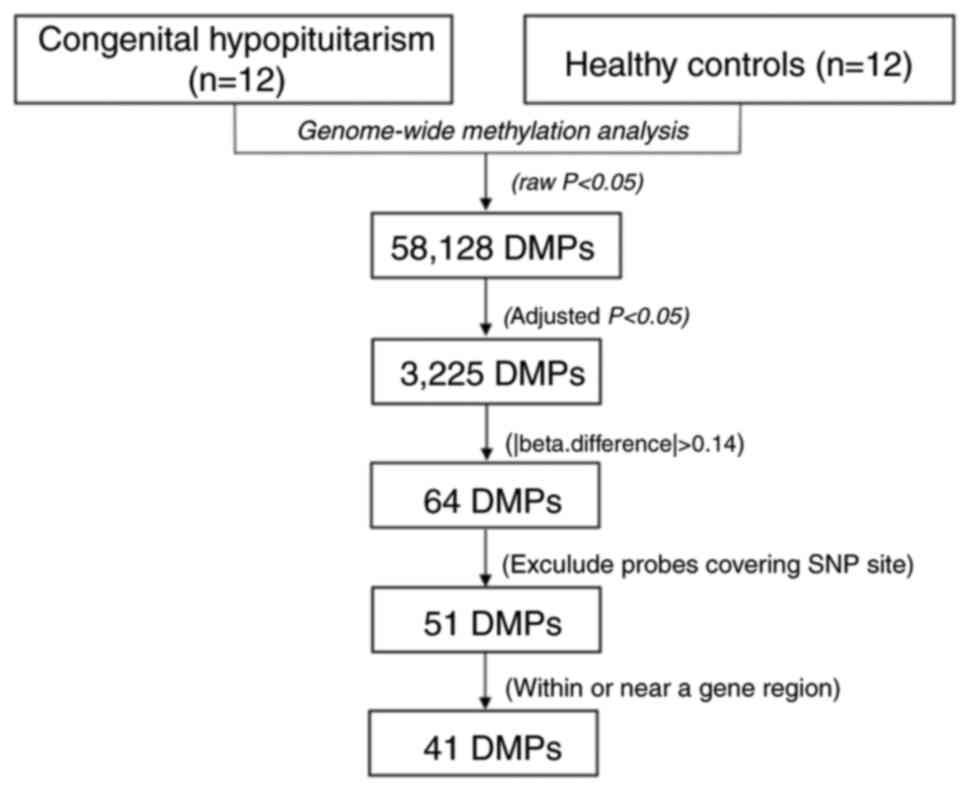
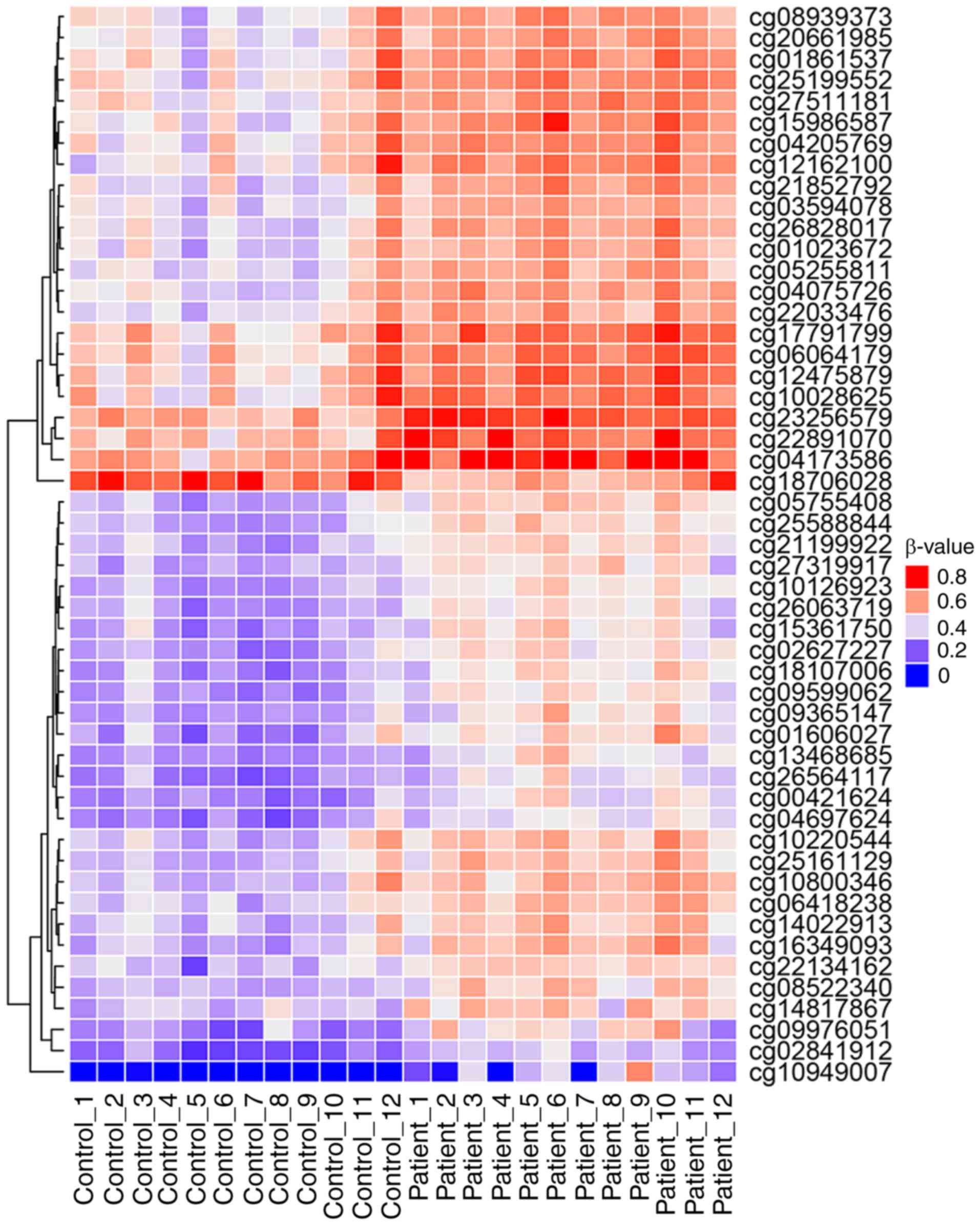
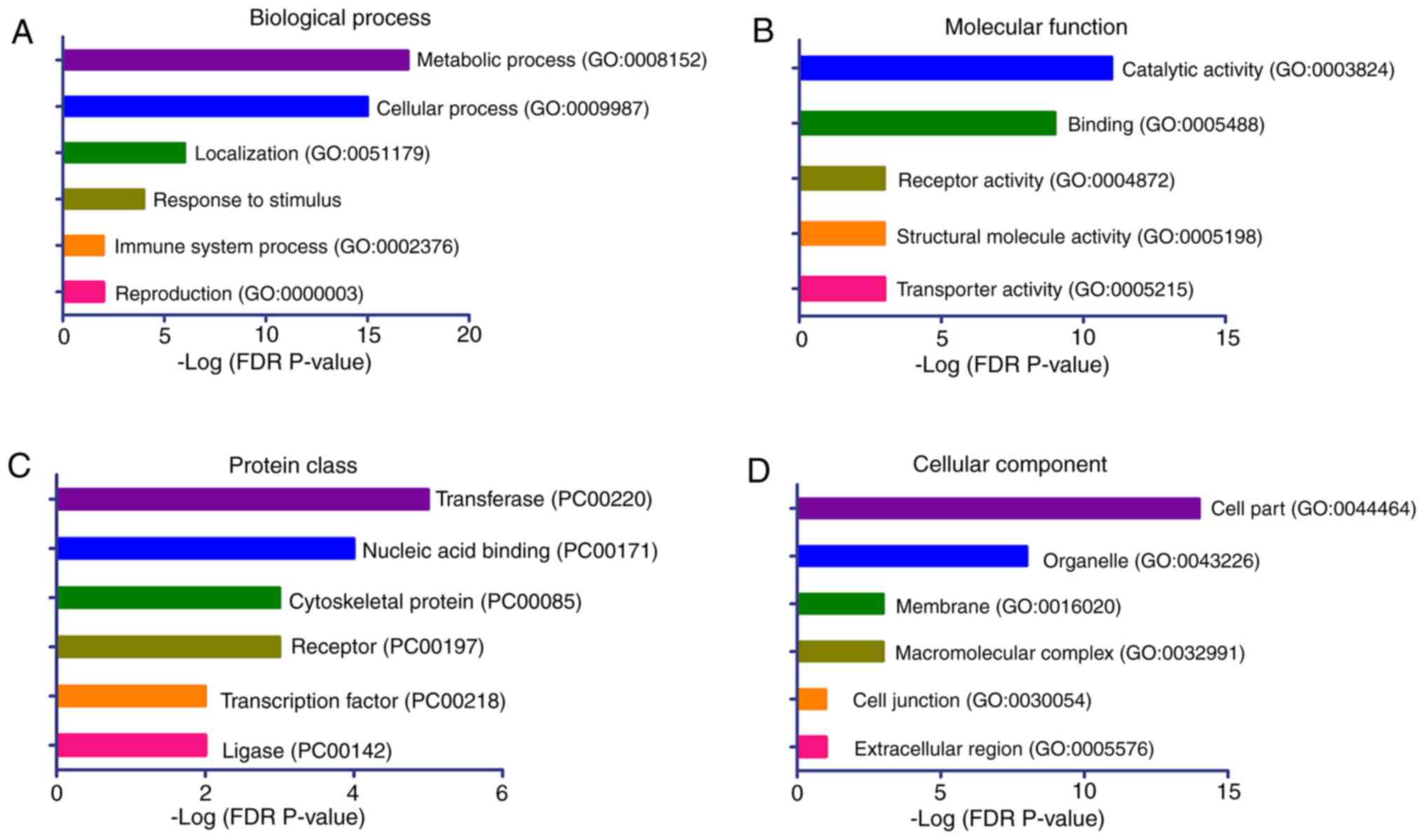
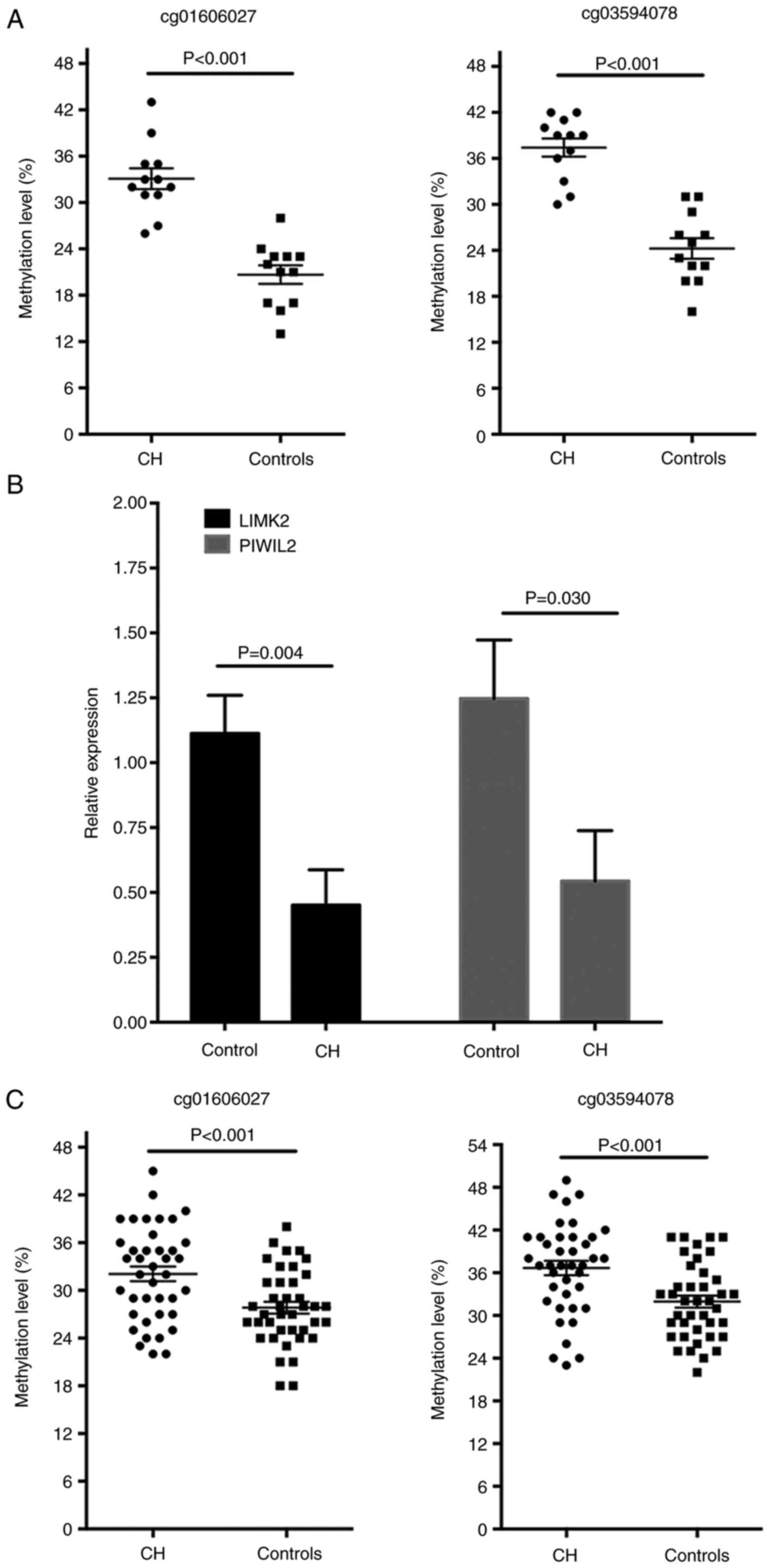
|
1
|
Regal M, Paramo C, Sierra SM and
Garcia-Mayor RV: Prevalence and incidence of hypopituitarism in an
adult Caucasian population in northwestern Spain. Clin Endocrinol
(Oxf). 55:735–740. 2001. View Article : Google Scholar
|
|
2
|
Agha A, Sherlock M, Brennan S, O’Connor
