Introduction
As percutaneous coronary intervention became widely
adopted in the 1980s, restenosis was reported in 30-60% of
patients, most presenting with recurrent symptoms 1-4 months
post-procedure (1,2). In recent years, with the increasing
prevalence of vascular stenosis, potential remedial strategies,
including medical therapy, surgical bypass and endovascular
revascularization, are becoming more widely applied in its clinical
treatment (3,4). Endovascular interventional therapy,
including percutaneous transluminal angioplasty (PTA) and stent
implantation, has demonstrated positive effects in the treatment of
vascular stenosis (5,6). However, the recurrence rate of
restenosis, a major drawback of PTA, is as high as 19.7 and 60%
within the first and fifth years following surgery, respectively
(7,8). Restenosis is a pathological outcome
of excessive oxidative stress and repair responses in injured local
blood vessels in response to interventional therapy (8), leading to a poor prognosis and
inferior quality of life in patients with vascular stenosis. The
post-operative occurrence of restenosis is one of the major
challenges associated with endovascular intervention and represents
a worldwide problem.
Vascular endothelial cell damage induced by PTA is
an essential factor in the occurrence of restenosis (9). Furthermore, injured endothelia
induce the production of reactive intermediates, including reactive
nitrogen species and reactive oxygen species (ROS), leading to
endothelial dysfunction (10). It
has previously been demonstrated that the level of ROS in local
blood vessels increases following PTA surgery or stent implantation
(11-13). Upon injury, endothelial
dysfunction promotes the excessive proliferation and migration of
vascular smooth muscle cells (VSMCs) and triggers neointimal
hyperplasia, resulting in narrowing of the lumen following
endovascular intervention (14,15).
Hydrogen sulfide (H2S), one of the
important gasotransmitters, is an endogenously produced gas signal
molecule that is synthesized and catalyzed in vivo through
cystathionine β-synthase and cystathionine γ-lyase, respectively
(16). H2S is involved
in a number of beneficial reactions in cardiovascular tissues,
thereby regulating multiple physiological functions, including
anti-oxidation, angiogenesis and vasodilation (17). According to the literature,
H2S has cardioprotective effects via the inhibition of
intimal hyperplasia after peripheral angioplasty (18,19) and antioxidative effects through
the elimination of hydrogen peroxide (H2O2)
and superoxide anion, thereby suppressing myocardial ischemic
injury (20). Nuclear
factor-E2-related factor 2 (Nrf2), a transcription factor with a
high sensitivity to oxidative stress, exerts antioxidative effects
by binding to antioxidant response elements (AREs) in the nucleus
and regulating the expression of downstream antioxidant genes,
including heme oxygenase (HO)-1 (21). A previous study demonstrated that
Nrf2 may be involved in the antioxidative activity of
H2S in H2S-mediated cardioprotection
(22). In addition,
hypoxia-inducible factor (HIF-1), a protein comprising HIF-1α and
HIF-1β subunits, has been revealed to serve an important role in
regulating angiogenesis, which is beneficial for wound healing
during peripheral angioplasty-induced blood vessel injury (23). Thus, it may be hypothesized that
the Nrf2 signaling pathway and HIF-1α serve roles in the
anti-restenosis effects of H2S.
Although the physiological and cardioprotective
effects of H2S have previously been documented, the
anti-restenosis effect and molecular mechanisms have not been fully
evaluated. Therefore, the purpose of the present study was to
investigate the anti-restenosis effect and signaling mechanisms
induced by H2S donor (NaHS) treatment using an in
vivo model of restenosis and in vitro cell culture.
Materials and methods
Animals
A total of 24 healthy adult male Sprague-Dawley (SD)
rats (8-9 weeks, 250±30 g) were purchased from the Hubei Provincial
Center for Disease Control and Prevention (Hubei, China). The rats
were housed under controlled conditions of 22±2°C and 55±5%
humidity under a 12-h light/12-h dark cycle and access to food and
water ad libitum. The rats were acclimatized for at least 1
week prior to being used in the experiments. All experiments
involving animal treatment were performed in accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals, and the protocol was approved by the Ethics
Committee of Union Hospital, affiliated to the Huazhong University
of Science and Technology (approval no. TJ-A20161216).
Construction of the balloon dilatation
restenosis model and NaHS treatment
All animals were randomly divided into three groups:
Normal, model and model+NaHS groups (n=8). The rats that did not
receive the balloon injury (sham-operated) were used as normal
controls. The rats in the model group (balloon injury) underwent
balloon catheter injury to construct the balloon dilatation
restenosis model as previously described (8,24)
and were allowed to recover for 4 weeks. In brief, the rats were
anesthetized by intraperitoneal injection of 40 mg/kg pentobarbital
and the right external carotid artery in the neck was isolated
using a midline incision. The distal end of the right external
carotid artery was ligated with a nylon suture, while the proximal
part was clamped with a bulldog clamp. Then, a small opening was
made in the distal end of the right external carotid artery using
eye scissors. A 2F balloon catheter (Edwards Lifesciences, Irvine,
CA, USA) was inserted into the right external carotid artery and
inflated with 200 µl saline. Next, the balloon catheter was
pulled back through the external carotid artery to break the blood
vessel endothelium. This manipulation was repeated thrice. Then,
the balloon catheter was removed after 20 min; the external carotid
artery was ligated and the incisions to the vessel and skin were
closed. Penicillin was used topically to prevent infection
following the surgery. The rats in the model+NaHS group underwent
the same balloon catheter injury and received treatment with
H2S in the form of sodium hydrosulfide (NaHS, a donor of
H2S; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Treatment with NaHS (30 µmol/kg) was performed by
intraperitoneal injection prior to the surgery and continued for 4
consecutive weeks following the surgery, once a day.
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were
obtained from American Type Culture Collection (Manassas, VA, USA)
and cultured in M200 (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Hyclone;
GE Healthcare Life Sciences, Logan, UT, USA). Human vascular smooth
muscle cells (HVSMCs) were obtained from American Type Culture
Collection and cultured in smooth muscle cell medium (ScienCell
Research Laboratories, Inc., San Diego, CA, USA) containing 10%
FBS. The two cell types were maintained at 37°C in a humidified
incubator with 5% CO2. Once 75% cell confluence was
achieved, they were treated with NaHS (0, 50, 100 or 200
µmol/l) for 48 h. The effect of NaHS on HVSMC proliferation
was detected using an MTT cell proliferation and cytotoxicity assay
kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
The untreated group was considered as the control group.
Furthermore, ~2×105 HUVECs were cultured in 6-well
plates and at 70-80% cell confluence, the cells were treated with
NaHS and then transfected with small interfering (si)RNA of target
sequences, including siNrf2 (5′-CAUUGAUGUUUCUGAUCUATT-3′; 100 nM,
48 h), siHIF-1α (5′-CUGAUAACGUGAACAAAUATT-3′; 100 nM, 48 h) or
siRNA negative control (siNC; 5′-GGUCUCACU CCCCAUAGAGTT-3′; 100 nM,
48 h; all Guangzhou RiboBio Co., Ltd., Guangzhou, China) using the
Lipofectamine RNAiMAX transfection reagent (Invitrogen; Thermo
Fisher Scientific Inc., Waltham, MA, USA). Cultured cells that had
not been transfected with siRNA or treated with NaHS were used as
controls.
Histological observation
After 4 weeks of treatment, the rats were euthanized
by overdose anesthetics (100 mg/kg pentobarbital). Then, the right
carotid arteries were isolated, placed in a cold saline solution
and the surrounding adipose tissue was dislodged from the carotid
artery samples. Subsequently, the artery samples were cut into
vessel segments (3.0 mm) and fixed in 4% paraformaldehyde for 1 h
at room temperature. The artery samples were embedded into paraffin
blocks using conventional procedures (25), including dehydration,
transparency, dipping and embedding. For morphological observation,
5.0-µm thick sections were stained with hematoxylin and
eosin (H&E) staining solution (0.2% hematoxylin solution and 1%
eosin solution; Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer's protocol. The areas occupied by the
external elastic lamina, the internal elastic lamina and the lumen
areas were measured by an optical microscope at a magnification of
×100. Other areas were calculated as follows: Neointimal area,
internal elastic lamina area - lumen area; medial area, external
elastic lamina - internal elastic lamina; and % of lumen
stenosis/normal, (lumen area in normal group - lumen area in model
or treatment group) / lumen area in normal group.
