, 1:1,000), Akt
(dilution, 1:1,000), phospho-Akt (dilution, 1:1,000), Erk1/2
(dilution, 1:1,000), p-Erk1/2 (dilution, 1:1,000), p38 MAPK
(dilution, 1:1,000), phospho-p38 MAPK (dilution, 1:1,000), AMPK
(dilution, 1:1,000), and p-AMPK. A secondary antibody incubation
followed, using goat anti-mouse or goat anti-rabbit IgG H&L
secondary antibodies (dilution, 1:2,000) for 2 h at room
temperature. An Odyssey CLx Infrared Imaging System (LI-COR, USA)
was used to acquire and analyze the blots. GAPDH used as an
internal control. The gray densities of the protein bands were
quantified using ImageJ (version 1.4.3.67; National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
GraphPad Prism (version 5.0; GraphPad Software,
Inc., CA, USA) was used for all statistical analyses. One-way
analysis of variance followed by Dunnett's test was used for
comparison of means. P<0.05 was considered to indicate a
statistically significant difference.
Results
Sch B modulates tBHP-induced HaCaT cell
death
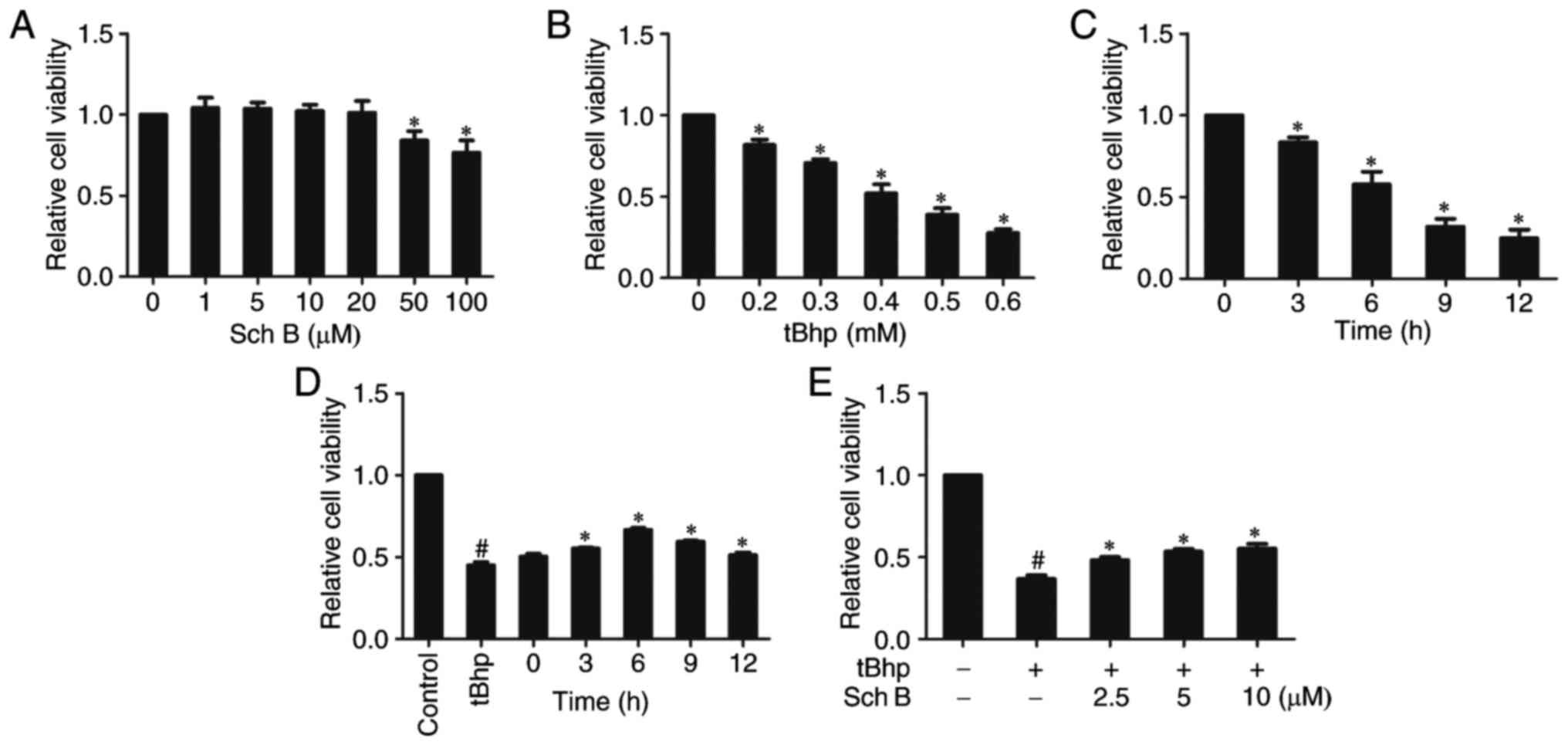
In order to evaluate the protective effect of Sch B
on tBHP-induced oxidative injury in HaCaT cells, we initially
evaluated the cytotoxic effects of Sch B alone (1, 5, 10, 20, 50
and 100 µM) on HaCaT cells via CCK-8 assay. When HaCaT cells
were treated with Sch B at <20 µM for 24 h, cell
viability did not decrease (Fig.
1A), indicating that Sch B exerted no cytotoxicity on HaCaT
cells at <20 µM.
To assess how HaCaT cells respond to oxidative
stress, viability was measured in response to tBHP treatment.
Viability decreased upon tBHP treatment (0.2-0.6 mM) for 6 h in a
dose-dependent fashion (Fig. 1B).
Furthermore, HaCaT-cell treatment with 0.4 mM tBHP for 3-12 h
further decreased viability in a time-dependent fashion (Fig. 1C). Since treatment with 0.4 mM
tBHP for 6 h resulted in ~50% viability, this dose was selected for
subsequent experiments regarding oxidative stress.
To determine how Sch B affects tBHP-induced
cytotoxicity, HaCaT cells were treated with 10 µM Sch B for
0, 3, 6, 9 and 12 h, and sequentially incubated with 0.4 mM tBHP
for 6 h. The CCK-8 assay demonstrated that pretreatment with Sch B
for 6 h provided the highest protective effect against tBHP-induced
cytotoxicity in HaCaT cells (Fig.
1D). Therefore, a pretreatment time of 6 h was selected for use
in subsequent experiments. HaCaT cells were treated with 2.5-10
µM Sch B for 6 h prior to 0.4 mM tBHP treatment to induce
oxidative stress. As shown in Fig.
