Introduction
Gefitinib, an epidermal growth factor receptor
(EGFR) tyrosine kinase inhibitor (TKI) has been frequently used in
targeted therapy for lung cancer (1). It is well known that EGFR has
widespread expression in normal skin tissues, including the
epidermis gland (2). Furthermore,
EGFR is important for the development and physiology of the normal
epidermis (2). However, the
clinical application of gefitinib is hampered by common skin
toxicities, including papulopustule destruction and skin
desquamation (3). These skin
toxicities originally occur due to damage to skin barrier function
(4). Therefore, in order to
optimize the application of gefitinib in patients with lung cancer,
the mechanisms underlying skin toxicities of gefitinib should be
investigated. Additionally, this field of skin and tumor research
is currently attracting substantial interest. In the present study,
the aim was to contribute to this field of research.
The epidermis creates a barrier to prevent water
loss and the invasion of allergens and infectious agents (5). Claudins (CLDNs) are the most
important components of tight junctions (TJs) (5). TJ dysfunction induces aberrant
stratum corneum issues by affecting the viability of normal human
epidermal keratinocytes (NHEKs) (5). Thus far, 24 CLDN gene family members
have been determined in human tissues (5). Previous studies reported that
abnormal expression levels of CLDN proteins may impair skin barrier
function (6,7). For example, the knockout of CLDN1 in
newborn mice resulted in mortality due to the effects of rapid
dehydration and apparent skin wrinkles, and the measurement
revealed that in these mice the epidermal barrier was severely
affected (8). Furthermore, CLDN1
gene mutations were determined in patients with neonatal sclerosing
cholangitis (a bile duct obstructive disease) and ichthyosis
(9). Additionally, previous study
indicated that CLDN2 and 4 were involved in the maintenance of the
epidermal barrier function (10).
Based on the aforementioned data and the commonly held hypothesis
that the EGFR pathway is important in regulating the skin barrier
function (11), it is reasonable
to speculate that the EGFR pathway may participate in regulating
CLDN proteins in skin tissues. This type of regulation may further
affect the skin barrier function, which may account for the skin
toxicities incurred by gefitinib.
Therefore, exogenous EGF and gefitinib were adopted
to interrupt the function of NHEKs by activating or inhibiting the
EGFR pathway. Additionally, the viability, transepithelial
electrical resistance (TER) and paracellular permeability (Pa%) in
NHEK were studied. Subsequently, the cell distributions and protein
levels of CLDN proteins during the intervention were detected in
NHEK. Finally, the potential pathways involved in gefitinib-induced
barrier function disruption in NHEKs were studied.
Materials and methods
Reagents
NHEKs were purchased from Cell Applications, Inc.,
(cat. no. 102K-05a; San Diego, CA, USA). Culture medium and
supplements were provided as following: Dulbecco's modified Eagle's
medium (DMEM)/F12 medium was purchased from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA) and fetal bovine serum (FBS)
was purchased from ExCell Biology, Inc. (Shanghai, China).
Phosphate buffered saline (PBS), 0.25% trypsin, 2 mM L-glutamine,
poly-D-Lysine coating solution and penicillin-streptomycin were
supplied by Hangzhou Best Biotechnology Co., Ltd. (Han Hangzhou,
China). EGF was purchased from Thermo Fisher Scientific, Inc. and
gefitinib was purchased from Selleck Chemicals (Houston, TX, USA).
PP2 (the inhibitor of Src signaling), U0126 [inhibitor of
extracellular-signal-regulated kinase (ERK)1/2 signaling] and
Stattic [inhibitor of signal transducer and activator of
transcription (STAT)3 signaling] were purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). AZD0530 (inhibitor of Src family
tyrosine kinases), GDC-0994 (inhibitor of ERK1/2) and SH-4-54 (STAT
inhibitor) were purchased from Selleck Chemicals.
MTT assay
NHEKs were cultured in high-glucose DMEM/F12 medium
containing 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin
at 37°C in an atmosphere containing 5% O2. A total of
5×104 cells/well were cultured on 6-well plates for 24 h
and then treated with EGF (0, 1, 2, 5, 10, 15 and 20 ng/ml) or
gefitinib (0, 0.1, 0.2, 0.5, 1, 1.5 and 2 µM) at 37°C for
another 24 h. Subsequently, the cells were collected and rinsed
twice with ice-cold PBS, and then incubated with 100 µl 0.5
mg/ml MTT solution for 3 h at 37°C. The resulting crystal was
dissolved in 150 µl dimethyl sulfoxide (DMSO) and the
optical density was measured at 570 nm wavelength using a
microplate reader.
TER and Pa% detection
A total of 2×105 NHEKs/well were seeded
on a 96-well Transwell plate. Gefitinib and/or EGF were added to
the apical or basal compartments of the Transwell inserts when a
cell confluence of 85% was obtained. TER was measured using a EVOM2
voltohmmeter with STX2 electrode (World Precision Instruments,
Sarasota, FL, USA) at 24 h. Results were expressed as
Ω·cm−2 and normalized as a percentage of the base-line
values.
To measure the paracellular flux of NHEKs, migration
experiments were conducted using a Transwell dish at 37°C. NHEKs
were seeded to the upper chamber in serum-free DMEM/F12; the lower
chamber contained DMEM/F12 with 10% FBS. Briefly, Transwells were
pre-incubated with Krebs Henselite Bicarbonate buffer (KHBB; pH
7.4; Thermo Fisher Scientific, Inc.) for 15 min at 37°C and washed
twice with fresh KHBB. After 24 h, fluorescein isothiocyanate
(FITC)-labeled-dextran (FD) dissolved in KHBB (0.1%) was loaded
into the apical or basal compartments of the Transwell inserts.
Cells on the upper surface of the filter were removed with a cotton
swab. After 2 h at 37°C, FD intensity of the medium in the apical
and basal compartments was determined with a fluorescence
spectrophotometer (Hitachi, Ltd., Tokyo, Japan). FITC flux was
expressed as the percentage of the apically-added FITC recovered in
the basal compartment after 2 h. The measurements aforementioned
were produced from four wells/experiment, and the experiments were
repeated four times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated using a Qiagen RNAeasy mini
kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's
protocols. Complementary DNA (cDNA) was then generated by reverse
transcription using a Takara PrimeScript RT Reagent kit (Takara
Bio, Inc., Otsu, Japan) according to the manufacturer's protocol.
