Introduction
Acute pulmonary embolism (APE) has been frequently
reported in critical care and emergency medicine departments and is
associated with a high mortality rate. Based on a patient’s
hemodynamic status and clinical characteristics, APE may be
classified as massive, submassive or non-massive (1). Massive pulmonary embolism is
life-threatening and is characterized by cardiogenic shock and
cardiac arrest (CA) upon presentation (2). The mortality of patients with APE
presenting with CA may be as high as 65% (3). Survivors of such acute events may be
more prone to heart failure (4).
Myocardial cell apoptosis is one of the underlying key mechanisms
associated with heart failure (5). Numerous studies have suggested that
the classic angiotensin (Ang)-converting enzyme (ACE)/Ang II/Ang II
type 1 receptor axis of the renin-angiotensin system (RAS)
regulates hemodynamics, as well as maintains ventricular
remodelling (6) and myocardial
cell apoptosis (7). By contrast,
a newly discovered ACE2/Ang(1-7)/Mas axis within the RAS has been
reported to prevent myocardial cell apoptosis caused by myocardial
infarction or ischemic reperfusion (8). A recent study by our group reported
a post-resuscitation imbalance in the serum ACE2/ACE axis that was
accompanied by regressive cardiac function, including lower left
heart stroke index, left cardiac work index, left ventricular
stroke work index, right cardiac output, right heart stroke volume,
right cardiac index and right heart stroke index, in a porcine
model of APE-CA (9). However, the
role of this imbalance in cardiac tissues during massive APE-CA has
remained to be fully elucidated. Therefore, the present study
further investigated the association between the imbalance of the
cardiac ACE2/ACE axis and myocardial apoptosis in a porcine APE-CA
model.
The previous study by our group indicated that an
ACE inhibitor, captopril, lowered the post-resuscitation mean right
ventricular pressure and pulmonary vascular resistance through
activating the serum ACE2/Ang(1-7)/Mas axis in APE (9). the ability of captopril to inhibit
myocardial cell apoptosis during myocardial ischemia/reperfusion
injury was previously reported (10). Studies have also suggested that
treatment with ACE2, Ang-(1-7)
and ACE inhibitors, or with angiotensin receptor blockers,
suppresses activation of the extracellular signal-regulated kinase
(ERK)1/2 pathway, which is known to have a role in myocardial cell
apoptosis (11,12). Therefore, the present study tested
the hypothesis that the ACE inhibitor captopril attenuates
myocardial cell apoptosis induced by massive APE.
Materials and methods
Ethical approval
The present study used 29 landrace pigs (age, 3
months; weight, 28±2 kg, 15 boars and 14 gilts) provided by the
Beijing Experimental Animal Center (Beijing, China; license no.
SCXK 11-00-002), as described previously (9). All animals were fed standard chow
and had free access to water and housed in their own cages with the
size of 80×80×90 cm in our facility. Room temperature was adjusted
to 26°C, and humidity was 60%. Appropriate care was taken to
minimize animal suffering. The Committee on the Ethics of Animal
Experiments of Capital Medical University approved the study
procedures (permit no. 2010-D-013). Experimental protocols were
designed in strict compliance with the Animal Care Guidelines of
the Institutional Animal Care and Use Committee of Capital Medical
University (Beijing, China) as well as the Utstein-style guidelines
(13).
Animal preparation
All animals received an intramuscular injection of
midazolam (0.2 mg/kg) followed by ear vein injection of propofol
(1.0 mg/kg). Pentobarbital (3%) was intravenously infused (8
mg/kg/h) to maintain anesthesia. Subsequently, all animals were
endotracheally intubated using a ventilator (Evita 4; Draeger
Medical, Lübeck, Germany). The ventilation mode was synchronized to
a tidal volume of 8 ml/kg and a respiratory frequency of 12-20
breaths per minute on room air. An inline infrared cacographic
(Respirometric Inc., Murrysville, PA, USA) was maintained between
30 and 40 mmHg to monitor the end-tidal pressure of CO2.
An arterial catheter was inserted into the aortic arch from the
left femoral artery for continuous measurement of the aortic
pressure. An A7-Fr Swan-Ganz catheter (Edwards Life Sciences,
Irvine, CA, USA) was placed into the pulmonary artery from the
right external jugular vein to monitor the mean pulmonary artery
pressure. Electrocardiograms and hemodynamics were also monitored
(M8001A; Philips Medizin Systeme GmbH, Boeblingen, Germany). A
triple lumen central venous catheter was placed in the left femoral
vein to monitor the central venous pressure. During the procedures,
normal saline was infused through the right femoral vein at 8
ml/kg/h to compensate for fluid loss, as well as maintain a steady
central venous pressure (5-12 mmHg). A large-bore catheter
(internal diameter, 1.0 cm) was inserted from the left external
jugular vein into the opening of the pulmonary artery by
computerized tomography guidance for infusion of blood clots
(9). All wounds were partially
sutured.
