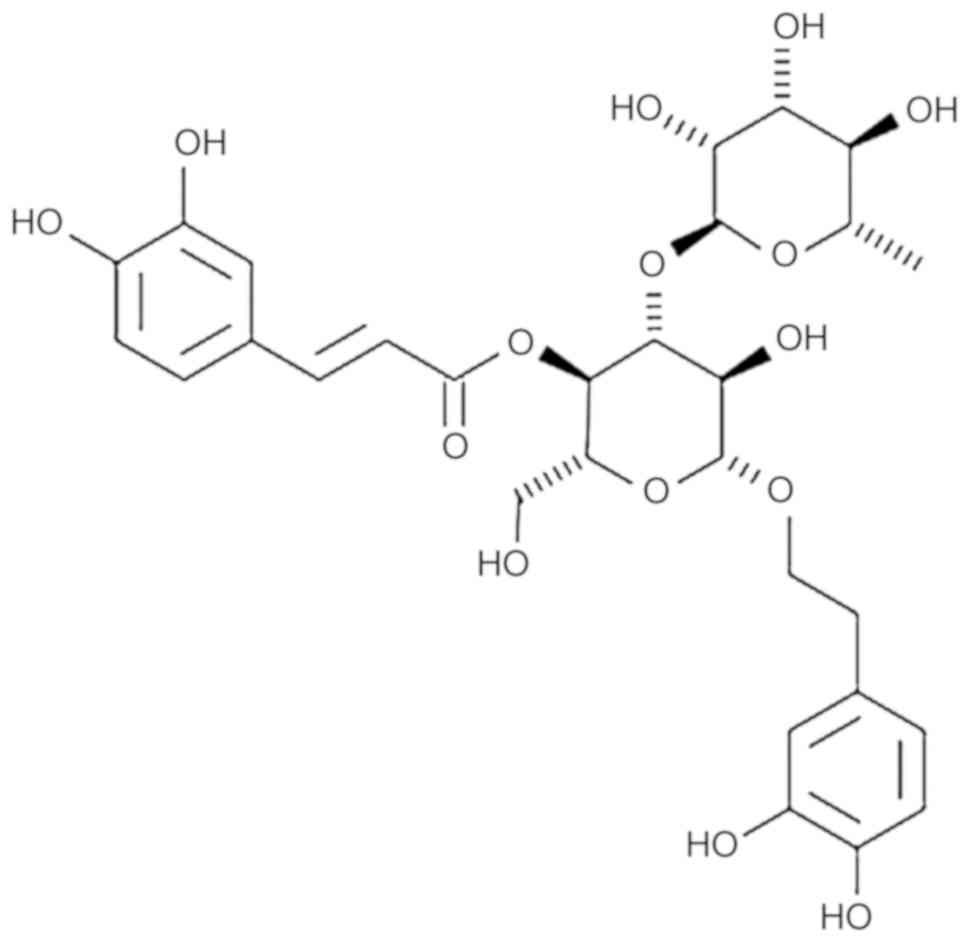
Introduction
Glioblastoma is the most common type of malignant
primary brain tumor and the most invasive and devastating primary
brain tumor (1,2), with a median survival rate of ~18
months with aggressive multimodality therapy. Current treatment
decisions are limited and include radiation, chemotherapy and
surgery in combination with the alkylating agent temozolomide (TMZ)
(3,4). Despite the availability of
aggressive therapies, patients with glioblastoma generally have a
poor prognosis (5). TMZ is the
major chemotherapeutic drug used in the clinical treatment of
malignant glioblastoma (6-8);
as an alkylating agent in the imidazotetrazine series, it is able
to penetrate the blood-brain barrier (9-11).
During malignant glioblastoma progression and extensive invasion
throughout the brain, steadily increasing drug resistance to TMZ
decreases its therapeutic efficacy (12,13). Accordingly, novel therapeutic
drugs to counter glioblastoma are urgently sought.
Previous studies have reported that glioma cells
undergo cell death via autophagy, or type II programmed cell death,
in response to TMZ (14,15). Autophagy can be inhibited by
3-methyladenine (3-MA) through the conversion of
micro-tubule-associated protein 1 light chain 3 (LC3)-I to LC3-II,
thus reducing glioblastoma cell death (16,17); however, the role of autophagy is
dependent on the cellular context. With the exception of a
cytotoxic role during TMZ action, the induction of autophagy by
cellular stress is a cytoprotective process that eliminates
stress-induced cytoplasmic aggregates, organelles and
macromolecules in mammalian cells through the lysosomal system. In
turn, cells involved in maintaining homeostasis receive energy
through these catabolic processes (18,19). The complex roles of autophagy
suggest that an autophagy activator may have anticancer effects in
combination with TMZ treatment by inducing glioblastoma cell
autophagy without exerting deleterious effects or providing
benefits to normal tissues. Of note, TMZ has exhibited synergistic
therapeutic effects in combination with vitamin D, including
suppressed cell viability and enhanced autophagy in glioblastoma
cells. Furthermore, the combination of TMZ and vitamin D has been
shown to exert anticancer effects in vivo, including reduced
tumor size and prolonged survival rate. These studies suggest that
combination treatment may have synergistic anticancer effects via
autophagic mechanisms in TMZ-based glioblastoma therapy (15).
Acteoside is a phenylethanoid glycoside that is
widely distributed in several tonified traditional Chinese herbal
medicines (20,21). A number of pharmacological
actions, including antioxidant, anticancer, anti-inflammatory,
antinephritic and antimetastatic actions, have been associated with
acteoside (22-24). Previous studies have demonstrated
that acteoside has protective effects against carbon tetrachloride-
and D-galactosamine-induced liver injury. The mechanisms underlying
the protective effects of acteoside are likely related to its
capacity to inhibit P450-mediated bioactivation and free radical
scavenging effects induced by carbon tetrachloride exposure
(25,26).
Therefore, evidence suggests that both acteoside and
TMZ induce anticancer effects through cell death. However, their
association and anticancer effects in the context of glioblastoma
remain to be elucidated. Therefore, the objective of the present
study was to verify the synergistic anticancer effects of acteoside
combined with TMZ in glioblastoma therapy. A cell viability assay
was performed to analyze the anticancer effects of the combination
treatment in C6 glioblastoma cells (a rat glioblastoma cell line),
and the results were compared with those following treatment with
TMZ alone. To further examine the mechanism of cell death caused by
cotreatment with acteoside and TMZ, apoptosis- and
autophagy-related genes in C6 cells were examined.