1E, after 6 h treatment with tBHP alone, cell viability
decreased by >50% compared with no-treatment control, however,
Sch B pretreatment mediated an increase in cell viability in
tBHP-injured cells in a dose dependent fashion (Fig. 1E), suggesting that Sch B protected
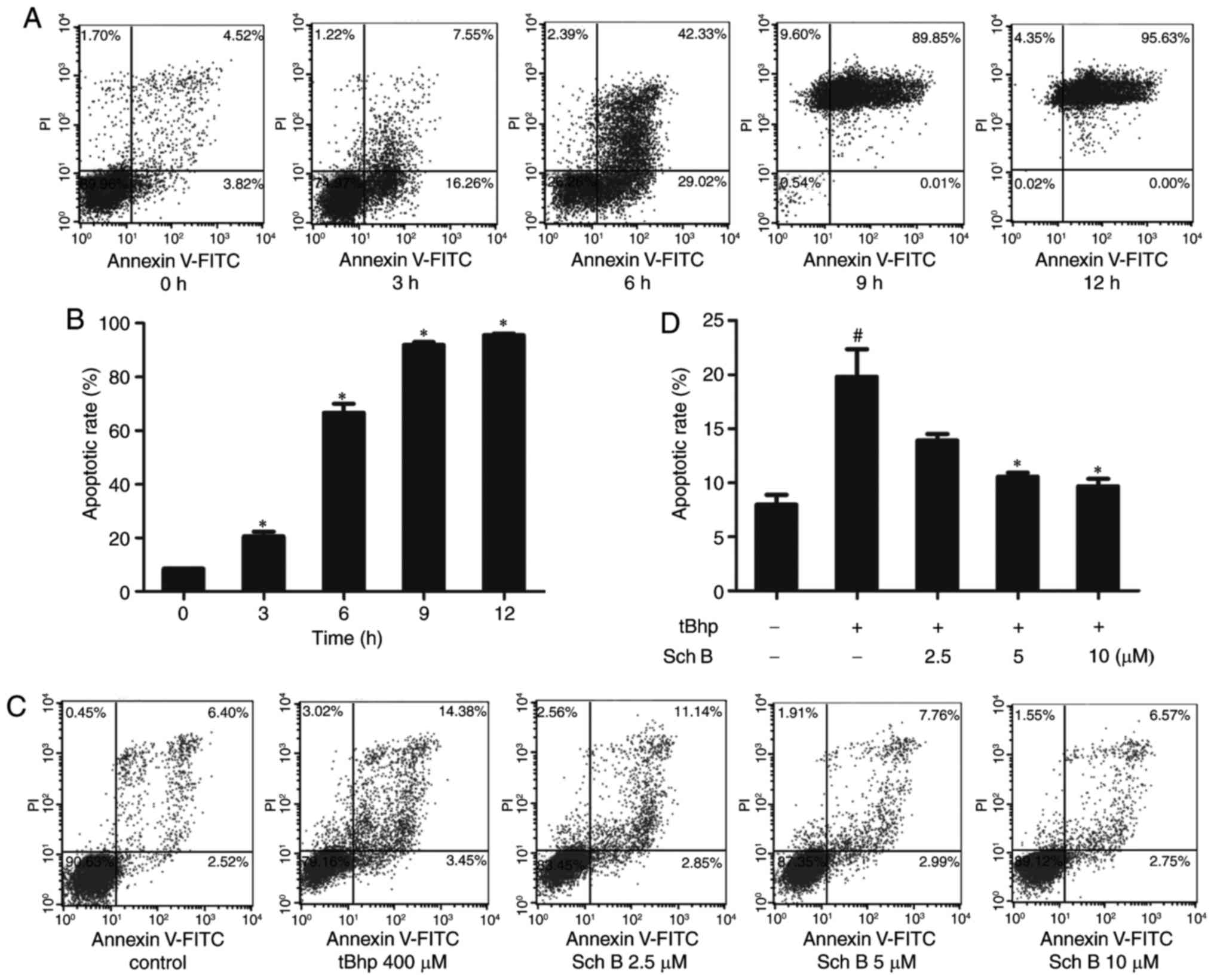
cells from tBHP-induced death. We further measured viability via
Annexin V-FITC/PI staining. The time course of cell death induced
by tBHP was examined, revealing that the number of apoptotic cells
stained positive for Annexin V was markedly increased after 3 h of
0.4 mM tBHP treatment, as compared with the untreated control.
Increased tBHP exposure time (6-12 h) led to an increased
proportion of cells undergoing late apoptosis
(Annexin-positive/PI-positive; Fig.
2A and B). To assess the protective effect of Sch B on
tBHP-induced apoptosis, HaCaT cells were pretreated with Sch B for
6 h prior to induction of apoptosis with tBHP for 3 h. The results
demonstrated that Sch B pretreatment resulted in a dose-dependent
decrease in apoptotic rate, indicating that Sch B has a marked
protective effect against tBHP-induced apoptosis (Fig. 2C and D). Together, these
experiments revealed that Sch B may prevent tBHP-induced HaCaT cell
death.
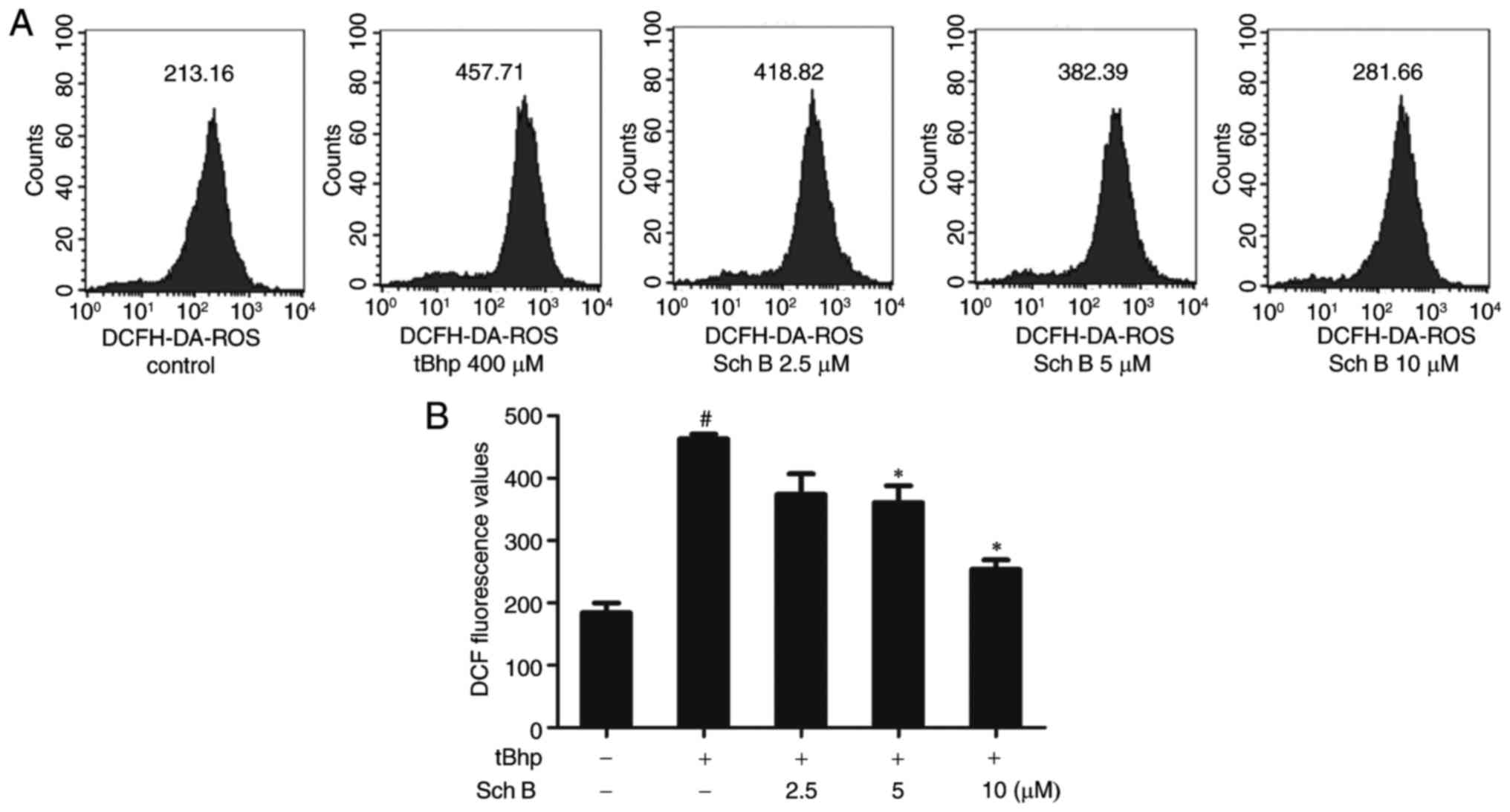
Sch B affects intracellular ROS levels in
tBHP-injured HaCaT cells
To assess the role of oxidative stress in
tBHP-induced injury in HaCaT cells, a DCFH-DA fluorescent probe was
employed to measure intracellular ROS levels over time. The results
demonstrated that tBHP treatment significantly increased the
fluorescence intensity in HaCaT cells, indicating intracellular ROS
generation. 2.5-10 µM Sch B inhibited ROS generation in a
dose-dependent fashion (Fig.
