Introduction
Hypopituitarism has been recognized as a rare
disorder with an estimated incidence of 4.2 cases per 100,000 per
year and an estimated prevalence of 45.5 cases per 100,000
worldwide (1). It is
characterized by the deficiency of one or more of the hormones
secreted by the pituitary gland. The disease may result from
distinct causes and it may be congenital or acquired. The most
common etiology factors for acquired hypopituitarism include
pituitary tumors (2), traumatic
brain injury (3-5), pituitary irradiation (6), congenital hypopituitarism (CH),
perinatal insults (7) and genetic
mutations (8). Although the
clinical symptoms of this disease are usually unspecific, it may
cause life-threatening events and lead to an increased risk of
cardiovascular diseases, poor pregnancy potential and infertility
for the patients (9,10).
As a heterogeneous entity with various types of
etiology factors, mutations in the genes encoding a number of
hypothalamo-pituitary transcription factors, including HESX
homeobox 1 (HESX1), LIM homeobox 1 (LHX1), PROP paired-like
homeobox 1 (PROP1), and POU class 1 homeobox 1 (POU1F1) may lead to
CH; however, only 10% of cases are estimated to be caused by
genetic factors (11). Prenatal
or birth trauma and/or asphyxia and specific types of midline
defect syndromes may additionally lead to CH (12); however, their association with CH
are usually hard to determine. The underlying mechanisms for the
clinical symptoms of patients with CH are due to the deficiency in
pituitary hormones, including the growth hormone,
adrenocorticotropin (ACTH), prolactin (PRL), thyroid stimulating
hormone (TSH), luteinising hormone (LH) and follicle-stimulating
hormone (FSH) (13). These
deficiencies result in recurrent hypoglycemia and acute adrenal
insufficiency with impairments of growth and neurodevelopment
(13). Furthermore, due to the
lack of gonadotropin hormones, spermatogenesis and/or testicular
development are abnormal, resulting in infertility for male
patients. In females, successful pregnancy is rare, as
hypopituitarism is associated with an increased risk of pregnancy
complications, including abortion, anemia, pregnancy-induced
hypertension, placental abruption, premature birth and postpartum
hemorrhage (14). In the clinical
setting, lifelong hormone replacement treatment (HRT) is applied to
patients with CH with corticosteroids, levothyroxine, sex hormones
and growth hormones; however, the treatments may only partially
recover the abnormal development of patients and therefore they
remain at high risk of infertility and mortality (15-17). Therefore, the underlying molecular
mechanisms for the pathogenesis of CH require further
examination.
DNA methylation is a crucial epigenetic modification
that influences the structure of the genome and regulates
subsequent gene transcription activities (18). Previous studies of targeted gene
methylation demonstrated the significant associations between DNA
methylation and hormone exposure in early-life, particularly in
hormonally-mediated sexual differentiation (19,20). As DNA methylation status may
affect gene expression in response to hormones during the lifespan
(20), it is possible that DNA
methylation may be modified in patients with CH with hormone
deficiency, and it may be involved in the development and
progression of the disease. Therefore, the epigenome-wide
methylation status of the whole blood DNA was detected in patients
with CH, in order to identify genes that may be aberrantly
methylated in patients with CH compared with control subjects. The
present results may provide further insight into the pathogenesis
of the disease and may provide novel therapeutic targets for CH
treatment and biomarkers for diagnosis.
Materials and methods
Participant recruitment
The discovery set included 12 male patients with CH
and 12 male healthy controls matched for age (±3 years) and
body-mass index (±1 kg/m2). The participants were
recruited at the Ruijin Hospital North (Shanghai, China) between
November 2015 and May 2016. Eligible patients were clinically
diagnosed with hypopituitarism based on clinical history, symptoms,
biochemical and brain magnetic resonance imaging (MRI) tests.
Patients with pituitary tumors, traumatic brain injury, pituitary
irradiation or a clear diction of dystocia history were excluded.
All CH patients had been diagnosed with panhypopituitarism with
multiple pituitary hormone deficiency at a young age (~6-12 years
old). The MRI scan of all the patients with CH suggested a presence
of a transected or interrupted hypothalamic-pituitary stalk or
pituitary hypoplasia (21). The
patients had received routine cortisone and levothyroxine
treatment; however, had not yet received the human chorionic
gonadotrophin treatment at the time of recruitment. Controls were
selected from local residents receiving annual physical
examination, were free of acute or chronic diseases (including
diabetes, cancer and cardiovascular diseases), and had no symptom
of growth and developmental abnormalities. The validation set
included another 40 male patients with CH and 40 age-matched male
controls. The recruitment of the validation set followed the same
protocols as described for the discovery set, and was conducted
between June 2016 and July 2017. Written informed consent was
obtained from all the participants, and the study was approved by
the Institutional Review Board of Ruijin Hospital North.
Endocrinological hormone tests
The serum hormonal levels were determined to
evaluate the hormone secretion activity of the pituitary gland
during the drug withdrawal period, using commercial
radioimmunoassay kits in the clinic. The blood levels of free
thyroxine 3, free thyroxine 4 (FT4), TSH, cortisol, ACTH, LH, FSH,
PRL, progesterone (PROG) and testosterone were determined with
Beckman DX I800 instruments (Beckman Coulter, Inc., Brea, CA, USA),
according to the manufacturer’s protocol in the clinical testing
lab.
Genome-wide DNA methylation level
analysis
In total, 5 ml blood was provided by each
participant during their drug withdrawal period and the genomic DNA
was isolated using the QIAamp DNA Blood Mini kit (Qiagen GmbH,
Hilden, Germany). Following quality assessment, bisulfite
conversion of the genomic DNA was performed using the Zymo EZ DNA
Methylation kit (Zymo Research Corp., Irvine, CA, USA). DNA
methylation levels of blood were quantified with the Human
Methylation450K BeadArray (Illumina, Inc., San Diego, CA, USA)
following the manufacturer’s protocol, and the samples of cases and
controls were processed simultaneously. The methylation level of a
specific CpG site was quantified as a β value ranging from 0 (no
methylation) to 1 (full methylation). The raw DNA methylation β
values were normalized using the Subset-quantile Within Array
Normalization method (22) with
the minfi Bioconductor (version 1.26.2) package (www.bioconductor.org). Probes with detection P>0.01
and/or covering potential single nucleotide polymorphism (SNP)
sites were excluded from analysis. Student’s t-test was performed
to identify the significantly methylated CpG loci between the
patients with CH and controls. The top candidate probes were
selected based on an adjusted P-value using the Benjamini-Hochberg
False Discovery Rate (FDR) procedure with adjusted P<0.05 and
β-difference >0.14 (23). The
DNA methylation data are available at the NCBI Gene Expression
Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession no.
GSE107737.
Pathway enrichment analysis of the differentially
methylated genes was performed with the publicly available tool
Protein Analysis Through Evolutionary Relationships (PANTHER;
http://www.pantherdb.org/) that provides access
to various gene set libraries (24). A pathway was considered as
significantly enriched if the FDR P-value was <0.05.
Pyrosequencing analysis
The pyrosequencing method was applied in order to
determine the methylation level of candidate CpG sites in the
validation set. The bisulfite conversion and cleanup of DNA samples
was performed with the EpiTect Fast Bisulfite Conversion kit
(Qiagen,GmbH). Fragments covering the target sites were amplified
from the bisulfite converted genomic DNA with the biotinylated
forward and normal reverse primers, which were designed with
PyroMark Assay Design Software (version 2.0; Qiagen, GmbH). The
biotinylated polymerase chain reaction (PCR) was performed with
EpiTap™ HS polymerase (Takara Biotechnology Co., Ltd., Dalian,
China) and the thermocycling conditions were as follows: 98°C for
10 sec, 55°C for 30 sec, 72°C for 30 sec with 35 cycles followed by
72°C for 45 sec and 4°C for hold. The amplicon product was purified
and made into single-stranded, which was used as the template in
the pyrosequencing reaction. The pyrosequencing was conducted using
the Pyro Mark Q24 Advanced (Qiagen, GmbH), according to the
manufacturer’s protocol, with the specific sequencing primers
(cg01606027, forward PCR primer 5′-TTT TGG TTA TTT GGA TAT GTT TTT
TGT GAT-3′, reverse PCR primer 5′-TCA ACC AAA CCA ACT AAT CCC TAA
TA-3′, and pyrosequencing primer 5′-GTA AGG AAA TGA TAA TTG TGA
A-3′; cg03594078, forward PCR primer 5′-TGT GTA AAT ATT TAA TGG TAG
TAA GTG TAG -3′, reverse PCR primer 5′-AAT TTA CTA AAT CCT ACC CCC
AAA A-3′ and the pyrosequencing primer 5′-AAT TTT TTG TTT TTT TAG
GAA TT-3′). The PyroMark CpG Software 1.0 (Qiagen, GmbH) was used
to calculate the CpG methylation levels based on the height of the
T and C peaks at the methylation site with the formula C/(C + T)
×100.