SA, O’Sullivan E, Rogers B, Faul C, Rawluk D, Tormey W and Thompson
CJ: Hypothalamic-pituitary dysfunction after irradiation of
nonpituitary brain tumors in adults. J Clin Endocrinol Metab.
90:6355–6360. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bondanelli M, De Marinis L, Ambrosio MR,
Monesi M, Valle D, Zatelli MC, Fusco A, Bianchi A, Farneti M and
degli Uberti EC: Occurrence of pituitary dysfunction following
traumatic brain injury. J Neurotrauma. 21:685–696. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herrmann BL, Rehder J, Kahlke S,
Wiedemayer H, Doerfler A, Ischebeck W, Laumer R, Forsting M, Stolke
D and Mann K: Hypopituitarism following severe traumatic brain
injury. Exp Clin Endocrinol Diabetes. 114:316–321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cuesta M, Hannon MJ, Crowley RK, Behan LA,
Tormey W, Rawluk D, Delargy M, Agha A and Thompson CJ: Symptoms of
gonadal dysfunction are more predictive of hypopituitarism than
nonspecific symptoms in screening for pituitary dysfunction
following moderate or severe traumatic brain injury. Clin
Endocrinol. 84:92–98. 2016. View Article : Google Scholar
|
|
6
|
Darzy KH and Shalet SM: Hypopituitarism
after cranial irradiation. J Endocrinol Invest. 28(Suppl 5):
S78–S87. 2005.