3).
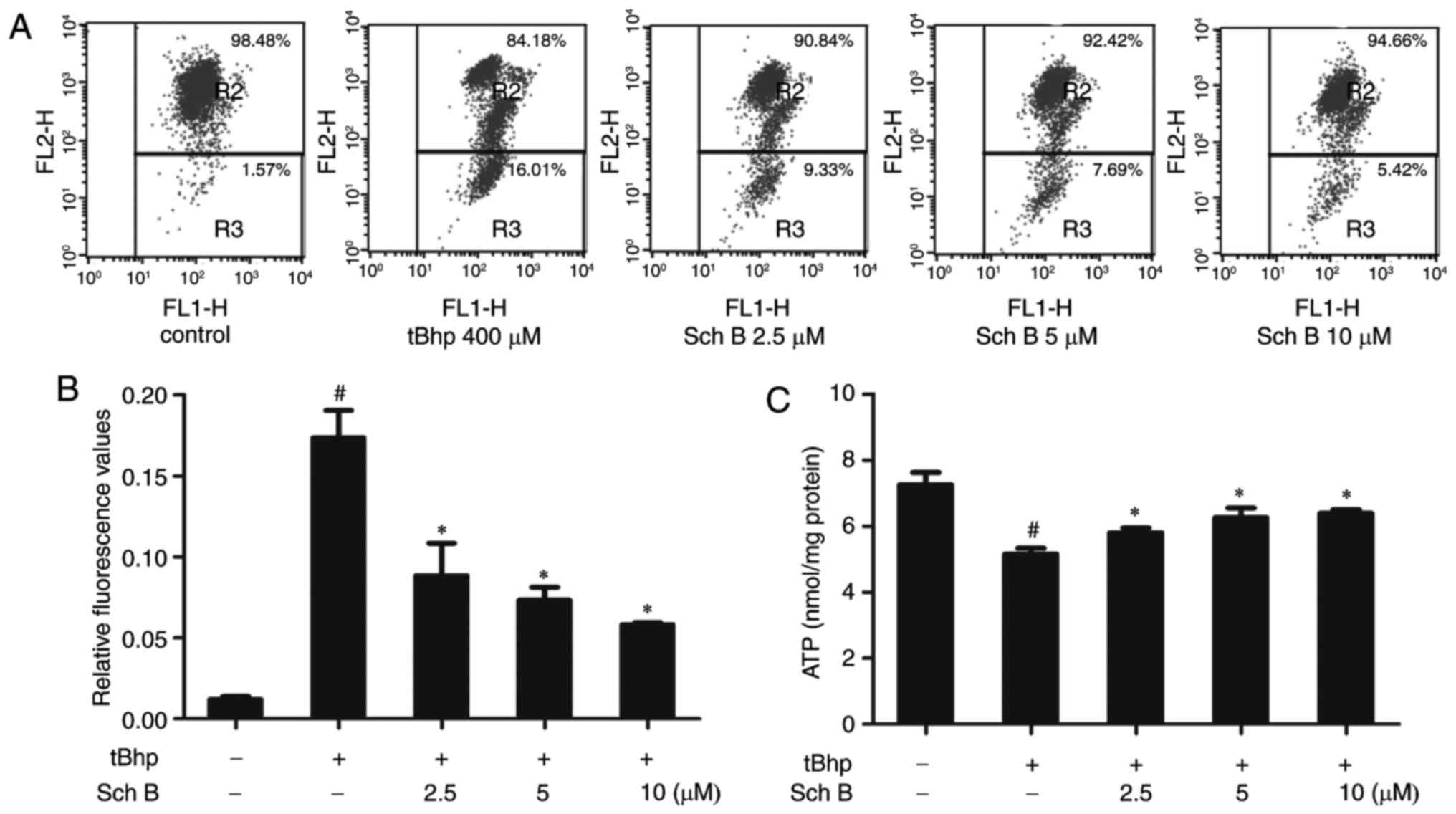
Sch B affects tBHP-induced mitochondrial
dysfunction in HaCaT cells
Oxidative stress can result in mitochondrial
dysfunction, a major cause of apoptosis. To evaluate mitochondrial
function, mitochondrial membrane potential (MMP) and intracellular
ATP levels were measured. Both a loss of MMP expression and
disruption in ATP energy supply are indicative of apoptosis. As
predicted, tBHP reduced MMP expression, as indicated by the
reduction in JC-1 fluorescence (Fig.
4A and B) and ATP production (Fig. 4C). Interestingly, Sch B treatment
rescued the effect on MMP levels and ATP production in tBHP-injured
HaCaT cells, indicating that Sch B can prevent mitochondrial
dysfunction induced by tBHP in HaCaT cells (Fig. 4A-C).