Reverse transcription-quantitative
(RT-q)PCR in whole blood cells
The white blood cells of the participants in the
training cohort were purified from whole blood collected in the
EDTA sodium tubes, by centrifugation at 2,000 x g for 15 min at
room temperature. The white blood cells were lysed with the
TRIzol® reagent (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and the total RNA was extracted with the
Direct-zol RNA kit (Zymo Research, Corp.). A total of 500 ng total
RNA were used to synthesize the cDNA using the PrimeScript 1st
strand cDNA Synthesis kit (Takara Biotechnology Co., Ltd.) and
incubated at 30°C for 10 min, 42°C for 30 min followed by 70°C for
15 min. The qPCR reaction was performed with the SYBR®
Premix Ex Taq II kit (Takara Biotechnology Co., Ltd.). LIM domain
kinase 2 (LIMK2) and piwi-like RNA-mediated gene silencing 2
(PIWIL2), The primers were: LIM kinase 2 (LIMK2), forward 5′-GGA
TTC CCT CAC CAA CTG GTA-3′ and reverse 5′-AGC CAC CAT AAA AGG CCC
TG-3′; piwi-like RNA-mediated gene silencing 2 (PIWIL2), forward
5′-AGC AGG TGG TAT CAG CAG AGA-3′ and reverse 5′-TGT ATT TTG ACG
AGG TTC AGT CC-3′; and GAPDH, forward 5′-AAG GTG AAG GTC GGA-3′ and
reverse 5′-AAT GAA GGG GTC ATT GAT GG-3′. qPCR was performed at
95°C for 30 sec followed by 95°C for 5 sec and 60°C for 30 sec with
40 cycles. Relative expression of the genes was calculated relative
to the mean expression level of LIMK2 and PIWIL2 in the control
group normalized by GAPDH with the 2-ΔΔCq method
(25).
Statistical analysis
Age, weight, height and body mass index (BMI) of the
participants are presented as mean ± standard deviation and the
differences between the groups were analyzed with the Student’s
t-test. The hormone levels of the participants were presented as
median level in together with the lowest to highest range of the
hormone, and the differences between the groups were evaluated with
the Mann-Whitney test. Differences for the methylation levels of
the CpG sites in pyrosequencing analysis and the relative
expression of the genes between the groups were evaluated with the
Student’s t-test. The receiver-operating characteristic (ROC) curve
analysis and the corresponding area under the curve (AUC) were
calculated to determine the diagnosis values for the methylation
levels of the candidate CpG sites. All analyses were performed with
GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Baseline characteristics of the
participants in the discovery set
To compare the genome-wide methylation levels of the
whole blood DNA, first, 12 patients with CH and 12 age-matched
controls (±3 years) were recruited as the discovery set. The basic
clinical characteristics of the participants are presented in
Table I. The mean age for the
patients with CH was 21.58±2.71 years and 24.00±3.67 years for the
controls (P=0.081). All patients with CH had received stan-dardized
cortisone and levothyroxine therapy at the time of enrollment with
the median treatment time of 6 years (range 2-10 years), and none
of them had received the human chorionic gonadotrophin treatment.
All patients had a symptom of oligospermia or azoospermia at the
time of recruitment with the Tanner stage between G1 to G3
(Table I). No significant
difference was identified between the patients with CH and controls
for height, weight and body mass index (Table I). Compared with the controls, the
patients with CH had significantly lower levels of the hormones
secreted by the pituitary gland. In addition, significantly lower
FT4, cortisol and ACTH levels were observed in the patients with CH
compared with the controls (Table
I). Furthermore, the hormones associated with gonadotropic
function, LH, FSH, PROG and TESTO, were additionally at lower
levels in the patients with CH compared with the controls (Table I).