Experimental protocol
Blood (~100 ml) was collected from the left femoral
vein and allowed to self-coagulate at room temperature (2-3 h) as
previously described (8). The
clots (10-15 ml) were cut into pieces (1.5×1×1 cm) and suspended in
normal saline in a large catheter tip syringe (1.0-cm internal
diameter, 50 ml). To obtain a model of APE-induced CA, the
suspension of blood clots was injected through the left external
jugular vein over ~2 min until a mean arterial pressure (MAP) of
<30 mmHg was reached (14).
The model of massive APE was prepared successfully and diagnosed by
chest-enhanced CT scanning. Subsequently, urokinase (15,000 U/kg,
Nanjing Nanda Pharmaceutical Co., Ltd. Nanjing, China) was infused
into the pulmonary artery using a Swan-Ganz catheter, and
cardiopulmonary resuscitation (CPR) was initiated immediately
according to the American Heart Association guidelines for CPR and
emergency cardiovascular care from 2010 (15). Return of the spontaneous
circulation (ROSC) was verified by a systolic blood pressure of
>50 mmHg maintained for 10 consecutive minutes (16). Animals were considered dead with
the absence of heart beat/breathing and eye reflex, and presence of
rigor mortis if ROSC was not restored within 30 min.
A total of 29 pigs were randomly assigned to three
groups. Group 1 (control group; n=5) was injected with a bolus of
propofol (100 mg) followed by 10 ml potassium chloride (15%) at 6 h
after initiation of the experiment. All animals in this group died
due to ventricular fibrillation. Group 2 (APE-CA group; n=5)
received a thrombus injection until the MAP reached <30 mmHg,
followed by CPR. All animals in this group died due to
electromechanical diastolic arrest or ventricular fibrillation.
Group 3 (APE-ROSC group; n=19) received a thrombus injection until
the MAP reached <30 mmHg, followed by urokinase treatment and
cardiopulmonary resuscitation, which was performed in accordance
with the 2010 American Heart Association guidelines for
cardiopulmonary resuscitation and emergency cardiovascular care
(15). In this group, 10 animals
were successfully resuscitated and randomly assigned to two
subgroups: The ROSC-Cap group (n=5) received an intravenous
injection of captopril (22.22 mg/kg; cat. no. C8856-1G;
Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) at 30 min after
ROSC, and the ROSC-SA group (n=5) received the same volume of
normal saline (SA) at 30 min after ROSC. Subsequently, all animals
in the two groups were euthanatized using an intravenous injection
of 10 ml potassium chloride (15%) following a bolus of propofol
(3.0 mg/kg) at 6 h after ROSC. The cardiac tissue was isolated,
immediately frozen in liquid nitrogen, and stored at −80°C
(17).
Histological evaluation
Myocardial tissue samples were fixed with 10%
buffered formalin, embedded in paraffin, and stained using
hematoxylin and eosin for further evaluation of pathological
changes. Scoring of cardiac pathological changes was based on the
criteria from the study of Rezkalla et al (18). In brief, five high-power visual
fields were obtained from each slice, the ratio of the area of
inflammatory cell infiltration and necrosis to the area of the
whole visual field in each visual field was calculated. No lesion
scored 0 points, lesion area <25% scored 1 points, lesion area
25-49% scored 2 points, 50 to 75% of lesion area scored 3 points,
lesion area > 75% scored 4 points (18).
