Introduction
Congenital long QT syndrome (LQTS) is a series of
conditions caused by cardiomyocyte ion channel mutations. Among the
15 types of LQTS, types 1, 2 and 3 are the most common and account
for 95% of all congenital LQTS cases (1,2).
Cardiac electrical activity disorders are the most notable
characteristics of LQTS, however, structural abnormalities of the
heart in patients with LQTS, which have been identified in several
previous studies, have long been ignored. In the early 1990s, Nador
et al found that ~55% of patients with LQTS had ventricle
contraction abnormalities (3). Of
216 patients with LQTS at the Mayo Clinic (Rochester, MN, USA), 25%
had echocardiography abnormalities (4). The high detectability of
LQTS-associated gene mutations in certain cardiomyopathies,
including arrhythmogenic right ventricular cardiomyopathy, dilated
cardiomyopathy, left ventricular noncompaction and ventricular
remodeling, show that the gene mutations of LQTS-associated
channels are important in these cardiomyopathies (5). Several types of infant congenital
heart defects are also accompanied by the detection of
LQTS-associated gene mutations (6,7).
These results demonstrate that LQTS structural abnormalities are
not individual cases but show prevalence in the LQTS patient
population. However, these abnormalities have not been recognized
by clinicians.
As reported in the aforementioned studies, the
structural changes of the heart in patients with LQTS cannot be
explained completely by abnormal electrical activity, such as a
prolonged repolarization time or ventricular tachyarrhythmia. The
mechanisms underlying structural changes of LQTS have been examined
previously. The most common accepted hypothesis is that LQTS gene
mutations can induce cell apoptosis. In 1993, James et al
observed abnormal cell apoptosis in biopsies of vascular
endothelium cells, vascular smooth muscle cells, sinoatrial node
cells and cardiomyocytes around the node obtained from patients
with LQTS (8). In a previous
in vivo study, N1325S-SCN5A transgenic mice were found to
manifest cardiac fibrosis and partial contraction disability, which
was mediated by ventricle cardiomyocyte apoptosis (9). Teng et al demonstrated that
N629D-hERG homozygous transgenic mice exhibited cardiomyocyte
apoptosis and cardiac deformity, and fetal mortality within 11 days
(10). These findings suggest
that the mechanism underlying the structural abnormalities of LQTS
may involve cell apoptosis caused by LQTS-related gene
mutations.
Endoplasmic reticulum stress (ERS) has a significant
role in defending against or adapting to cellular damage in order
to restore homeostasis. The unfolded protein response (UPR) is the
most widely investigated pathway in ERS. The UPR can be triggered
by large quantities of unfolded or misfolded proteins that have
accumulated in the ER; this results in ERS-associated proteins,
such as glucose regulated protein 78 (GRP78), being upregulated,
decreased whole-cell scale protein expression, or ER-associated
degradation (11). The UPR is
composed of three downstream signal transduction pathways: Protein
kinase R-like endoplasmic reticulum kinase (PERK), activating
transcription factor 6 (ATF6) and inositol-requiring enzyme 1
(IRE1). When ERS occurs, the expression of GRP78 increases, and it
dissociates from PERK, ATF6 or IRE1 so it can recognize and assist
in the folding of any misfolded proteins or in degrading the
misfolded protein. If the ERS is persistent or excessive, and the
cell cannot be rescued from damage, then programed cell death,
particularly cell apoptosis, is initiated. PERK-eukaryotic
translation-initiation factor-2α (eIF2α)-C/EBP homologous protein
(CHOP) is a significant ERS-mediated apoptotic pathway. PERK can be
phosphorylated to activate eIF2α and promote the expression of
CHOP/GADD153, which is an important apoptosis-inducing
transcription factor (12).
B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) are
a pair of molecules that have anti-and pro-apoptotic regulatory
effects, respectively (13). They
are also involved in the regulation of ERS-mediated cell death
(14). One of the caspase
members, caspase-12, is an ER-specific protein that can be
activated under ERS conditions. Cleaved caspase-12 can activate and
initiate downstream enzyme reactions, ultimately starting the
process of apoptosis (15). The
activation of caspase-3 is the terminal step of cell apoptosis; it
can be cleaved to its activated form to complete apoptosis
(16).
The human ether-à-go-go-related gene (hERG) encodes
the hERG channel, which produces the important repolarization
current IKr. Mutations of this channel lead to channel
dysfunction and result in LQTS type 2. The majority of the hERG
mutations are characterized by channel protein transfer deficiency,
with protein accumulating in endoplasmic reticulum (ER) and a
failure of the channel to anchor in the cell membrane as a
functional ion channel (17). It
has been found that mutated I539R-hERG protein accumulates in the
ER, activating ERS through the ATF6 pathway (18). The same effect occurs with
unfolded E637R-hERG and G572R-hERG proteins, which are degraded by
activating ERS-induced proteasome degradation (19). Mutations of hERG can cause protein
retention in the ER and evoke the UPR; whether this is the
mechanism of LQTS 2-induced cardiomyocyte apoptosis requires
further investigation.
Based on the evidence described above, it was
hypothesized that LQTS 2 hERG mutations cause cell apoptosis by
inducing the ERS pathway. In the present study, the LQTS 2 family
mutation L539fs/47-hERG was used. It was found that L539fs/47-hERG