 | Table ICharacteristics of the paired
patients congenital with hypopituitarism and controls in the
discovery set. |
Table I
Characteristics of the paired
patients congenital with hypopituitarism and controls in the
discovery set.
|
Characteristics | Congenital
hypopituitarism, n=12 | Controls, n=12 | P-value |
|---|
| Basic
information | | | |
| Age, years | 21.58±2.71 | 24.00±3.67 | 0.081 |
| Height, cm | 169.0±8.72 | 173.0±5.17 | 0.185 |
| Weight, kg | 65.13±16.94 | 67.75±3.93 | 0.606 |
| BMI,
kg/m2 | 22.42±3.95 | 22.70±1.92 | 0.827 |
| Tanner stage | G1/G2/G3
(3/6/3) | - | - |
| Thyroid
function | | | |
| FT3, pmol/l | 3.70
(2.69-6.34) | 4.22
(3.51-5.06) | 0.434 |
| FT4, pmol/l | 9.67
(6.86-17.19) | 13.52
(11.67-17.08) | 0.018 |
| TSH,
µIU/l | 0.69
(0.00-5.94) | 1.80
(0.56-2.76) | 0.369 |
| Corticotropic
function | | | |
| Cortisol,
µg/dl | 0.465
(0.01-5.55) | 12.41
(7.10-21.00) | <0.001 |
| ACTH, pg/ml | 9.09
(5.27-10.96) | 42.00
(20.00-55.00) | <0.001 |
| Gonadotropic
function | | | |
| LH, mIU/l | 0.11
(0.01-1.26) | 4.05
(1.47-5.29) | <0.001 |
| FSH,
µIU/ml | 0.18
(0.02-1.70) | 4.97
(1.80-7.98) | <0.001 |
| PRL, ng/ml | 16.81
(2.21-49.13) | 8.58
(4.27-11.62) | 0.065 |
| E2, pg/ml | 20.00
(20.00-28.60) | 26.46
(20.00-35.40) | 0.087 |
| PROG, ng/ml | 0.10
(0.10-0.20) | 0.12
(0.10-0.79) | 0.017 |
| TESTO, ng/ml | 0.10
(0.10-2.00) | 4.75
(2.25-7.01) | <0.001 |
Genome-wide methylation analysis of whole
blood DNA in CH patients
Using the Human Methylation450K BeadArray, the CpG
methylation levels of the whole blood DNA in patients with CH and
controls were determined. The Manhattan plot demonstrated the CpG
sites on each chromosome and their corresponding unadjusted
P-values for the differences of meth-ylation levels between
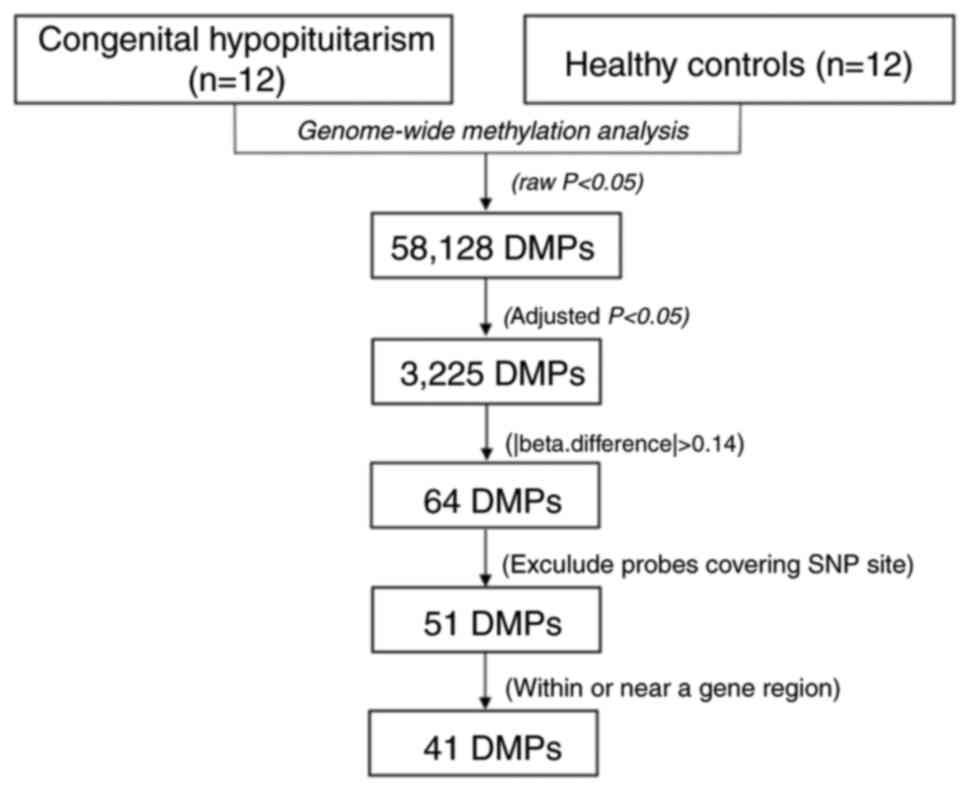
patients with CH and the controls (Fig. 1). The selection criteria for the
candidate CpG sites are presented in Fig. 2. Following the epigenome-wide
methylation analysis of the discovery set, 58,128 differentially
methylated probes (DMPs) were identified between the patients with
CH and the controls (unadjusted P-value <0.05). Of these, 3,225
DMPs had an FDR adjusted P<0.05 and 64 probes had a β-difference
>0.14. Subsequent to excluding probes covering potential SNP
sites, 51 significantly aberrant methylated CpG sites were selected
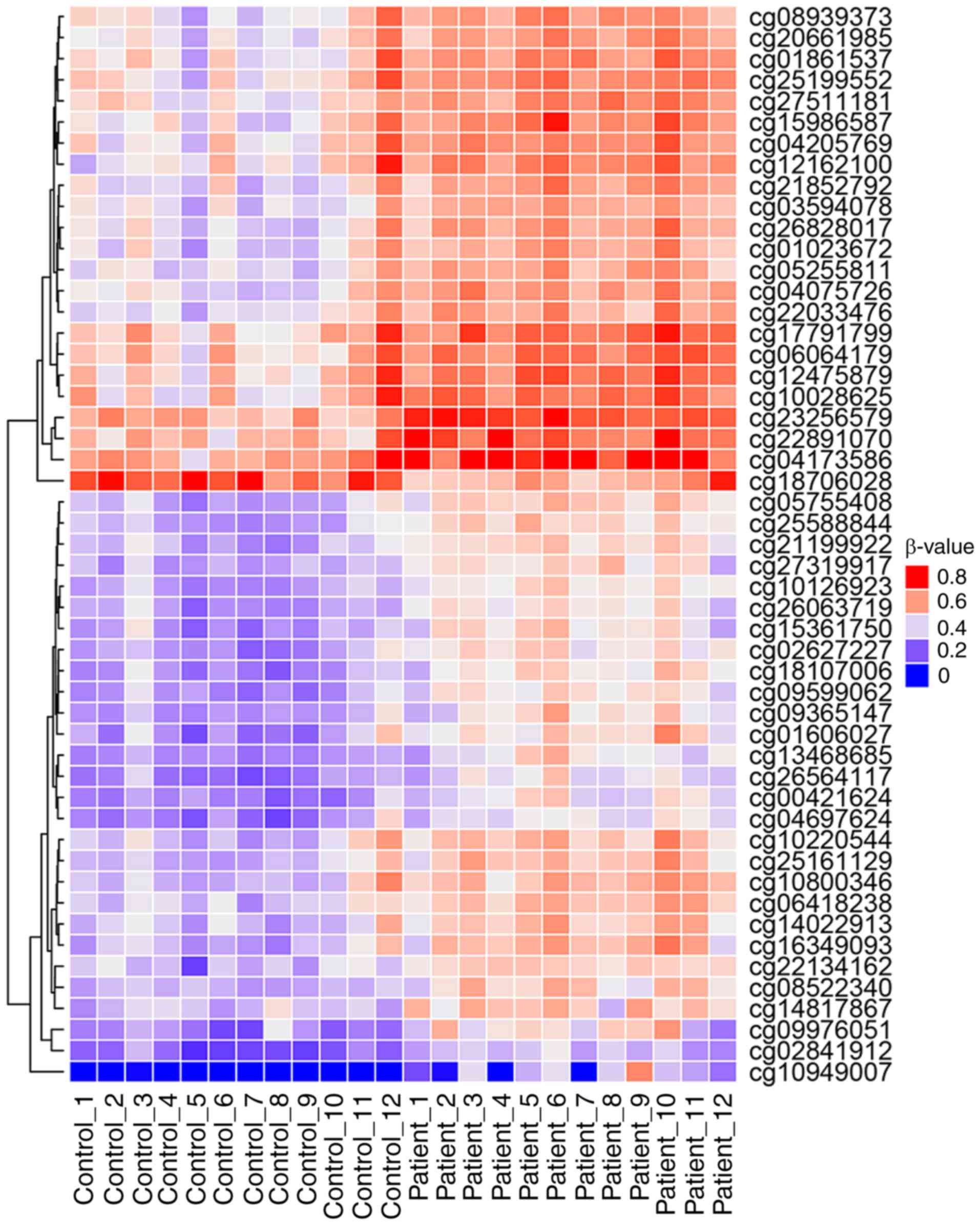
for further analysis (Fig. 3). Of
these, 50 sites were hypermethylated, whereas, only one CpG site,
cg18706028, was hypomethylated in patients with CH (Fig. 3). These 51 DMPs were clustered
into five regions according to gene annotation, including 10 DMPs
located the in TSS1500Ind, one DMP in TSS200Ind, 27 DMPs located on
gene body, three in the 5′-untranslated region (UTR), one in the
3′-UTR region of the gene, and the other 10 DMPs located in the
intergenic region (Table
II).
| Table IIDifferentially methylated positions
in the peripheral blood DNA of 12 paired patients with congenital
hypopituitarism and controls in the discovery set. |
Table II
Differentially methylated positions
in the peripheral blood DNA of 12 paired patients with congenital
hypopituitarism and controls in the discovery set.
| Gene/CpG
region | Gene symbol | CpG site | P-value | FDR P-value |
β-differencea | Chr | Region |
|---|
| TSS1500Ind | C11orf41 | cg08939373 | 0.0002 | 0.0391 | −0.1453 | 11 | - |
| GPR77 | cg15361750 | <0.0001 | 0.0170 | −0.1517 | 19 | - |
| LIMK2 | cg01606027 | 0.0001 | 0.0277 | −0.1715 | 22 | N_Shore |
| NEIL3 | cg14022913 | 0.0001 | 0.0255 | −0.1429 | 4 | N_Shore |
| SLC27A3 | cg00421624 | <0.0001 | 0.0052 | −0.1515 | 1 | N_Shore |
| PRR4 | cg23256579 | <0.0001 | 0.0011 | −0.1650 | 12 | - |
| GLRX | cg10949007 | 0.0001 | 0.0281 | −0.2245 | 5 | - |