The cDNA was used for RT-qPCR using the SYBR® Premix
Ex-Taq™ kit (Takara Bio, Inc.) on a CFX96 real time PCR system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's protocol. The thermocycling conditions were as
follows: Firstly, 94°C for 5 min; secondly, 94°C for 45 sec and
55°C for 30 sec; and finally, 72°C for 30 sec. In total there were
40 thermal cycles. The 2−ΔΔCq method was used to
calculate the relative gene expression (12). All expression data were normalized
to human β-actin (ACTB). Primers sequences were provided as
follows: EGFR forward, 5′-TGACTGAGGACAGCATAGACGA-3′ and reverse,
5′-GGGCTGGACAGTGTTGAGATAC-3′; CLDN1 forward,
5′-CATTGGTGTCTGGAGACCTG-3′ and reverse, 5′-AATGCCTTGCTCAAACACAG-3′;
CLDN2 forward, 5′-TAAGAAGCCAGGTGGATGTG-3′ and reverse, 5′-CGC
CTGAAGAGTTTCTAGGG-3′; CLDN4 forward, 5′-AAC CCTGACTTTGGGATCTG-3′
and reverse, 5′-AGATGC AGGCAGACAGAGTG-3′; ERK1 forward, 5′-TCCATC
GACATCTGGTCTGT-3′ and reverse, 5′-TGAGCT GATCCAGGTAGTGC-3′; ERK2
forward, 5′-CCGTGACCT CAAGCCTTC-3′ and reverse, 5′-GCCAGGCCAAAG
TCACAG-3′; and ACTB forward, 5′-TCCTTCCTGGGC ATGGAGT-3′ and
reverse, 5′-CAGGAGGAGCAATGATCT TGAT-3′.
Immunoblotting
A total of 5×104 NHEKs/well were cultured
on 6-well plates for 24 h at 37°C, and then treated with EGF or
gefitinib at 37°C for another 24 h. Next, cultured cells were
rinsed with ice-cold PBS and then lysed in
radioimmuno-precipitation assay buffer at 4°C for 10 min (Cell
Signaling Technology, Inc., Danvers, MA, USA) containing complete
protease inhibitors and phosphatase inhibitors (Roche Diagnostics,
Indianapolis, IN, USA), 5 mM dithiothreitol and 1 mM
phenylmethylsulfonyl fluoride (Sigma Aldrich; Merck KGaA). Protein
concentrations in the resulting supernatants were determined using
a Bio-Rad protein assay (Bio-Rad Laboratories, Inc.). Aliquots
containing 40 µg total proteins were loaded and separated by
8% SDS-PAGE, and then transferred to a polyvinylidene fluoride
membrane (PVDF; EMD Millipore, Billerica, MA, USA). Subsequently,
the PVDF membrane was blocked using 5% skim milk in tris-buffered
saline with 0.5% Tween-20 (Sigma-Aldrich; Merck KGaA) at room
temperature for 1 h, the membranes were incubated overnight at 4°C
with primary antibodies. The primary antibodies were as follows:
Anti-phospho (p)-EGFR (1:1,000; cat. no. ab134005), anti-Src
(1:1,000; cat. no. ab47405), anti-p-Src (1:1,000; cat. no.
ab40660), anti-STAT3 (1:1,000; cat. no. ab119352), anti-p-STAT3
(1:1,000; cat. no. ab76315), anti-ERK1/2 (1:1,000; cat. no.
ab17942), anti-p-ERK1/2 (1:1,000; cat. no. ab214362), anti-CLDN 1
(1:1,000; cat. no. ab15098), anti-CLDN 2 (1:1,000; cat. no.
ab53032), anti-CLDN 4 (1:1,000; cat. no. ab53156) and anti-ACTB
(1:1,000; cat. no. ab8227) were provided by Abcam (Cambridge, UK).
Next, the membranes were incubated with goat anti-mouse horseradish
peroxidase-conjugated secondary antibodies (1:5,000; cat. no.
ab97040) at room temperature for 1 h. Finally, the PVDF membranes
were incubated with enhanced chemiluminescence reagent (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) to detect the blots. The
images from the western blot analysis assay were analyzed using
Quantity One 1-D (Bio-Rad Laboratories, Inc.).
Immunohistochemistry
A total of 5×104 NHEKs/well were cultured
on 6-well plates for 24 h and then treated with EGF or gefitinib at
37°C for another 24 h. After that, cultured cells were plated on
poly-D-lysine-coated coverslips were rinsed twice with PBS and
fixed with 4% paraformaldehyde for 30 min at room temperature.
Cells were then washed 3 times with PBS, permeabilized in 0.4%
Triton X-100 for 10 min and blocked for 1 h at room temperature in
PBS with 0.5% Tween-20 containing 4% bovine serum albumin (Bio-Rad
Laboratories, Inc.). Following overnight incubation at 4°C with the
indicated primary antibodies (Abcam): Anti-CLDN 1 (1:1,000; cat.
no. ab15098), anti-CLDN 2 (1:1,000; cat. no. ab53032) and anti-CLDN
4 (1:1,000; cat. no. ab53156), cells were washed with PBS three
times for 10 min each and then incubated for 2 h at room
temperature with FITC/tetramethylrhodamine-conjugated goat
anti-mouse secondary antibodies (1:5,000; cat. no. ab97040; Abcam).
Cells were exposed to 0.5 µg/ml DAPI (Sigma-Aldrich; Merck
KGaA) for 5 min at 37°C. The coverslips were mounted using
Fluoromount Aqueous Mounting medium (Sigma Aldrich; Merck KGaA) and
imaged using an Olympus Fluoview FV1000 confocal laser scanning
microscope. Raw images were analyzed using the Olympus FV10-ASW 2.1
Viewer software (magnification ×400; Olympus Corporation, Tokyo,
Japan).
Cell signalling pathways
In order to further investigate the potential
pathways involved in NHEK endothelial barrier function, NHEKs were
treated at 37°C for 24 h with different treatments as follows: i) 5
ng/ml EGF; ii) 5 ng/ml EGF + 10 µM PP2 (the inhibitor of the
Src pathway); iii) 5 ng/ml EGF + 10 µM U0126 (the inhibitor
of the ERK1/2 pathway); iv) 5 ng/ml EGF + 20 µM Stattic (the
inhibitor of the STAT3 pathway); and v) 5 ng/ml EGF + 1 µM
gefitinib; or vi) DMSO (as a control). According to the
manufacturer, 10 µM PP2, 10 µM U0126 or 20 µM
Stattic exert an inhibitory effect on Src, ERK or STAT3,
respectively. Following incubation with these inhibitors, the cells
were collected and the total protein was extracted, and then a
western blot assay was applied as described above to detect the
expression of following proteins: p-EGFR, EGFR, Src, p-Src (Y418),
STAT3, p-STAT3 (Y705), ERK1/2, p-ERK1/2, CLDN1, CLDN2 and
CLDN4.
Statistical analysis
Unless indicated otherwise, results are presented as
the mean ± standard deviation. Statistical analyses were conducted
using a one-way analysis of variance followed by a Dunnett's test
using the software SPSS v.19.0 (IBM Corp., Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of EGF or/and gefitinib on NHEK
cell viability
It has been reported that EGFR signaling is involved
in the function of the skin barrier (11). In order to determine the functions
of the EGFR pathway on the skin barrier function, NHEKs were
treated with EGF or gefitinib and the cell viability and the
expression of p-EGFR were detected. As depicted in Fig. 1A and B, the cell viability was
significantly increased by 2, 5 and 10 ng/ml EGF compared with the
control group (P<0.05) with 5 ng/ml EGF inducing peak cell
viability (P<0.01), while >1 µM gefitinib demonstrated
a significant dose-dependent inhibitory effect on cell viability
(P<0.05). Additionally, the results of the western blot analysis
demonstrated that the protein levels of p-EGFR in NHEKs were
significantly increased with 2 and 5 ng/ml EGF compared with the
control group (P<0.05), and peaked at 5 ng/ml EGF (P<0.01;
Fig. 1C and D). By contrast, 2
µM gefitinib exerted the most significant inhibitory effect
on p-EGFR levels in the NHEK compared with the control group
(P<0.01; Fig. 1C and E);
therefore, 5 ng/ml EGF and 2 µM gefitinib were selected for
subsequent experiments. Furthermore, EGF was able to significantly
partly reverse gefitinib-induced NHEK cell growth inhibition
(P<0.05; Fig. 1F).