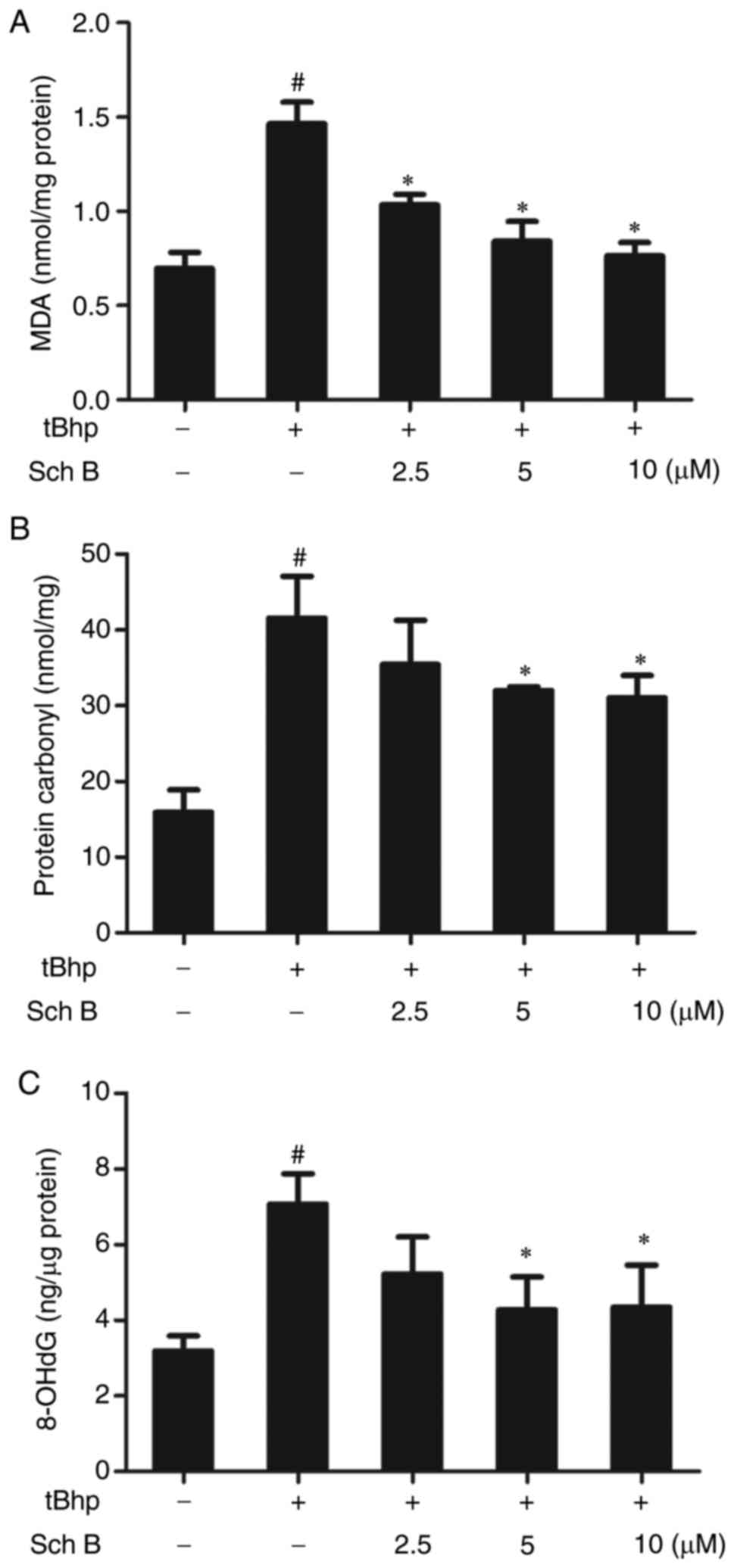
Sch B affects the tBHP-induced oxidation
of biomolecules in HaCaT cells
Oxidative stress was further analyzed by evaluating
oxidative damage to cellular biomolecules such as lipids, proteins
and DNA. Sch B significantly reduced tBHP-induced MDA production in
HaCaT cells, indicating a reduction in lipid peroxidation in
response to this compound (Fig.
5A). Carbonylated protein levels were used to assess protein
oxidation, and the results are presented in Fig. 5B: tBHP induced protein oxidation,
whereas Sch B significantly inhibited this induction. Similarly,
based on measurements of the damage marker 8-oxo-2'-deoxyguanosine
(8-oxo-dG), Sch B also significantly attenuated tBHP-induced DNA
damage (Fig. 5C). Thus, Sch B
effectively attenuated tBHP-induced oxidative damage to
biomolecules in HaCaT cells.
Sch B affects the expression of
antioxidant enzymes in tBHP-injured HaCaT cells
To determine whether Sch B protects HaCaT cells from
tBHP-induced oxidative damage through inducing the expression of
endogenous antioxidant enzymes, the effects of Sch B pretreatment
on mRNA expression levels of antioxidant enzymes, including HO-1,
SOD, GPx and CAT, were examined in tBHP-injured HaCaT cells. Our
Figs. 1 and 2 indicate the rapid induction of
cellular death by tBHP in HaCaT cells, and that exposure of HaCaT
cells to 4 mM tBHP for 3 h was enough to induce a significant
increase in apoptotic rate. In order to demonstrate that the
induction of antioxidant enzymes is responsible for the protective
effects of Sch B, its effects on the expression levels of
antioxidant enzymes after exposure to tBHP for 2 h were analyzed.
As shown in Fig. 6, exposure to
tBHP for 2 h did not significantly alter HO-1, SOD or GPx
expression in HaCaT cells, but led to a significant decline in that
of CAT. Notably, Sch B treatment prior to tBHP exposure
significantly increased expression of HO-1, SOD and GPx, and
restored tBHP-decreased CAT mRNA levels in HaCaT cells. These
results suggest that Sch B can fortify the antioxidant system via
increasing the expression of various antioxidant enzymes.