|
|
7
|
Craft WH, Underwoood LE and Van Wyk JJ:
High incidence of perinatal insult in children with idiopathic
hypopituitarism. J Pediatr. 96:397–402. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nystrom HF, Saveanu A, Barbosa EJ, Barlier
A, Enjalbert A, Glad C, Palming J, Johannsson G and Brue T:
Detection of genetic hypopituitarism in an adult population of
idiopathic pituitary insufficiency patients with growth hormone
deficiency. Pituitary. 14:208–216. 2011. View Article : Google Scholar
|
|
9
|
Tomlinson JW, Holden N, Hills RK, Wheatley
K, Clayton RN, Bates AS, Sheppard MC and Stewart PM: Association
between premature mortality and hypopituitarism. West Midlands
Prospective Hypopituitary Study Group. Lancet. 357:425–431. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kohno H, Kuromaru R, Ueyama N and Miyako
K: Premature mortality and hypopituitarism. Lancet. 357:1973–1974.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fernandez-Rodriguez E, Quinteiro C,
Barreiro J, Marazuela M, Pereiro I, Peino R, Cabezas-Agricola JM,
Dominguez F, Casanueva FF and Bernabeu I: Pituitary stalk
dysgenesis-induced hypopituitarism in adult patients: Prevalence,
evolution of hormone dysfunction and genetic analysis.
Neuroendocrinology. 93:181–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geffner ME: Hypopituitarism in childhood.
Cancer Control. 9:212–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Braslavsky D, Mendez MV, Prieto L,
Keselman A, Enacan R, Gruneiro-Papendieck L, Jullien N, Savenau A,
Reynaud R, Brue T, et al: Pilot neonatal screening program for
central congenital hypothyroidism: Evidence of significant
detection. Horm Res Paediatr. 88:274–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du X, Yuan Q, Yao Y, Li Z and Zhang H:
Hypopituitarism and successful pregnancy. Int J Clin Exp Med.
7:4660–4665. 2014.
|
|
15
|
Clayton RN: Mortality, cardiovascular
events and risk factors in hypopituitarism. Growth Horm IGF Res.
8(Suppl A): S69–S76. 1998. View Article : Google Scholar
|
|
16
|
Kung AW, Zhong YY, Lam KS and Wang C:
Induction of spermatogenesis with gonadotrophins in Chinese men
with hypogonadotrophic hypogonadism. Int J Androl. 17:241–247.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bulow B, Hagmar L, Eskilsson J and Erfurth
EM: Hypopituitary females have a high incidence of cardiovascular
morbidity and an increased prevalence of cardiovascular risk
factors. J Clin Endocrinol Metab. 85:574–584. 2000.PubMed/NCBI
|
|
18
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCarthy MM and Nugent BM: Epigenetic
contributions to hormonally-mediated sexual differentiation of the
brain. J Neuroendocrinol. 25:1133–1140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nugent BM, Schwarz JM and McCarthy MM:
Hormonally mediated epigenetic changes to steroid receptors in the
developing brain: Implications for sexual differentiation. Horm
Behav. 59:338–344. 2011. View Article : Google Scholar
|
|
21
|
Pinto G, Netchine I, Sobrier ML, Brunelle
F, Souberbielle JC and Brauner R: Pituitary stalk interruption
syndrome: A clinical-biological-genetic assessment of its
pathogenesis. J Clin Endocrinol Metab. 82:3450–3454.
1997.PubMed/NCBI
|
|
22
|
Maksimovic J, Gordon L and Oshlack A:
SWAN: Subset-quantile Within Array Normalization for illumina
Infinium HumanMethylation450 BeadChips. Genome Biol. 13:R442012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benjamini Y and Hochberg Y: The adaptive
control of the false discovery rate in multiple hypotheses testing.