Materials and methods
Cell culture
The C6 rat glioblastoma cell line was purchased from
American Type Culture Collection (Rockville, MD, USA). The cells
were cultured under sterile conditions at 37°C in a humid
environment with 5% of CO2 in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum
(FBS), and 1% antibiotics and antimycotics (all Welgene, Daegu,
Korea).
Plant material
Abeliophyllum distichum Nakai [voucher no.
Park1001(ANH)] was collected in Misunhyang Theme park,
Seongbul-Mountain Recreation Forest, 78, Chungmin-rogigok-gil,
Goesan-eup, Goesan-gun, Chungcheongbuk-do, Korea.
Callus induction
To induce callus formation (27), 1-cm2 leaf explants were
isolated from the fresh plants. The explant was cultured on
Murachige and Skoog medium (4% sucrose, 0.9% agar supplemented with
1 mg/l naphthalene acetic acid and 1 mg/l 2,4-dichlorophenoxyacetic
acid, pH 5.7) at 25°C. The callus was induced 20 days later. A
sufficient quantity of acteoside was obtained through subculture to
separate and purify acteoside from the callus (Fig. 1). Different batches of purified
acteoside were used in each experiment. Each experiment was
performed three times using a different batch.
Isolation and purification
The ethylacetate fractions were concentrated in
vacuo and used as a sample for the purification of acteoside.
Acteoside was isolated and purified by the Accelerated
Chromatographic Isolation system (Isolera™ Spektra, Biotage,
Uppsala, Sweden) using SNAP KPHOSPHO-SIL and SNAP Ultra Cartridges
(Biotage). The acteoside purified from the callus was
quantitatively analyzed using the standard acteoside by HPLC-PDA
analysis. Finally, acteoside of ≥95% purity was obtained from the
callus and used as a sample for chemotherapy of temozolomide-based
glioblastoma.
3-(4,5-Dimethylthiazol-2-yl)-,5-diphenyltetrazolium bromide (MTT)
assay
To identify the TMZ half maximal inhibitory
concentration (IC50 value) against C6 cells,
1×104 cells in single cell suspensions were seeded into
individual wells of 96-well plates and incubated for 24 h at 37°C
prior to TMZ treatment at the indicated concentrations (1, 5, 10,
and 20 mM) at 37°C for 24 h. Following determination of the TMZ
IC50 value as 5 mM, the cells were treated for 24 h with
5 mM TMZ, 50 µM acteoside, or a combination of the two (5 mM
TMZ and 50 µM acteoside). MTT solution (5 mg/ml) was added
to each well (10 µl) and cultured at 37°C for 2 h.
Subsequently, the medium was removed and DMSO was added at a volume
of 200 µl each and reacted at room temperature for 30 min.
The absorbance at 595 nm was measured to determine cell
viability.
Wound-healing assay
A C6 cell suspension in 1 ml was cultured in a
12-well plate and grown to confluence. The cells were treated with
TMZ, acteoside or TMZ + acteoside and then scratched with a sterile
10-µl pipette tip to create an artificial wound. At 0 and 24
h post-wounding, digital images of the wound healing process were
captured using an inverted microscope. Cell migration was
quantified by measuring the size of the scar at 0 and 24 h using
the image analyzing software, ImageJ 1.43u/Java1.6.0_22 software
(NIH, Bethesda, Maryland, USA). Each experiment was performed three
times and the measurements were performed in triplicate.
Immunoblotting
The C6 cells were treated with TMZ, acteoside or TMZ
+ acteoside for 24 h. Cells were lysed on ice by the RIPA (25 mM
Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 1% sodium
deoxycholate, 0.1% SDS) lysis buffer supplemented with protease and
phosphatase inhibitors cocktail (Roche, Basel, Switzerland) for 30
min. After centrifugation at 18,341 × g for 15 min at 4°C,
supernatant was obtained. Protein concentration was determined
using Bradford Protein Assays (Bio-rad, Hercules, CA, USA). Equal
amounts of total proteins (30 µg) were loaded into single
wells and fractionated via electrophoresis via a 10-15% SDS-PAGE.
The proteins were electrotransferred to polyvinylidene difluoride
membranes (EMD Millipore, Bedford, MA, USA). Following incubation
in blocking solution containing 5% nonfat milk in Tween/Tris-buffer
saline for 30 min at room temperature. The blots were probed with
1:1,000-diluted primary antibodies (cat. no. 9662, caspase 3; Cell
Signaling Technology, Inc., Danvers, MA, USA), B0-cell lymphoma 2
(sc-7382, Bcl-2), Bcl-2-associated X protein (cat. no. 2772, Bax),
phosphorylated (p-)p53 (sc-101762), total-p53 (sc-6243), β-actin
(cat. no. 4970; all; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), LC-3 (L8918, Sigma; Merck KGaA, Darmstadt, Germany), Rab7
(cat. no. 9367, Cell Signaling Technology, Inc.), p62 (cat. no.
114), p-p38 (cat. no. 9211), total-p38 (cat. no. 9212), p-c-Jun
N-terminal kinase (cat. no. 9251, p-JNK), total-JNK (cat. no.
9252), p-extracellular signal-regulated kinase (cat. no. 9101,
p-ERK), total-ERK (cat. no. 9102; all Cell Signaling Technology,
Inc.) overnight at 4°C. This was followed by incubation with
horseradish peroxidase-conjugated secondary antibodies (1:2,000;
LF-SA8001A, Goat Anti-Mouse IgG-HRP) or LF-SA8002A (Goat
Anti-Rabbit IgG-HRP), AB Frontier, Seoul, Korea.) for 2 h at room
temperature. The proteins were then visualized by exposure to
Chemi-Doc (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Immunofluorescence staining
Lysosomal-associated membrane protein 1 (LAMP1) and
LC3 are markers for lysosomal and autophagy. The cells
(1×105 cells/well) were prepared on sterilized glass
coverslips (BD Biosciences, Franklin Lakes, NJ, USA) in triplicate.