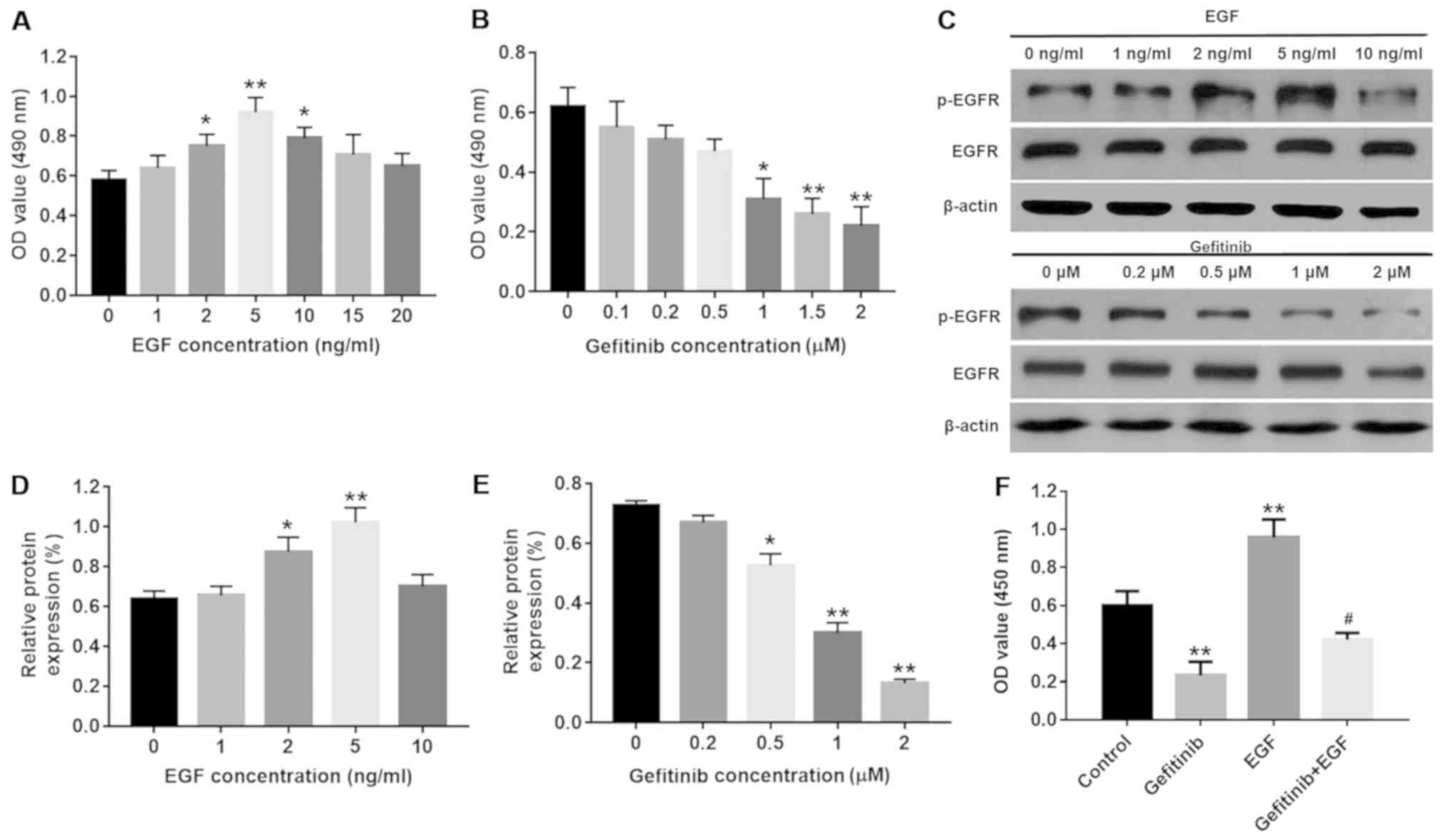 | Figure 1Effects of EGF or/and gefitinib on
NHEK cell viability. (A) NHEK were treated with 0, 1, 2, 5, 10, 15
or 20 nM EGF for 24 h and the cell viability was detected with an
MTT assay. *P<0.05 and **P<0.01 vs. the
control group, n=3. (B) NHEK were treated with 0, 0.1, 0.2, 0.5, 1,
1.5 or 2 µM gefitinib for 24 h. *P<0.05 and
**P<0.01 vs. the control group, n=3. (C) Protein
expression of p-EGFR was investigated by western blot analysis in
NHEK following treatment with EGF or gefitinib for 24 h. (D)
Relative expression levels of p-EGFR were quantified following
treatment with EGF. *P<0.05 and
**P<0.01 vs. the control group, n=3. (E) Relative
expression levels of p-EGFR were quantified following treatment
with gefitinib. *P<0.05 and **P<0.01
vs. the control group, n=3. (F) NHEK were treated with 2 µM
gefitinib and/or 5 nM EGF for 24 h and the cell viability was
detected with an MTT assay. **P<0.01 vs. the control
group, #P<0.05 vs. gefitinib group, n=3. NHEK, normal
human epidermal keratinocytes; EGF, epidermal growth factor; EGFR,
epidermal growth factor receptor; p-, phosphorylated. |
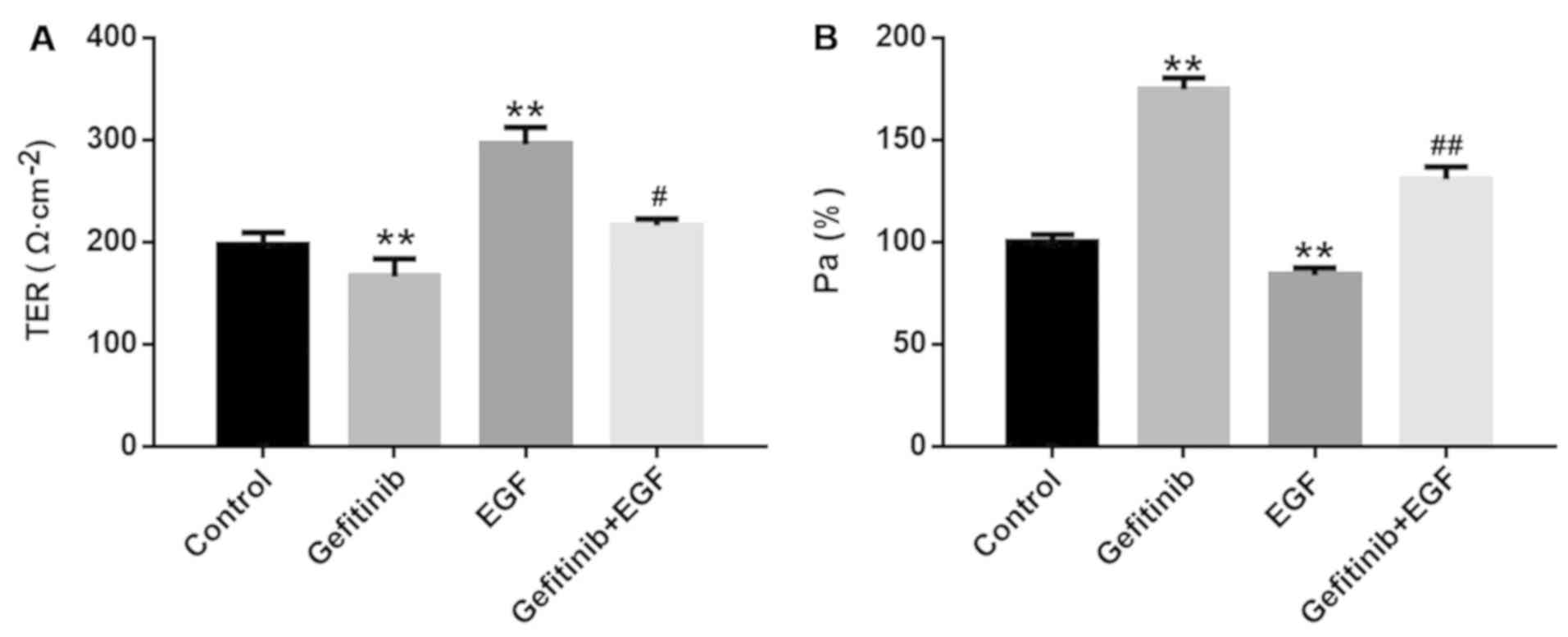
Effects of gefitinib on the cell barrier
functions of NHEK
Subsequently, the cell barrier functions of NHEK
were monitored by detecting TER and Pa%. Compared with the control
group, gefitinib significantly reduced cell resistance from
195.58±7.84 to 147.36±21.94 (P<0.01; Fig. 2A) and significantly increased Pa%
from 1.00±0.03 to 1.78±0.06 (P<0.01; Fig. 2B) in NHEK. In contrast, EGF
notably increased TER (292.62±20.54 vs 195.58±7.84; P<0.01;
Fig. 2A) and significantly
decreased Pa% (0.85±0.03 vs 1.00±0.03; P<0.01; Fig 2B) in NHEK compared with the control
group. Furthermore, the effects of gefitinib on TER (P<0.05) and
Pa% (P<0.01) were significantly reversed by EGF treatment in
NHEK (Fig. 2A and B).
Effects of gefitinib on the expression
levels and cellular distri- butions of CLDNs in NHEK