| HNRPLL | cg18107006 | <0.0001 | 0.0043 | −0.1682 | 2 | S_Shore |
| PIWIL2 | cg03594078 | <0.0001 | 0.0132 | −0.1414 | 8 | N_Shore |
| TSS200Ind | HIF3A | cg22891070 | 0.0001 | 0.0227 | −0.1683 | 19 | S_Shore |
| GENEBODY | AGA | cg09976051 | 0.0001 | 0.0264 | −0.1792 | 4 | N_Shore |
| C20orf3 | cg20661985 | 0.0001 | 0.0267 | −0.1460 | 20 | N_Shelf |
| CCKBR | cg18706028 | 0.0001 | 0.0305 | 0.1487 | 11 | Island |
| DOT1L | cg04173586 | 0.0001 | 0.0212 | −0.1632 | 19 | S_Shore |
| ECT2 | cg10028625 | 0.0003 | 0.0455 | −0.1628 | 3 | - |
| FLJ13197 | cg21199922 | <0.0001 | 0.0039 | −0.1433 | 4 | - |
| KCNK5 | cg05255811 | <0.0001 | 0.0113 | −0.1495 | 6 | - |
| N4BP1 | cg27319917 | <0.0001 | 0.0041 | −0.1410 | 16 | - |
| NKG7 | cg10126923 | <0.0001 | 0.0013 | −0.1487 | 19 | S_Shelf |
| PLEK | cg13468685 | <0.0001 | 0.0045 | −0.1444 | 2 | - |
| PLEKHM1P | cg06064179 | 0.0001 | 0.0335 | −0.1433 | 17 | N_Shelf |
| PRPSAP2 | cg14817867 | <0.0001 | 0.0096 | −0.1473 | 17 | - |
| RASA3 | cg10800346 | 0.0001 | 0.0221 | −0.1600 | 13 | - |
| REV3L | cg09365147 | <0.0001 | 0.0156 | −0.1423 | 6 | - |
| TAF1B | cg25588844 | <0.0001 | 0.0021 | −0.1572 | 2 | - |
| THOC7 | cg22134162 | <0.0001 | 0.0067 | −0.1404 | 3 | - |
| TRIM35 | cg05755408 | <0.0001 | 0.0045 | −0.1580 | 8 | - |
| VIM | cg26063719 | <0.0001 | 0.0033 | −0.1440 | 10 | S_Shore |
| WIPI1 | cg25161129 | <0.0001 | 0.0123 | −0.1550 | 17 | N_Shore |
| FGGY | cg10220544 | 0.0003 | 0.0482 | −0.1429 | 1 | - |
| KDM1A | cg25199552 | 0.0001 | 0.0333 | −0.1476 | 1 | S_Shelf |
| RPTOR | cg27511181 | <0.0001 | 0.0035 | −0.1568 | 17 | - |
| RPTOR | cg06418238 | <0.0001 | 0.0050 | −0.1449 | 17 | - |
| THADA | cg22033476 | <0.0001 | 0.0198 | −0.1500 | 2 | - |
| CTSC | cg08522340 | <0.0001 | 0.0038 | −0.1503 | 11 | N_Shelf |
| DPH5 | cg04205769 | 0.0001 | 0.0333 | −0.1481 | 1 | N_Shelf |
| PROM1 | cg17791799 | 0.0003 | 0.0456 | −0.1434 | 4 | - |
| UTR5Ind | AGPAT4 | cg02627227 | <0.0001 | 0.0033 | −0.1600 | 6 | - |
| CPNE3 | cg12162100 | 0.0003 | 0.0493 | −0.1583 | 8 | S_Shelf |
| SYNE1 | cg02841912 | <0.0001 | 0.0051 | −0.1408 | 6 | N_Shore |
| UTR3Ind | PITPNB | cg26564117 | <0.0001 | 0.0117 | −0.1436 | 22 | - |
| Intergenic | - | cg15986587 | <0.0001 | 0.0159 | −0.1789 | 19 | N_Shelf |
| - | cg16349093 | <0.0001 | 0.0158 | −0.1776 | 5 | - |
| - | cg21852792 | <0.0001 | 0.0171 | −0.1654 | 2 | N_Shelf |
| - | cg04075726 | <0.0001 | 0.0079 | −0.1627 | 2 | - |
| - | cg26828017 | <0.0001 | 0.0192 | −0.1584 | 16 | - |
| - | cg01861537 | 0.0001 | 0.0342 | −0.1544 | 11 | - |
| - | cg09599062 | <0.0001 | 0.0050 | −0.1521 | 3 | - |
| - | cg12475879 | 0.0002 | 0.0394 | −0.1518 | 17 | S_Shelf |
| - | cg01023672 | 0.0001 | 0.0383 | −0.1461 | 12 | S_Shelf |
| - | cg04697624 | <0.0001 | 0.0189 | −0.1409 | 12 | N_Shelf |
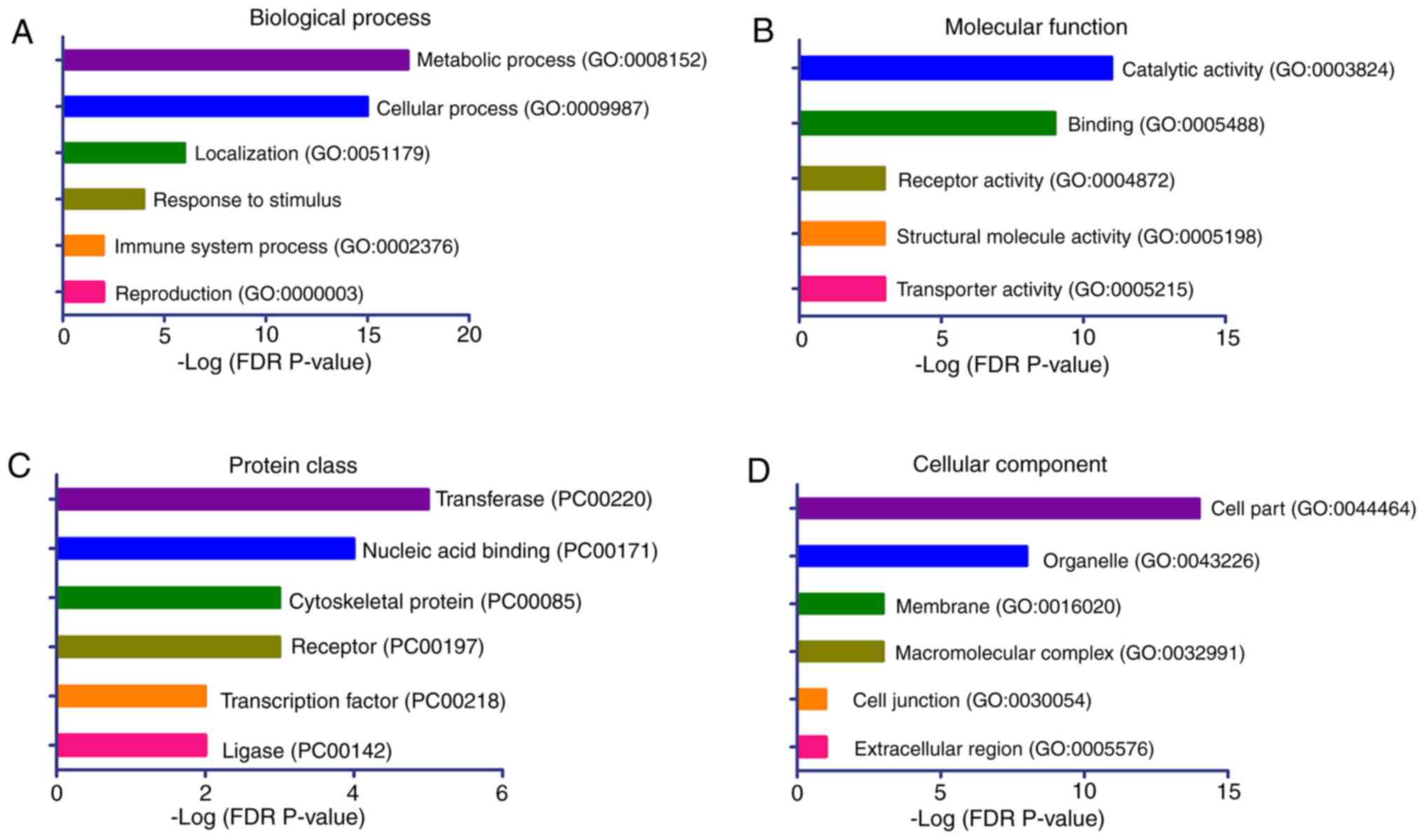
Gene set enrichment analysis of the
aberrant methylated genes
To provide further insights into the biological
processes associated with the aberrant methylated genes in the
patients with CH, a gene set enrichment analysis was conducted for
these genes using the PANTHER classification system. The 41
candidate genes with significant DMPs were included in the analyses
(Table II). The results
demonstrated that the genes were significantly enriched in the
biological processes of ‘metabolic process’, ‘cellular process’,
‘localization’, ‘response to stimulus’, ‘immune system process’ and
‘reproduction’ (Fig. 4A). The
significantly enriched molecular functions of these genes were
‘catalytic activity’, ‘binding’, ‘receptor’ and ‘transporter
activity’ (Fig. 4B). The top
enriched protein classes were ‘transferase’, ‘nucleic acid
binding’, ‘cytoskeletal protein’ and ‘receptor’ (Fig. 4C), while the top ranked cellular
components were ‘cell part’ and ‘organelle’ (Fig. 4D).