Following treatment with TMZ, acteoside or TMZ + acteoside for 24
h, the cells were fixed in 4% paraformaldehyde for 30 min, blocked
with 5% normal chicken serum (S-3000; Vector Laboratories, Inc.,
Burlingame, CA, USA), 0.1% Triton X-100 and then incubated 1 h for
permeabilization and to block non-specific protein-protein
interactions. The cells were then incubated with the anti-LAMP1
(1:200; sc-19992, Santa Cruz Biotechnology, Inc.) and anti-LC3
(1:400; PM036, MBL International, Woburn, MA, USA) overnight at
4°C. The cells were then washed twice with PBS and incubated for an
additional 2 h in the dark with a mixture of Alexa Fluor 488 and
594 secondary antibodies (1:200; Alexa Fluor 488 (A-11008) and 594
(A-11007), Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
washed again with PBS. The nuclei were stained using
4,6-diamidino-2-phenylindole (Sigma; Merck KGaA), and then washed
twice with PBS. The slides were then mounted and images were
captured under an LSM-700 laser confocal microscope.
Flow cytometry
The cells were analyzed for Mitotracker Green and
MitoSOX by flow cytometry using a FACSCanto II flow cytometer, as
indicated by the manufacturer (BD Biosciences). Following two
washes with PBS, the cells were fixed in 4% paraformaldehyde for 30
min at room temperature and permeabilized with 0.25% Triton X-100
in PBS for 20 min. The cells were stained with primary antibodies
for overnight at 4°C (1:200) and then with secondary antibodies for
1 h on ice. Following two washes with PBS, the cells were fixed in
4% paraformaldehyde and assayed immediately. The flow cytometry
data were collected using 10,000 cells and were analyzed using
FlowJo 7.6.1 software (Tree Star, Inc., Ashland, OR, USA).
Statistical analysis
All data were analyzed using GraphPad Prism 5.0 and
are presented as the mean ± standard error of the mean. Data were
analyzed with one-way analysis of variance with Tukey's
multiple-comparisons post hoc test for the comparison of mean
values among multiple groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
Cotreatment with TMZ and acteoside
suppresses glioblastoma cell proliferation and migration
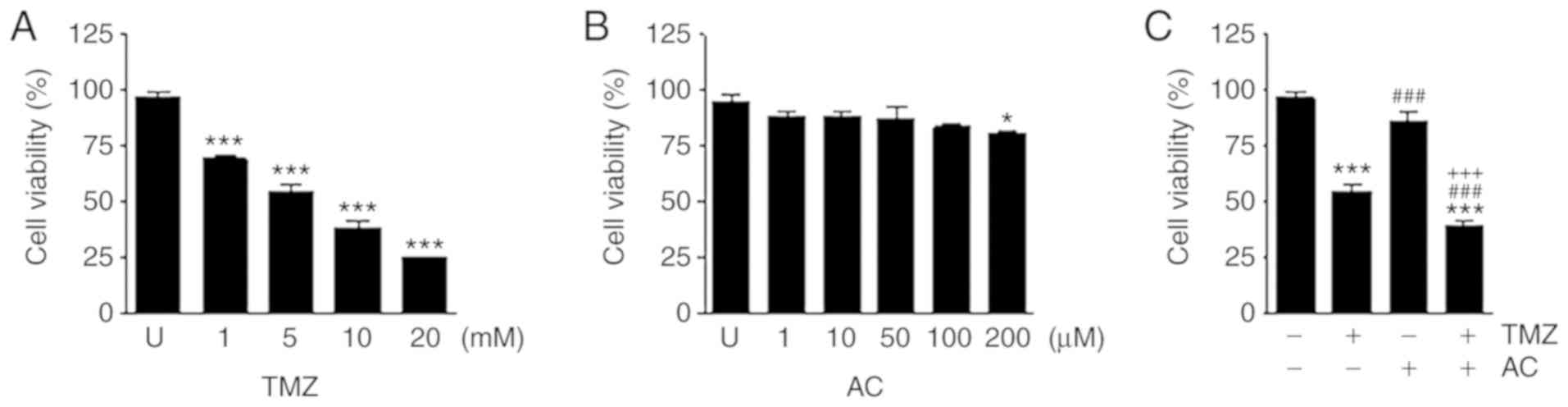
Combined treatment with TMZ and acteoside inhibited
the viability of C6 cells. TMZ reduced cell viability in a
dose-dependent manner (Fig. 2A),
whereas acteoside alone had no significant effect at any treatment
level (Fig. 2B). Following
incubation in culture medium containing TMZ with or without
acteoside for 24 h, an MTT assay was performed to compare the
cytotoxicities of different treatment levels. TMZ significantly
suppressed cell viability, whereas acteoside did not significantly
suppress cell growth. Combined treatment with TMZ and acteoside
markedly reduced cell viability (Fig.