Since CLDNs are important components of cell TJ
(13), their expression and
localization were analyzed in NHEK. The RT-qPCR and western blot
analysis results demonstrated that EGF significantly increased
CLDN1 and 4 expression (P<0.01), and decreased CLDN2
(P<0.01), compared with the controls. By contrast, gefitinib
significantly downregulated the levels of CLDN1 and 4 (P<0.01)
and significantly upregulated the levels of CLDN2 (P<0.01)
compared with the controls (Fig. 3A
and B). Furthermore, the localization of CLDNs in NHEK
demonstrated the corresponding changes (Fig. 3C). CLDN1 became more enriched in
the nucleus of NHEK following EGF treatment (Fig. 3C-2), whereas CLDN4 accumulated in
the cytoplasm (Fig. 3C-5). By
contrast, the fluorescent intensities of CLDN1 and 4 were
diminished, while CLDN2 was enhanced in the nucleus and cytoplasm
(Fig. 3C-3, C-6 and C-9) in
gefitinib-treated NHEK. These changes may be associated with
gefitinib-induced barrier function disruption in NHEK.
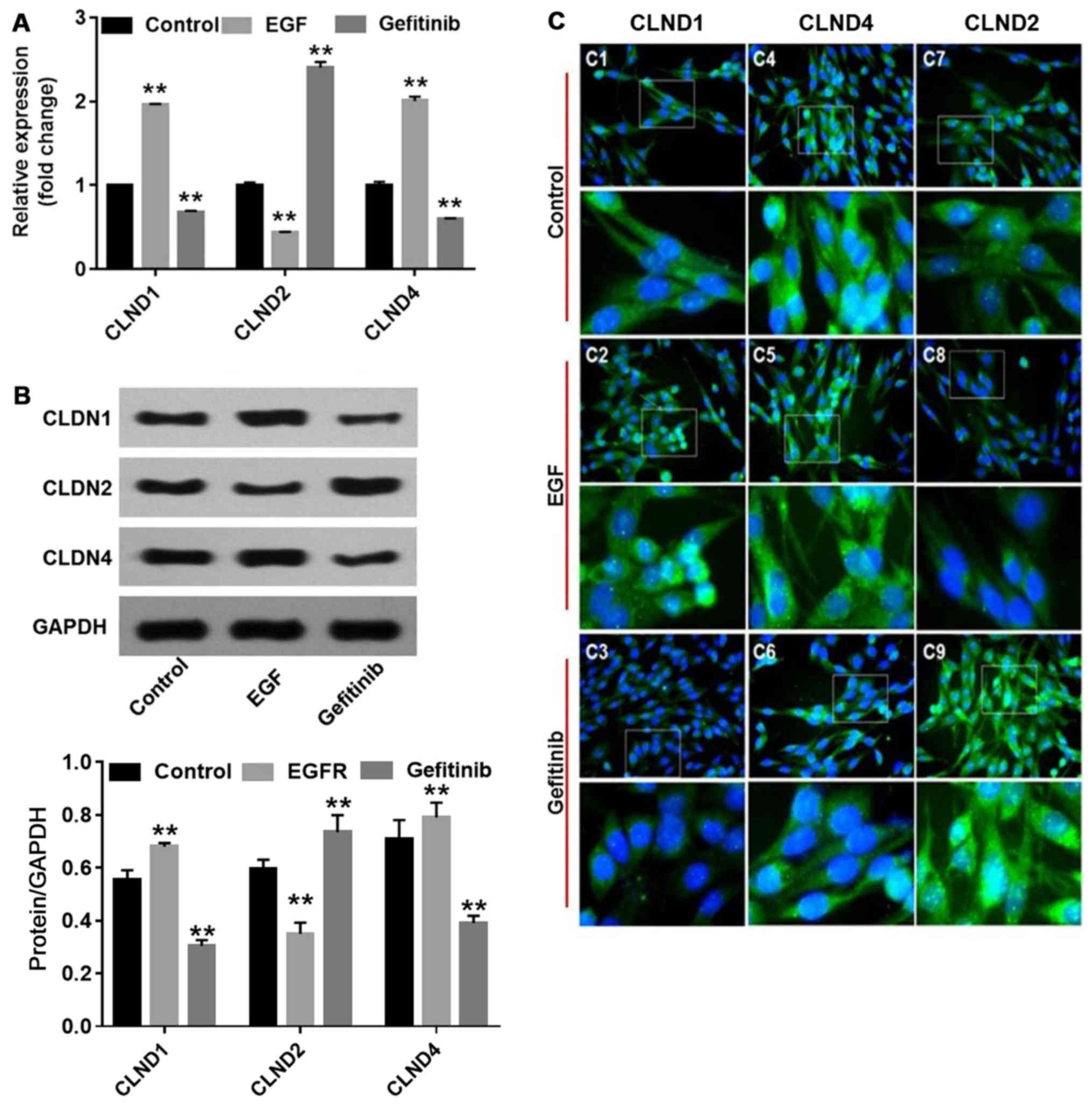 | Figure 3Effects of EGF or gefitinib on the
expression levels, cellular distributions and activation of CLDN1,
2 and 4. NHEK were treated with gefitinib or EGF for 24 h. (A) mRNA
levels of CLDN1, 2 and 4 were detected with reverse
transcription-quantitative polymerase chain
reaction.**P<0.01 vs. the control group, n=3. (B)
Protein levels of CLDN1, 2 and 4 were detected with western blot
analysis. **P<0.01 vs. the control group, n=3. (C)
Cellular distributions of CLDN1, 2 and 4 were measured using an
immunohistochemistry assay. EGF, epidermal growth factor; NHEK,
normal human epidermal keratinocytes; CLDN, claudin |
Investigation of the potentially involved
pathways
It has been reported that Src, ERK and STAT3 may
serve a function as regulators of the endothelial barrier function
(14,15). In order to further investigate if
these pathways were involved, different specific inhibitors were
applied. As depicted in Fig.