J Behav Educ Statist. 25:60–83. 2000. View Article : Google Scholar
|
|
24
|
Mi H, Muruganujan A and Thomas PD: PANTHER
in 2013: Modeling the evolution of gene function, and other gene
attributes, in the context of phylogenetic trees. Nucleic Acids
Res. 41(Database Issue): D377–D386. 2013. View Article : Google Scholar :
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Prabhakar VK and Shalet SM: Aetiology,
diagnosis, and management of hypopituitarism in adult life.
Postgrad Med J. 82:259–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pfaffle R and Klammt J: Pituitary
transcription factors in the aetiology of combined pituitary
hormone deficiency. Best Pract Res Clin Endocrinol Metab. 25:43–60.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kelberman D, Rizzoti K, Lovell-Badge R,
Robinson IC and Dattani MT: Genetic regulation of pituitary gland
development in human and mouse. Endocr Rev. 30:790–829. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takagi M, Nagasaki K, Fujiwara I, Ishii T,
Amano N, Asakura Y, Muroya K, Hasegawa Y, Adachi M and Hasegawa T:
Heterozygous defects in PAX6 gene and congenital hypopituitarism.
Eur J Endocrinol. 172:37–45. 2015. View Article : Google Scholar
|
|
30
|
Romero CJ, Nesi-Franca S and Radovick S:
The molecular basis of hypopituitarism. Trends Endocrinol Metab.
20:506–516. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xie W, Ren M, Li L, Zhu Y, Chu Z, Zhu Z,
Ruan Q, Lou W, Zhang H, Han Z, et al: Perinatal testosterone
exposure potentiates vascular dysfunction by ERβ suppression in
endothelial progenitor cells. PLoS One. 12:e01829452017. View Article : Google Scholar
|
|
32
|
Huen K, Harley K, Kogut K, Rauch S,
Eskenazi B and Holland N: DNA methylation of LINE-1 and Alu
repetitive elements in relation to sex hormones and pubertal timing
in Mexican-American children. Pediatr Res. 79:855–862. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pantalone KM and Faiman C: Male
hypogonadism: more than just a low testosterone. Cleve Clin J Med.
79:717–725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bramble MS, Roach L, Lipson A, Vashist N,
Eskin A, Ngun T, Gosschalk JE, Klein S, Barseghyan H, Arboleda VA
and Vilain E: Sex-specific effects of testosterone on the sexually
dimorphic transcriptome and epigenome of embryonic neural
stem/progenitor cells. Sci Rep. 6(36916): 2016
|
|
35
|
Ouni M, Belot MP, Castell AL, Fradin D and
Bougneres P: The P2 promoter of the IGF1 gene is a major epigenetic
locus for GH responsiveness. Pharmacogenomics J. 16:102–106. 2016.
View Article : Google Scholar :
|
|
36
|
Takahashi H, Funakoshi H and Nakamura T:
LIM-kinase as a regulator of actin dynamics in spermatogenesis.
Cytogenet Genome Res. 103:290–298. 2003. View Article : Google Scholar
|
|
37
|
Croft DR, Crighton D, Samuel MS, Lourenco
FC, Munro J, Wood J, Bensaad K, Vousden KH, Sansom OJ, Ryan KM, et
al: p53-mediated transcriptional regulation and activation of the
actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway
promotes cell survival. Cell Res. 21:666–682. 2011. View Article : Google Scholar :
|
|
38
|
Sumi T, Hashigasako A, Matsumoto K and
Nakamura T: Different activity regulation and subcellular
localization of LIMK1 and LIMK2 during cell cycle transition. Exp
Cell Res. 312:1021–1030. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heng YW, Lim HH, Mina T, Utomo P, Zhong S,
Lim CT and Koh CG: TPPP acts downstream of RhoA-ROCK-LIMK2 to
regulate astral microtubule organization and spindle orientation. J
Cell Sci. 125:1579–1590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li X, Zhu Y, Cao Y, Wang Q, Du J, Tian J,
Liang Y and Ma W: LIM kinase activity is required for microtubule
organising centre positioning in mouse oocyte meiosis. Reprod
Fertil Dev. 29:791–804. 2017. View Article : Google Scholar
|
|
41
|
Takahashi T, Koshimizu U, Abe H, Obinata T
and Nakamura T: Functional involvement of Xenopus LIM kinases in
progression of oocyte maturation. Dev Biol. 229:554–567. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takahashi H, Koshimizu U, Miyazaki J and
Nakamura T: Impaired spermatogenic ability of testicular germ cells
in mice deficient in the LIM-kinase 2 gene. Dev Biol. 241:259–272.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yuan ZH and Zhao YM: The regulatory
functions of piRNA/PIWI in spermatogenesis. Yi Chuan. 39:683–691.