LIMK2 and PIWIL2 are hypermethylated in
patients with CH
CH in male patients is typically associated with
decreased spermatogenesis and testicle development latency
(26). The methylation analysis
demonstrated that the genes LIMK2 and PIWIL2, which have vital
roles in the reproductive system development, were hypermethylated
in the TSS1500Ind region (Table
II). As the hypermethylation of CpG sites in the promoter may
reduce the transcription activities of the target genes, it was
hypothesized that the aberrant methylation of these two genes may
be involved in the infertility of the male patients with CH. With
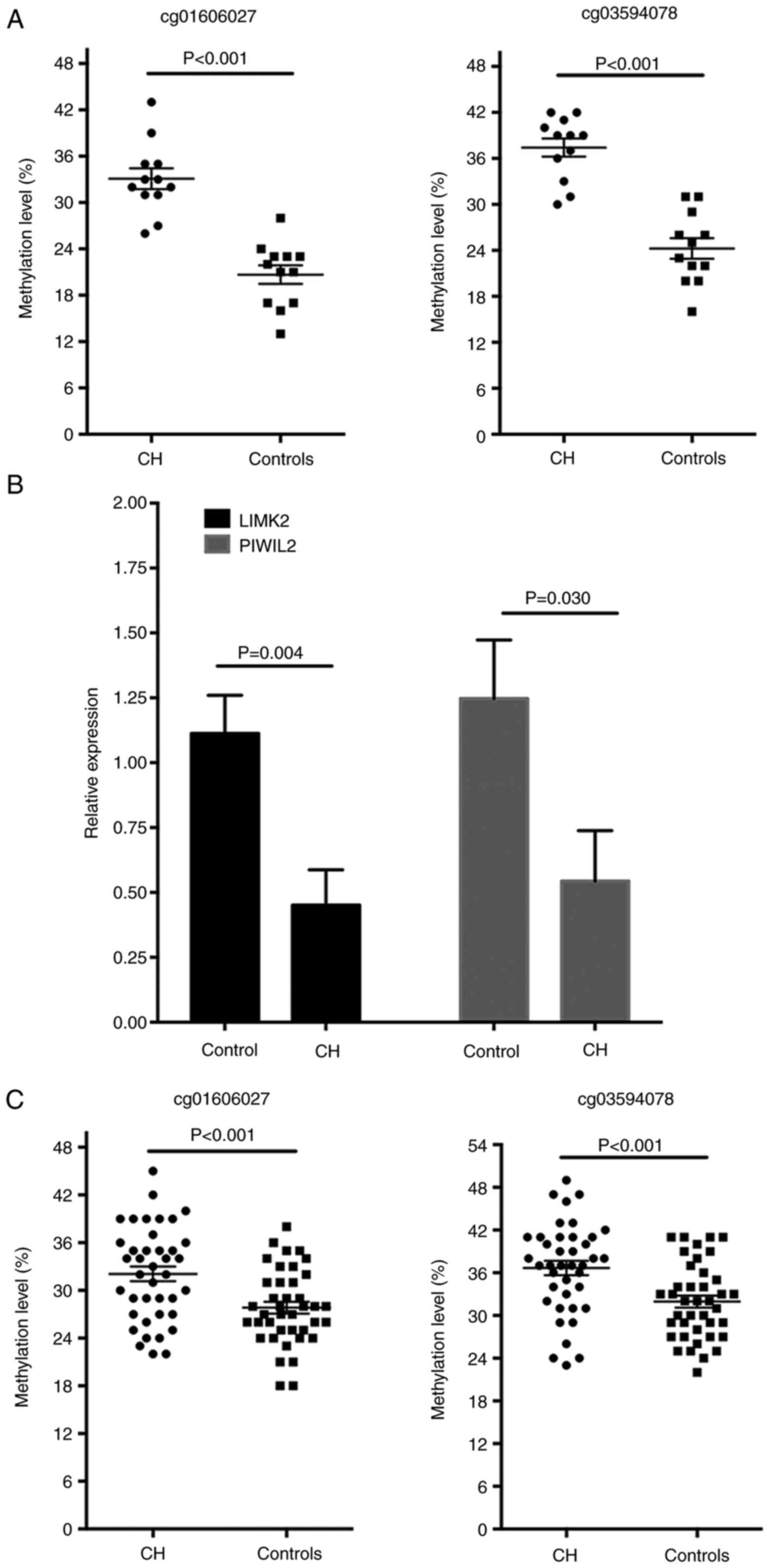
the pyrosequencing methods, the hypermethylated levels of
cg01606027 on LIMK2 and cg03594078 on PIWIL2 of the CH patients in
the discovery cohort were validated and it was identified that the
methylation level of the two CpG sites was significantly increased
in patients with CH (P<0.001 for the two sites; Fig. 5A). In addition, the mRNA
expression levels of LIMK2 and PIWIL2 in white blood cells were
significantly decreased in the patients with CH compared with the
normal controls, as detected by RT-qPCR analysis (Fig. 5B). The methylation levels of these
two loci were further determined in another validation set with 40
pairs of patients with CH and controls. The baseline
characteristics of the validation set are presented in Table III. No significant difference
was observed for the basic characteristics in patients with CH and
controls, whereas, the blood levels of TSH, cortisol, ACTH, LH,
FSH, PRL and TESTO were significantly lower in the patients with CH
compared with controls (Table
III). Compared with the controls, the methylation of the
cg01606027 on LIMK2 and cg03594078 on PIWIL2 was increased in the
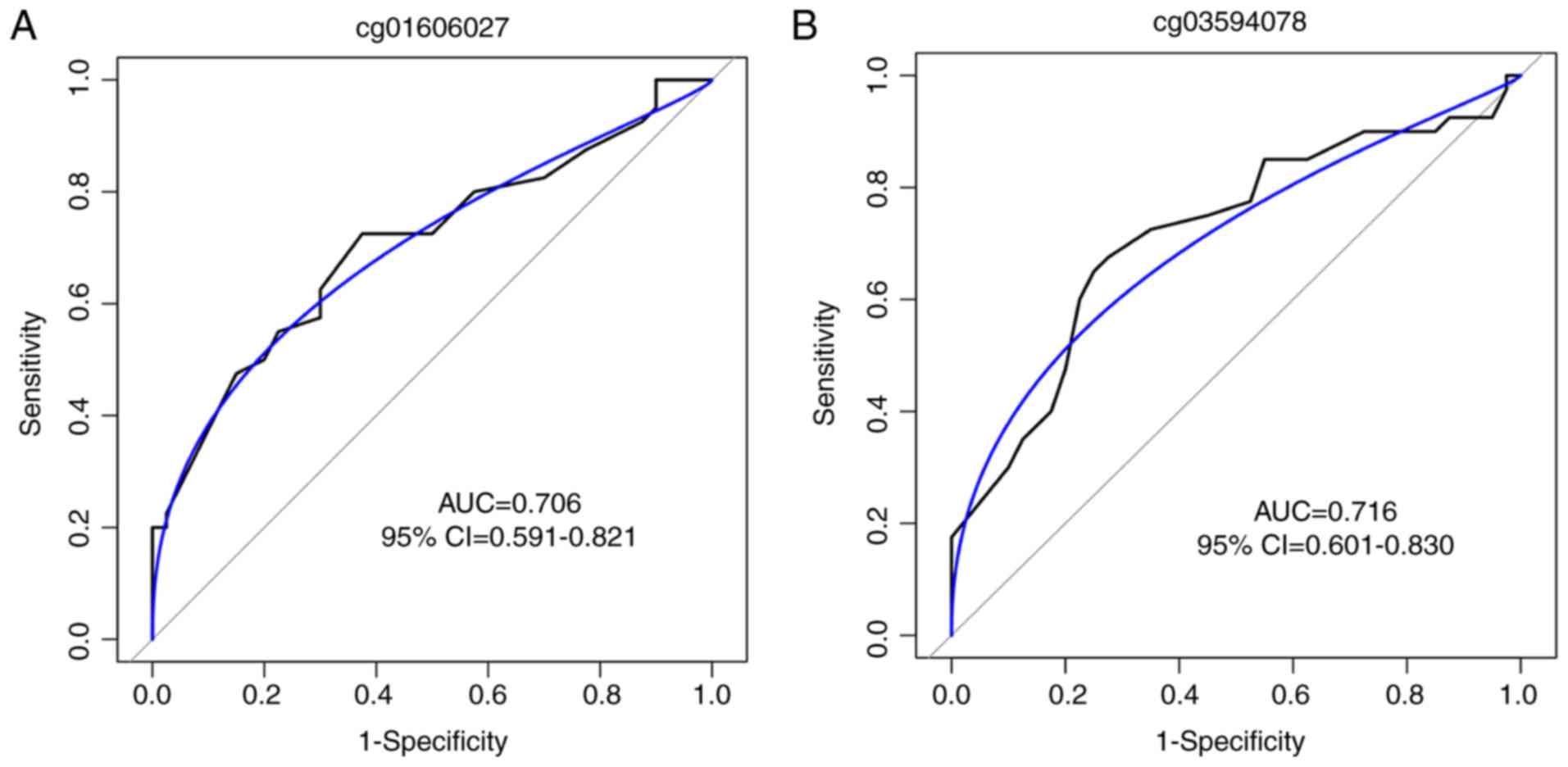
patients with CH (P<0.001 for the two sites; Fig. 5C). To estimate the potential
diagnostic power of these two methylated CpG sites with regards to
differentiating CH samples from controls, the predictive
performance of the methylation levels of the two CpG sites for CH
disease was examined with the ROC test in the validation cohort.