Measurement of endogenous H2S
production
Carotid artery tissue and plasma H2S
production was measured using the H2S content assay kit
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) according to the manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Carotid arteries were ground using liquid nitrogen
(−196°C). Total RNA was extracted from carotid arteries using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
dissolved in RNA-free water. Next, cDNA was synthesized using the
superscript™ II reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.) and diluted into the concentration of 100
ng/µl for RT-qPCR. The thermal cycling conditions for
reverse transcription were as follows: 70°C for 10 min, 42°C for 50
min and 70°C for 15 min. RT-qPCR was performed using the QuantiFast
SYBR-Green PCR kit (Qiagen, Inc., Valencia, CA, USA). RT-qPCR was
performed at 95°C for 10 min, followed by 40 cycles at 95°C for 10
sec, 60°C for 45 sec and 72°C for 30 sec; and a last step at 72°C
for 10 min. The primers were purchased from Sangon Biotech Co.,
Ltd., (Shanghai, China). The primer sequences used in present study
as follows: Nrf2 forward, 5′-ATTCAAGCCGATTAGAGG-3′ and reverse,
5′-ATTGCTCCTTGGACATCA-3′; HO-1 forward, 5′-ATGTCCCAGGATTTGTC-3′ and
reverse, 5′-CTGCTT GTTTCGCTCT-3′; HIF-1α forward, 5′-GGCTTTGTTATG
GTGCTA-3′ and reverse, 5′-TTGTTCTTTCCCCTTTCT-3′; vascular
endothelial growth factor (VEGF) forward,
5′-AGGGCAGAATCATCACGAAGT-3′ and reverse, 5′-AGG
GTCTCGATTGGATGGCA-3′; GAPDH forward, 5′-CAA GTTCAACGGCACAG-3′ and
reverse, 5′-CCAGTAGACTCC ACGACAT-3′. The quantified results were
normalized to those of GADPH using the 2−ΔΔCq method
(26).
Western blot analysis
Nuclear and cytoplasmic proteins of tissue samples
were extracted using Nuclear and Cytoplasmic Protein Extraction kit
(Beyotime Institute of Biotechnology, Shanghai, China) according to
the manufacturer's protocol. The total protein samples from HUVECs
treated with different concentrations of NaHS (0, 50, 100 or 200
µmol/l), or transfected with siNrf2, siHIF-1α or si-NC were
extracted using RIPA Lysis buffer containing a protease inhibitor
cocktail (Roche Applied Science, Madison, WI, USA). The
concentration of protein was measured using a bicinchoninic acid
protein assay kit. Subsequently, 15 µg of protein/sample was
subjected to 12% SDS-PAGE and separated proteins were transferred
to polyvinylidene difluoride membranes. The membranes were blocked
with 5% skim milk [dissolved in PBS containing 0.1% Tween-20
(PBST)] for 1 h at room temperature. Then, the membranes were
incubated at 4°C overnight with the following primary antibodies:
Rabbit monoclonal anti-Nrf2 (1:2,000; cat. no. ab62352); mouse
monoclonal anti-HO-1 (1:1,000; cat. no. ab13248); mouse monoclonal
anti-HIF-1α (1:2,000; cat. no. ab16066); rabbit polyclonal
anti-VEGF (1:2,000; cat. no. ab32152); rabbit polyclonal
anti-Histone H3 (1:800; cat. no. ab61251); mouse monoclonal
anti-GAPDH (1:2,000; cat. no. ab8245) (all from Abcam, Cambridge,
UK). Following washing with PBST thrice, the membranes were
incubated with the goat anti-rabbit IgG H&L (1:2,000; cat. no.
ab6702) or goat anti-mouse IgG H&L (1:5,000; cat. no. ab6708)
secondary antibodies (both from Abcam) for 1.5 h at room
temperature. The protein band signals were detected using BeyoECL
Plus [ultra-sensitive electrochemical luminescence (ECL) kit;
Beyotime Institute of Biotechnology] and an automatic
chemiluminescence analyzer (Tanon-5200; Tanon Science and
Technology Co., Ltd., Shanghai, China). Gray values of bands were
quantified using ImageJ software (version 1.42; National Institutes
of Health, Bethesda, MD, USA) and normalized to GAPDH or Histone
H3.
Immunofluorescence
HUVECs were divided into five groups: Control, NaHS
treated, NaHS treated + siRNA-NC, siNrf2 transfected and NaHS
treated + siNrf2. After 48 h, ~1×105 HUVECs cultured on
slides were fixed in 4% paraformaldehyde for 10 min at room
temperature, treated with 0.5% Triton X-100 at 37°C for 15 min and
blocked with 1% bovine serum albumin (Gibco; Thermo Fisher
Scientific, Inc.) at room temperature for 1 h. Subsequently, the
cells were incubated with the mouse monoclonal anti-HIF-1α (1:200;
cat. no. ab16066; Abcam) and rabbit monoclonal anti-Nrf2 (1:300;
cat. no. ab62352; Abcam) primary antibodies at room temperature for
1 h. Next, the slides were incubated with the goat anti-mouse IgG
H&L (Alexa Fluor 488) (1:1,000; cat. no. ab150113; Abcam) and
goat anti-rabbit IgG H&L (Alexa Fluor 647) (1:1,000; cat. no.
ab150079; Abcam) fluorescein-conjugated secondary antibodies. The
fluorescence signals were detected using an inverted fluorescence
microscope at a magnification of ×200.
Tube formation assay
HUVECs were divided into five groups: Control, NaHS
treated, NaHS treated + siRNA-NC, NaHS treated + siHIF-1α and VEGF
treated. Matrigel (BD Biosciences; Becton, Dickinson and Company,
San Jose, CA, USA) was diluted with FBS-free and growth factor-free
M200 medium at a ratio of 1:1. Then, 50 µl of the mixture
was placed into 96-well plates and incubated at 37°C for 30 min.
Subsequently, a cell suspension containing 1×104 HUVECs
were added into the 96-well plates coated with Matrigel. Next, 50
ng/ml of recombinant human VEGF (Gibco; Thermo Fisher Scientific,
Inc.) was added to the medium as the positive control group (the
VEGF group). Following culturing for 12 h, tube formation was
observed under a light microscope at a magnification ×100 and total
tube area was evaluated using ImageJ software (v.1.42).
Wound scratch assay
HUVECs were divided into six groups: Control, NaHS
treated, siNrf2, NaHS treated + siNrf2, siHIF-1α, NaHS treated +
siHIF-1α. HVSMCs were divided into four groups: Control, 50
µmol/l NaHS treated, 100 µmol/l NaHS treated and 200
µmol/l NaHS treated. A cell suspension containing
2×105 HUVECs or 2×105 HVSMCs was added into
6-well plates and cultured for 12 h. Subsequently, the medium was
replaced with fresh medium containing 2% FBS and 1 µg/ml
mitomycin C to suppress cell proliferation for 12 h. Wounds were
made to the cell monolayer using 10-µl pipette tips, and the
detached cells were removed following washing with PBS heated to
37°C. The cells were further cultured, and wound closure area was
monitored for 8 h. The migrative rate was evaluated using ImageJ
software (v.1.42).