2C). Treatment with TMZ or acteoside applied alone reduced
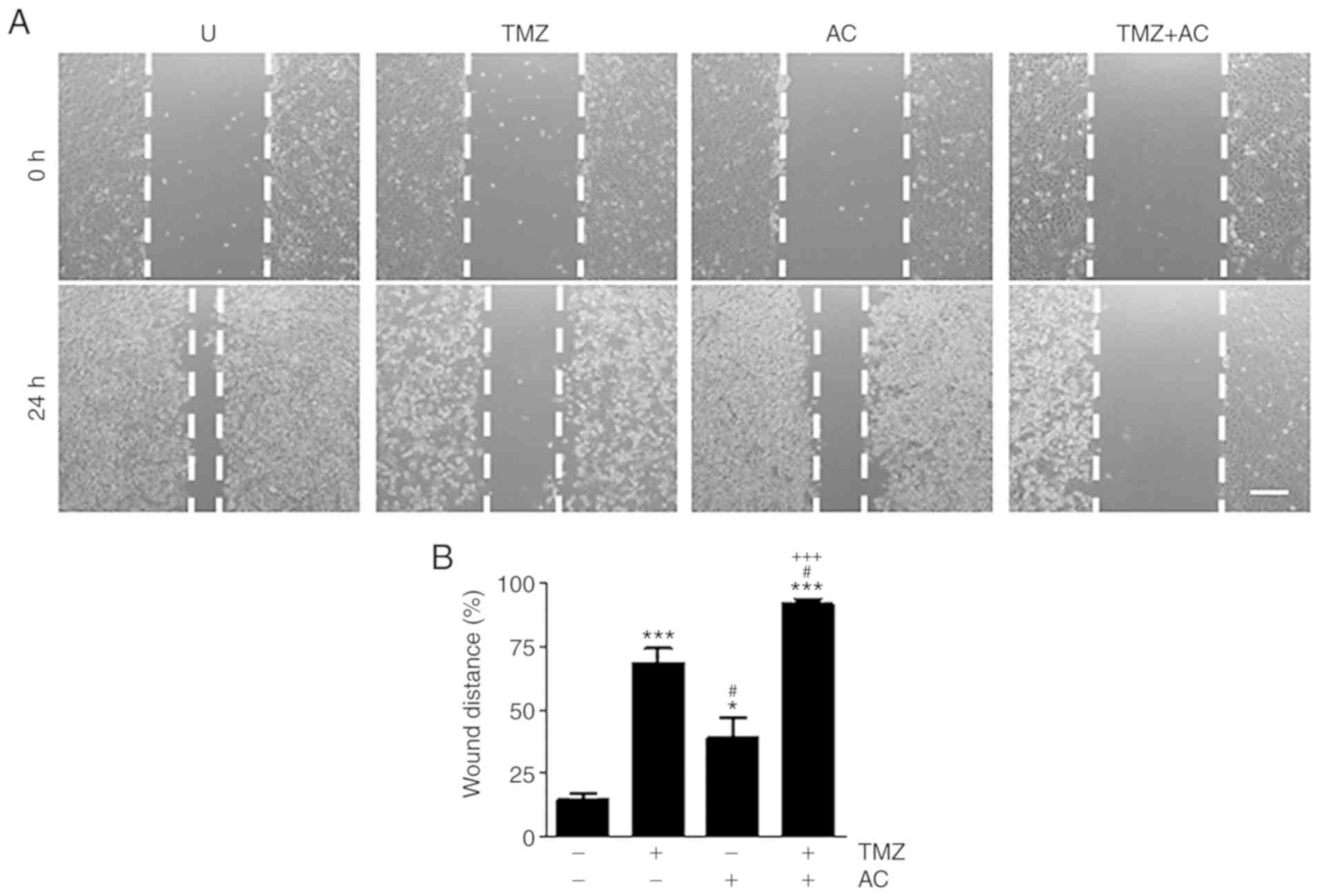
wound-healing ability (Fig. 3A).
The wound distance (%) was measured using the results shown in
Fig. 3A with ImageJ software. The
wound distance increased significantly following combination
treatment (Fig. 3B), compared
with either individual treatment. These results indicate that
cotreatment with TMZ and acteoside was an effective inhibitor of
glioblastoma cell proliferation and migration.
Acteoside and TMZ affect apoptosis
synergistically in C6 cells
The results of the western blot analysis revealed
that the levels of cleaved caspase-3, Bax and phosphorylated p53
were higher and the levels of Bcl-2 were lower following
combination treatment with TMZ and acteoside than following
treatment with TMZ alone (Fig.
4A). Acteoside-mediated mitochondrial function and the
generation of reactive oxygen species (ROS), which are key
mediators of apoptotic signaling, were then observed in the
TMZ-treated C6 cells. To verify mitochondrial mass,
fluorescence-activated cell sorting analysis was performed using
MitoTracker Green. Cotreatment with TMZ and acteoside resulted in a
higher mitochondrial mass than treatment with TMZ alone (Fig. 4B). ROS generation was
significantly higher following cotreatment with TMZ and acteoside
than treatment with TMZ alone (Fig.
4C). These results suggest that ROS generation and
mitochondrial function are important in apoptosis induced by
treatment with TMZ + acteoside.
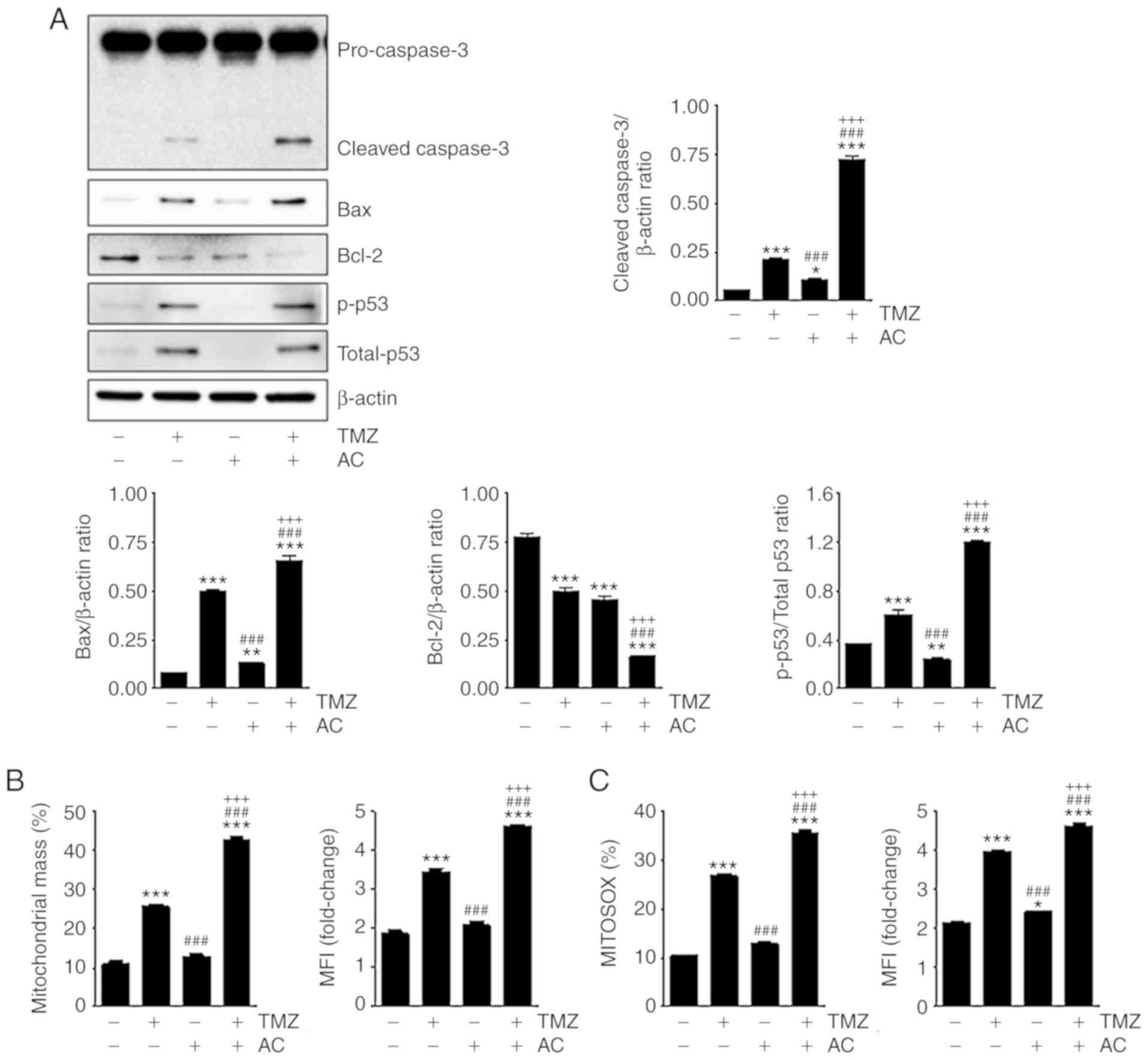 | Figure 4Acteoside and TMZ affect apoptosis
synergistically in C6 cells. (A) Western blot analysis revealed the
levels of cleaved caspase-3, Bax, Bcl-2, p-p53, total-p53 and