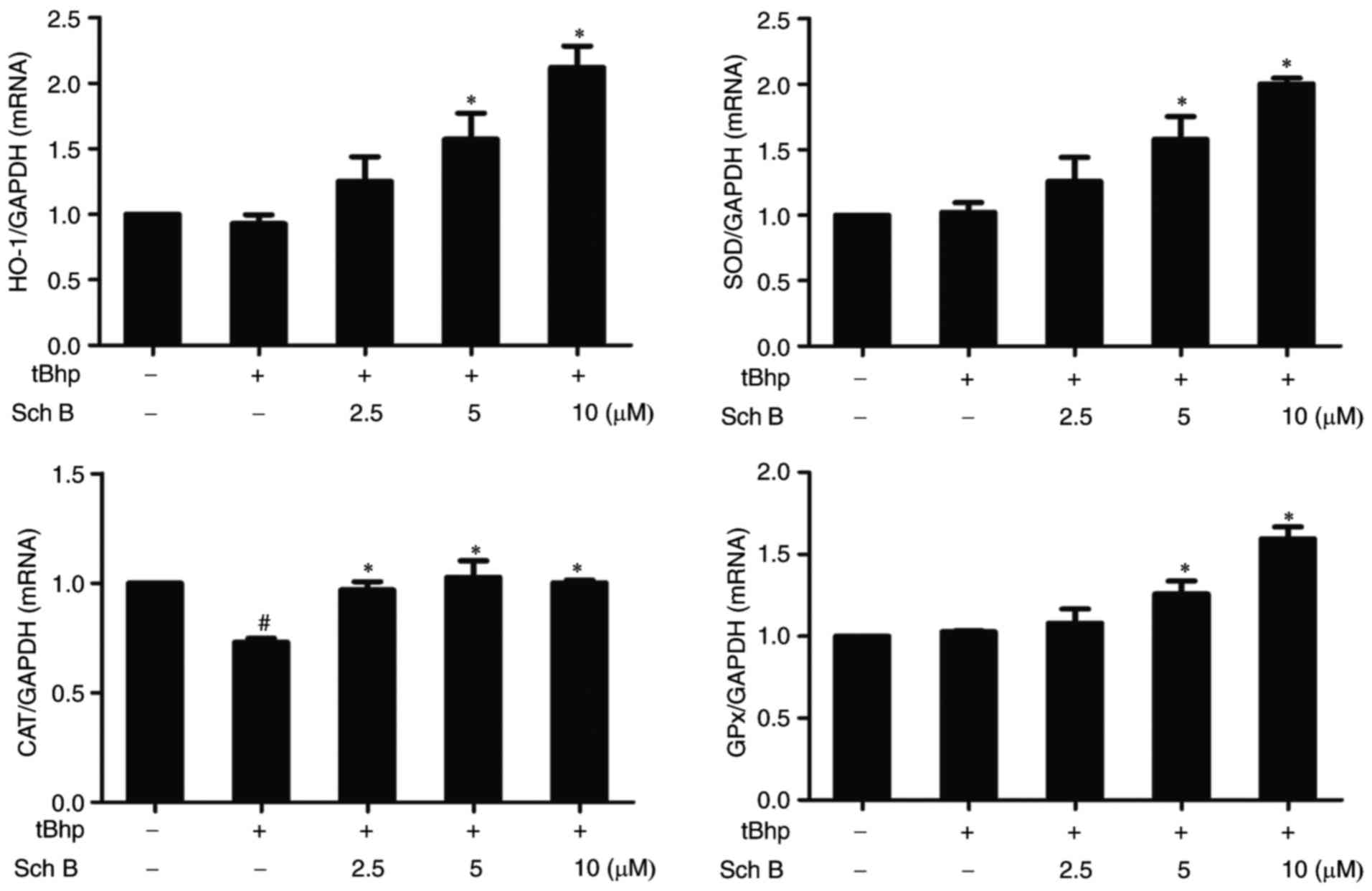 | Figure 6Sch B affects the expression of
antioxidant enzymes in tBHP-injured cells. HaCaT cells were
pre-treated with the indicated amount of Sch B s for 6 h, then
treated with 0.4 mM of tBHP for 2 h. HO-1, SOD, CAT, and GPx
expression was then measured by reverse transcription-quantitative
polymerase chain reaction. Data are presented as the mean ±
standard error of the mean of three different experiments
#P<0.05 vs. untreated controls and
*P<0.05 vs. tBHP only control. Sch B, Schisandrin B;
tBHP, tert-Butyl hydroperoxidesuperoxide dismutase; HO-1, heme
oxygenase 1; SOD, superoxide dismutase; CAT, catalase; GPx,
glutathione peroxidase. |
The effects of Sch B on Nrf2
activation
As the Nrf2/Keap1/ARE pathway is considered a master
regulatory pathway of the induction of antioxidant enzymes under
oxidative stress, we examined whether Sch B exerted anti-oxidant
activity by activating the Nrf2 signaling pathway. tBHP alone
resulted in a slight decrease in both nuclear and total Nrf2
protein levels compared with untreated cells, while Sch B treatment
prior to tBHP stimulation further increased nuclear and total Nrf2
protein levels relative to the tBHP-only group in a dose-dependent
fashion (Fig. 7A and B). This
indicates that Sch B treatment resulted in Nrf2 accumulation and
nuclear translocation. Sch B treatment did not alter Keap1
expression levels, but did markedly increased the phosphorylation
level of Nrf2 (Fig. 7),
suggesting that Sch B may activate Nrf2 via modulating its
phosphorylation rather than acting on negative regulators. As AMPK,
Akt and MAPKs (including Erk1/2, JNK and p38) have been proposed to
function as upstream kinases that mediate the phosphorylation and
activation of Nrf2, it was next examined how Sch B affects AMPK,
Akt, and MAPK signaling, in order to investigate their role in the
modulatory effect of Sch B on Nrf2. It was demonstrated that Sch B
pretreatment followed by tBHP stimulation led to increased
phosphorylation levels of AMPK, Akt, Erk1/2, JNK and p38 relative
to tBHP treatment only (Fig. 7B and
C). This suggests that the modulatory effect of Sch B on Nrf2
is mediated by multiple upstream kinases, including AMPK, Akt and
MAPKs.
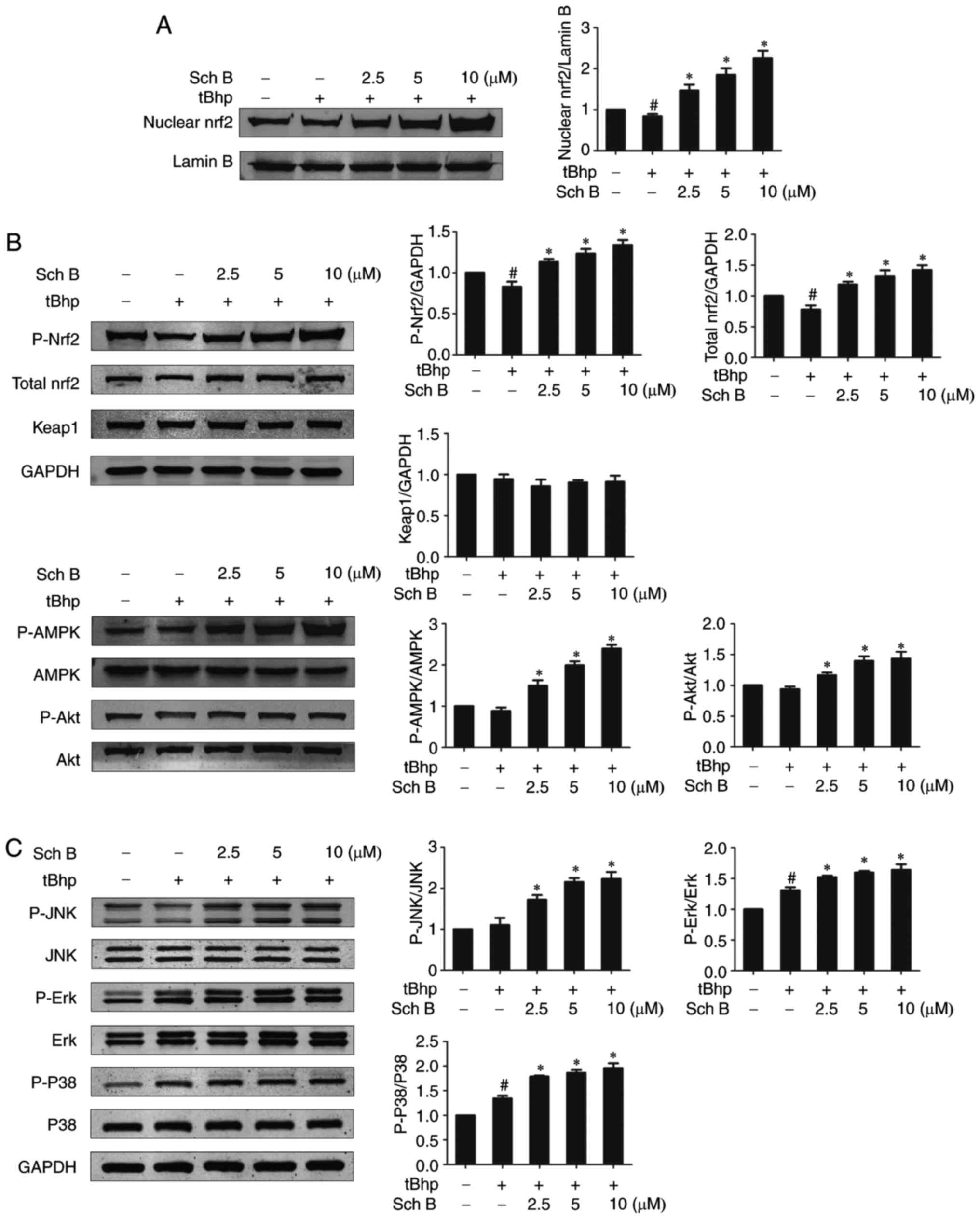 | Figure 7Sch B affects Nrf2 activation. HaCaT
cells were pre-treated with the indicated amounts of Sch B for 6 h,
then treated with 0.4 mM of tBHP for 1 h. (A) Nuclear Nrf2 protein
levels were assessed by western blotting and quantified by
densitometric analysis of (n=3). (B) The levels of total Nrf2,
Keap1, Akt, AMPK and as well as phosphorylated levels were assessed
via western blotting, and quantified by densitometric analysis. (C)
The levels of MAPKs (including JNK, Erk1/2 and p38) and their
phosphorylated levels were assessed via western blotting and
quantified by densitometric analysis (n=3). Data are presented as
the mean ± standard error of the mean of three different
experiments. #P<0.05 vs. untreated controls.