Statistical analysis
The data in the present study are expressed as the
mean ± standard deviation. Differences among multiple groups were
evaluated using one-way analysis of variance followed by the
Tukey's post hoc test and differences between two groups were
evaluated using the Student's t-test using the statistical software
SPSS v.19.0 (IBM, Corp., Armonk, NY, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
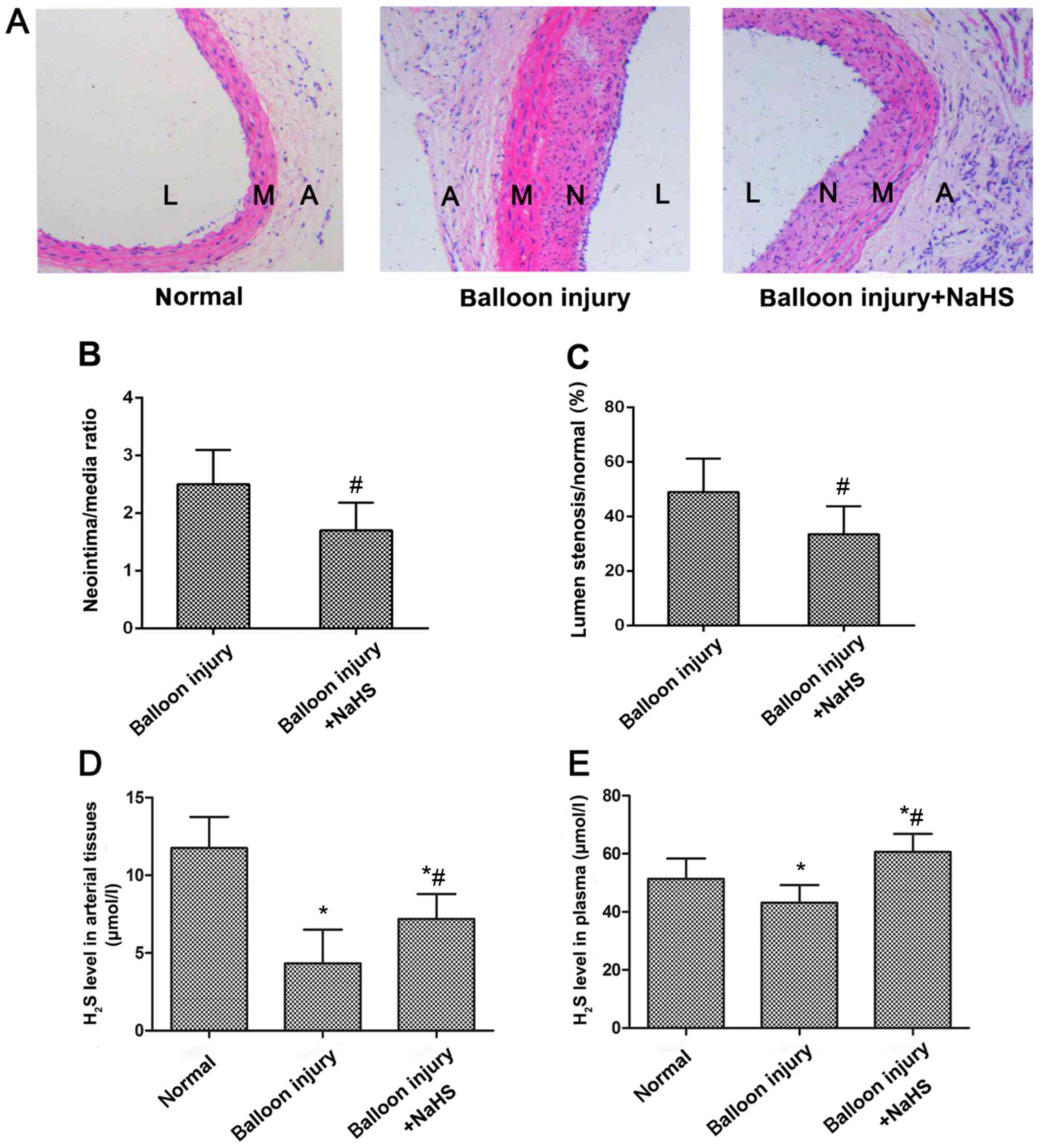
Increased H2S production
prevents neointimal hyperplasia in SD rats with carotid artery
restenosis
A restenosis model was established to explore
whether H2S was able to inhibit carotid artery
restenosis. As shown in Fig. 1A,
neointimal hyperplasia was evident in the carotid arteries of the
model group rats following balloon catheter injury. However,
neointimal hyperplasia was reduced following NaHS treatment
compared with the model group, as demonstrated by the significant
reduction in the luminal stenosis ratio, and the ratio between the
neointima and media areas (Fig. 1B
and C). Measurement of H2S production in the
arterial tissues and the plasma indicated that restenosis
significantly reduced H2S expression levels compared
with the sham control group (Fig. 1D
and E). However, following treatment with NaHS for 4 weeks,
H2S levels in the arterial tissues and the plasma in the
treatment group were significantly increased compared with the
model group.
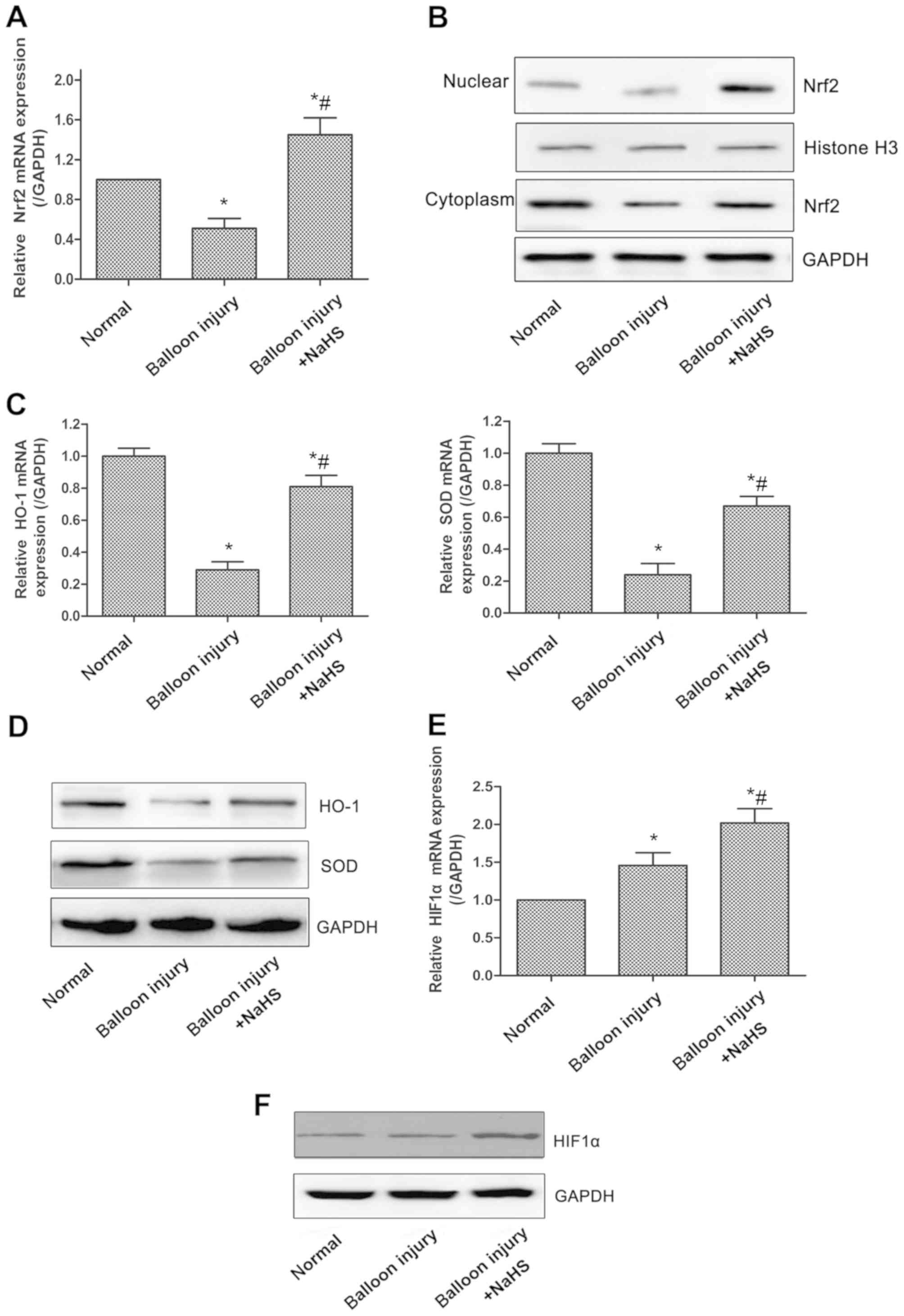
Increased H2S production
enhances antioxidative effects through Nrf2/HIF-1α signaling
pathway activation in vivo
Oxidative stress is associated with the progression
of restenosis following percutaneous coronary intervention
(27), and the Nrf2 signaling
system has been regarded as perhaps the most important cellular
defense against oxidative stress (28). Initially, the mRNA and protein
expression levels of Nrf2 were examined, and it was revealed that
the total mRNA expression of Nrf2 was significantly reduced in the
model group compared with the sham group, whereas compared with the
model group, significantly increased expression levels of Nrf2 were
observed in rats treated with NaHS (Fig. 2A). Nrf2 expression in the
nucleoprotein and plasmocin of the arterial tissues demonstrated
similar changes (Fig. 2B). The
expression levels of HO-1 and the antioxidative enzyme superoxide
dismutase (SOD), two of the downstream genes of the Nrf2 signaling
pathway, were also significantly decreased in the model group,
whereas NaHS significantly upregulated HO-1 and SOD expression
(Fig. 2C and D). Furthermore,
HIF-1α appeared to be involved in the oxidative stress response, as
compared with the model group, the expression of HIF-1α was
significantly increased in the carotid artery tissues of rats in
the model + NaHS group, whereas the expression of HIF-1α was
increased slightly in the model group compared with the sham group
(Fig. 2E and F).