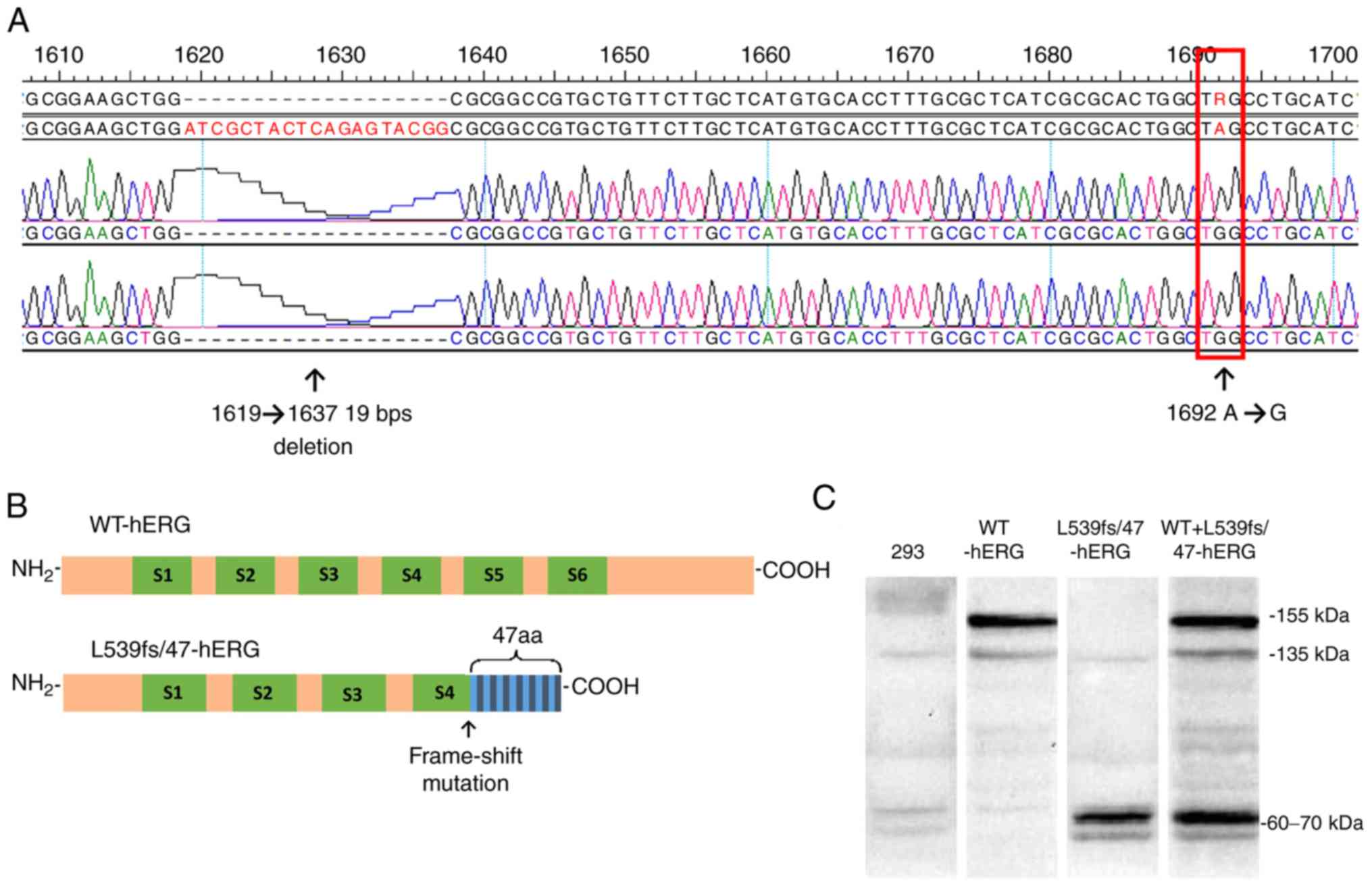
is a complex mutation consisting of a 19-bp deletion at site
1619-1637 (CCGTACTCTGAGTAGCGAT) together with an A→G point mutation
at 1692 bp, resulting in a frame-shift after the 539th amino acid
and a premature stop at the 47th amino acid after this. The mutated
hERG protein translation ends at the 4th transmembrane section. It
was also found that this mutation results in truncated protein
transfer deficiency, protein retention in the ER and dysfunction of
the hERG channel (20). The
L539fs/47-hERG plasmid was used to construct an LQTS 2 cell model
in the present study. This was used to investigate cell apoptosis
and detect the expression levels of ERS-associated factors to
identify possible molecular pathways involved in ERS-induced
apoptosis.
Materials and methods
Plasmid propagation and DNA
sequencing
The wild-type (WT)-hERG and L539fs/47-hERG plasmids
were previously constructed at the Department of Cardiovascular
Medicine, First Affiliated Hospital of Xi'an Jiaotong University
(Xi'an, China); the mutation sequence was obtained from the LQTS 2
family proband. WT or L539fs/47 hERG-pEGFP-C2 plasmids were
constructed containing green fluorescent protein (GFP) and the
target gene. The L539fs/47-hERG plasmid was sequenced by Sangon
Biotech Co., Ltd. (Shanghai, China). The following two primers were
used: 5′-AGCGAACCCACAATGTCACT-3′ and 5′-GAGTAGCGATCCAGCTTCCG-3′.
The two plasmids were amplified by transformation into
Escherichia coli (CB105-02; Tiangen Biotech Co., Ltd.,
Beijing, China), and were extracted using a Roche Genopure Plasmid
Maxi kit (Roche Diagnostics GmbH, Mannheim, Germany). The plasmids
were sequenced and verified using BLAST [Nucleotide collection
(nr/nt) database; https://blast.ncbi.nlm.nih.gov/Blast.cgi] with the
WT-hERG cDNA sequence and SeqMan software (version 7.1.0).
Cell culture and model construction
As 293 cells have no endogenous protein expression
of hERG, 293 cells were selected for transfection. The 293 cells
(American Type Culture Collection, Manassas, VA, USA) were
incubated in high-glucose DMEM (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA) with 10% fetal bovine serum (Sijiqing
Biological Engineering Materials, Hangzhou, China) at 5%
CO2 and 37°C. The cells were digested with 0.25% trypsin
(HyClone; GE Healthcare Life Sciences) when they reached ~80%
confluence.
The cells were divided into three groups to
establish the different cell models. A total of 4 µg of
plasmid DNA and 10 µl of X-tremeGENE HP DNA transfection
reagent (Roche Diagnostics GmbH) were incubated in 200 µl
DMEM for 20 mins prior to being added to the medium. Following
incubation in 35-mm dishes (cat. no. 430165; Corning Inc., Corning,
NY, USA) at 5% CO2 and 37°C for 12 h (70-90%
confluence), the 293 cells were ready to be transfected. The first
group was transfected with 4 µg of WT-hERG plasmid to
construct the WT cell model. The second group was transfected with
4 µg of L539fs/47-hERG plasmid to construct the homozygous
mutation cell type. The third group was transfected with 2
µg of WT-hERG plasmid and 2 µg of L539fs/47-hERG
plasmid simultaneously to construct the heterozygous mutation cell
model. Following incubation at 5% CO2 and 37°C for 4-6
h, the culture medium was replaced to minimize damage from the
transfection agent. The subsequent experiments were conducted at
least 24 h following transfection, when the hERG channel was amply
expressed on the cytomembrane.
Confocal laser scanning microscopy
analysis
To verify the distribution of hERG channel proteins
in the cell, a fluorescein-labeled hERG plasmid was used together
with the pDsRed2-ER plasmid to transfect the 293 cells. The day
before transfection, the three groups of cells were subcultured in
35-mm glass bottom dishes (Nest Biotechnology, Wuxi, China) for
confocal visualization at a density of 1×105 cells per
dish. The cells were then transfected with 4 µg of
WT-hERG-pEGFP-C2, 4 µg of L539fs/47-hERG-pEGFP-C2 or 2
µg of WT-hERG-pEGFP-C2 and 2 µg of
L539fs/47-hERG-pEGFP-C2. Each plate was also simultaneously
transfected with 4 µg of pDsRed2-ER plasmid. Following
incubation overnight, the cells were observed using a two-channel
confocal laser scanning microscope (Nikon C2; Xi'an Jiaotong
University, registration no. 21207771).
Treatment with 4-phenyl butyric acid
(4-PBA)
The chemical chaperone 4-PBA is an ERS inhibitor.
4-PBA (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) was dissolved
in dimethyl sulfoxide (DMSO) and diluted to 1 mol/l for storage.
The 4-PBA was added to the cell culture medium immediately
following transfection. An MTT assay was used to identify the
interference concentration of 4-PBA that did not affect cell
activity (data not shown). The concentration of 50 µmol/l
was selected as the 4-PBA interference concentration.
Hoechst 33342 staining
Hoechst 33342 (Wanlei Bio Co., Ltd., Shanghai,
China) staining is a typical cell apoptosis detection method.
Sterile coverslips were placed in the bottom of 6-well plates prior
to seeding with 293 cells. Following transfection with plasmids and
overnight incubation, the culture medium was discarded and the
cells were rinsed twice with PBS. The coverslips were covered with
4% paraformaldehyde for 5 mins and subsequently washed three times
with PBS. Hoechst 33342 was then added to the dishes and incubated
in the dark for 5 mins. The wells were rinsed twice using PBS prior
to observation with a fluorescence microscope (Olympus BX51;
Olympus Corporation, Tokyo, Japan). The numbers of apoptotic cells
in each group were counted in at least three microscopic visual
fields.