The AUC was 0.706 [95% confidence interval (CI), 0.591-0.821;
Fig. 6A] for cg01606027 and 0.716
(95% CI, 0.601-0.830; Fig. 6B)
for cg03594078, suggesting that the two loci may hold promise in CH
disease diagnosis.
| Table IIICharacteristics of the patients with
congenital hypopituitarism and controls in the validation set. |
Table III
Characteristics of the patients with
congenital hypopituitarism and controls in the validation set.
|
Characteristics | Congenital
hypopituitarism, n=40 | Controls, n=40 | P-value |
|---|
| Basic
information | | | |
| Age, years | 26.49±3.42 | 25.08±3.56 | 0.143 |
| Height, cm | 166.7±8.71 | 171.5±6.34 | 0.093 |
| Weight, kg | 63.28±14.92 | 65.31±7.06 | 0.068 |
| BMI,
kg/m2 | 23.10±4.19 | 22.23±2.31 | 0.768 |
| Thyroid
function | | | |
| FT3, pmol/l | 3.77
(2.57-5.07) | 4.19
(2.32-5.43) | 0.196 |
| FT4, pmol/l | 11.28
(6.96-16.92) | 12.45
(7.34-17.08) | 0.090 |
| TSH,
µIU/l | 0.15
(0.00-4.42) | 1.53
(0.56-3.25) | <0.001 |
| Corticotropic
function | | | |
| Cortisol,
µg/dl | 3.01
(0.13-19.13) | 13.69
(7.10-21.21) | <0.001 |
| ACTH, pg/ml | 20.54
(4.65-60.15) | 43.00
(20.00-55.00) | <0.001 |
| Gonadotropic
function | | | |
| LH, mIU/l | 0.18
(0.01-3.98) | 3.82
(1.27-8.26) | <0.0001 |
| FSH,
µIU/ml | 0.72
(0.05-6.16) | 5.04
(1.45-10.36) | <0.0001 |
| PRL, ng/ml | 15.79
(1.63-54.57) | 8.32
(3.56-11.67) | <0.0001 |
| E2, pg/ml | 24.30
(20.00-68.90) | 23.00
(20.00-45.40) | 0.8898 |
| PROG, ng/ml | 0.10
(0.10-0.49) | 0.10
(0.10-0.89) | 0.1731 |
| TESTO, ng/ml | 2.59
(0.10-10.67) | 4.31
(2.25-7.02) | 0.0429 |
Discussion
In the present study, the genome-wide DNA
methylation levels in the peripheral blood of male patients with CH
were determined and compared with age-matched controls. A total of
51 DMPs, that may influence gene expression levels involved in CH
development and progression, were identified. Pathway enrichment
analysis of the hypermethylated and hypomethylated DMP-associated
genes demonstrated that these were involved in ‘metabolic process’,
‘immune system process’ and ‘reproduction’. In addition, two
hypermethylated CpG loci located on the promoter regions of LIMK2
and PIWIL2, which are closely associated with testiculal
development and spermatogenesis, were observed in the patients with
CH. These results suggested that epigenetic alterations may
contribute to the pathogenesis of CH, and may provide novel
therapeutic targets in the future. Furthermore, the methylation
levels of the two loci had moderate predictive abilities in
patients with CH, suggesting that these may serve as potential
diagnostic or therapeutic markers for CH in the future.
CH is a heterogeneous disease with complex causes,
which may be acquired or are idiopathic (27,28). Previous studies observed that the
most common mutated genes in patients with CH are PROP1, POU1F1,
OTX2, HESX1, LHX3, GLI2, CDON, TGIF, SHH and LHX412; however, the
prevalence of mutations on these genes was low (29,30). Prenatal and birth trauma or
asphyxia may additionally lead to CH; however, their associations
with CH are not yet well established. Therefore, the exact
pathogenesis of CH remains unclear.
DNA methylation, which is the addition of a methyl
group to the cytosine in a CpG dinucleotide, has vital roles in
development and cellular differentiation. Aberrant methylation of
genes has been demonstrated to contribute to the development and
progression of various diseases. Due to the severe deficiency of
the hormones secreted by the pituitary gland, HRT is an effective
treatment method for patients with CH; however, they still present
with increased risk for hypoglycemia, cardiovascular diseases,
microphallus, infertility and hepatitis (12). Previous studies demonstrated that
the hormone deficiency is significantly associated with DNA
methylation levels (31,32). Gonadotropin deficiency results in
lower levels of testosterone in male patients with CH (33). With a mouse embryonic neural stem
cell (eNSC) model, Bramble et al (34) demonstrated that transcripts in
response to testosterone-propionate (TP) treatment are enriched in
genes that affect histone modification and DNA methylation. The TP
treatment results in a global decrease in 5-methylcytosine
abundance in the genomic DNA, and the DNA hypomethylation is
maintained in the daughter cell linages of eNSCs post-exposure
(34). These results suggested
that testosterone deficiency in early life may lead to the aberrant
hypermethylation of genes, which contributes to the pathogenesis of
neural diseases (34). In
addition, DNA methylation levels additionally influence the
response to hormone treatment in the clinic. Ouni et al
(35) demonstrated that the P2
promoter of the IGF1 gene mediated the responsiveness to growth
hormone in short children. Therefore, aberrant epigenetic gene
modulation may contribute to the pathogenesis of CH and
additionally influence the responsiveness of patients to hormone
treatment.
LIMK2 belongs to the serine LIM kinase family
(including LIMK1 and LIMK2) and it serves as a regulator of actin
dynamics (36). Previous studies
demonstrated that LIMK2 has important roles in cell movement,
division and structure formation (37,38). A previous study observed that
LIMK2 stimulates pathologic cancer cell division by regulating
actin filaments (39). In
addition, LIMK2 is not only involved in mitosis; however,
additionally meiosis, and LIMK2 activity is essential for
microtubule center organization and distribution in mouse oocyte
meiosis (40,41). Using a mouse knockout model,
Takahashi et al (42)
identified that LIMK2−/− mice did not demonstrated
embryonic lethality or any phenotypic abnormalities in the
postnatal growth and development, except for the lack of
spermatogenesis in the testis. Compared with the wide-type mice,
LIMK2−/− mice were smaller in size and had partial
degeneration of spermatogenic cells in the seminiferous tubules
(42). The viability of the
LIMK2−/− spermatogenic cells was significantly
diminished under stress conditions and the potential for germ cells
to differentiate in a regenerative state was additionally impaired
in the LIMK2−/− testis (42). These results suggested that LIMK2
is critical for the proper progression of spermatogenesis. The
present study demonstrated that LIMK2 was hypermethylated and
downregulated in the white blood cells of patients with CH compared
with the controls, which may partially explain the impaired
spermatogenic ability of the patients. LIMK2 may serve as a
potential therapeutic target for CH and treatment with testosterone
may decrease the methylation of LIMK2 in testicular germ cells. In
addition, the methylation levels of LIMK2 may influence the outcome
of HRT in the clinic; however, these considerations require further
examination in future studies.
PIWIL2, additionally termed MILI, belongs to the
PIWI family, a group of argonaute proteins that interact with a
class of small piwi-interacting (pi)RNAs, specifically expressed in
the testes during spermatogenesis (43). In the mammalian testes, PIWIL2 is
involved in the early phases of spermatogenesis, maintaining genome
stability by regulating the retrotranspo-sons through DNA
methylation silencing in germline cells (44). PIWIL2 is required for germline
stem cell self-renewal (45), and
PIWIL2 deficiency leads to increased transposon expression, and
defects in meiosis and germ cell survival (46). Heyn et al (47) observed that PIWIL2 was
hypermethylated in the semen DNA of infertile males, which resulted
in the defective production of piRNAs and the hypomethylation of
the LINW-1 repetitive sequence. Friemel et al (48) identified that the PIWIL2 was
hypermethylated in in the peripheral blood DNA of patients with
idiopathic male infertility compared with control males, which is
consistent with the results of the present study. These results
suggested that DNA methylation of PIWIL2 may regulate gene
expression in spermatogenesis and result in abnormal sperm cell
development, which may partially contribute to the infertility
disorders of male patients with CH in the clinic.
Besides these two loci, CpG sites located in other
genes, including hypoxia-inducible factor (HIF)-3α and
regulatory-associated protein of mammalian target of rapamycin
(mTOR) complex 1 (RPTOR), were additionally noted in the present
study; however, their roles in CH development and progression
remain unknown. Hypermethylation of HIF-3α has been observed in
children with obesity, and a positive association between BMI and
DNA methylation level of HIF-3α was identified in a previous study
(49). HIF-3α is highly expressed
in adipocytes and has vital roles in adipogenesis (50). Pfeiffer et al (51) demonstrated that the methylation
levels of CpG site cg22891070, which was hypermethylated in
patients with CH, was significantly higher in visceral adipose
tissue (VAT) compared with subcutaneous adipose tissue (SAT), and
cg22891070 methylation levels were significantly correlated with
BMI, and abdominal SAT and VAT area. Therefore, aberrant HIF-3α
methylation may contribute to adipose tissue dysfunction, which may
lead to the increased risk of obesity-associated diseases in
patients with CH. Two CpG sites (cg27511181 and cg06418238) were
identified to be hypermethylated in the gene body of RPTOR, which
is an important scaffolding protein that recruits the mTOR
substrates to rapamycin-senstitive mTOR complex 1 (mTORC1)
(52). RPTOR is required for the
suppressive function of regulatory T cells (53). Tang et al (54) demonstrated that the CpG site
cg06418238 was hypomethylated in the peripheral blood DNA of
patients with breast cancer compared with controls. However, the
role of RPTOR and its hypermethylation in the development and
progression of CH remain unclear. In the future, novel identified
epigenetically regulated genes and their roles in CH development
and progression require further investigation, which may provide
further insight for the mechanisms for the pathogenesis of CH.