*P<0.05 vs. tBHP only control. Sch B, Schisandrin B;
Nrf2, nuclear factor-erythroid 2-related factor 2; tBHP, tert-Butyl
hydroperoxidesuperoxide dismutase; Keap1, Kelch-ECH associated
protein 1; AMPK, monophosphate-activated protein kinase; MAPK,
mitogen acctivatd protein kinase; Erk, extracellular
signal-regulated kinase; p-, phosphorylated. |
Discussion
In the present study, the ability of Sch B to shield
cells from oxidative damage induced by tBHP was investigated in
HaCaT cells. It was demonstrated that Sch B significantly decreased
tBHP-induced cytotoxicity and apoptotic cell death. Furthermore,
Sch B efficiently prevented tBHP-induced mitochondrial dysfunction
and biomolecule oxidation in HaCaT cells. Notably, Sch B induced
Nrf2 accumulation and promoted the transcription of its major
target antioxidant enzymes, while inhibiting tBHP-induced ROS
overproduction in HaCaT cells. These findings imply that Sch B can
protect HaCaT cells from tBHP-induced cell damage via enforcing its
endogenous enzymatic antioxidant defense systems.
Mitochondria are major ROS producers within cells,
and yet are themselves sensitive to oxidative stress. Redox
impairment can easily cause mitochondrial dysfunction by inhibiting
mitochondrially located enzymes associated with ATP production
(2,40). Disruption of oxidative
phosphorylation directly affects respiratory chain electron flux,
causing reduced MMP expression (41). This may be accompanied by
increased electron leakage in the mitochondrial respiratory chain,
enhancing ROS production. This can create a destructive feedback
loop, which may drive activation of the intrinsic apoptotic pathway
(42). Mitochondrial dysfunction,
overproduction of ROS, and enhanced rates of cell death are
important factors in aging and other human pathological processes
(43,44). In the present study, loss of the
MMP, decreased mitochondrial ATP production, and overproduction of
ROS were observed following tBHP challenge in HaCaT cells. These
negative effects of tBHP on mitochondrial dysfunction were
attenuated by Sch B pretreatment, which disrupted the tBHP-induced
reduction in viability and apoptosis. These results indicate that
Sch B may enhance or maintain mitochondrial function, thus allowing
HaCaT cells to better resist tBHP-induced oxidative damage.
ROS can damage proteins, lipids, nucleic acids, and
carbohydrates. In the context of weakening of antioxidant systems,
oxidative damage may result in permanent changes in the redox state
of these biomolecules, disrupting normal physiology. For example,
lipid peroxidation in the cellular membrane can disrupt cell
surface or mitochondrial membranes (45). Mitochondrial membrane damage in
turn leads to cytochrome c release and intrinsic apoptosis
(46). Numerous products derived
from biomolecule oxidation are used as oxidation biomarkers to
assess oxidative changes. The most widely used biomarkers include
MDA for lipid peroxidation, carbonylated proteins for protein
oxidation and 8-OHdG for DNA oxidation (47,48). It was found that the levels of
MDA, carbonylated protein and 8-OHdG were all substantially
elevated in HaCaT cells exposed to tBHP, indicating a state of
oxidative stress. Treatment of cells with Sch B reduced the levels
of all oxidation biomarkers, confirming its ability to overcome
tBHP-induced oxidative injury.
Antioxidant enzymes, including HO-1, SOD, GPx and
CAT, are vital for the protection of cells against oxidative
stressors. HO-1 is inducible and metabolizes heme to generate
carbon monoxide (CO), biliverdin, and iron in a rate-limiting
fashion. These heme derivatives possess antioxidant effects
(49). SOD, GPx and CAT are
responsible for the inactivation and elimination of superoxide and
hydrogen peroxide (50).
Therefore, these antioxidant enzymes are essential cytoprotective
agents, defending cells from the toxic effects of ROS by
maintaining relatively low intracellular ROS levels. In the present
study, it was demonstrated that Sch B treatment increased
antioxidant enzyme expression in tBHP-damaged HaCaT cells,
indicating that the induction of endogenous antioxidant enzymes
mediates the protective effects of Sch B, at least in part.
Nrf2 is the key transcription factor regulating
antioxidant enzyme expression. Its activity is primarily regulated
via interaction with Keap1, which sequesters Nrf2 in the cytoplasm
and directs it for degradation by the proteasome. Nrf2
phosphorylation also regulates the activity of Keap1 by
facilitating its translocation into the nucleus (51). In the present study, Sch B
treatment resulted in an increase in total and nuclear Nrf2 in
tBHP-challenged HaCaT cells, indicating that the Nrf2 pathway was
activated by Sch B, which is involved in the Sch B-mediated
induction of antioxidant enzymes. Sch B also induced Nrf2
phosphorylation without altering levels of Keap1, suggesting that
Sch B most likely activates Nrf2 via modulating its
phosphorylation. A number of protein kinases, such as PI3K/Akt,
MAPKs and AMPK, serve as upstream signals mediating Nrf2
phosphorylation under oxidative stress (52-54). Phosphorylation of these kinases
was assessed as a readout for their activation, and it was
demonstrated that Sch B treatment markedly induced the
phosphorylation of multiple kinases, including 3 members of MAPKs
(Erk1/2, JNK and p38), Akt and AMPK, suggesting that the activation
of Akt, MAPKs and AMPK may contribute to Sch B-induced Nrf2
phosphorylation and its subsequent transcriptional activation. This
is partially consistent with previous studies that determined that
MAPK signaling upon Sch B treatment of hepatocyte and cardiomyocyte
cell lines led to Nrf2 activation (19,22). Together, these results suggest
that Sch B may activate multiple upstream kinases by inducing their
phosphorylation, and that they, in turn, phosphorylate Nrf2 and
facilitate its nuclear translocation, leading to the expression of
antioxidant enzymes.