4A–E, PP2, U0126, Stattic and gefitinib exerted a significant
inhibitory effect on their respective targets in NHEK compared with
the control groups (P<0.01). Furthermore, EGF-induced CLDN1 and
CLDN4 upregulation and CLDN2 downregulation may be partially
reversed by PP2, U0126 or Stattic, compared with the EGF treatment
group. (Fig. 4A and F–H).
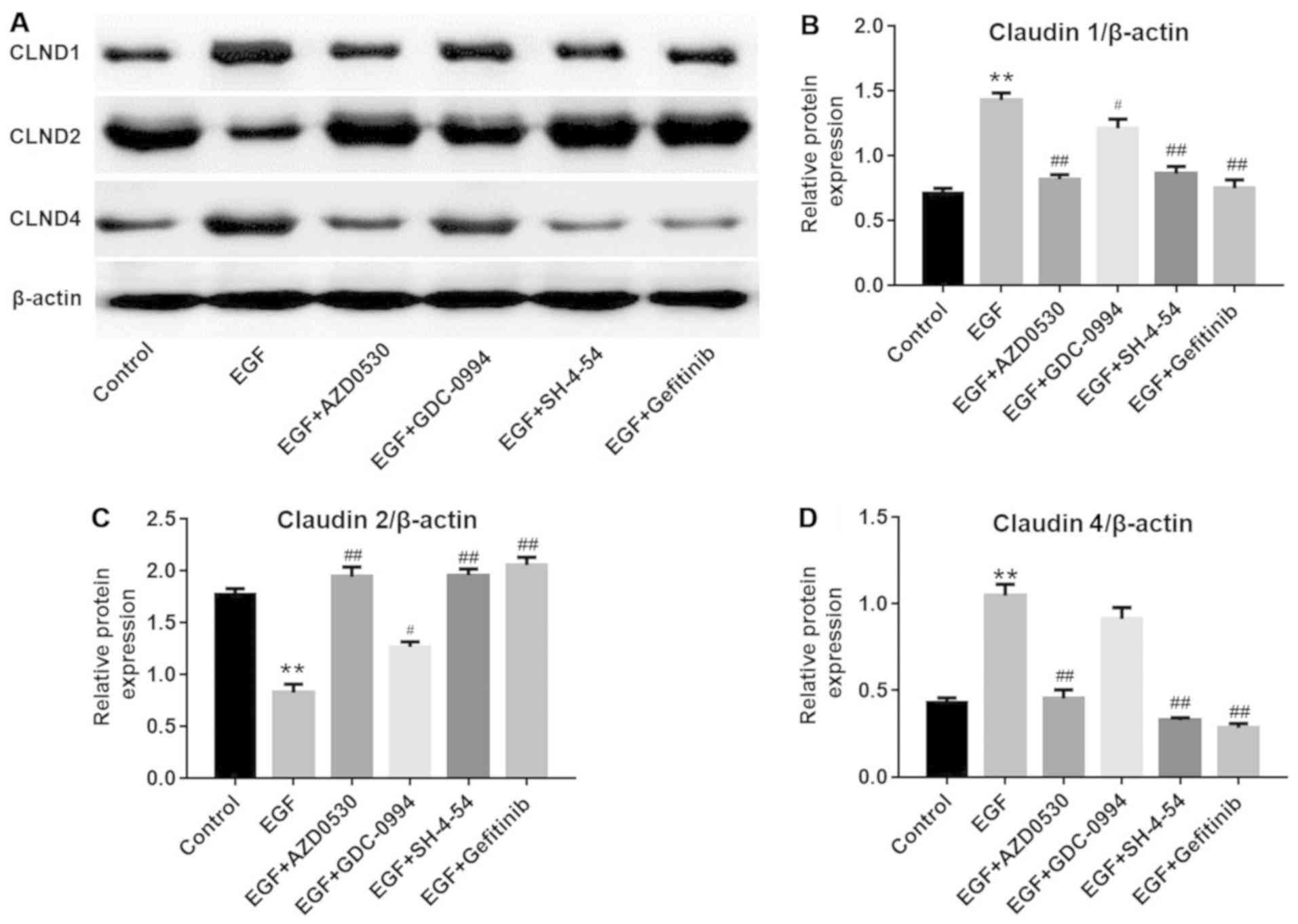
Consistently, western blot analysis (Fig. 5) demonstrated that EGF-induced
CLDN1 (Fig. 5B) and CLDN4
upregulation (Fig. 5D) and CLDN2
downregulation (Fig. 5C) was
reversed by another Src inhibitor (AZD0530), ERK1/2 inhibitor
(GDC-0994) or STAT3 inhibitor (SH-4-54), respectively.