β-actin in C6 cells treated with TMZ (5 mM), acteoside (50
µM), and TMZ (5 mM) + acteoside (50 µM) for 24 h. Bar
graphs indicated the density of cleaved caspase-3, Bax, Bcl2,
total-p53, and p-p53. (B) MitoTracker Green flucorescence was
measured by FACS analysis for mitochondrial mass. MFI indicates
mitochondrial mass. (C) C6 cells were stained for MitoSOX to detect
ROS by FACS analysis. Bar graph shows ROS fluorescence intensity.
*P<0.05, **P<0.01 and
***P<0.001 vs. untreated group;
###P<0.001 vs. TMZ group; +++P<0.001
vs. AC group. TMZ, temozolomide; AC, acteoside; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein; p-, phosphorylated;
FACS, fluorescence-activated cell sorting; MFI, mean fluorescence
intensity; ROS, reactive oxygen species. |
Cotreatment with TMZ and acteoside
induces autophagy in C6 cells
TMZ has been reported to induce autophagy (28,29); therefore, the present study
examined the extent to which autophagy was induced by TMZ +
acteoside treatment. The C6 cells were treated with TMZ with or
without acteoside; after 24 h, and the cells were stained for
immunofluorescence to detect LAMP1 and LC3 as markers of autophagy.
Higher expression levels of LAMP1 and LC3 were observed following
the combination treatment (Fig.
5A). Autophagy-related protein expression was also examined,
and it was found that TMZ induced the conversion of LC3-I to
LC3-II, which is a signature of autophagosome generation. A higher
rate of conversion of LC3-I to LC3-II was observed following
cotreatment with TMZ and acteoside than treatment with TMZ alone.
The expression levels of Rab7 and p62 indicated autophagy induction
in the TMZ-treated cells (Fig.
5B). These findings suggest that cotreatment with TMZ and