Western blot analysis
Following incubation for 24 h, the cells were washed
twice with ice-cold PBS. RIPA lysis buffer (150 µl, Genshare
Biological, Xi'an, China) mixed with 1% PMSF (Roche Diagnostics,
Basel Switzerland) and 2% phosphatase inhibitor (Genshare
Biological) was added to the cells and the homogenate was collected
using a cell scraper. The samples were centrifuged at 12,000 × g
and 4°C for 30 mins prior to collection of the supernatant
whole-cell protein. The protein extracts were quantified using a
BCA protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and boiled for 5 mins prior to being applied to SDS-PAGE gels
(quantity of protein loaded, 20 µg). The separation gels
range from 8-12% according to the target protein weight. The
proteins were separated by electrophoresis at 80 mV for 30 mins and
120 mV for 60-90 mins. The target protein bands were then cut and
set in the transmembrane unit. Following electrophoresis at 200 mA
for 60-80 mins, the proteins were transferred onto a PVDF membrane.
The membrane was then submerged in blocking buffer (TBST with 5%
milk) at room temperature for 1 h, followed by incubation in
diluted primary antibody at 4°C overnight. The membrane was
subsequently incubated in diluted HRP-labeled secondary antibody
for 1 h at room temperature. The bands were detected using a
chemiluminescence detector (Bio-Rad ChemiDoc XRS, Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and were analyzed using
Quantity One software (version, 4.6.7).
The primary antibodies were as follows: hERG
(sc-377388; 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), GRP78 (cat. no. WL0781; 1:1,000; Wanleibio Co., Ltd.,
Shanghai, China), phosphorylated (p-)PERK (cat. no. bs-23340R;
1:500; Bioss, Beijing, China), p-eIF2α (cat. no. 3398; 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), CHOP (cat. no.
WL00880; 1:1,000), Bax (cat. no. WL01637; 1:500), Bcl-2 (cat. no.
WL01556; 1:500), cleaved-caspase-3 (cat. no. WL01857; 1:500; all
Wanleibio Co., Ltd.), caspase-12 (cat. no. 2202; 1:1,000) and GAPDH
(cat. no. sc-25778; 1:1,000; both Santa Cruz Biotechnology, Inc.).
The secondary antibodies were goat anti-rabbit antibody (cat. no.
WL023a; 1:5,000; Wanleibio Co, Ltd.) and rabbit anti-mouse antibody
(cat. no. 31430; 1:10,000; Thermo Fisher Scientific, Inc.).
Statistical analysis
All data are presented as the mean ± standard
deviation and were analyzed using SPSS version 21.0 software (IBM
Corp., Armonk, NY, USA). One-way analysis of variance was used to
compare the means of more than two groups when the data were
distributed according to normality and homogeneity of variance.
Pairwise comparisons were conducted using the LSD test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Cell model construction and
verification
The results of DNA sequencing showed that the
L539fs/47-hERG plasmid contained a mutated hERG segment
characterized by the absence of a 19-bp segment at the 1619-1637
site (CCGTACTCTGAGTAGCGAT) and an A→G mutation at 1692 site
(Fig. 1A). Protein translation
ceased at the 4th transmem-brane section of the hERG channel
protein (Fig. 1B).
At 24 h post-transfection, the hERG protein was
sufficiently expressed to be detectable by western blot analysis. A
mouse primary antibody targeted the hERG N-terminus. This antibody
also detected the truncated L539fs/47-hERG protein. The WT-hERG
showed two bands, one at 135 kDa and another at 155 kDa, which
represented the non-glycosylated and glycosylated protein,
respectively. The L539fs/47-hERG homozygous mutant protein only
exhibited a band at 60-70 kDa, which was attributed to protein
truncation. The heterozygous mutant cell type had the 135 and 155
kDa bands but also the truncated protein band at 60-70 kDa
(Fig. 1C).
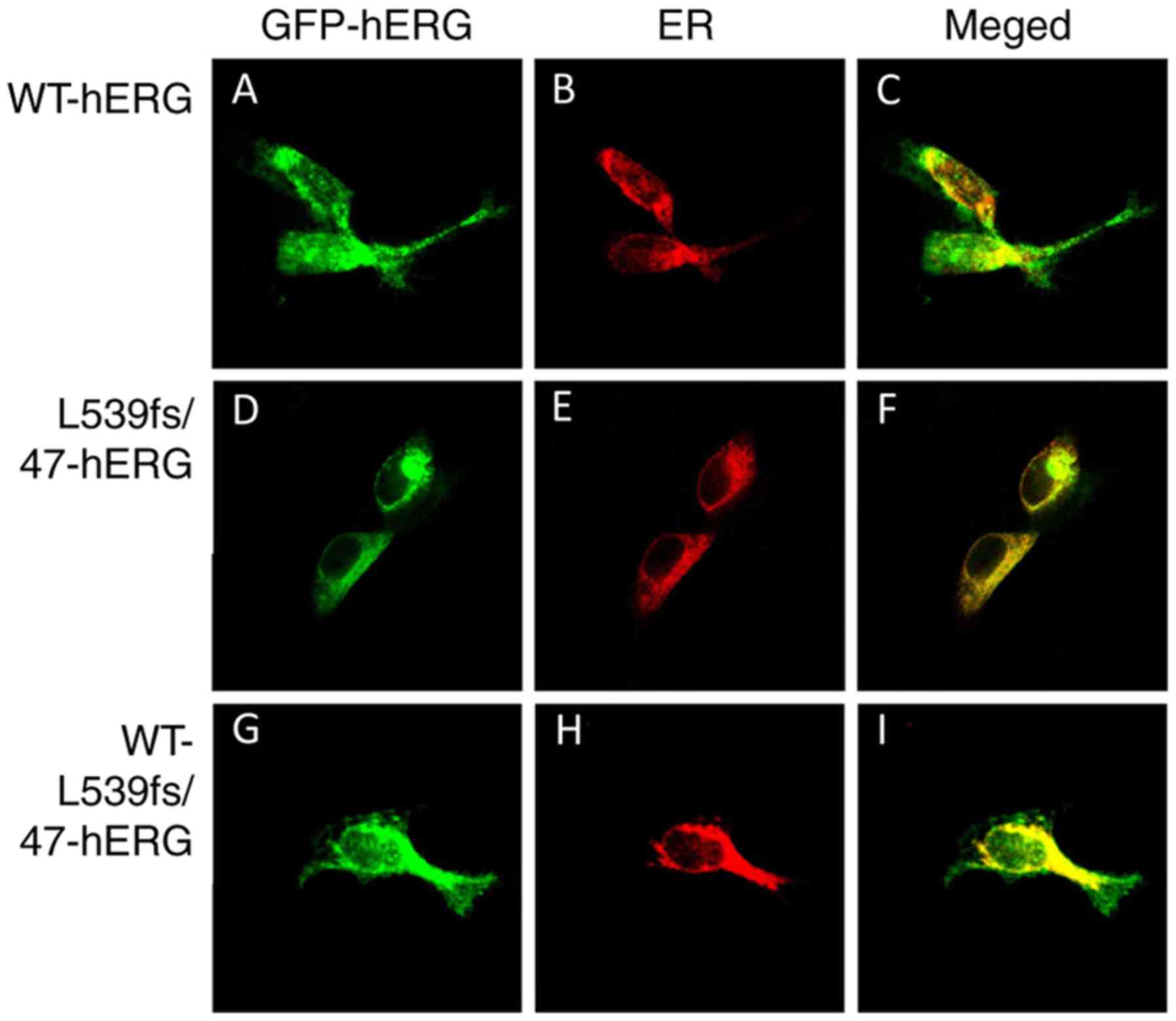
The distribution of the hERG channel protein was
detected by confocal laser scanning microscopy. The red florescence
label calreticulin marked the endoplasmic reticulum, and the green
florescence indicated the location of the hERG protein. In the
WT-hERG cells, the green florescence from the WT-hERG protein was
located predominantly at the cell outer membrane (Fig. 2A-C). The middle row in Fig. 2 indicates that the L539fs/47-hERG
homozygous mutant cells tended to have channel proteins mainly
located in the inner cell structure (Fig. 2D-F), particularly in the ER, which
is shown as yellow florescence in the merged image. The bottom row
in Fig. 2 shows the
WT-L539fs/47-hERG heterozygous mutation. The channel protein was
present in the ER and at the cell membrane, and the merged image
shows yellow fluorescence inside the cell and green fluorescence
around the cell (Fig. 2G-I). The
results demonstrate that the mutated L539fs/47-hERG protein was
recognized and retained in the ER and was not expressed on the cell
membrane. These results confirm our previous results regarding
L539fs/47-hERG (20).