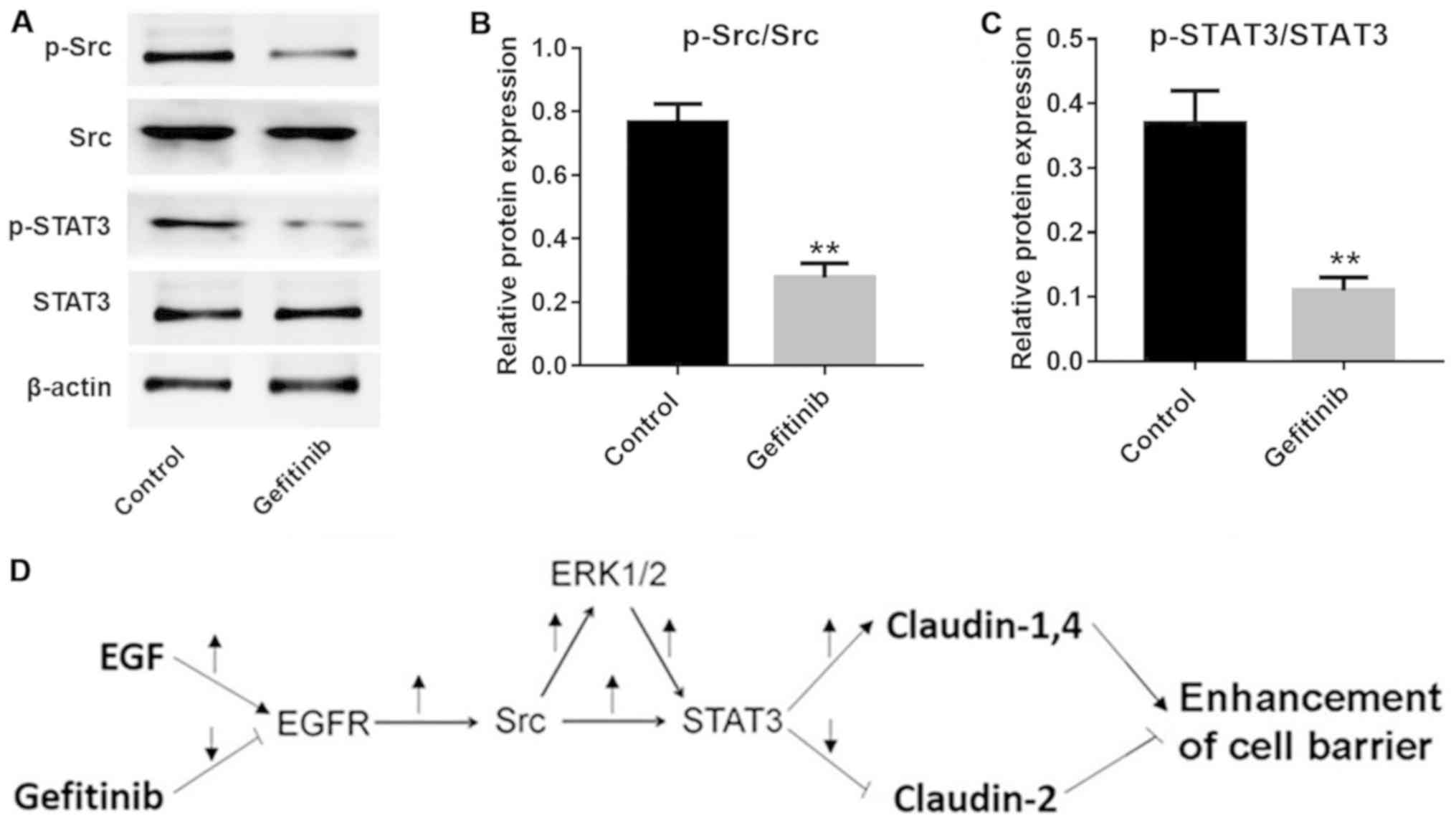
Additionally, the expression levels of p-Src and p-STAT3 were
significantly inhibited by gefitinib in NHEK compared with the
control cells (P<0.01; Fig.
6A–C). These data indicated that the Src and STAT3 pathways
were involved in gefitinib-induced barrier function disruption in
NHEK (Fig. 6D).
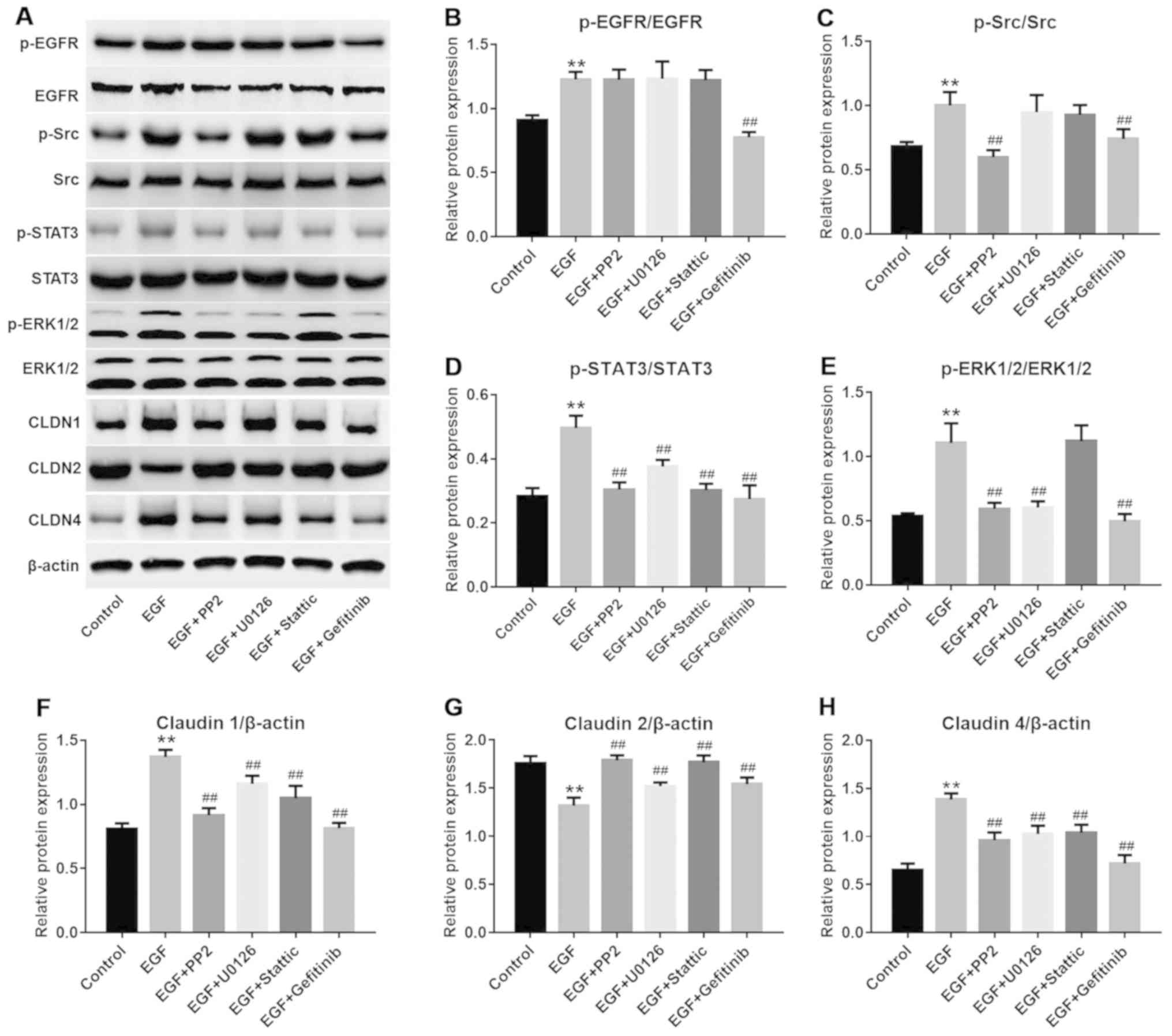 | Figure 4EGF-induced Src and STAT3 pathway
activation were reversed by gefitinib. (A) Representative images of
western blot analysis for proteins in different groups. (B) Protein
levels of p-EGFR were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. (C) Protein levels
of p-Src were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. (D) Protein levels
of p-STAT3 were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. (E) Protein levels
of p-ERK were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01, vs the EGF group, n=3. (F) Protein levels
of CLND1 were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. (G) Protein levels
of CLND2 were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. (H) Protein levels
of CLND4 were detected with western blot analysis.