H2S donor promotes the
activation of the Nrf2/HIF-1α signaling pathway in HUVECs
Oxidative stress leads to endothelial dysfunctions,
which are a key factor for intimal hyperplasia (29). In order to assess the effects and
its mechanisms of H2S on oxidative stress, the
expression of Nrf2 and HO-1 was detected in HUVECs following
treatment with different dosage of NaHS (0, 50, 100 or 200
µmol/l). As shown in Fig.
3A, the transcriptional levels of total Nrf2 and HO-1 increased
in a dose-dependent manner. The nuclear protein level of Nrf2 and
total protein level of HO-1 also demonstrated an increasing trend,
while the cytoplasmic protein level of Nrf2 reduced with increased
concentrations of NaHS (Fig. 3B).
HIF-1α mRNA levels significantly increased in a dose-dependent
manner, which was also confirmed at the protein level (Fig. 3C and D). These data indicated that
the increased H2S production, induced by NaHS, promoted
activation of the Nrf2/HIF-1α signaling pathway in HUVECs. The
comparison among groups demonstrated significant differences
between the NaHS and control groups, whereas no significant
differences were observed for Nrf2 mRNA expression and HIF-1α
protein expression between the 100 and 200 µmol/l NaHS
groups. Thus, HUVECs was treated with 100 µmol/l NaHS in the
subsequent experiments.
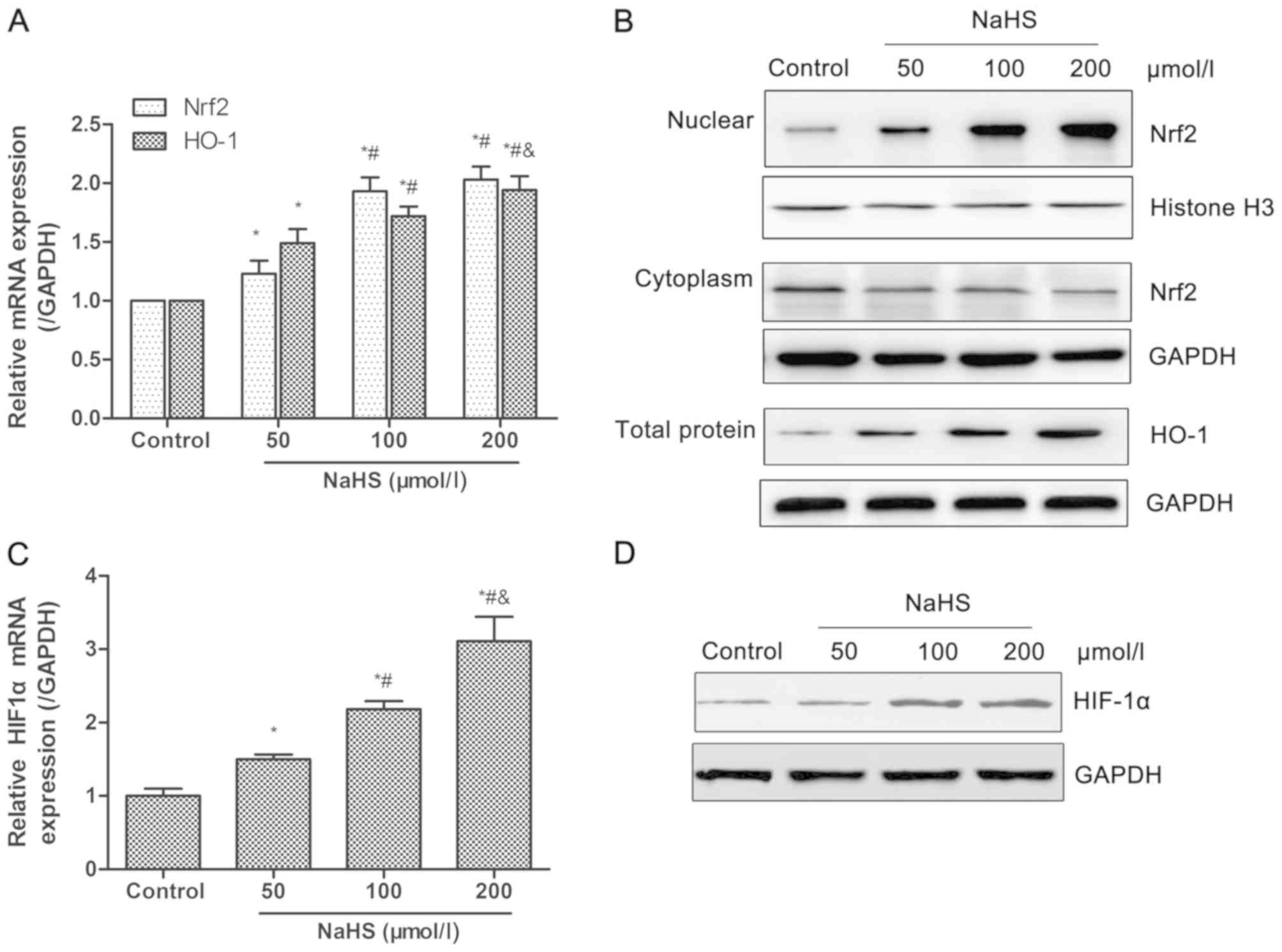 | Figure 3Effects of NaHS on the activation of
the Nrf2/HIF-1α signaling pathway in HUVECs. HUVECs were treated
with different dosage of NaHS (0, 50, 100, 200 µmol/l) for
48 h. Then, the transcriptional and protein levels of Nrf2, HO-1
and HIF-1α were detected by RT-qPCR and western blot analysis,
respectively. (A and B) Treatment with NaHS increased (A) mRNA
levels, and (B) nuclear accumulation of Nrf2 and expression of
HO-1. Following treatment with NaHS, (C) mRNA and (D) protein
expression levels of HIF-1α increased. Representative data from
three independent experiments are presented. *P<0.05
vs. the control group; #P<0.05 vs. the 50
µmol/l NaHS treated group; &P<0.05 vs. the
100 µmol/l NaHS treated group. NaHS, sodium hydrosulfide;
Nrf2, nuclear factor-E2-related factor 2; HIF-1α, hypoxia-inducible
factor-1α; HO-1, heme oxygenase-1; GAPDH, glyceraldehyde-phosphate