In conclusion, the present study demonstrated that,
in human keratinocyte-derived HaCaT cells, Sch B exhibited a
protective ability against tBHP-induced cytotoxicity and apoptotic
cell death by preventing mitochondrial dysfunction, suppressing
intracellular ROS generation, and mitigating biomolecule oxidation.
These findings further suggest that Sch B activates Nrf2 via
phosphorylation of its upstream kinases, including Akt, MAPKs
(Erk1/2, JNK and p38) and AMPK, leading to transcription of
antioxidant enzymes, including HO-1, SOD, GPx, and CAT, which
protect cells against oxidative stress. These results support the
use of Sch B to protect skin against oxidative stress, and
associated diseases and conditions including skin aging.
Acknowledgements
Not applicable.
Funding
The preset study was supported by the Opening
Project of Zhejiang Provincial Top Key Discipline of Pharmaceutical
Sciences (grant no. YKFJ3-010).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZH and MD conceived and designed the experiments.
MD, FW, YC and YG performed the experiments. ZH, TL, PS and GH
analyzed the data. ZH, SG, WD prepared the manuscript. ZH and MD
revised the manuscript. All authors read and approved the final
manuscript.
Patient consent for publication
Not applicable.
Ethics approval and consent to
participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kudryavtseva AV, Krasnov GS, Dmitriev AA,
Alekseev BY, Kardymon OL, Sadritdinova AF, Fedorova MS, Pokrovsky
AV, Melnikova NV, Kaprin AD, et al: Mitochondrial dysfunction and
oxidative stress in aging and cancer. Oncotarget. 7:44879–44905.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valko M, Leibfritz D, Moncol J, Cronin MT,
Mazur M and Telser J: Free radicals and antioxidants in normal
physiological functions and human disease. Int J Biochem Cell Biol.
39:44–84. 2007. View Article : Google Scholar
|
|
3
|
de Magalhães JP and Church GM: Cells
discover fire: Employing reactive oxygen species in development and
consequences for aging. Exp Gerontol. 41:1–10. 2006. View Article : Google Scholar
|
|
4
|
Shindo Y, Witt E, Han D, Epstein W and
Packer L: Enzymic and non-enzymic antioxidants in epidermis and
dermis of human skin. J Invest Dermatol. 102:122–124. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Crane D, Haussinger D, Graf P and Sies H:
Decreased flux through pyruvate dehydrogenase by thiol oxidation
during t-butyl hydroperoxide metabolism in perfused rat liver.
Hoppe Seylers Z Physiol Chem. 364:977–987. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bellomo G, Thor H and Orrenius S: Increase
in cytosolic Ca2+ concentration during t-butyl
hydroperoxide metabolism by isolated hepatocytes involves NADPH
oxidation and mobilization of intracellular Ca2+ stores.
FEBS Lett. 168:38–42. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kucera O, Endlicher R, Rousar T, Lotkova
H, Garnol T, Drahota Z and Cervinkova Z: The effect of tert-butyl
hydroperoxide-induced oxidative stress on lean and steatotic rat
hepatocytes in vitro. Oxid Med Cell Longev. 2014:7525062014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vessey DA, Lee KH and Blacker KL:
Characterization of the oxidative stress initiated in cultured
human keratinocytes by treatment with peroxides. J Invest Dermatol.
99:859–863. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wikramanayake TC, Simon J, Mauro LM, Perez
CI, Roberts B, Elgart G, Alvarez-Connelly E, Schachner LA and
Jimenez JJ: Tert-butyl hydroperoxide, an organic peroxide, causes
temporary delay in hair growth in a neonatal rat model. Clin Exp
Dermatol. 36:661–664. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Niture SK, Khatri R and Jaiswal AK:
Regulation of Nrf2-an update. Free Radic Biol Med. 66:36–44. 2014.
View Article : Google Scholar
|
|
11
|
Surh YJ, Kundu JK and Na HK: Nrf2 as a
master redox switch in turning on the cellular signaling involved
in the induction of cytoprotective genes by some chemopreventive
phytochemicals. Planta Med. 74:1526–1539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hybertson BM and Gao B: Role of the Nrf2
signaling system in health and disease. Clin Genet. 86:447–452.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bosch R, Philips N, Suárez-Pérez JA,
Juarranz A, Devmurari A, Chalensouk-Khaosaat J and Gonzalez S:
Mechanisms of photo-aging and cutaneous photocarcinogenesis, and
photoprotective strategies with phytochemicals. Antioxidants
(Basel). 4:248–268. 2015. View Article : Google Scholar
|
|
14
|
Holmström KM, Kostov RV and
Dinkova-Kostova AT: The multifaceted role of Nrf2 in mitochondrial
function. Curr Opin Toxicol. 1:80–91. 2016. View Article : Google Scholar
|
|
15
|
Hawkins KE, Joy S, Delhove JM, Kotiadis
VN, Fernandez E, Fitzpatrick LM, Whiteford JR, King PJ, Bolanos JP,
Duchen MR, et al: NRF2 orchestrates the metabolic shift during
induced pluripotent stem cell reprogramming. Cell Rep.
14:1883–1891. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu GT: Pharmacological actions and
clinical use of fructus schizandrae. Chin Med J (Engl).
102:740–749. 1989.
|
|
17
|
Panossian A and Wikman G: Pharmacology of
Schisandra chinensis Bail: An overview of Russian research and uses
in medicine. J Ethnopharmacol. 118:183–212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lam PY and Ko KM: Schisandrin B as a
hormetic agent for preventing age-related neurodegenerative
diseases. Oxid Med Cell Longev. 2012:2508252012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leong PK, Chiu PY, Chen N, Leung H and Ko
KM: Schisandrin B elicits a glutathione antioxidant response and
protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway
in AML12 hepatocytes. Free Radic Res. 45:483–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lam PY, Leong PK, Chen N and Ko KM:
Schisandrin B enhances the glutathione redox cycling and protects
against oxidant injury in different types of cultured cells.
Biofactors. 37:439–446. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chiu PY, Leung HY, Poon MK, Mak DH and Ko
KM: Effects of schisandrin B enantiomers on cellular glutathione
and mena-dione toxicity in AML12 hepatocytes. Pharmacology.
77:63–70. 2006. View Article : Google Scholar
|
|
22
|
Chiu PY, Chen N, Leong PK, Leung HY and Ko
KM: Schisandrin B elicits a glutathione antioxidant response and
protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway
in H9c2 cells. Mol Cell Biochem. 350:237–250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiu PY and Ko KM: Schisandrin B-induced
increase in cellular glutathione level and protection against
oxidant injury are mediated by the enhancement of glutathione
synthesis and regeneration in AML12 and H9c2 cells. Biofactors.