L539fs/47-hERG triggers ERS
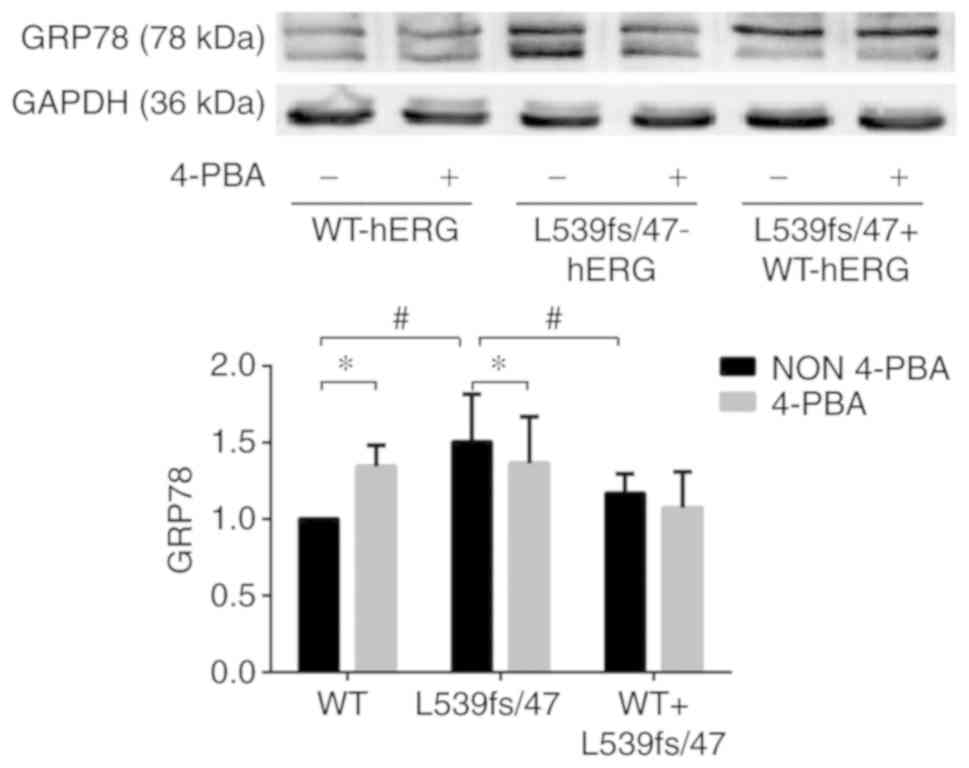
GRP78 is a chaperone anchored in the ER that can
recognize unfolded proteins and assist in their complete folding or
lead them to degradation. Therefore, it is the most representative
marker of ERS activation. Increasing the expression of GRP78 under
ERS conditions tends to relieve the ER burden. The L539fs/47-hERG
protein has been demonstrated to be a truncated protein that is
retained in the ER. To ascertain whether the abundant misfolded
protein triggered ERS, whole-cell protein was extracted from the
WT-hERG, L539fs/47-hERG and WT-L539fs/47-hERG cells to evaluate the
expression levels of GRP78 by western blot analysis. These samples
were compared with extracted whole-cell protein of the three groups
in which the ERS inhibitor 4-PBA was added. The 4-PBA concentration
was 50 µmol/l and was determined according to prior
experiments.
The three groups of cells showed significant
differences in expression levels of GRP78. The expression of GRP78
was significantly higher in the L539fs/47 cells compared with that
in the WT cells (Fig. 3A and B).
Compared with the WT cells, the L539fs/47 cells showed
significantly higher expression of GRP78 (P=0.004), whereas
expression in the WT-L539fs/47 cells was similar to that in the WT
cells, showing no significant difference in the expression of GRP78
(P=0.120). The ERS inhibitor 4-PBA significantly reduced the
expression of GRP78 in the L539fs/47-hERG cells (P=0.039), showing
that the mutation-induced ERS was reversed by 4-PBA. Furthermore,
following 4-PBA treatment, the WT-L539fs/47-hERG cells exhibited a
small decrease in expression of GRP78 (P=0.573), whereas the
WT-hERG cells exhibited higher expression of GRP78 (P=0.041),
indicating that ERS was not activated in these two cells.
Therefore, retention of the homozygous hERG mutant protein in the
ER was able to trigger ERS.
L539fs/47-hERG causes cell apoptosis
It is well known that patients with LQTS exhibit
classical cardiomyocyte electrical activity disorder. In addition,
multiple studies have reported that the hERG channel mutation can
induce cell apoptosis, not only in heart biopsies from patients,
but also in studies in vitro (8,9,10).
Therefore, it was hypothesized that the hERG missense mutation,
L539fs/47-hERG, also causes cell apoptosis. To verify this, 293
cells were divided into three groups: WT-hERG, L539fs/47-hERG and
WT-L539fs/47-hERG. Each of these groups was then divided into two
sub-groups, with or without 4-PBA treatment. Cell apoptosis was
then detected by Hoechst 33342 apoptosis staining.