dehydrogenase. |
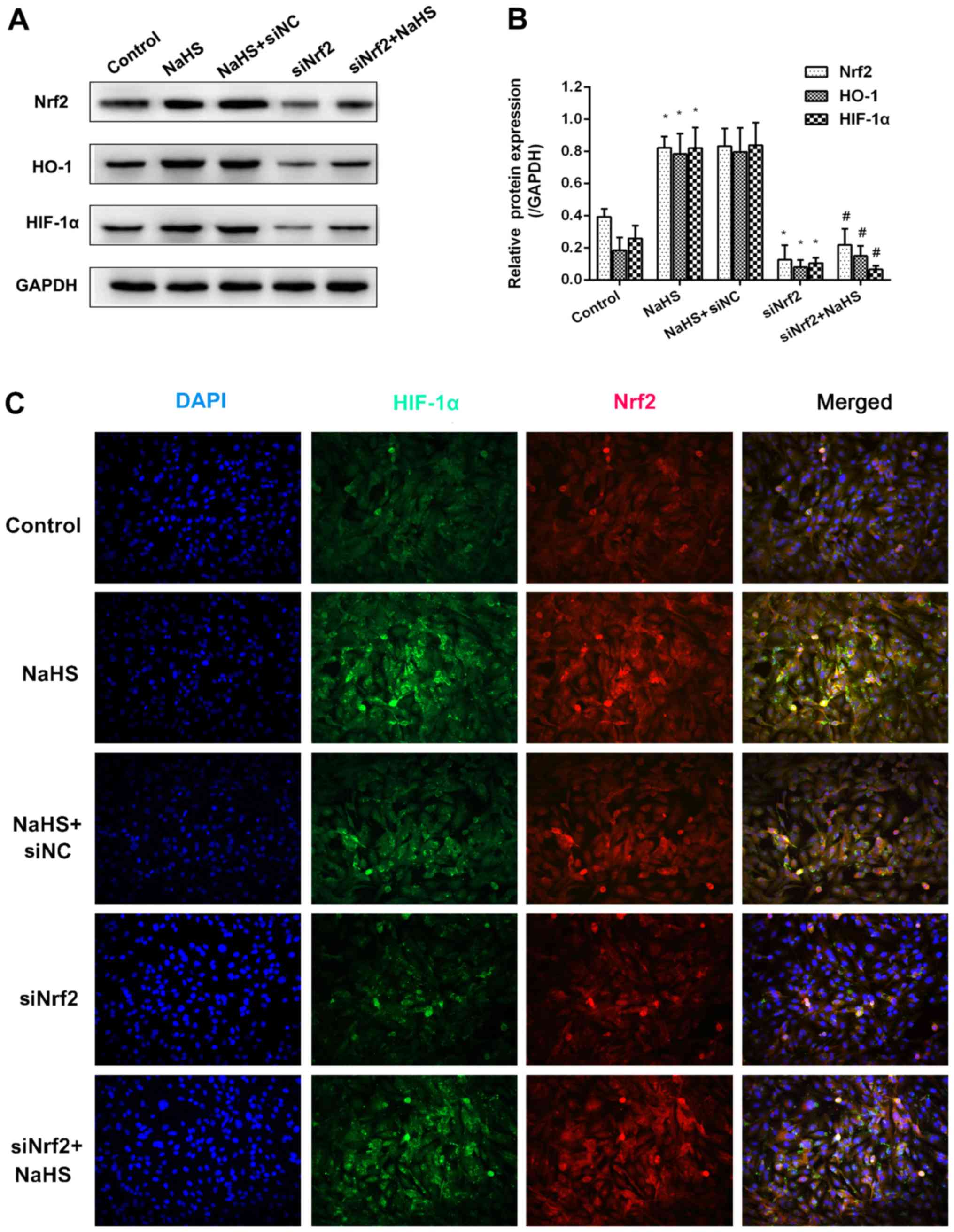
H2S donor-induced HIF-1α
expression is dependent on the Nrf2 signaling pathway in
HUVECs
In order to explore the association between HIF-1α
and Nrf2, Nrf2 expression was inhibited using siNrf2. As shown in
Fig. 4A and B, NaHS treatment
significantly increased Nrf2 mRNA expression and markedly increased
Nrf2 protein expression compared with the control group. Similarly,
the mRNA and protein expression levels of downstream HO-1 and
HIF-1α increased following treatment with NaHS. The use of siNrf2
resulted in a significant reduction in all three mRNA levels
compared with the control group. However, the effects of NaHS on
the expression of HO-1 and HIF-1α were not observed when Nrf2
expression was suppressed using siNrf2 when compared with the
siNrf2 alone group. Furthermore, the results of immunofluorescence
revealed that the fluorescence signal of Nrf2 and HIF-1α evidently
increased in the NaHS group, whereas the fluorescence signals of
the two were markedly attenuated in the siNrf2+NaHS group compared
with the NaHS group (Fig. 4C).
These suggested that the H2S-induced HIF-1α accumulation
was dependent on the expression of Nrf2.
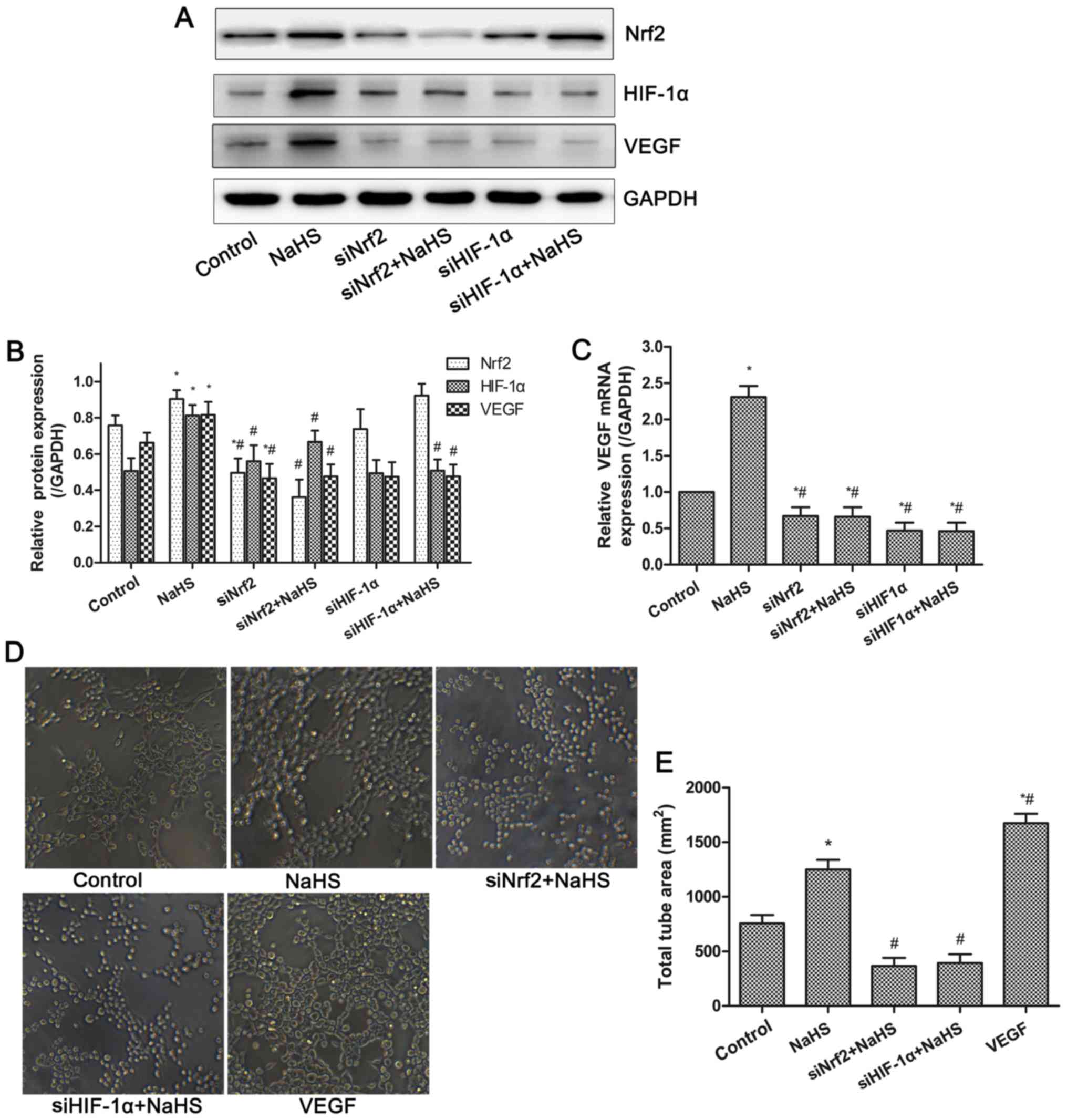
H2S donor increases VEGF
expression and tube formation via Nrf2/HIF-1α activation in
HUVECs
VEGF is a well-known pro-angiogenic factor. The mRNA
and protein expressions of VEGF in HUVECs were examined by RT-qPCR
and western blot analysis. As shown in Fig. 5, the mRNA and protein expression
levels of VEGF were significantly increased in the NaHS group.
However, inhibition of Nrf2 or HIF-1α expression significantly
suppressed the increased VEGF expression induced by NaHS treatment.
Thus, NaHS-induced VEGF expression was dependent on the activation
of the Nrf2/HIF-1α signaling pathway in HUVECs. Furthermore, the
tube formation assay demonstrated that compared with the control
group, NaHS or VEGF treatment significantly promoted tube formation
of HUVECs, whereas this effect was inhibited following the
suppression of Nrf2 and HIF-1α expression. These results indicated
that NaHS treatment increased VEGF expression and tube formation
via activation of Nrf2/HIF-1α in HUVECs.