26:221–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lam PY and Ko KM: (-)Schisandrin B
ameliorates paraquat-induced oxidative stress by suppressing
glutathione depletion and enhancing glutathione recovery in
differentiated PC12 cells. Biofactors. 37:51–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ba Q, Cui C, Wen L, Feng S, Zhou J and
Yang K: Schisandrin B shows neuroprotective effect in
6-OHDA-induced Parkinson's disease via inhibiting the negative
modulation of miR-34a on Nrf2 pathway. Biomed Pharmacother.
75:165–172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lai Q, Luo Z, Wu C, Lai S, Wei H, Li T,
Wang Q and Yu Y: Attenuation of cyclosporine A induced
nephrotoxicity by schisandrin B through suppression of oxidative
stress, apoptosis and autophagy. Int Immunopharmacol. 52:15–23.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dong Q, Hou H, Wu J and Chen Y: The
Nrf2-ARE pathway is associated with Schisandrin b attenuating
benzo(a)pyrene-Induced HTR cells damages in vitro. Environ Toxicol.
31:1439–1449. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Q, Zhang H, Cao Y, Li Y, Sun S, Zhang
J and Zhang G: Schisandrin B attenuates CCl4-induced liver fibrosis
in rats by regulation of Nrf2-ARE and TGF-β/Smad signaling
pathways. Drug Des Devel Ther. 11:2179–2191. 2017. View Article : Google Scholar :
|
|
29
|
Chiu PY, Leung HY, Poon MK and Ko KM:
Chronic schisandrin B treatment improves mitochondrial antioxidant
status and tissue heat shock protein production in various tissues
of young adult and middle-aged rats. Biogerontology. 7:199–210.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chiu PY, Tang MH, Mak DH, Poon MK and Ko
KM: Hepatoprotective mechanism of schisandrin B: Role of
mitochondrial glutathione antioxidant status and heat shock
proteins. Free Radic Biol Med. 35:368–380. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thandavarayan RA, Giridharan VV, Arumugam
S, Suzuki K, Ko KM, Krishnamurthy P, Watanabe K and Konishi T:
Schisandrin B prevents doxorubicin induced cardiac dysfunction by
modulation of DNA damage, oxidative stress and inflammation through
inhibition of MAPK/p53 signaling. PLoS One. 10:e01192142015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen N, Chiu PY and Ko KM: Schisandrin B
enhances cerebral mitochondrial antioxidant status and structural
integrity, and protects against cerebral ischemia/reperfusion
injury in rats. Biol Pharm Bull. 31:1387–1391. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiu PY, Leung HY and Ko KM: Schisandrin B
enhances renal mitochondrial antioxidant status, functional and
structural integrity, and protects against Gentamicin-Induced
nephrotoxicity in rats. Biol Pharm Bull. 31:602–605. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ko KM and Lam BY: Schisandrin B protects
against tert-butylhy-droperoxide induced cerebral toxicity by
enhancing glutathione antioxidant status in mouse brain. Mol Cell
Biochem. 238:181–186. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ko KM, Chen N, Leung HY, Leong EP, Poon MK
and Chiu PY: Long-term schisandrin B treatment mitigates
age-related impairments in mitochondrial antioxidant status and
functional ability in various tissues, and improves the survival of
aging C57BL/6J mice. Biofactors. 34:331–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chiu PY, Lam PY, Yan CW and Ko KM:
Schisandrin B protects against solar irradiation-induced oxidative
injury in BJ human fibroblasts. Fitoterapia. 82:682–691. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hou W, Gao W, Wang D, Liu Q, Zheng S and
Wang Y: The protecting effect of deoxyschisandrin and schisandrin B
on HaCaT cells against UVB-induced damage. PLoS One.
10:e01271772015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gao C, Chen H, Niu C, Hu J and Cao B:
Protective effect of Schizandrin B against damage of UVB irradiated
skin cells depend on inhibition of inflammatory pathways.
Bioengineered. 8:36–44. 2017. View Article : Google Scholar
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
40
|
Nulton-Persson AC and Szweda LI:
Modulation of mitochondrial function by hydrogen peroxide. J Biol
Chem. 276:23357–23361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lieven CJ, Vrabec JP and Levin LA: The
effects of oxidative stress on mitochondrial transmembrane
potential in retinal ganglion cells. Antioxid Redox Signal.
5:641–646. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang CH, Wu SB, Wu YT and Wei YH:
Oxidative stress response elicited by mitochondrial dysfunction:
Implication in the pathophysiology of aging. Exp Biol Med
(Maywood). 238:450–460. 2013. View Article : Google Scholar
|
|
43
|
Naidoo K, Hanna R and Birch-Machin MA:
What is the role of mitochondrial dysfunction in skin photoaging?
Exp Dermatol. 27:124–128. 2018. View Article : Google Scholar
|
|
44
|
Tulah AS and Birch-Machin MA: Stressed out
mitochondria: The role of mitochondria in ageing and cancer
focussing on strategies and opportunities in human skin.
Mitochondrion. 13:444–453. 2013. View Article : Google Scholar
|
|
45
|
Girotti AW: Photosensitized oxidation of
membrane lipids: Reaction pathways, cytotoxic effects, and
cytoprotective mechanisms. J Photochem Photobiol B. 63:103–113.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chipuk JE, Bouchier-Hayes L and Green DR:
Mitochondrial outer membrane permeabilization during apoptosis: The
innocent bystander scenario. Cell Death Differ. 13:1396–1402. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hawkins CL, Morgan PE and Davies MJ:
Quantification of protein modification by oxidants. Free Radic Biol
Med. 46:965–988. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Marrot L and Meunier JR: Skin DNA
photodamage and its biological consequences. J Am Acad Dermatol.
58(5 Suppl 2): S139–S148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Motterlini R and Foresti R: Heme
oxygenase-1 as a target for drug discovery. Antioxid Redox Signal.
20:1810–1826. 2014. View Article : Google Scholar
|
|
50
|
Goyal MM and Basak A: Hydroxyl radical
generation theory: A possible explanation of unexplained actions of
mammalian catalase. Int J Biochem Mol Biol. 3:282–289.
2012.PubMed/NCBI
|
|
51
|
Bryan HK, Olayanju A, Goldring CE and Park
BK: The Nrf2 cell defence pathway: Keap1-dependent and -independent
mechanisms of regulation. Biochem Pharmacol. 85:705–717. 2013.
View Article : Google Scholar
|
|
52
|
Chen HH, Chen YT, Huang YW, Tsai HJ and
Kuo CC: 4-Ketopinoresinol, a novel naturally occurring ARE
activator, induces the Nrf2/HO-1 axis and protects against
oxidative stress-induced cell injury via activation of PI3K/AKT
signaling. Free Radic Biol Med. 52:1054–1066. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun Z, Huang Z and Zhang DD:
Phosphorylation of Nrf2 at multiple sites by MAP kinases has a
limited contribution in modulating the Nrf2-dependent antioxidant
response. PLoS One. 4:e65882009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shen G, Hebbar V, Nair S, Xu C, Li W, Lin
W, Keum YS, Han J, Gallo MA and Kong AN: Regulation of Nrf2
transactivation domain activity. The differential effects of
mitogen-activated protein kinase cascades and synergistic
stimulatory effect of Raf and CREB-binding protein. J Biol Chem.
279:23052–23060. 2004. View Article : Google Scholar : PubMed/NCBI
|