There were a number of limitations of the present
study. The sample size in the discovery set was relatively small.
Other CpG sites that may be aberrantly methylated in patients with
CH may not have been identified due to the statistical power
limitations. The methylation status of the genomic DNA was derived
from the whole blood, and its consistence with the DNA methylation
levels in the target cells of the pituitary gland or testes are
largely unknown. Whether LIMK2 and PIWIL2 were hypermethylated in
sperm cells and their roles in the infertility of patients with CH
require examination in the future. The mechanisms for the aberrant
methylation levels of the target genes remain unknown. Whether the
hypermethylation or hypomethylation of the CpG sites was caused by
perinatal damage or hormone deficiency, and the molecular
mechanisms for the epigenetic regulation of these genes under
stress require further investigation in future studies. The effect
of HRT in the epigenetic regulation of the aberrantly methylated
genes in patients with CH and whether the methylated loci may
regulate the outcome of HRT requires further study.
In conclusion, the present study determined the
genome-wide DNA methylation levels of the peripheral whole blood
DNA in male patients with CH and demonstrated that 51 CpG sites
were significantly hypermethylated or hypomethylated in patients
with CH compared with normal controls. Two CpG sites cg01606027 and
cg03594078 located at the coding region of the genes LIMK2 and
PIWIL2, that are essential for spermatogenesis, were
hypermethylated, which may contribute to azoospermia or
oligozoospermia in male patients with CH. These results suggested
that the aberrant epigenetic regulated genes may contribute to the
pathogenesis of CH, and that they may serve as novel therapeutic
targets or diagnosis biomarkers for CH in the future.
Acknowledgments
Not applicable.
Funding
The present study was financially supported by
grants from the National Key R&D Program of China (grant nos.
2017YFC0907001 and 2018ZX10302205), the Science and Technology
Commission of Shanghai Municipality (grant no. 16411966800;
Shanghai, China), the Shanghai Municipal Commission of Health and
Family Planning (grant nos. 20164Y0250 and 201840160; Shanghai,
China), the Science and Technology Commission of Jiading District
(grant nos. JDKW-2017-W09 and JDKW-2017-W11; Shanghai, China), the
interdisciplinary funding of Shanghai Jiao Tong
University (grant no. YG2017QN57; Shanghai, China)
and the Ruijin Hospital North for Young Talents (grant nos.
2017RCPY-A01 and 2017RCPY-C01; Shanghai, China).
Availability of data and materials
The Human Methylation 450K DNA methylation data are
available at the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession
no. GSE107737.
Authors’ contributions
XF and PC drafted the manuscript and coordinated the
study. JC and JL performed the array analysis. XF, CC, EX and PC
conducted the validation experiments. JL and PC performed the
bioinformatics analysis. XF and PC performed the polymerase chain
reaction experiments. XF, CC, EX and JC acquired the samples and
information of the participants. XF, JL and PC designed and
supervised the study, and revised the manuscript. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all the
participants, and the study was approved by the Institutional
Review Board of Ruijin Hospital North (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations
Abbreviations:
|
ACTH
|
adrenocorticotropin
|
|
AUC
|
area under the curve
|
|
BMI
|
body mass index
|
|
CI
|
confidence interval
|
|
CH
|
congenital hypopituitarism
|
|
DMP
|
differentially methylated probe
|
|
FDR
|
false discovery rate
|
|
FSH
|
follicle-stimulating hormone
|
|
HRT
|
hormone replacement treatment
|
|
LH
|
luteinising hormone
|
|
MRI
|
magnetic resonance imaging
|
|
PRL
|
prolactin
|
|
PROG
|
progesterone
|
|
ROC
|
receiver-operating characteristic
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
SAT
|
subcutaneous adipose tissue
|
|
TSH
|
thyroid stimulating hormone
|
|
VAT
|
visceral adipose tissue
|
References
|
1
|
Regal M, Paramo C, Sierra SM and
Garcia-Mayor RV: Prevalence and incidence of hypopituitarism in an
adult Caucasian population in northwestern Spain. Clin Endocrinol
(Oxf). 55:735–740. 2001. View Article : Google Scholar
|
|
2
|
Agha A, Sherlock M, Brennan S, O’Connor
SA, O’Sullivan E, Rogers B, Faul C, Rawluk D, Tormey W and Thompson
CJ: Hypothalamic-pituitary dysfunction after irradiation of
nonpituitary brain tumors in adults. J Clin Endocrinol Metab.
90:6355–6360. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bondanelli M, De Marinis L, Ambrosio MR,
Monesi M, Valle D, Zatelli MC, Fusco A, Bianchi A, Farneti M and
degli Uberti EC: Occurrence of pituitary dysfunction following
traumatic brain injury. J Neurotrauma. 21:685–696. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herrmann BL, Rehder J, Kahlke S,
Wiedemayer H, Doerfler A, Ischebeck W, Laumer R, Forsting M, Stolke
D and Mann K: Hypopituitarism following severe traumatic brain
injury. Exp Clin Endocrinol Diabetes. 114:316–321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cuesta M, Hannon MJ, Crowley RK, Behan LA,
Tormey W, Rawluk D, Delargy M, Agha A and Thompson CJ: Symptoms of
gonadal dysfunction are more predictive of hypopituitarism than
nonspecific symptoms in screening for pituitary dysfunction
following moderate or severe traumatic brain injury. Clin
Endocrinol. 84:92–98. 2016. View Article : Google Scholar
|
|
6
|
Darzy KH and Shalet SM: Hypopituitarism
after cranial irradiation. J Endocrinol Invest. 28(Suppl 5):
S78–S87. 2005.
|
|
7
|
Craft WH, Underwoood LE and Van Wyk JJ:
High incidence of perinatal insult in children with idiopathic
hypopituitarism. J Pediatr. 96:397–402. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nystrom HF, Saveanu A, Barbosa EJ, Barlier
A, Enjalbert A, Glad C, Palming J, Johannsson G and Brue T:
Detection of genetic hypopituitarism in an adult population of
idiopathic pituitary insufficiency patients with growth hormone
deficiency. Pituitary. 14:208–216. 2011. View Article : Google Scholar
|
|
9
|
Tomlinson JW, Holden N, Hills RK, Wheatley
K, Clayton RN, Bates AS, Sheppard MC and Stewart PM: Association
between premature mortality and hypopituitarism. West Midlands
Prospective Hypopituitary Study Group. Lancet. 357:425–431. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kohno H, Kuromaru R, Ueyama N and Miyako
K: Premature mortality and hypopituitarism. Lancet. 357:1973–1974.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fernandez-Rodriguez E, Quinteiro C,
Barreiro J, Marazuela M, Pereiro I, Peino R, Cabezas-Agricola JM,
Dominguez F, Casanueva FF and Bernabeu I: Pituitary stalk
dysgenesis-induced hypopituitarism in adult patients: Prevalence,
evolution of hormone dysfunction and genetic analysis.
Neuroendocrinology. 93:181–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geffner ME: Hypopituitarism in childhood.
Cancer Control. 9:212–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Braslavsky D, Mendez MV, Prieto L,
Keselman A, Enacan R, Gruneiro-Papendieck L, Jullien N, Savenau A,
Reynaud R, Brue T, et al: Pilot neonatal screening program for
central congenital hypothyroidism: Evidence of significant
detection. Horm Res Paediatr. 88:274–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du X, Yuan Q, Yao Y, Li Z and Zhang H:
Hypopituitarism and successful pregnancy. Int J Clin Exp Med.
7:4660–4665. 2014.
|
|
15
|
Clayton RN: Mortality, cardiovascular
events and risk factors in hypopituitarism. Growth Horm IGF Res.
8(Suppl A): S69–S76. 1998. View Article : Google Scholar
|
|
16
|
Kung AW, Zhong YY, Lam KS and Wang C:
Induction of spermatogenesis with gonadotrophins in Chinese men
with hypogonadotrophic hypogonadism. Int J Androl. 17:241–247.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bulow B, Hagmar L, Eskilsson J and Erfurth
EM: Hypopituitary females have a high incidence of cardiovascular
morbidity and an increased prevalence of cardiovascular risk
factors. J Clin Endocrinol Metab. 85:574–584. 2000.PubMed/NCBI
|
|
18
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCarthy MM and Nugent BM: Epigenetic
contributions to hormonally-mediated sexual differentiation of the
brain. J Neuroendocrinol. 25:1133–1140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nugent BM, Schwarz JM and McCarthy MM:
Hormonally mediated epigenetic changes to steroid receptors in the
developing brain: Implications for sexual differentiation. Horm
Behav. 59:338–344. 2011. View Article : Google Scholar
|
|
21
|
Pinto G, Netchine I, Sobrier ML, Brunelle
F, Souberbielle JC and Brauner R: Pituitary stalk interruption
syndrome: A clinical-biological-genetic assessment of its
pathogenesis. J Clin Endocrinol Metab. 82:3450–3454.