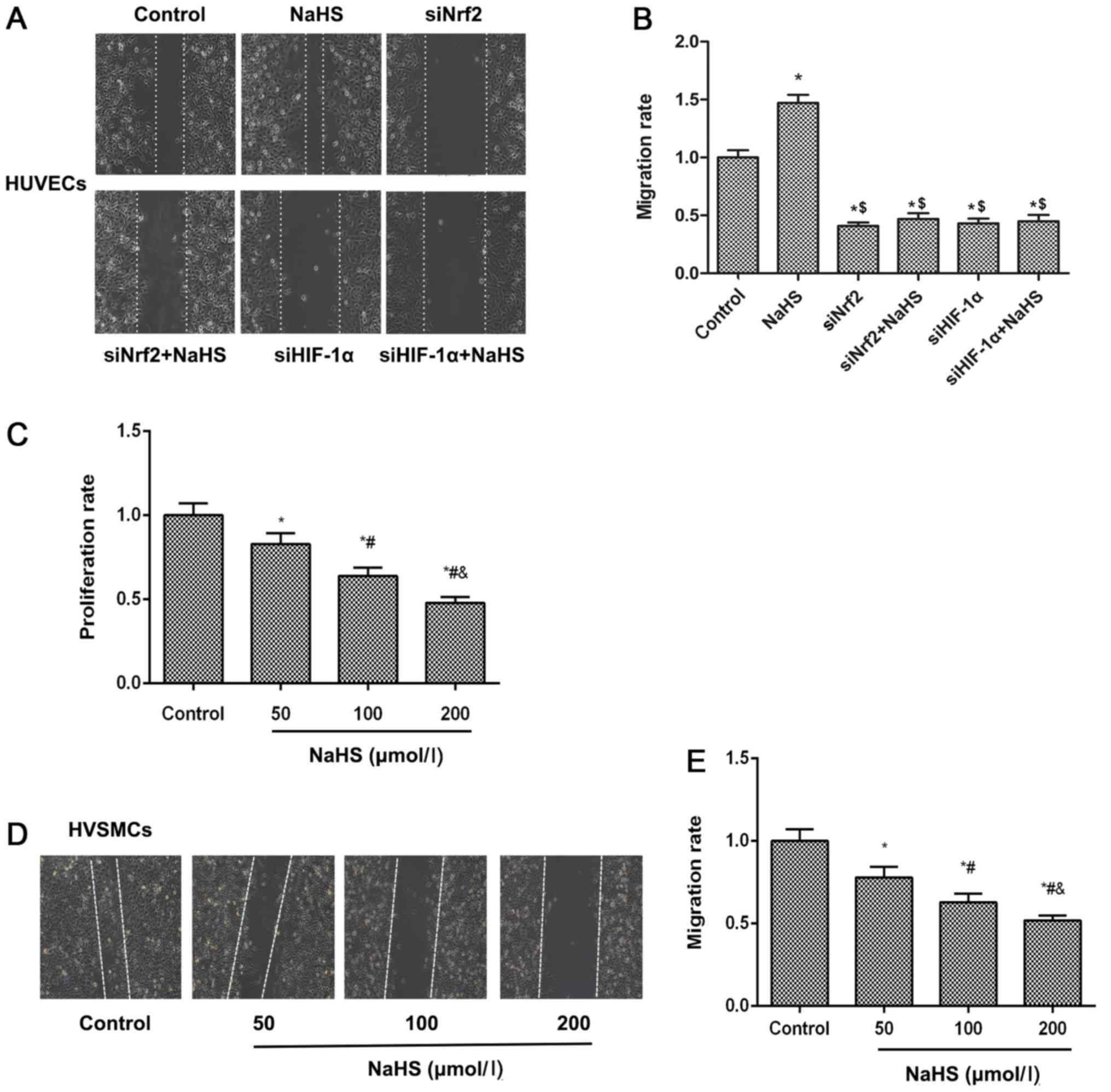
H2S donor induces cell
migration via Nrf2/HIF-1α activation in HUVECs but inhibits HVSMC
proliferation and migration
Cell migration of vascular endothelial cells to
impaired region is an essential process for the repair of damaged
blood vessels. The cell migratory abilities of HUVECs and HVSMCs
were examined using a wound scratch migration assay. The results
demonstrated that NaHS treatment accelerated cell migration of
HUVECs, meanwhile cell migration ability was inhibited when Nrf2 or
HIF-1α expression levels were suppressed by siNrf2 and siHIF-1α,
respectively (Fig. 6A and B).
These results suggested that NaHS treatment induced cell migration
by activating the Nrf2/HIF-1α signaling pathway in HUVECs. For the
HVSMC proliferation assay, as shown in Fig. 6C, NaHS inhibited HVSMC
proliferation in a concentration-dependent manner. Similarly, this
phenomenon was observed on the migratory ability of HVSMCs
(Fig. 6D and E), suggesting that
NaHS treatment suppressed HVSMC migration and proliferation.
Discussion
It is well known that intimal hyperplasia induced by
endothelial dysfunction and excessive VSMC proliferation is the
central feature associated with the development of luminal stenosis
diseases. During the treatment of these diseases, endovascular
interventional therapies, including balloon catheter angioplasty,
may induce exfoliation of endothelial cells, which triggers
platelet activation and aggregation on vascular walls, leading to
endothelial dysfunction (19). In
response to these events, VSMCs switch to a proliferative
phenotype. HVSMCs migrate through the endothelium layer and
continue to grow on the inner surface of vessels, resulting in
neointimal tissue formation. Accumulating evidence indicates that
H2S diminishes the progression of intimal hyperplasia
and suppresses restenosis following balloon angioplasty (20,30,31). In present study, a balloon injury
model was used to investigate the molecular mechanisms underlying
the anti-restenosis effects of H2S. Consistent with the
hypothesis, the contents of H2S in the plasma and
arterial tissues were increased in the balloon injury model
following NaHS (a donor of H2S) treatment, whereas
intimal hyperplasia was suppressed. Previously, using a rabbit
model with atherosclerotic-like lesions, it was demonstrated that
treatment with NaHS significantly inhibited arterial restenosis by
reducing the intimal area and the intima/media ratio, accompanied
by reduced SMC proliferation and elevated SMC apoptosis in the
neointima (32). These results
are similar to those obtained in the present study, and thereby
indicate that H2S derived from exogenous NaHS is
effective in treating experimentally induced vascular stenosis.
Furthermore, with respect to the pathogenesis of
restenosis, increased ROS production has been observed to occur at
an early stage following angioplasty (33). ROS are considered to serve a role
as universal regulators in vascular biology, including in
endothelial and VSMC homeostasis, the inflammatory response,
vascular remodeling, and endothelial dysfunction (24). Increased ROS production may
trigger increased oxidative stress, resulting in vascular damage by
accelerating endothelial dysfunction and macrophage activation
(33). Oxidative stress may
aggravate endothelial dysfunction through a variety of pathways,
including oxidation of low-density lipoproteins, upregulation of
adhesion molecules and monocyte chemoattractant protein 1
chemokines, activation of matrix metalloproteinases, and
inactivation of nitric oxide. In turn, these cytokines and growth
factors stimulate VSMC proliferation and extracellular matrix
remodeling (33). In addition to
direct interaction, increased ROS or oxidative stress promotes VSMC
proliferation and migration, eventually leading to the initiation
and development of restenosis following PTA (34,35).
Within cells, a variety of physiological antioxidant
defense mechanisms have evolved to ameliorate oxidative
stress-induced damage. Activation of the Nrf2 signal pathway serves
an important role in antioxidant effects through promoting the
binding of AREs and upregulation of antioxidant genes. Nrf2, a
well-known Cap-N-Collar transcription factor, is essential for
ARE-mediated transcription, including that of NADPH quinone
oxidoreductase 1 (NQO1) and HO-1 (36). Previous studies have indicated
that Nrf2 protects against tissue fibrosis, diabetic nephropathy,
and non-alcoholic fatty liver, presumably through an enhancement of
cellular antioxidant capacity, such as via increased expression of
NQO1 and HO-1 (37-39). In vitro experiments have
indicated that the transcriptional activity and nuclear
localization of Nrf2 are inhibited in various ROS-mediated cell
damage models involving HUVECs and human coronary artery
endothelial cells, accompanied by increases in cell apoptosis
(40). Furthermore, several
studies have revealed that overexpression of Nrf2 prevents
neointimal hyperplasia by inhibiting the proliferation of VSMCs
following vascular injury through HO-1-dependent antioxidant and
anti-inflammatory effects (41,42). The results obtained in present
study indicate that the mRNA levels of Nrf2 and its nuclear
accumulation are markedly decreased in rats with restenosis, and
that the mRNA and protein levels of HO-1 and SOD are also reduced.