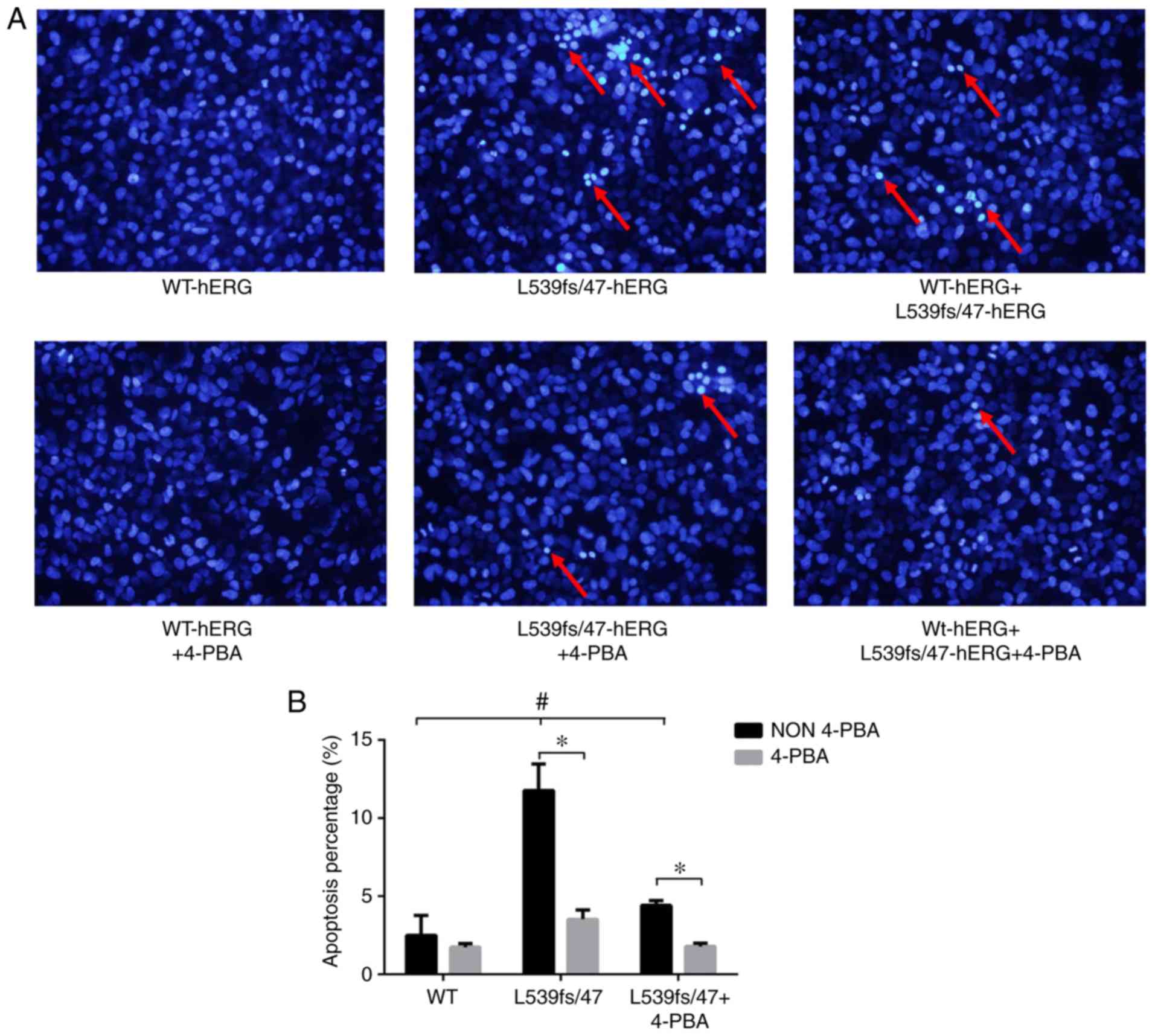
The three groups of cells were stained with Hoechst
33342, in which bright blue florescence indicates chromatin
gathering and karyopyknosis in the process of cell apoptosis. As
shown in the upper row of Fig.
4A, the apoptotic rates of the three groups were significantly
different (2.50±1.26, 11.78±1.69 and 4.41±0.30% respectively,
P<0.01; Fig. 4B). The
L539fs/47-hERG cells exhibited a greater extent of apoptosis (red
arrow) compared with the WT-hERG cells (P<0.01). The
WT-L539fs/47-hERG heterozygous mutant cells exhibited staining
indicating sporadic apoptosis compared with the other two groups
(Fig. 4B). These results
indicated that the L539fs/47-hERG homozygous mutation led to cell
apoptosis.
ERS inhibitor 4-PBA can reverse the
apoptosis induced by mutation
Long-term or intense ERS promotes apoptosis. The
results showed that the channel gene mutation L539fs/47-hERG
triggered ERS, which subsequently induced cell apoptosis. To
further verify this, the ERS inhibitor 4-PBA was introduced to
interrupt ERS in the L539fs/47-hERG cells and observe the state of
apoptosis. The results, shown in the bottom row of Fig. 4A, indicate that treatment with 50
µmol/l 4-PBA notably reduced apoptosis in the homozygous and
heterozygous mutated cells (3.53±0.59 and 1.79±0.22%, P=0.014 and
0.003, respectively) but had no effect on WT-hERG cells
(1.75±0.22%, P=0.437) (Fig. 4B).
These findings revealed that the L539fs/47-hERG-induced apoptosis
was a result of ERS activation, and that ERS inhibition by 4-PBA
prevented apoptosis in the mutated cells.
Examination of the mechanisms of
L539fs/47-hERG-induced apoptosis
The results demonstrated that L539fs/47-hERG can
cause cell apoptosis by inducing the activation of ERS. Therefore,
the present study focused on elucidating which ERS molecular
pathways involving the hERG mutation were active in inducing the
progression of apoptosis. The 293 cells were divided into three
groups and transfected with hERG plasmids. Pathway-associated
protein expression was detected by western blot analysis. The
results are shown in Fig. 5A and
B. Accordingly, the PERK-eIF2α-CHOP pathway was activated in
L539fs/47 cells. Following transfection, the activation of PERK to
p-PERK was significantly increased in the L539fs/47 cells compared
with the WT and heterozygous cells (P=0.007 and 0.014,
respectively). The downstream molecule of p-PERK, eIF2α, is an
important regulator mediating apoptosis in ERS. The expression of
eIF2α was elevated in the homozygous mutant L539fs/47 cells
(P=0.005 and 0.037 compared with WT and WT-L539fs/47,
respectively). The PERK-eIF2α-CHOP pathway is essential in
upregulating CHOP-mediated cell apoptosis. Significantly higher
levels of CHOP were observed in the L539fs/47 cells (P=0.005), with
only marginal elevation observed in the WT-L539fs/47 cells
(P=0.089), compared with the WT cells. Bax/Bcl-2 regulation was
also indicated to be involved in L539fs/47-induced cell apoptosis.
The L539fs/47 cells exhibited a significantly higher Bax/Bcl-2
ratio compared with that in the WT cells (P=0.009). However, the
expression levels of p-PERK, eIF2α, CHOP and Bax/Bcl-2 in the
WT-L539fs/47 heterozygous mutant cells were not significantly
elevated compared with those in the WT cells (P=0.569, 0.147, 0.059
and 0.058, respectively).