acteoside enhanced the autophagic process in glioblastoma
cells.
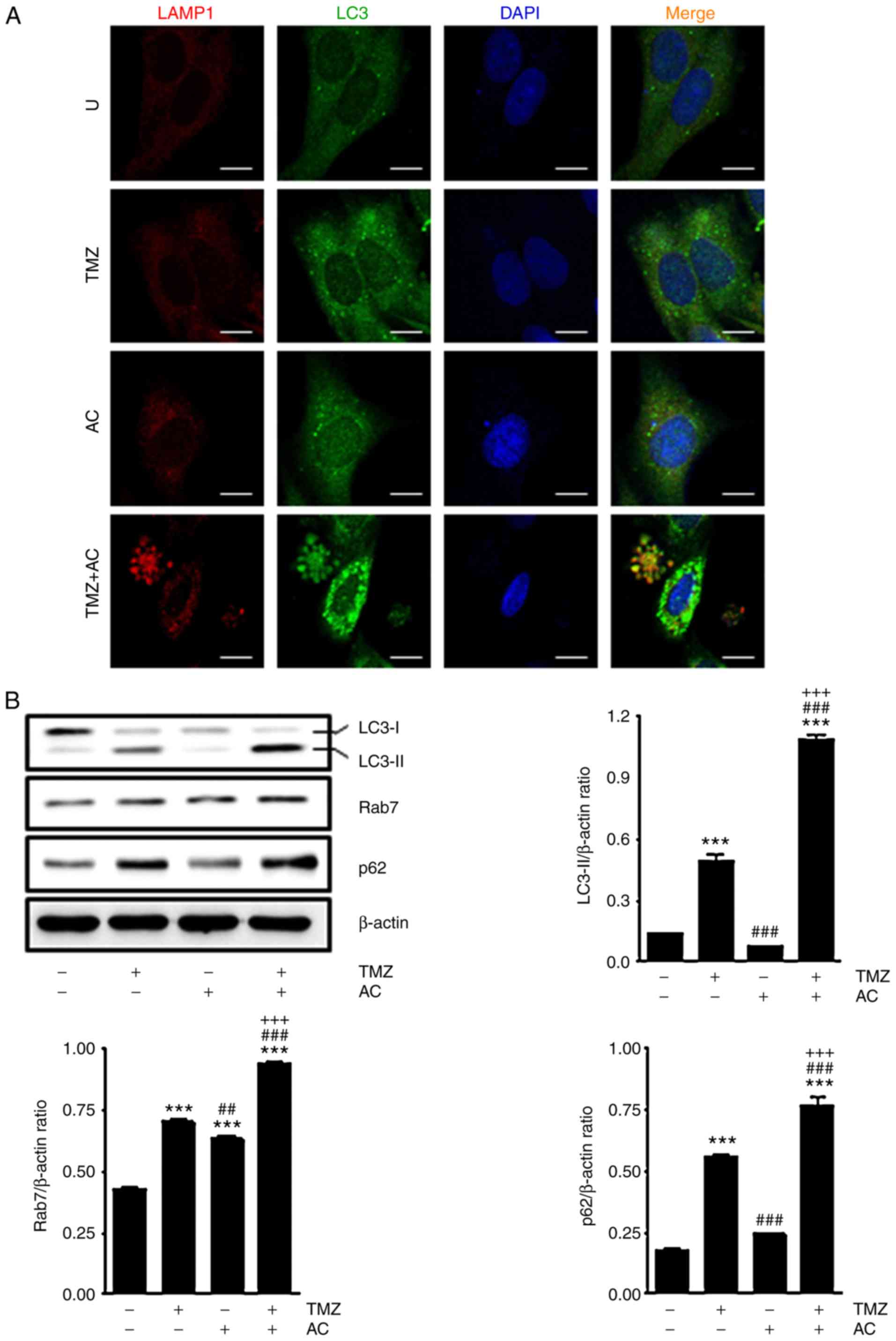 | Figure 5Cotreatement with TMZ + acteoside
induces autophagy in C6 cells. (A) Confocal microscopy shows
expression of autophagosomes with anti-LAMP1 and anti-LC3
antibodies in C6 cells treated for 24 h with TMZ (5 mM) + acteoside
(50 µM). Red, LAMP1; Green, LC3; Blue, DAPI. Scale bar, 10
µm. (B) Western blot analysis showing the levels of LC3,
Rab7, p62 and β-actin in cells treated with acteoside with or
without TMZ for 24 h. Band densities of LC3-Ⅱ, Rab7 and p63 are
indicated as bar graphs. ***P<0.001 vs. Untreated
group; ##P<0.01 and ###P<0.001 vs. TMZ
group; +++P<0.001 vs. AC group. U, Untreated; TMZ,
temozolomide; AC, acteoside. LC3, microtubule-associated protein 1
light chain 3; LAMP1, lysosomal-associated membrane protein 1;
4,6-diamidino-2-phenylindole. |
Acteoside induces MAPK pathway gene
expression in TMZ-based treatment
Previous studies have demonstrated that TMZ
regulates the MAPK pathway, including p38, JNK and ERK (30). Therefore, the present study
investigated whether acteoside affects TMZ-related MAPK gene
expression. The C6 cells were treated with TMZ with or without
acteoside. After 24 h, TMZ treatment resulted in higher expression
levels of p-p38, p-JNK and p-ERK. The expression of TMZ-regulated
genes was significantly higher following TMZ + acteoside treatment
than treatment with TMZ alone (Fig.
6). These observations suggest that acteoside affects TMZ-based
therapeutic mechanisms via the MAPK pathway.
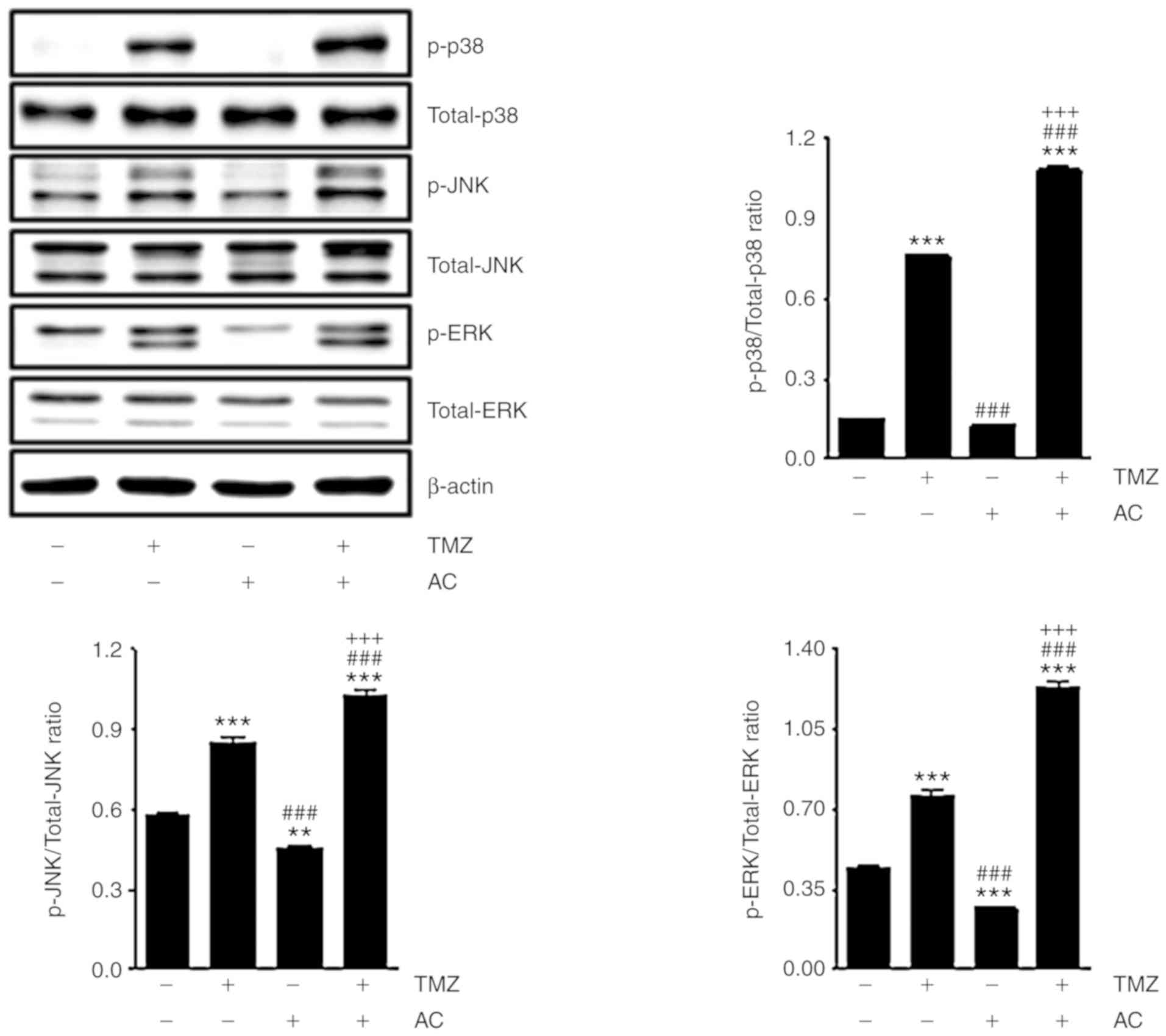 | Figure 6Acteoside induces mitogen-activated
protein kinase pathway gene expression in TMZ-based treatment.