Increasing evidence has indicated that activation of the Nrf2
signal pathway suppresses neointimal hyperplasia by increasing the
expression of antioxidant genes, including HO-1 (43,44). Other studies have demonstrated
that Nrf2 may be involved in the antioxidant activity of
H2S during H2S-mediated cardioprotection
(22). As one of the well-known
target genes stimulated by Nrf2, the by-products of HO-1 have been
reported to inhibit proliferation and induce apoptosis of VSMCs
(45). In the present study, it
was revealed NaHS treatment significantly prevented neointimal
hyperplasia in rats with restenosis through increasing
H2S levels and the nuclear accumulation of Nrf2 protein.
Furthermore, on the basis of its effects on HUVEC migration through
increasing Nrf2 levels, NaHS treatment is also effective at
inhibiting the proliferation and migration of human VSMCs. A
previous in vitro experiment reported that exogenous
H2S inhibits VSMC proliferation in a hyperglycemic state
via modulation of mitochondrial fusion-fission (46).
ROS production is involved in the regulation of VEGF
and HIF-1α expression, and angiogenesis (47). Abnormal activation of the HIF-1α
signaling pathway stimulates the upregulation of VEGF expression,
which promotes angiogenesis (48). The results of the current study
revealed that NaHS treatment increased the expression of HIF-1α and
VEGF, whereas inhibition of Nrf2 or HIF-1α expression significantly
suppressed VEGF expression, and decreased the tube formation
ability of HUVECs. These results suggest that the Nrf2/HIF-1α
signaling pathway is involved in NaHS-induced VEGF expression. In a
follicle-stimulating hormone (FSH)-induced ovarian epithelial
cancer cell (OEC) model, it was previously reported that FSH
induces ROS production and activation of Nrf2 signaling, whereas
the elimination of ROS or knockdown of Nrf2 blocks FSH-induced VEGF
expression (49). In addition,
the knockdown of Nrf2 has been revealed to impair HIF-1α signaling
activation, indicating that ROS and the aberrant expression of
Nrf2/HIF-1α serve important roles in FSH-induced angiogenesis in
OECs (49). The findings of a
further study indicated that inhibition of Nrf2 expression
restrains angiogenesis in colon cancer through suppression of the
HIF-1α signaling pathway (50),
indicating that the HIF-1α signaling pathway is regulated by Nrf2.
Additionally, it has been reported that H2S induces
angiogenesis in endothelial cells and promotes damage repair
(18). The proliferation and
migration of endothelial cells also contribute to damage repair.
The current data also indicated that NaHS treatment promoted the
tube formation and migration of HUVECs in vitro, whereas the
knockdown of Nrf2 or HIF-1α attenuated NaHS-induced tube formation
and migration in HUVECs.
In summary, the findings of the current study
indicate that NaHS-derived H2S production prevented
neointimal hyperplasia in a restenosis rat model, and promoted tube
formation and migration of HUVECs to accelerate damage repair.
These results also demonstrate that these protective effects may be
mediated via the activation of the Nrf2/HIF-1α signaling pathway.
Furthermore, it has been suggested that the regulation of
H2S production through the administration of
H2S donors or targeting of Nrf2/HIF-1α signaling pathway
may be potential strategies to prevent the development of
cardiovascular narrowing diseases, including restenosis.
Funding
The present study was supported by grants from the
Science Foundation of Wuhan Science and Technology Bureau (grant
no. 02.07.040555) and the Science Foundation of Wuhan Union
Hospital (grant no. 02.03.2017-335).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
KL and WW conceived of the study and drafted the
manuscript together. AX designed the study and performed the
statistical analysis. YC participated in the study design and
constructed the balloon dilatation restenosis model. XC and YL
performed the experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experiments involving animal treatment were
performed in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory Animals, and the protocol
was approved by the Ethics Committee of Union Hospital, affiliated
to the Huazhong University of Science and Technology (approval no.
TJ-A20161216).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Holmes DR Jr, Vlietstra RE, Smith HC,
Vetrovec GW, Kent KM, Cowley MJ, Faxon DP, Gruentzig AR, Kelsey SF,
Detre KM, et al: Restenosis after percutaneous transluminal
coronary angioplasty (PTCA): A report from the PTCA Registry of the
National Heart, Lung, and Blood Institute. Am J Cardiol.
53:77C–81C. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RJ, Gunn J, Ahsan A, Fishbein MC,
Bowes RJ, Oakley D, Wales C, Steffen W, Campbell S and Nita H: Use
of therapeutic ultrasound in percutaneous coronary angioplasty.
Experimental in vitro studies and initial clinical experience.
Circulation. 89:1587–1592. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klein AJ and Ross CB: Endovascular
treatment of lower extremity peripheral arterial disease. Trends
Cardiovasc Med. 26:495–512. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller AJ, Takahashi EA, Harmsen WS, Mara
KC and Misra S: Treatment of Superficial Femoral Artery Restenosis.
J Vasc Interv Radiol. 28:1681–1686. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alazzaz A, Thornton J, Aletich VA, Debrun
GM, Ausman JI and Charbel F: Intracranial percutaneous transluminal
angioplasty for arteriosclerotic stenosis. Arch Neurol.
57:1625–1630. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Christidou FP, Kalpakidis VI, Iatrou KD,
Zervidis IA, Bamichas GI, Gionanlis LC, Natse TA and Sombolos KJ:
Percutaneous trans-luminal angioplasty (PTA) and venous stenting in
hemodialysis patients with vascular access-related venous stenosis
or occlusion. Radiography. 12:127–133. 2006. View Article : Google Scholar
|
|
7
|
Schmidt A, Ulrich M, Winkler B, Klaeffling
C, Bausback Y, Bräunlich S, Botsios S, Kruse HJ, Varcoe RL, Kum S,
et al: Angiographic patency and clinical outcome after
balloon-angioplasty for extensive infrapopliteal arterial disease.
Catheter Cardiovasc Interv. 76:1047–1054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meng Z, Gao P, Chen L, Peng J, Huang J, Wu
M, Chen K and Zhou Z: Artificial zinc-finger transcription factor
of A20 suppresses restenosis in Sprague Dawley rats after carotid
injury via the PPARα pathway. Mol Ther Nucleic Acids. 8:123–131.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Losordo DW, Isner JM and Diaz-Sandoval LJ:
Endothelial recovery: The next target in restenosis prevention.
Circulation. 107:2635–2637. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kugiyama K, Kerns SA, Morrisett JD,
Roberts R and Henry PD: Impairment of endothelium-dependent
arterial relaxation by lysolecithin in modified low-density
lipoproteins. Nature. 344:160–162. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dussault S, Dhahri W, Desjarlais M,
Mathieu R and Rivard A: Elsibucol inhibits atherosclerosis
following arterial injury: Multifunctional effects on cholesterol
levels, oxidative stress and inflammation. Atherosclerosis.
237:194–199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kochiadakis GE, Arfanakis DA, Marketou ME,
Skalidis EI, Igoumenidis NE, Nikitovic D, Giaouzaki A, Chlouverakis
G and Vardas PE: Oxidative stress changes after stent implantation:
A randomized comparative study of sirolimus-eluting and bare metal
stents. Int J Cardiol. 142:33–37. 2010. View Article : Google Scholar
|
|
13
|
Misra P, Reddy PC, Shukla D, Caldito GC,
Yerra L and Aw TY: In-stent stenosis: Potential role of increased
oxidative stress and glutathione-linked detoxification mechanisms.