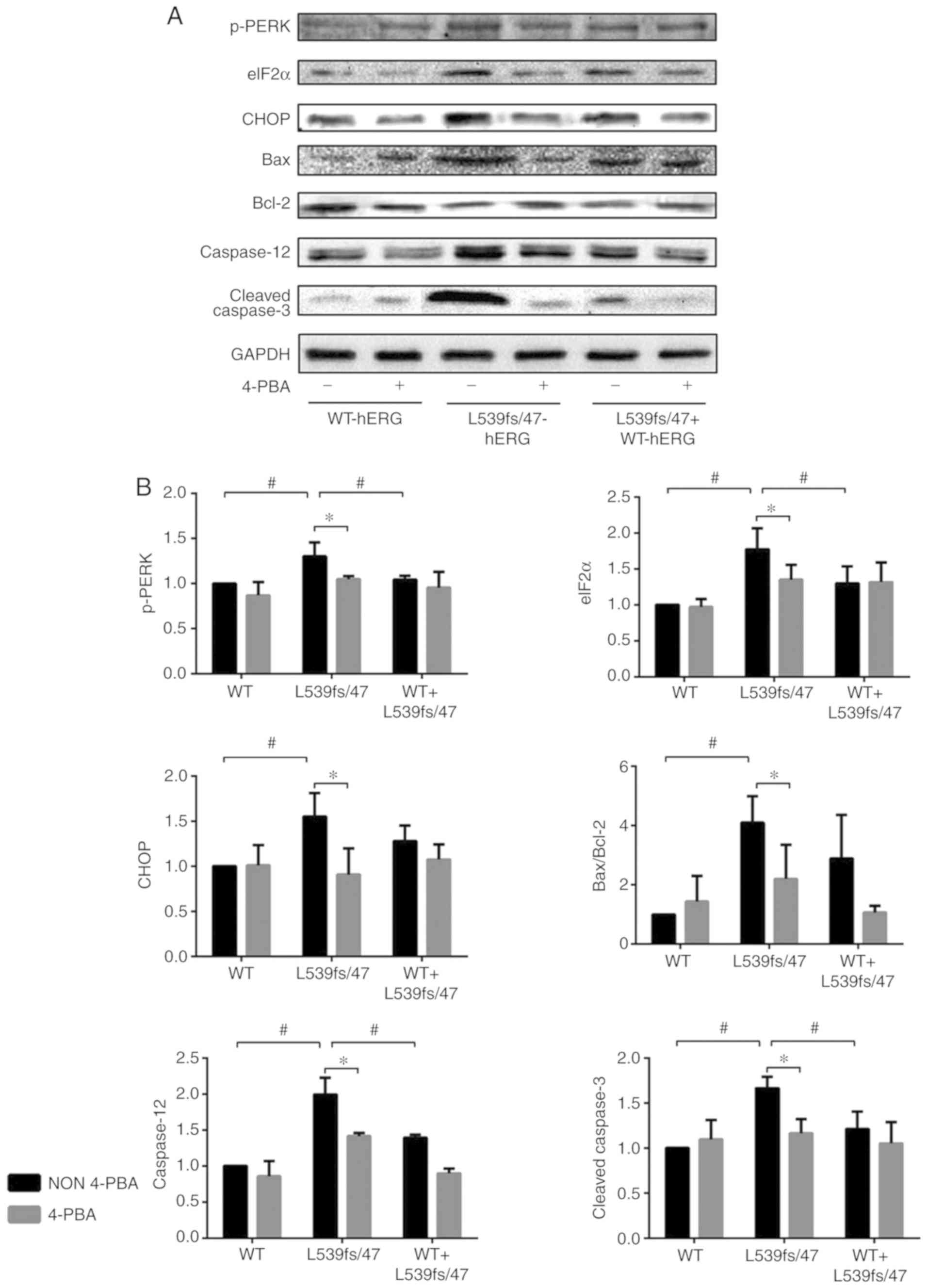 | Figure 5Western blot analysis results for
ERS-associated proteins. (A) The representative blotting images and
(B) quantified data demonstrate elevated p-PERK, eIF2a, CHOP,
Bax/Bcl-2, Caspase-12 and cleaved Caspase-3 level in L539fs/47-hERG
cells, which can be reversed by 4-PBA. Each experiment was repeated
at least three times. #P<0.05 between the three model
cell groups; *P<0.05 between two subgroups treated
with or without 4-PBA intervention. P<0.05 was considered to
indicate a statistically significant difference. ERS, endoplasmic
reticulum stress; hERG, human ether-à-go-go-related gene; WT,
wild-type; 4PBA, 4-phenyl butyric acid; p-PERK, phosphorylated
protein kinase R-like endoplasmic reticulum kinase; eIF2a;
eukaryotic translation-initiation factor-2α; CHOP, C/EBP homologous
protein; Bcl2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein. |
As shown in Fig. 5A
and B, the results showed increased expression of caspase-12 in
the L539fs/47-hERG-transfected cells compared with that in the WT
(P<0.05) and WT-L539fs/47 (P<0.05) cells. The expression of
caspase-12 in the heterozygous mutant cells was also higher
compared with that in the WT cells (P=0.013). The active form of
caspase-3, cleaved-caspase-3, was significantly increased in the
L539fs/47 mutation group compared with that in the other two groups
(P=0.001 and 0.006). However, no significant difference in
expression was observed between the WT-L539fs/47 cells and WT cells
(P=0.099).
Treatment with 4-PBA decreased the PERK-eIF2α-CHOP
pathway, Bax/Bcl-2 ratio, and expression levels of caspase-12 and
cleaved-caspase-3 in the L539fs/47 homozygous mutant cells (all
P<0.05; Fig. 5A and B) but not
in the WT or WT-L539fs/47 cells. These results support the results
of GRP78 described above and suggest that cell apoptosis mediated
by different activated pathways in L539fs/47 cells is the result of
ERS.
Discussion
Historically, LQTS has been defined by abnormalities
in electrical activity due to its unusual ECG appearance. As early
recognition and intervention has become more effective, patients
with LQTS have a longer average life expectancy; therefore,
structural abnormalities that were hidden by early onset sudden
mortality are now being revealed to clinicians and researchers. As
mentioned above, several heart structural abnormalities are
associated with LQTS gene mutations, however, these cases have
received less attention. Almost all previous studies and clinical
guidelines have focused on abnormal ECG activities and not on
structural defects; a limited number of studies have investigated
the mechanisms of LQTS-induced structural abnormalities. Cell
apoptosis is an important natural procedure in the development of
the cardiovascular system (21).
However, extensive apoptosis in the fetal heart can induce heart
defects. In the mature heart, apoptosis is an initiation factor of
fibrosis, ventricle remodeling or contraction abnormality (22,23). A series of studies have shown
marked apoptosis in cells with LQTS gene mutations using human
heart biopsies and in vivo and in vitro methods. In
other areas, including cancer research, the hERG channel has been
found to be an important component in the regulation of apoptosis.
In colon carcinoma, esophageal squamous cell carcinoma, leukemic
cells, endometrial cancer cells and cervical squamous cell
carcinoma cells, hERG has a higher expression level and exhibits
anti-apoptotic behaviors. The application of an hERG inhibitor can
reverse cancer cell apoptosis (24).
LQTS 2 is the most common type of LQTS in China, and
it accounts for 54.5% of all congenital LQTS cases (25). Our previous study examined a
family diagnosed with the L539fs/47-hERG mutation. The proband was
a 20-year-old female who experienced her first syncope upon waking.
The proband's ECGs showed a QTc of 0.60 sec with paroxysmal
polytypic ventricular tachycardia. DNA sequencing of the family
members revealed that the proband's father and one brother had the
KCNH2 gene mutation L539fs/47-hERG. L539fs/47 is a complex
mutation, as is previously described. According to the results of
our previous study of mutated hERG channel function, the
L539fs/47-hERG protein does not have normal channel function
(20). The results of the present
study using confocal laser microscopy verified the results of the
previous study, indicating that the mutated protein can be
recognized by the ER and be retained in it.
The in vitro experiments performed in the
present study also identified that the homozygous mutated
L539fs/47-hERG protein can lead to cell apoptosis. Similar trends
were identified on investigating the molecular pathway; elevated
expression levels of GRP78 were found in L539fs/47-hERG homozygous
cells. It was also found that the PERK-eIF2α-CHOP pathway was
involved in L539fs/47-hERG-induced ERS-mediated cell apoptosis.
Heterozygous mutations of L539fs/47 did not provoke ERS to induce
apoptosis. The same results were also identified in the analyses of
Bax/Bcl-2 and caspase-12 (Fig.