1997.PubMed/NCBI
|
|
22
|
Maksimovic J, Gordon L and Oshlack A:
SWAN: Subset-quantile Within Array Normalization for illumina
Infinium HumanMethylation450 BeadChips. Genome Biol. 13:R442012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benjamini Y and Hochberg Y: The adaptive
control of the false discovery rate in multiple hypotheses testing.
J Behav Educ Statist. 25:60–83. 2000. View Article : Google Scholar
|
|
24
|
Mi H, Muruganujan A and Thomas PD: PANTHER
in 2013: Modeling the evolution of gene function, and other gene
attributes, in the context of phylogenetic trees. Nucleic Acids
Res. 41(Database Issue): D377–D386. 2013. View Article : Google Scholar :
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Prabhakar VK and Shalet SM: Aetiology,
diagnosis, and management of hypopituitarism in adult life.
Postgrad Med J. 82:259–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pfaffle R and Klammt J: Pituitary
transcription factors in the aetiology of combined pituitary
hormone deficiency. Best Pract Res Clin Endocrinol Metab. 25:43–60.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kelberman D, Rizzoti K, Lovell-Badge R,
Robinson IC and Dattani MT: Genetic regulation of pituitary gland
development in human and mouse. Endocr Rev. 30:790–829. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takagi M, Nagasaki K, Fujiwara I, Ishii T,
Amano N, Asakura Y, Muroya K, Hasegawa Y, Adachi M and Hasegawa T:
Heterozygous defects in PAX6 gene and congenital hypopituitarism.
Eur J Endocrinol. 172:37–45. 2015. View Article : Google Scholar
|
|
30
|
Romero CJ, Nesi-Franca S and Radovick S:
The molecular basis of hypopituitarism. Trends Endocrinol Metab.
20:506–516. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xie W, Ren M, Li L, Zhu Y, Chu Z, Zhu Z,
Ruan Q, Lou W, Zhang H, Han Z, et al: Perinatal testosterone
exposure potentiates vascular dysfunction by ERβ suppression in
endothelial progenitor cells. PLoS One. 12:e01829452017. View Article : Google Scholar
|
|
32
|
Huen K, Harley K, Kogut K, Rauch S,
Eskenazi B and Holland N: DNA methylation of LINE-1 and Alu
repetitive elements in relation to sex hormones and pubertal timing
in Mexican-American children. Pediatr Res. 79:855–862. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pantalone KM and Faiman C: Male
hypogonadism: more than just a low testosterone. Cleve Clin J Med.
79:717–725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bramble MS, Roach L, Lipson A, Vashist N,
Eskin A, Ngun T, Gosschalk JE, Klein S, Barseghyan H, Arboleda VA
and Vilain E: Sex-specific effects of testosterone on the sexually
dimorphic transcriptome and epigenome of embryonic neural
stem/progenitor cells. Sci Rep. 6(36916): 2016
|
|
35
|
Ouni M, Belot MP, Castell AL, Fradin D and
Bougneres P: The P2 promoter of the IGF1 gene is a major epigenetic
locus for GH responsiveness. Pharmacogenomics J. 16:102–106. 2016.
View Article : Google Scholar :
|
|
36
|
Takahashi H, Funakoshi H and Nakamura T:
LIM-kinase as a regulator of actin dynamics in spermatogenesis.
Cytogenet Genome Res. 103:290–298. 2003. View Article : Google Scholar
|
|
37
|
Croft DR, Crighton D, Samuel MS, Lourenco
FC, Munro J, Wood J, Bensaad K, Vousden KH, Sansom OJ, Ryan KM, et
al: p53-mediated transcriptional regulation and activation of the
actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway
promotes cell survival. Cell Res. 21:666–682. 2011. View Article : Google Scholar :
|
|
38
|
Sumi T, Hashigasako A, Matsumoto K and
Nakamura T: Different activity regulation and subcellular
localization of LIMK1 and LIMK2 during cell cycle transition. Exp
Cell Res. 312:1021–1030. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heng YW, Lim HH, Mina T, Utomo P, Zhong S,
Lim CT and Koh CG: TPPP acts downstream of RhoA-ROCK-LIMK2 to
regulate astral microtubule organization and spindle orientation. J
Cell Sci. 125:1579–1590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li X, Zhu Y, Cao Y, Wang Q, Du J, Tian J,
Liang Y and Ma W: LIM kinase activity is required for microtubule
organising centre positioning in mouse oocyte meiosis. Reprod
Fertil Dev. 29:791–804. 2017. View Article : Google Scholar
|
|
41
|
Takahashi T, Koshimizu U, Abe H, Obinata T
and Nakamura T: Functional involvement of Xenopus LIM kinases in
progression of oocyte maturation. Dev Biol. 229:554–567. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takahashi H, Koshimizu U, Miyazaki J and
Nakamura T: Impaired spermatogenic ability of testicular germ cells
in mice deficient in the LIM-kinase 2 gene. Dev Biol. 241:259–272.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yuan ZH and Zhao YM: The regulatory
functions of piRNA/PIWI in spermatogenesis. Yi Chuan. 39:683–691.
2017.PubMed/NCBI
|
|
44
|
Kuramochi-Miyagawa S, Watanabe T, Gotoh K,
Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri
TW, et al: DNA methylation of retrotransposon genes is regulated by
Piwi family members MILI and MIWI2 in murine fetal testes. Genes
Dev. 22:908–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Unhavaithaya Y, Hao Y, Beyret E, Yin H,
Kuramochi-Miyagawa S, Nakano T and Lin H: MILI, a PIWI-interacting
RNA-binding protein, is required for germ line stem cell
self-renewal and appears to positively regulate translation. J Biol
Chem. 284:6507–6519. 2009. View Article : Google Scholar :
|
|
46
|
Houwing S, Kamminga LM, Berezikov E,
Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz
E, Moens CB, et al: A role for Piwi and piRNAs in germ cell
maintenance and transposon silencing in Zebrafish. Cell. 129:69–82.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Heyn H, Ferreira HJ, Bassas L, Bonache S,
Sayols S, Sandoval J, Esteller M and Larriba S: Epigenetic
disruption of the PIWI pathway in human spermatogenic disorders.
PLoS One. 7:e478922012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Friemel C, Ammerpohl O, Gutwein J,
Schmutzler AG, Caliebe A, Kautza M, von Otte S, Siebert R and Bens
S: Array-based DNA methylation profiling in male infertility
reveals allele-specific DNA methylation in PIWIL1 and PIWIL2.
Fertil Steril. 101:1097–1103.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Demerath EW, Guan W, Grove ML, Aslibekyan
S, Mendelson M, Zhou YH, Hedman AK, Sandling JK, Li LA, Irvin MR,
et al: Epigenome-wide association study (EWAS) of BMI, BMI change
and waist circumference in African American adults identifies
multiple replicated loci. Hum Mol Genet. 24:4464–4479. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hatanaka M, Shimba S, Sakaue M, Kondo Y,
Kagechika H, Kokame K, Miyata T and Hara S: Hypoxia-inducible
factor-3alpha functions as an accelerator of 3T3-L1 adipose
differentiation. Biol Pharm Bull. 32:1166–1172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pfeiffer S, Kruger J, Maierhofer A,
Bottcher Y, Kloting N, El Hajj N, Schleinitz D, Schon MR, Dietrich
A, Fasshauer M, et al: Hypoxia-inducible factor 3A gene expression
and methylation in adipose tissue is related to adipose tissue
dysfunction. Sci Rep. 6:279692016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kim DH, Sarbassov DD, Ali SM, King JE,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeng H, Yang K, Cloer C, Neale G, Vogel P
and Chi H: mTORC1 couples immune signals and metabolic programming
to establish T(reg)-cell function. Nature. 499:485–490. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang Q, Holland-Letz T, Slynko A, Cuk K,
Marme F, Schott S, Heil J, Qu B, Golatta M, Bewerunge-Hudler M, et
al: DNA methylation array analysis identifies breast cancer
associated RPTOR, MGRN1 and RAPSN hypomethylation in peripheral
blood DNA. Oncotarget. 7:64191–641202. 2016. View Article : Google Scholar : PubMed/NCBI
|