Angiology. 59:469–474. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tardif JC, Grégoire J and L'Allier PL:
Prevention of restenosis with antioxidants: Mechanisms and
implications. Am J Cardiovasc Drugs. 2:323–334. 2002. View Article : Google Scholar
|
|
15
|
Strauss BH, Chisholm RJ, Keeley FW,
Gotlieb AI, Logan RA and Armstrong PW: Extracellular matrix
remodeling after balloon angioplasty injury in a rabbit model of
restenosis. Circ Res. 75:650–658. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kamoun P: Endogenous production of
hydrogen sulfide in mammals. Amino Acids. 26:243–254. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheung SH and Lau JYW: Hydrogen sulfide
mediates athero-protection against oxidative stress via
S-sulfhydration. PLoS One. 13:e01941762018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Altaany Z, Moccia F, Munaron L, Mancardi D
and Wang R: Hydrogen sulfide and endothelial dysfunction:
Relationship with nitric oxide. Curr Med Chem. 21:3646–3661. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma B, Liang G, Zhang F, Chen Y and Zhang
H: Effect of hydrogen sulfide on restenosis of peripheral arteries
after angioplasty. Mol Med Rep. 5:1497–1502. 2012.PubMed/NCBI
|
|
20
|
Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang
Y, Du J and Tang C: Endogenous hydrogen sulfide regulation of
myocardial injury induced by isoproterenol. Biochem Biophys Res
Commun. 318:756–763. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jian Z, Li K, Liu L, Zhang Y, Zhou Z, Li C
and Gao T: Heme oxygenase-1 protects human melanocytes from
H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest
Dermatol. 131:1420–1427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Calvert JW, Jha S, Gundewar S, Elrod JW,
Ramachandran A, Pattillo CB, Kevil CG and Lefer DJ: Hydrogen
sulfide mediates cardioprotection through Nrf2 signaling. Circ Res.
105:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schwacha MG, Nickel E and Daniel T: Burn
injury-induced alterations in wound inflammation and healing are
associated with suppressed hypoxia inducible factor-1alpha
expression. Mol Med. 14:628–633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patel VI, Daniel S, Longo CR, Shrikhande
GV, Scali ST, Czismadia E, Groft CM, Shukri T, Motley-Dore C,
Ramsey HE, et al: A20, a modulator of smooth muscle cell
proliferation and apoptosis, prevents and induces regression of
neointimal hyperplasia. FASEB J. 20:1418–1430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Noro T, Takehara N, Sumitomo K, Takeuchi
T, Ishii Y, Kato J, Kawabe J and Hasebe N: Initial reduction of
oxidative stress by angiotensin receptor blocker contributes long
term outcomes after percutaneous coronary intervention. Am J
Cardiovasc Dis. 4:159–167. 2014.
|
|
28
|
Zhang H, Davies KJA and Forman HJ:
Oxidative stress response and Nrf2 signaling in aging. Free Radic
Biol Med. 88(Pt B): 314–336. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang F, Drummond GR and Dusting GJ:
Suppression of oxidative stress in the endothelium and vascular
wall. Endothelium. 11:79–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin Z, Chan H, Ning J, Lu K and Ma D: The
role of hydrogen sulfide in pathologies of the vital organs and its
clinical application. J Physiol Pharmacol. 66:169–179.
2015.PubMed/NCBI
|
|
31
|
Baskar R, Sparatore A, Del Soldato P and
Moore PK: Effect of S-diclofenac, a novel hydrogen sulfide
releasing derivative inhibit rat vascular smooth muscle cell
proliferation. Eur J Pharmacol. 594:1–8. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma B, Liang G, Zhang F, Chen Y and Zhang
H: Effect of hydrogen sulfide on restenosis of peripheral arteries
after angioplasty. Mol Med Rep. 5:1497–1502. 2012.PubMed/NCBI
|
|
33
|
Chen Q, Wang Q, Zhu J, Xiao Q and Zhang L:
Reactive oxygen species: Key regulators in vascular health and
diseases. Br J Pharmacol. 175:1279–1292. 2018. View Article : Google Scholar
|
|
34
|
Raaz U, Toh R, Maegdefessel L, Adam M,
Nakagami F, Emrich FC, Spin JM and Tsao PS: Hemodynamic regulation
of reactive oxygen species: Implications for vascular diseases.
Antioxid Redox Signal. 20:914–928. 2014. View Article : Google Scholar :
|
|
35
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oh CJ, Kim JY, Choi YK, Kim HJ, Jeong JY,
Bae KH, Park KG and Lee IK: Dimethylfumarate attenuates renal
fibrosis via NF-E2-related factor 2-mediated inhibition of
transforming growth factor-β/Smad signaling. PLoS One.
7:e458702012. View Article : Google Scholar
|
|
37
|
Cho HY, Reddy SP, Yamamoto M and
Kleeberger SR: The transcription factor NRF2 protects against
pulmonary fibrosis. FASEB J. 18:1258–1260. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zheng H, Whitman SA, Wu W, Wondrak GT,
Wong PK, Fang D and Zhang DD: Therapeutic potential of Nrf2
activators in streptozotocin-induced diabetic nephropathy.
Diabetes. 60:3055–3066. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu W, Shao L, Zhou C, Wang H and Guo J:
Upregulation of Nrf2 expression in non-alcoholic fatty liver and
steatohepatitis. Hepatogastroenterology. 58:2077–2080. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen B, Lu Y, Chen Y and Cheng J: The role
of Nrf2 in oxidative stress-induced endothelial injuries. J
Endocrinol. 225:R83–R99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JY, Cho HJ, Sir JJ, Kim BK, Hur J,
Youn SW, Yang HM, Jun SI, Park KW, Hwang SJ, et al: Sulfasalazine
induces haem oxygenase-1 via ROS-dependent Nrf2 signalling, leading
to control of neointimal hyperplasia. Cardiovasc Res. 82:550–560.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee HJ, Seo M and Lee EJ: Salvianolic acid
B inhibits atherogenesis of vascular cells through induction of
Nrf2-dependent heme oxygenase-1. Curr Med Chem. 21:3095–3106. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ashino T, Yamamoto M, Yoshida T and
Numazawa S: Redox-sensitive transcription factor Nrf2 regulates
vascular smooth muscle cell migration and neointimal hyperplasia.
Arterioscler Thromb Vasc Biol. 33:760–768. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oh CJ, Park S, Kim JY, Kim HJ, Jeoung NH,
Choi YK, Go Y, Park KG and Lee IK: Dimethylfumarate attenuates
restenosis after acute vascular injury by cell-specific and
Nrf2-dependent mechanisms. Redox Biol. 2:855–864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Moraes JA, Barcellos-de-Souza P, Rodrigues
G, Nascimento-Silva V, Silva SV, Assreuy J, Arruda MA and
Barja-Fidalgo C: Heme modulates smooth muscle cell proliferation
and migration via NADPH oxidase: A counter-regulatory role for heme
oxygenase system. Atherosclerosis. 224:394–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun A, Wang Y, Liu J, Yu X, Sun Y, Yang F,
Dong S, Wu J, Zhao Y, Xu C, et al: Exogenous H2S
modulates mitochondrial fusion-fission to inhibit vascular smooth
muscle cell proliferation in a hyperglycemic state. Cell Biosci.
6:362016. View Article : Google Scholar
|
|
47
|
Song S, Xiao X, Guo D, Mo L, Bu C, Ye W,
Den Q, Liu S and Yang X: Protective effects of Paeoniflorin against
AOPP-induced oxidative injury in HUVECs by blocking the
ROS-HIF-1α/VEGF pathway. Phytomedicine. 34:115–126. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Y, Liu J, Wang S, Luo X, Li Y, Lv Z,
Zhu J, Lin J, Ding L and Ye Q: The DEK oncogene activates VEGF
expression and promotes tumor angiogenesis and growth in
HIF-1α-dependent and -independent manners. Oncotarget.
7:23740–23756. 2016.PubMed/NCBI
|
|
49
|
Zhang Z, Wang Q, Ma J, Yi X, Zhu Y, Xi X,
Feng Y and Jin Z: Reactive oxygen species regulate FSH-induced
expression of vascular endothelial growth factor via Nrf2 and HIF1α
signaling in human epithelial ovarian cancer. Oncol Rep.
29:1429–1434. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D,
Lee YM, Ku SK, Jung Y and Kwak MK: NRF2 blockade suppresses colon
tumor angiogenesis by inhibiting hypoxia-induced activation of
HIF-1α. Cancer Res. 71:2260–2275. 2011. View Article : Google Scholar : PubMed/NCBI
|