6). The mutation caused apoptosis and molecular changes, which
were reversed by intervention with the ERS inhibitor 4-PBA. These
results indicate that hERG homozygous mutation-induced cell
apoptosis may be the underlying mechanism of LQTS 2 structural
abnormalities and that ERS, which is provoked by the mutated
protein retained in the ER lumen, is the mechanism of apoptosis. In
clinical practice, homozygous mutations of hERG always cause severe
symptoms and can lead to mortality during the embryonic period;
however, individuals with heterozygous mutations can survive and
can be asymptomatic. For this reason, the majority of patients with
LQTS 2 carry a heterozygous mutation. The results of the present
study showed that the L539fs/47-hERG homozygous mutation resulted
in more marked effects on cell apoptosis and molecular biology,
with similar manifestations to LQTS 2. This is likely to explain
why the cardiovascular structural abnormalities of patients with
LQTS remained undetected for so long. In addition, as shown in
Fig. 3, 4-PBA reduced the expression of GRP78 in
the L539fs/47-hERG and WT-L539fs/47-hERG cells, but resulted in
higher expression of GRP78 (P=0.041) in the WT-hERG cells. This
result indicated that ERS was not activated in WT-hERG cells. The
elevation of GRP78 may be the result of other factors, including
the 4-PBA solvent DMSO or other random errors.
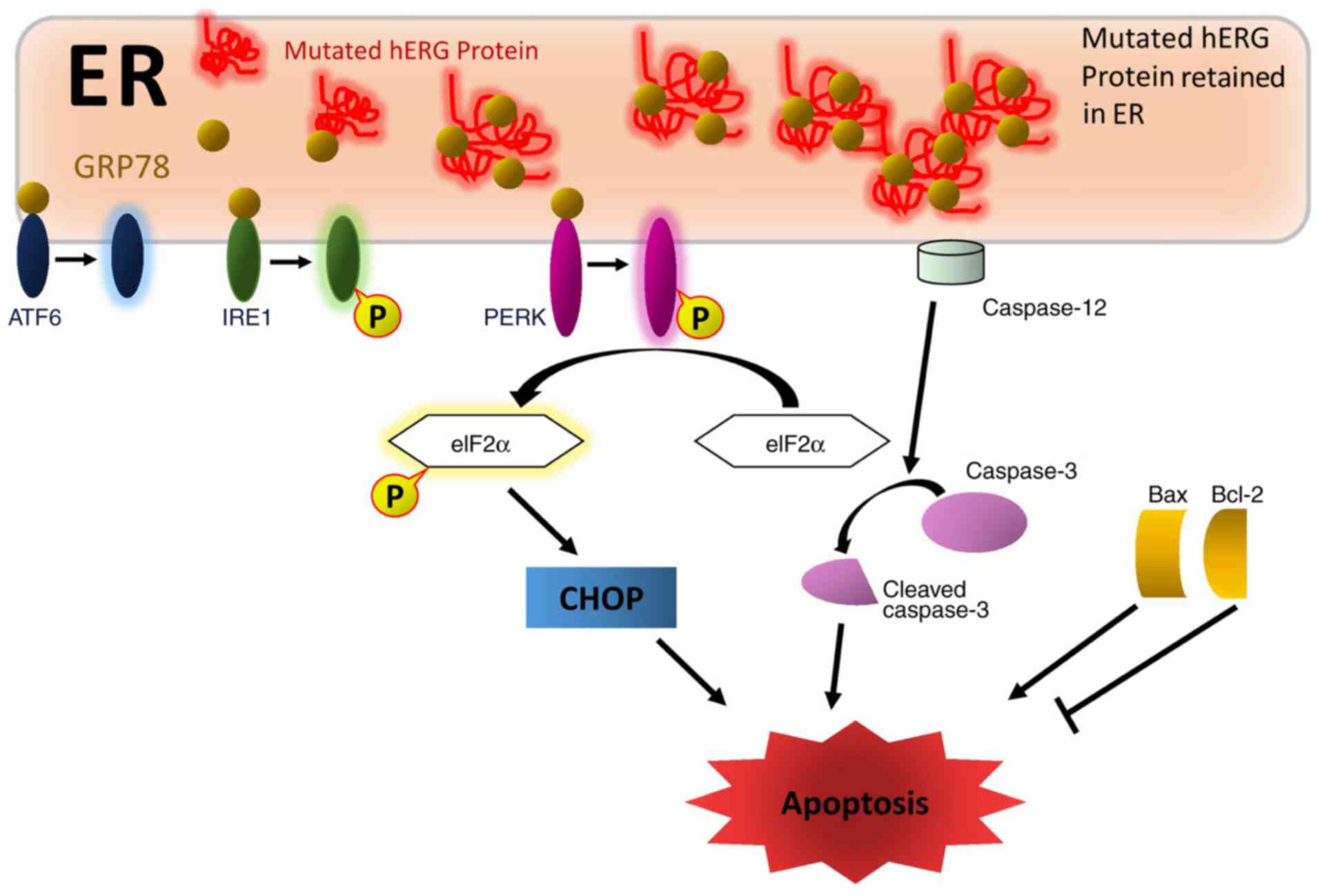 | Figure 6Proposed mechanisms of mutated
hERG-induced cell apoptosis. Mutated L539fs/47-hERG protein
retained in the ER lumen provokes ERS. Elevated expression levels
of GRP78 activate the PERK-eIF2α-CHOP pathway, which is important
in the mechanism of L539fs/47-hERG-induced ERS-mediated cell
apoptosis. ER-specific caspase-12 is involved in the apoptotic
mechanisms caused by the hERG mutation through cleaving and
activating caspase-3. The pair of apoptosis factors Bax/Bcl-2 also
regulate L539fs/47-hERG-induced cell apoptosis. hERG, human
ether-à-go-go-related gene; WT, wild-type; 4PBA, 4-phenyl butyric
acid; ER, endoplasmic reticulum; ERS, ER stress; ATF6, activating
transcription factor 6; IRE1, inositol-requiring enzyme 1; p-PERK,
phosphorylated protein kinase R-like endoplasmic reticulum kinase;
eIF2a; eukaryotic translation-initiation factor-2α; CHOP, C/EBP
homologous protein; Bcl2, B-cell lymphoma 2; Bax, Bcl-2-associated
X protein. |
To the best of our knowledge, the present study is
the first to demonstrate the role of the ERS pathway
PERK-eIF2α-CHOP in LQTS 2-induced cell apoptosis. An additional
molecule, ATF6, can also be activated in LQTS mutation-induced ERS.
In a study by Wang et al, the hERG mutations G572R and E637K
activated the expression of ATF6 and induced mutated protein
degradation through the ubiquitin-mediated proteasome pathway
(19). Keller et al found
that NF-κB signaling is activated by the I539R-hERG mutation
(18). Whether the two ERS
molecules ATF6 and IRE1, or other pathways, such as NF-κB, are
involved in L539fs/47-hERG-induced cell apoptosis requires further
investigation.
The L539fs/47-hERG mutation is a functional deletion
mutation that disables the potassium ion channel function of the
protein; therefore, the balance of ions, such as Ca2+
and Na+, between the cytoplasm and the extracellular
matrix is significantly altered by decreasing the K+
outflow. This change may trigger cell apoptosis through multiple
mechanisms. Under ERS conditions, multiple factors are involved in
the regulation of ER Ca2+ release; for example, CHOP and
Bcl-2 lead to the accumulation of Ca2+ in the cytoplasm
(26,27). High Ca2+ concentrations
can cause cell apoptosis via the activation of calpain. Calpain is
a widely distributed protease in mammalian cells. It can be
activated by Ca2+ and subsequently induce caspase and
non-caspase cell apoptotic pathways. An increased calpain has been
found in apoptotic L539fs/47-hERG cells (28). Additional pathways may also be
mechanisms of LQTS 2-related gene mutations that cause cell
apoptosis, including calcium overload or oxidative stress.