2017.PubMed/NCBI
|
|
44
|
Kuramochi-Miyagawa S, Watanabe T, Gotoh K,
Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri
TW, et al: DNA methylation of retrotransposon genes is regulated by
Piwi family members MILI and MIWI2 in murine fetal testes. Genes
Dev. 22:908–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Unhavaithaya Y, Hao Y, Beyret E, Yin H,
Kuramochi-Miyagawa S, Nakano T and Lin H: MILI, a PIWI-interacting
RNA-binding protein, is required for germ line stem cell
self-renewal and appears to positively regulate translation. J Biol
Chem. 284:6507–6519. 2009. View Article : Google Scholar :
|
|
46
|
Houwing S, Kamminga LM, Berezikov E,
Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz
E, Moens CB, et al: A role for Piwi and piRNAs in germ cell
maintenance and transposon silencing in Zebrafish. Cell. 129:69–82.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Heyn H, Ferreira HJ, Bassas L, Bonache S,
Sayols S, Sandoval J, Esteller M and Larriba S: Epigenetic
disruption of the PIWI pathway in human spermatogenic disorders.
PLoS One. 7:e478922012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Friemel C, Ammerpohl O, Gutwein J,
Schmutzler AG, Caliebe A, Kautza M, von Otte S, Siebert R and Bens
S: Array-based DNA methylation profiling in male infertility
reveals allele-specific DNA methylation in PIWIL1 and PIWIL2.
Fertil Steril. 101:1097–1103.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Demerath EW, Guan W, Grove ML, Aslibekyan
S, Mendelson M, Zhou YH, Hedman AK, Sandling JK, Li LA, Irvin MR,
et al: Epigenome-wide association study (EWAS) of BMI, BMI change
and waist circumference in African American adults identifies
multiple replicated loci. Hum Mol Genet. 24:4464–4479. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hatanaka M, Shimba S, Sakaue M, Kondo Y,
Kagechika H, Kokame K, Miyata T and Hara S: Hypoxia-inducible
factor-3alpha functions as an accelerator of 3T3-L1 adipose
differentiation. Biol Pharm Bull. 32:1166–1172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pfeiffer S, Kruger J, Maierhofer A,
Bottcher Y, Kloting N, El Hajj N, Schleinitz D, Schon MR, Dietrich
A, Fasshauer M, et al: Hypoxia-inducible factor 3A gene expression
and methylation in adipose tissue is related to adipose tissue
dysfunction. Sci Rep. 6:279692016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kim DH, Sarbassov DD, Ali SM, King JE,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeng H, Yang K, Cloer C, Neale G, Vogel P
and Chi H: mTORC1 couples immune signals and metabolic programming
to establish T(reg)-cell function. Nature. 499:485–490. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang Q, Holland-Letz T, Slynko A, Cuk K,
Marme F, Schott S, Heil J, Qu B, Golatta M, Bewerunge-Hudler M, et
al: DNA methylation array analysis identifies breast cancer
associated RPTOR, MGRN1 and RAPSN hypomethylation in peripheral
blood DNA. Oncotarget. 7:64191–641202. 2016. View Article : Google Scholar : PubMed/NCBI
|