**P<0.01 vs. the control group;
##P<0.01 vs. the EGF group, n=3. EGF, epidermal
growth factor; NHEK, normal human epidermal keratinocytes; STAT3,
signal transducer and activator of transcription 3. CLDN, claudin;
p-, phosphorylated; EGFR, epidermal growth factor receptor. |
Discussion
Since NHEK are frequently used as model cells to
study the functions of the skin cell barrier (16-20), they were also used in the present
study. In the present study, it was firstly observed that exogenous
gefitinib was able to damage the cell barrier function via
inhibiting the EGFR, Src and STAT3 pathways, accompanied by
regulating the expression of CLDN proteins. Furthermore, all these
effects, caused by gefitinib, may be reversed by treatment with
EGF. EGF has been previously known to increase the TER of
epithelial LLCPK1 cells (21).
The effects of EGF on TER or regulation of CLDN proteins have
previously been investigated in renal carcinoma (MDCK cells).
Flores-Benitez et al (22), reported that CLDN1, 3 and 4
proteins may be upregulated in MDCK cells by EGF, and that the
downstream ERK signaling pathway served a notable role in the
process of regulating the kidney Pa%. The present results are in
agreement with these data.
EGF or gefitinib regulate the changes in the
composition of TJ (notably, affecting CLDN1, 2 and 4) through a
number of mechanisms (23,24).
CLDN2 is necessary for TJ strand formation (25). It is able to form cation and
water-selective channels, and is necessary for the uptake of
Na+, water and Ca2+ (26-28). Therefore, CLDN2 is responsible for
the low TER phenotype of cells (29). In contrast, CLDN1 and 4 are
involved in the structure formation of epidermal TJ (10). CLDN4 was demonstrated to confer a
high resistance phenotype in epithelial cells (30,31). Consequently, the enhancement of
the cell barrier function may be achieved by reducing CLDN2 and
augmenting CLDN1 and 4 levels (32).
The present results demonstrated that gefitinib may
disrupt cell barrier function by decreasing the expression of CLDN1
and 4 and increasing the expression of CLDN2 (Fig. 3). In terms of the mechanism,
previous studies have reported that EGF activated ERK1/2, which in
turn may downregulate CLDN2 and upregulate CLDN1, 3 and 4 at TJ
(33-35). A similar change in ERK1/2 activity
was observed following treatment with gefitinib in NHEK.
Notably, the present study was performed in NHEK
in vitro. Considering the sophisticated environment in
vivo, further experiments are required to evaluate the effect
of gefitinib on the skin barrier function and the potential
involvement of signaling molecules, including Src or STAT3, in
animal models (36,37). Additionally, the present study
indicated that gefitinib was capable of damaging the skin cell
barrier function by regulating the protein levels of CLDN1, 2 and
4. The present study will be notably beneficial for the continued
investigation into issues regarding skin toxicities and the
clinical application of gefitinib. Furthermore, the
gefitinib-induced barrier function disruption in NHEK was indicated
that it may partially be due to Src and STAT3 pathway inhibition.
Therefore, a novel potential EGFR-Src-STAT3-ERK signaling cascade
was proposed. These novel mechanisms are in accordance with
previous reports (36,37); however, they provide novel insight
into the prevention of skin barrier dysfunction caused by
EGFR-TKIs. Since gefitinib belongs to the family of EGFR-TKIs, the
research on gefitinib may additionally indicate the involvement of
other members in this family (38). It is noteworthy that PP2 and
AZD0530 inhibit various Src family kinases including c-Src, Lck,
c-YES, Lyn, Fyn, Fgr and Blk. Thus, the other Src family pathways
that may be involved in gefitinib-induced skin toxicities require
further investigations in the future.
In conclusion, it was determined that the mechanisms
underlying the skin toxicities of gefitinib may involve CLDN1 and 4
downregulation and CLDN2 upregulation in NHEK. Additionally, the
Src and STAT3 pathways were identified to be involved in
gefitinib-induced barrier function disruption in the NHEK. The
present data may provide a novel strategy for improving skin
toxicity of gefitinib in patients with lung cancer.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81271743).
Availability of data and materials
All datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YWa and LX conducted NHEK cell culture, performed
the MTT assay and assisted with manuscript preparation. SZ and JB
executed TER and Pa% detection. YWu and JQ prepared RNA samples and
conducted RT-qPCR. XJ was responsible for immunoblotting. DZ
completed the immunohistochemical assay. YD analyzed and
interpreted the experiment data. HF designed the study,
investigated the potential cell signaling pathways and was the
major contributor of this manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Ehmann LM, Ruzicka T and Wollenberg A:
Cutaneous side-effects of EGFR inhibitors and their management.
Skin Therapy Lett. 16:1–3. 2011.PubMed/NCBI
|
|
2
|
Baas JM, Krens LL, Guchelaar HJ, Ouwerkerk
J, de Jong FA, Lavrijsen AP and Gelderblom H: Recommendations on
management of EGFR inhibitor-induced skin toxicity: A systematic
review. Cancer Treat Rev. 38:505–514. 2012. View Article : Google Scholar
|
|
3
|
Pomerantz RG, Mirvish ED and Geskin LJ:
Cutaneous reactions to epidermal growth factor receptor inhibitors.
J Drugs Dermatol. 9:1229–1234. 2010.PubMed/NCBI
|
|
4
|
Reck M and Gutzmer R: Management of the
cutaneous side effects of therapeutic epidermal growth factor
receptor inhibition. Onkologie. 33:470–479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Overgaard CE, Daugherty BL, Mitchell LA
and Koval M: Claudins: Control of barrier function and regulation
in response to oxidant stress. Antioxid Redox Signal. 15:1179–1193.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morita K, Miyachi Y and Furuse M: Tight
junctions in epidermis: From barrier to keratinization. Eur J
Dermatol. 21:12–17. 2011.PubMed/NCBI
|
|
7
|
Kirschner N, Bohner C, Rachow S and
Brandner JM: Tight junctions: Is there a role in dermatology? Arch
Dermatol Res. 302:483–493. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Furuse M, Hata M, Furuse K, Yoshida Y,
Haratake A, Sugitani Y, Noda T, Kubo A and Tsukita S: Claudin-based
tight junctions are crucial for the mammalian epidermal barrier: A
lesson from claudin-1-deficient mice. J Cell Biol. 156:1099–1111.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hadj-Rabia S, Baala L, Vabres P,
Hamel-Teillac D, Jacquemin E, Fabre M, Lyonnet S, De Prost Y,
Munnich A, Hadchouel M, et al: Claudin-1 gene mutations in neonatal
sclerosing cholangitis associated with ichthyosis: A tight junction
disease. Gastroenterology. 127:1386–1390. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Turksen K and Troy TC: Barriers built on
claudins. J Cell Sci. 117:2435–2447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tran QT, Kennedy LH, Leon Carrion S,
Bodreddigari S, Goodwin SB, Sutter CH and Sutter TR: EGFR
regulation of epidermal barrier function. Physiol Genomics.