Western blot analysis showed the levels of p-p38, total-p38, p-JNK,
total-JNK, p-ERK and total-ERK in cells treated with TMZ (5 mM)
with or without acteoside (50 µM) for 24 h. The ratio of
p-p38, p-JNK and p-ERK compared with total-p38, total-JNK and
total-ERK, respectively, are shown in bar graphs.
**P<0.01 and ***P<0.001 vs. untreated
group; ###P<0.001 vs. TMZ group;
+++P<0.001 vs. AC group. TMZ, temozolomide; AC,
acteoside; JNK, c-Jun N-terminal kinase; ERK, exrtacellular
signal-regualted kinase; p-, phosphorylated. |
Discussion
The results of the present study establish that the
combined treatment with TMZ with acteoside offers therapeutic
potential for glioblastoma treatment, and provide evidence that
chemosensitization to a combination of TMZ and acteoside can occur
through autophagy enhancement. As the mutation of glioblastoma
cells can lead to apoptotic pathway inactivation, the induction of
autophagy by TMZ + acteoside cotreatment may represent an
alternative method for glioblastoma therapy. However, whether
autophagy is the sole mechanism for glioblastoma therapy with TMZ +
acteoside cotreatment remains to be elucidated.
Autophagy is an essential cellular mechanism for the
degradation of proteins and cytoplasmic organelles. The catabolic
advantage of induced autophagy may be important in stressful
conditions; therefore, the induction of autophagy may be an
adaptive mechanism for cell death prevention (31-33). Previous studies have reported that
reduced autophagy is correlated with cancer (34-36). In addition, several proteins and
signaling pathways associated with autophagy are deregulated during
malignant transformation, resulting in a reduction in autophagic
activity. High expression levels of LC3 have also been associated
with increased survival rates in patients with glioblastoma with
poor performance scores (37,38). Similarly, the results of the
present study indicate that the restoration of normal autophagy may
be a latent strategy for glioblastoma therapy, and may serve as a
mechanism for the restriction of abnormal tumor cell growth.
In normal autophagy, particular cytoplasmic
components are isolated within autophagosomes which then fuse with
a lysosome to be degraded and recycled (39,40). When autophagy is upregulated,
autophagosome formation rates surpass lysosomal degradation rates;
this condition is termed autophagic stress. If stress or abnormal
autophagy continues, cell death can occur via energy depletion or
changes in the beclin-1/Bcl-2 balance. Apoptosis can also be
triggered by autophagosome hyperactivity, engulfing cytoplasmic
organelles including the mitochondria or endoplasmic reticulum
(41). It has been suggested that
general autophagy can induce cell death during normal cytoplasmic
and organelle turnover in healthy cells. In this process, the cell
'cannibalizes' itself from the inside, a key characteristic of type
II programmed cell death. Although the mechanisms by which
acteoside enhances the therapeutic effect of TMZ-based glioblastoma
therapy remain to be fully elucidated, the anticancer effects
exhibited by the combination treatment examined in the present
study may be associated with these autophagic mechanisms.
Acteoside is a phenyl-ethanoid glycoside derived
from plants (22,26). Previous studies have reported on
the various biological activities of acteoside. The changes in the
expression levels of cleaved caspase-3 and LC3 in the present study
demonstrate that treatment with a combination of TMZ and acteoside
induced apoptosis and autophagy in C6 cells (Figs. 4 and 5). The combination treatment was shown
to have a synergistic effect on glioblastoma cells. Although other
possible mechanisms of these synergistic effects remain to be
elucidated, the present study identified the induction of autophagy
as a crucial tumoricidal mechanism of acteoside chemosensitization
during TMZ-based glioblastoma therapy.
Funding
This study was supported by the National Research
Foundation of Korea (NRF) grants funded by the Korean government
(MSIP; grant nos. 2016R1C1B1015811 and 2017R1D1A3B03036420).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
TWH and JJK drafted the manuscript and performed the
experiments. TWH, DHK, DBK, TWJ, and GHK performed the experiments
and data interpretation. KAY contributed materials/analysis tools
and helped in data analysis. MM, DEC, JHP, and JJK designed the
experiments, provided critical suggestions for the manuscript, and
reviewed and revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Friedman HS, Kerby T and Calvert H:
Temozolomide and treatment of malignant glioma. Clin Cancer Res.
6:2585–2597. 2000.PubMed/NCBI
|
|
2
|
Mrugala MM and Chamberlain MC: Mechanisms
of disease: Temozolomide and glioblastoma-look to the future. Nat
Clin Pract Oncol. 5:476–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Agarwala SS and Kirkwood JM: Temozolomide,
a novel alkylating agent with activity in the central nervous
system, may improve the treatment of advanced metastatic melanoma.
Oncologist. 5:144–151. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stevens MF, Hickman JA, Stone R, Gibson
NW, Baig GU, Lunt E and Newton CG: Antitumor imidazotetrazines. 1.
Synthesis and chemistry of
8-carbamoyl-3-(2-chloroethyl)imidazo[5,1-d]-1,2,3, 5-tetrazin-4(3
H)-one, a novel broad-spectrum antitumor agent. J Med Chem.
27:196–201. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fulda S: Cell death-based treatment of
glioblastoma. Cell Death Dis. 9:1212018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen PH, Shen WL, Shih CM, Ho KH, Cheng
CH, Lin CW, Lee CC, Liu AJ and Chen KC: The CHAC1-inhibited Notch3
pathway is involved in temozolomide-induced glioma cytotoxicity.
Neuropharmacology. 116:300–314. 2017. View Article : Google Scholar
|
|
7
|
Oshiro S, Tsugu H, Komatsu F, Ohmura T,
Ohta M, Sakamoto S, Fukushima T and Inoue T: Efficacy of
temozolomide treatment in patients with high-grade glioma.