The results of the present study were obtained based
on in vitro experiments. Under in vivo conditions,
multiple ion channels and several biological factors are involved
in the process of LQTS-induced cell apoptosis. Furthermore, the
protein expression of hERG is differential in different regions of
the heart, which may evoke ER stress to varying degrees. The
variability of cardiomyocyte apoptosis is a potential mechanism in
the pathological process of LQTS. Therefore, to identify other
mechanisms involved in hERG mutation-induced cell apoptosis, an
LQTS type 2 transgenic animal is to be used in future
investigations.
In conclusion, the present study is the first, to
the best of our knowledge, to investigate the mechanisms of
cardiomyo-cyte apoptosis caused by hERG channel mutations. It was
found that L539fs/47-hERG mutated protein accumulates in the ER and
induces ERS, and ERS-associated cell apoptosis is the mechanism by
which the mutations are able to cause apoptosis. These mechanisms
of LQTS heart structural abnormalities may be future targets of
intervention in the treatment of patients with LQTS. The results
also provide novel strategies for the investigation of heart
diseases in children, including congenital heart disease, fetal
mortality and cardiomyopathies.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270236).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SM and CS designed the experiments; SM and YZ
performed the experiments; SM and MC analyzed the experimental
data. SM, YZ and CS wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr G. Michael
Vincent (LDS Hospital, Salt Lake City, UT, USA) and Dr Blake D.
Anson (University of Wisconsin, Madison, WI, USA) for providing the
WT-hERG plasmid, and Dr Aifeng Zhang for constructing the
L539fs/47-hERG plasmid.
References
|
1
|
Ackerman MJ, Priori SG, Willems S, Berul
C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R,
et al: HRS/EHRA expert consensus statement on the state of genetic
testing for the channelopathies and cardiomyopathies: This document
was developed as a partnership between the Heart Rhythm Society
(HRS) and the european Heart Rhythm association (EHRA). Europace.
13:1077–1109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
John RM, Tedrow UB, Koplan BA, Albert CM,
Epstein LM, Sweeney MO, Miller AL, Michaud GF and Stevenson WG:
Ventricular arrhythmias and sudden cardiac death. Lancet.
380:1520–1529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nador F, Beria G, De Ferrari GM,
Stramba-Badiale M, Locati EH, Lotto A and Schwartz PJ: Unsuspected
echocardiographic abnormality in the long QT syndrome. Diagnostic,
prognostic, and pathogenetic implications. Circulation.
84:1530–1542. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haugaa KH, Amlie JP, Berge KE, Leren TP,
Smiseth OA and Edvardsen T: Transmural differences in myocardial
contraction in long-QT syndrome: Mechanical consequences of ion
channel dysfunction. Circulation. 122:1355–1363. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zaklyazminskaya E and Dzemeshkevich S: The
role of mutations in the SCN5A gene in cardiomyopathies. Biochim
Biophys Acta. 1863:1799–1805. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Walls J, Sanatani S and Hamilton R:
Post-hoc diagnosis of congenital long QT syndrome in patients with
tetralogy of Fallot. Pediatr Cardiol. 26:107–110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murugan SJ, Parsons JM and Bennett C: A
case of long QT syndrome associated with familial occurrence of
persistent patency of the arterial duct. Cardiol Young. 15:309–311.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
James TN, Terasaki F, Pavlovich ER and
Vikhert AM: Apoptosis and pleomorphic micromitochondriosis in the
sinus nodes surgically excised from five patients with the long QT
syndrome. J Lab Clin Med. 122:309–323. 1993.PubMed/NCBI
|
|
9
|
Zhang T, Yong SL, Drinko JK, Popović ZB,
Shryock JC, Belardinelli L and Wang QK: LQTS mutation N1325S in
cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis,
cardiac fibrosis and contractile dysfunction in mice. Int J
Cardiol. 147:239–245. 2011. View Article : Google Scholar
|
|
10
|
Teng GQ, Zhao X, Lees-Miller JP, Quinn FR,
Li P, Rancourt DE, London B, Cross JC and Duff HJ: Homozygous
missense N629D hERG (KCNH2) potassium channel mutation causes
developmental defects in the right ventricle and its outflow tract
and embryonic lethality. Circ Res. 103:1483–1491. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang S and Kaufman RJ: The impact of the
unfolded protein response on human disease. J Cell Biol.
197:857–867. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bohnen MS, Peng G, Robey SH, Terrenoire C,
Iyer V, Sampson KJ and Kass RS: Molecular pathophysiology of
congenital long QT syndrome. Physiol Rev. 97:89–134. 2017.
View Article : Google Scholar :
|
|
18
|
Keller SH, Platoshyn O and Yuan JX: Long
QT syndrome-associated I593R mutation in HERG potassium channel
activates ER stress pathways. Cell Biochem Biophys. 43:365–377.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Huang X, Zhou J, Yang X, Li D, Mao
H, Sun HH, Liu N and Lian J: Trafficking-deficient G572R-hERG and
E637K-hERG activate stress and clearance pathways in endoplasmic
reticulum. PLoS One. 7:e298852012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang A, Sun C, Zhang L, Lv Y, Xue X, Li
G, Cui C and Yan GX: L539 fs/47, a truncated mutation of human
ether-a-go-go-related gene (hERG), decreases hERG ion channel
currents in HEK 293 cells. Clin Exp Pharmacol Physiol. 40:28–36.
2013. View Article : Google Scholar
|
|
21
|
Abdelwahid E, Pelliniemi LJ and Jokinen E:
Cell death and differentiation in the development of the
endocardial cushion of the embryonic heart. Microsc Res Tech.
58:395–403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang
H, Wei Y, Cavener DR, Hu X, Hall J, et al: Endoplasmic reticulum
stress sensor protein kinase R-like endoplasmic reticulum kinase
(PERK) protects against pressure overload-induced heart failure and
lung remodeling. Hypertension. 64:738–744. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jehle J, Schweizer PA, Katus HA and Thomas
D: Novel roles for hERG K(+) channels in cell proliferation and
apoptosis. Cell Death Dis. 2:e1932011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu J, Dayi HU, Liu W, Li C, Qin X, Li Y,
Li Z, Li L, Dong W, Qi Y and Wang Q: Clinical characters and
mutation analysis of potassium channel genes KCNH2 in 77 pedigrees
of Chinese with long QT syndrome. Sci Technol Eng. 11:1529–1533.
2006.In Chinese.
|
|
26
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lam M, Dubyak G, Chen L, Nunez G, Miesfeld
RL and Distelhorst CW: Evidence that BCL-2 represses apoptosis by
regulating endoplasmic reticulum-associated Ca2+ fluxes.
Proc Natl Acad Sci USA. 91:6569–6573. 1994. View Article : Google Scholar
|
|
28
|
Ma S, Zhao Y, Wei Y and Sun C: GW28-e0322
increasing calpain expression regulates hERG mutation L539fs/47
induced cardiomyocyte apoptosis. J Am Coll Cardiol. 70:C6–C7. 2017.
View Article : Google Scholar
|