44:455–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
13
|
Zhang J, Ni C, Yang Z, Piontek A, Chen H,
Wang S, Fan Y, Qin Z and Piontek J: Specific binding of Clostridium
perfringens enterotoxin fragment to Claudin-b and modulation of
zebrafish epidermal barrier. Exp Dermatol. 24:605–610. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adam AP: Regulation of endothelial
adherens junctions by tyrosine phosphorylation. Mediators Inflamm.
2015:2728582015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alsaffar H, Martino N, Garrett JP and Adam
AP: Interleukin-6 promotes a sustained loss of endothelial barrier
function via Janus kinase-mediated STAT3 phosphorylation and de
novo protein synthesis. Am J Physiol Cell Physiol. 314:C589–C602.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharma V, Singh SK, Anderson D, Tobin DJ
and Dhawan A: Zinc oxide nanoparticle induced genotoxicity in
primary human epidermal keratinocytes. J Nanosci Nanotechnol.
11:3782–3788. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mikami D, Sakai S, Sasaki S and Igarashi
Y: Effects of asterias amurensis-derived sphingoid bases on the de
novo ceramide synthesis in cultured normal human epidermal
keratinocytes. J Oleo Sci. 65:671–680. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szöllősi AG, Gueniche A, Jammayrac O,
Szabó-Papp J, Blanchard C, Vasas N, Andrási M, Juhász I, Breton L
and Bíró T: Bifidobacterium longum extract exerts
pro-differentiating effects on human epidermal keratinocytes, in
vitro. Exp Dermatol. 26:92–94. 2017. View Article : Google Scholar
|
|
19
|
Dang NN, Pang SG, Song HY, An LG and Ma
XL: Filaggrin silencing by shRNA directly impairs the skin barrier
function of normal human epidermal keratinocytes and then induces
an immune response. Braz J Med Biol Res. 48:39–45. 2015. View Article : Google Scholar :
|
|
20
|
Woo SW, Rhim DB, Kim C and Hwang JK:
Effect of standardized boesenbergia pandurata extract and its
active compound panduratin A on skin hydration and barrier function
in human epidermal keratinocytes. Prev Nutr Food Sci. 20:15–21.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mullin JM, Laughlin KV, Ginanni N, Marano
CW, Clarke HM and Peralta Soler A: Increased tight junction
permeability can result from protein kinase C
activation/translocation and act as a tumor promotional event in
epithelial cancers. Ann N Y Acad Sci. 915:231–236. 2000. View Article : Google Scholar
|
|
22
|
Flores-Benitez D, Rincon-Heredia R,
Razgado LF, Larre I, Cereijido M and Contreras RG: Control of tight
junctional sealing: Roles of epidermal growth factor and
prostaglandin E2. Am J Physiol Cell Physiol. 297:C611–C620. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flores-Benítez D, Ruiz-Cabrera A,
Flores-Maldonado C, Shoshani L, Cereijido M and Contreras RG:
Control of tight junctional sealing: Role of epidermal growth
factor. Am J Physiol Renal Physiol. 292:F828–F836. 2007. View Article : Google Scholar
|
|
24
|
Kojima T, Yamamoto T, Lan M, Murata M,
Takano K, Go M, Ichimiya S, Chiba H and Sawada N: Inhibition of MAP
kinase activity moderates changes in expression and function of
Cx32 but not claudin-1 during DNA synthesis in primary cultures of
rat hepatocytes. Med Electron Microsc. 37:101–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Furuse M, Fujita K, Hiiragi T, Fujimoto K
and Tsukita S: Claudin-1 and -2: Novel integral membrane proteins
localizing at tight junctions with no sequence similarity to
occludin. J Cell Biol. 141:1539–1550. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amasheh S, Meiri N, Gitter AH, Schöneberg
T, Mankertz J, Schulzke JD and Fromm M: Claudin-2 expression
induces cation-selective channels in tight junctions of epithelial
cells. J Cell Sci. 115:4969–4976. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosenthal R, Milatz S, Krug SM, Oelrich B,
Schulzke JD, Amasheh S, Günzel D and Fromm M: Claudin-2, a
component of the tight junction, forms a paracellular water
channel. J Cell Sci. 123:1913–1921. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Muto S, Hata M, Taniguchi J, Tsuruoka S,
Moriwaki K, Saitou M, Furuse K, Sasaki H, Fujimura A, Imai M, et
al: Claudin-2-deficient mice are defective in the leaky and
cation-selective paracellular permeability properties of renal
proximal tubules. Proc Natl Acad Sci USA. 107:8011–8016. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Furuse M, Furuse K, Sasaki H and Tsukita
S: Conversion of zonulae occludentes from tight to leaky strand
type by introducing claudin-2 into Madin-Darby canine kidney I
cells. J Cell Biol. 153:263–272. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Colegio OR, Van Itallie CM, McCrea HJ,
Rahner C and Anderson JM: Claudins create charge-selective channels
in the paracellular pathway between epithelial cells. Am J Physiol
Cell Physiol. 283:C142–C147. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Colegio OR, Van Itallie C, Rahner C and
Anderson JM: Claudin extracellular domains determine paracellular
charge selectivity and resistance but not tight junction fibril
architecture. Am J Physiol Cell Physiol. 284:C1346–C1354. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou J, Gomes AS, Paul DL and Goodenough
DA: Study of claudin function by RNA interference. J Biol Chem.
281:36117–36123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ikari A, Takiguchi A, Atomi K and Sugatani
J: Epidermal growth factor increases clathrin-dependent endocytosis
and degradation of claudin-2 protein in MDCK II cells. J Cell
Physiol. 226:2448–2456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Singh AB and Harris RC: Epidermal growth
factor receptor activation differentially regulates claudin
expression and enhances transepithelial resistance in Madin-Darby
canine kidney cells. J Biol Chem. 279:3543–3552. 2004. View Article : Google Scholar
|
|
35
|
Findley MK and Koval M: Regulation and
roles for claudin-family tight junction proteins. IUBMB Life.
61:431–437. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
García-Hernández V, Flores-Maldonado C,
Rincon-Heredia R, Verdejo-Torres O, Bonilla-Delgado J,
Meneses-Morales I, Gariglio P and Contreras RG: EGF regulates
claudin-2 and -4 expression through Src and STAT3 in MDCK cells. J
Cell Physiol. 230:105–115. 2015. View Article : Google Scholar
|
|
37
|
Singh AB, Sharma A and Dhawan P: Claudin-1
expression confers resistance to anoikis in colon cancer cells in a
Src-dependent manner. Carcinogenesis. 33:2538–2547. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ogawa Y, Kiba T, Nakano K, Fujiwara K,
Taniguchi H, Hosokawa A, Nakashima T, Kimoto S, Kajiume S, Okada Y,
et al: Prospective study of biotin treatment in patients with
erythema due to gefitinib or erlotinib. Gan To Kagaku Ryoho.
41:517–522. 2014.In Japanese. PubMed/NCBI
|