Anticancer Res. 29:911–917. 2009.PubMed/NCBI
|
|
8
|
van den Bent MJ: Adjuvant treatment of
high grade gliomas. Ann Oncol. 17(Suppl 10): x186–x190. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen W, Hu JA and Zheng JS: Mechanism of
temozolomide-induced antitumour effects on glioma cells. J Int Med
Res. 42:164–172. 2014. View Article : Google Scholar
|
|
10
|
Barciszewska AM, Gurda D, Głodowicz P,
Nowak S and Naskręt-Barciszewska MZ: A new epigenetic mechanism of
temozolomide action in glioma cells. PLoS One. 10:e01366692015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai X and Sughrue ME: Glioblastoma: New
therapeutic strategies to address cellular and genomic complexity.
Oncotarget. 9:9540–9554. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
13
|
Johannessen TC and Bjerkvig R: Molecular
mechanisms of temozolomide resistance in glioblastoma multiforme.
Expert Rev Anticancer Ther. 12:635–642. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan Y, Xu Z, Dai S, Qian L, Sun L and Gong
Z: Targeting autophagy to sensitive glioma to temozolomide
treatment. J Exp Clin Cancer Res. 35:232016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bak DH, Kang SH, Choi DR, Gil MN, Yu KS,
Jeong JH, Lee NS, Lee JH, Jeong YG, Kim DK, et al: Autophagy
enhancement contributes to the synergistic effect of vitamin D in
temozolomide-based glioblastoma chemotherapy. Exp Ther Med.
11:2153–2162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li C, Liu Y, Liu H, Zhang W, Shen C, Cho
K, Chen X, Peng F, Bi Y, Hou X, et al: Impact of autophagy
inhibition at different stages on cytotoxic effect of autophagy
inducer in glioblastoma cells. Cell Physiol Biochem. 35:1303–1316.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ni H, Gong Y, Yan JZ and Zhang LL:
Autophagy inhibitor 3-methyladenine regulates the expression of
LC3, Beclin-1 and ZnTs in rat cerebral cortex following recurrent
neonatal seizures. World J Emerg Med. 1:216–223. 2010.PubMed/NCBI
|
|
18
|
Xie Z and Klionsky DJ: Autophagosome
formation: Core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gutierrez MG, Master SS, Singh SB, Taylor
GA, Colombo MI and Deretic V: Autophagy is a defense mechanism
inhibiting BCG and Mycobacterium tuberculosis survival in infected
macrophages. Cell. 119:753–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu YT, Wu MT, Lin CC, Chien CF and Tsai
TH: Pharmacokinetic studies of chinese medicinal herbs using an
automated blood sampling system and liquid chromatography-mass
spectrometry. J Tradit Complement Med. 2:33–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inoue M, Sakuma Z, Ogihara Y and Saracoglu
I: Induction of apoptotic cell death in HL-60 cells by acteoside, a
phenylpropanoid glycoside. Biol Pharm Bull. 21:81–83. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohno T, Inoue M, Ogihara Y and Saracoglu
I: Antimetastatic activity of acteoside, a phenylethanoid
glycoside. Biol Pharm Bull. 25:666–668. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee KJ, Woo ER, Choi CY, Shin DW, Lee DG,
You HJ and Jeong HG: Protective effect of acteoside on carbon
tetrachloride-induced hepatotoxicity. Life Sci. 74:1051–1064. 2004.
View Article : Google Scholar
|
|
24
|
Peerzada KJ, Faridi AH, Sharma L, Bhardwaj
SC, Satti NK, Shashi B and Tasduq SA: Acteoside-mediates
chemoprevention of experimental liver carcinogenesis through STAT-3
regulated oxidative stress and apoptosis. Environ Toxicol.
31:782–798. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao J, Liu T, Ma L, Yan M, Zhao Y, Gu Z
and Huang Y: Protective effect of acteoside on immunological liver
injury induced by Bacillus Calmette-Guerin plus lipopolysaccharide.
Planta Med. 75:1463–1469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiong Q, Hase K, Tezuka Y, Tani T, Namba T
and Kadota S: Hepatoprotective activity of phenylethanoids from
Cistanche deserticola. Planta Med. 64:120–125. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koh DS, Seo BS and Lee CH: Studies on the
in vitro induction of callus from anther culture of Abeliophyllum
distichum. J Chonbuk Natl Univ. 31:153–159. 1989.In Korean.
|
|
28
|
Würstle S, Schneider F, Ringel F, Gempt J,
Lämmer F, Delbridge C, Wu W and Schlegel J: Temozolomide induces
autophagy in primary and established glioblastoma cells in an EGFR
independent manner. Oncol Lett. 14:322–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koukourakis MI, Mitrakas AG and
Giatromanolaki A: Therapeutic interactions of autophagy with
radiation and temozolomide in glioblastoma: Evidence and issues to
resolve. Br J Cancer. 114:485–496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Y, Gao F, Jiang R, Liu H, Hou J, Yi
Y, Kang L, Liu X, Li Y and Yang M: Down-regulation of AQP4
expression via p38 MAPK signaling in temozolomide-induced glioma
cells growth inhibition and invasion impairment. J Cell Biochem.
118:4905–4913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bento CF, Renna M, Ghislat G, Puri C,
Ashkenazi A, Vicinanza M, Menzies FM and Rubinsztein DC: Mammalian
autophagy: How does it work? Annu Rev Biochem. 85:685–713. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fulda S: Autophagy in Cancer Therapy.
Front Oncol. 7:1282017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aoki H, Kondo Y, Aldape K, Yamamoto A,
Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K,
Shinojima N, et al: Monitoring autophagy in glioblastoma with
antibody against isoform B of human microtubule-associated protein
1 light chain 3. Autophagy. 4:467–475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cj P, Hv E, Vijayakurup V, R Menon G, Nair
S and Gopala S: High LC3/beclin expression correlates with poor
survival in glioma: A definitive role for autophagy as evidenced by
in vitro autophagic flux. Pathol Oncol Res. Oct 11–2017.PubMed/NCBI
|
|
39
|
Monastyrska I and Klionsky DJ: Autophagy
in organelle homeostasis: Peroxisome turnover. Mol Aspects Med.
27:483–494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Farre JC and Subramani S: Peroxisome
turnover by micropexophagy: An autophagy-related process. Trends
Cell Biol. 14:515–523. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.
View Article : Google Scholar : PubMed/NCBI
|