Western blot analysis
Using a radioimmunoprecipitation assay (RIPA) buffer
and protease inhibitors (Roche, Basel, Switzerland), proteins from
the left and right myocardium were extracted and isolated according
to the manufacturer’s protocol. Each tissue sample (20 mg) was
chopped into fragments and homogenized in 200 µl RIPA
buffer. Tissue lysates were centrifuged (13,000 × g, 20 min, 4°C),
and the supernatant was immediately collected and stored at −80°C
until further use. The protein concentration in the supernatants
was measured using a Bicinchoninic Acid Protein Assay kit (Pierce;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The centrifuged
proteins were mixed with the same volume of 5X SDS buffer [2.5 ml
0.5 mol/l Tris-HCl (pH 6.8), 0.5 g SDS, 0.39 g dithiothreitol, 2.5
ml glycerol and 0.025 g bromophenol blue] to obtain a mixture at a
final concentration of 3 µg/µl. The homogenates were
heated at 95°C for 5 min. Total protein (40 µg) was resolved
by 8, 10 and 15% SDS-PAGE using a standard biotinylated molecular
weight marker (Biomed, Beijing, China). The samples were
wet-transferred to a poly-vinylidene difluoride membrane (0.45
µm; EMD Millipore, Billerica, MA, USA) and stained using
Ponceau-S Stain to assess equal loading of protein. The blots were
blocked using 5% non-fat dry milk for 2 h in a Tris-buffered saline
solution with Tween-20 [TBST; 1 mol/l Tris-HCl (pH 7.5), 100 mM
NaCl and 20% Tween-20] and incubated overnight at 4°C with a
mixture of ACE2 (cat. no. SC-390851), ACE (cat. no. SC-2079), Ang
II receptor type 1 (AT1; cat. no. SC-81671), B-cell lymphoma 2
(Bcl-2)-associated X protein (Bax; cat. no. SC-6236) and Bcl-2
antibodies (1:500 dilution; cat. no. SC-492, Santa Cruz
Biotechnology, Dallas, TX, USA), Mas receptor antibody (1:200
dilution; cat. no. AAR-013, Alomone Labs, Jerusalem, Israel),
1:1,000 dilution of cleaved caspase-3 (cat. no. 9664, Cell
Signaling Technology, Inc., Danvers, MA, USA) and caspase-3
antibodies (cat. no. SC-7272, Santa Cruz Biotechnology, Inc.) and
β-actin antibody (1:1,000 dilution; cat. no. ab8226, Abcam,
Cambridge, MA, USA). After washing with TBST, the blots were
incubated with horseradish peroxidase-conjugated anti-rabbit or
anti-mouse immunoglobulin G (1:10,000; cat. no. 111-035-003;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) as
the secondary antibody for 40 min at room temperature. After
washing the blots with TBST again, the bands were visualized using
enhanced Immobilon™ Western chemiluminescent HRP substrate (cat.
no. WBKLSO500, Millipore, Billerica, MA, USA) and analyzed using a
Gel Image System version 4.00 (Tanon Science & Technology Co.,
Ltd., Shanghai, China) was used for densitometric quantification of
the blots.
Immunohistochemistry
Myocardial tissues were fixed in 10% buffered
formalin for 48 h at room temperature, dehydrated in a sequence of
100, 95, 70 and 50% graded alcohol solutions, sliced at 4 µm
thickness, and embedded in paraffin. The sections were rinsed in
histosol, rehydrated in a graded series of ethanols and blocked
with 5% bovine serum albumin (Sigma-Aldrich; Merck KGaA) for 4 h.
Sections were incubated with anti-Mas receptor antibody (1:200
dilution; cat. no. AAR-013, Alomone Labs, Jerusalem, Israel) and
anti-phosphorylated (p)-ERK1/2 antibody (1:1,000 dilution; cat. no.
4376S, Cell Signaling Technology, Inc.) at 4°C overnight. After
rinsing with PBS, sections were incubated in
polyperoxidase-conjugated secondary antibody (1:200 dilution;
PV-9001, ZSGB-Biotechnology, Beijing, China) for 2 h at room
temperature. Diaminobenzidine tetrahydrochloride (Sigma-Aldrich;
Merck KGaA) was then used to stain the sections. After
counterstaining with hematoxylin, sections were viewed under an
inverted phase microscope (IX80 microscope, Olympus Corporation,
Tokyo, Japan). Image-Pro Insight 6.0 (Media Cybernetics, Rockville,
MD, USA) was used for image analysis.
Ultrastructural analysis
Electron microscopy was employed to evaluate
ultrastructural markers of myocardial cell apoptosis. Samples of
the left and right myocardium were collected from pigs within 1-2
min of being euthanized. The myocardium was sliced (1 mm) on ice,
fixed in 2.5% (v/v) glutaraldehyde at 4°C for 30 min, post-fixed in
1% (v/v) osmic acid for 1 h, and dehydrated via a graded ethanol
series and embedded in Epon 812 at 40°C for 4 h, 50°C for 2 h and
90°C for 12 h. Thinner sections (0.5-1 µm) were prepared,
stained with 0.5% (w/v) toluidine blue and viewed under a light
microscope (Olympus). Ultra-thin sections (~60 nm) were cut,
double-stained with 2.0% (w/v) uranyl acetate for 20 min and 2.0%
(w/v) lead citrate for 15 min, and viewed under a transmission
electron microscope (Hitachi HT7700; Hitachi Ltd., Tokyo,
Japan).
Statistical analysis
Values are expressed as the mean ± standard
deviation. SPSS 16.0 (SPSS, Inc., Chicago, IL) was used for data
analyses, and one-way analysis of variance coupled with
Bonferroni’s correction (Newman-Keuls test) was used for post-hoc
comparisons. Continuous variables were assessed for normal
distribution by using the Kolmogorov-Smirnov test and equal
variances by the homogeneity of variance test. Pearson’s
correlation coefficient was calculated to determine the
correlations between the ratios of ACE2/ACE, as well as between
caspase-3, Bcl-2, Bax and Bcl-2/Bax. P<0.05 was considered to
indicate a statistically significant difference.
Results
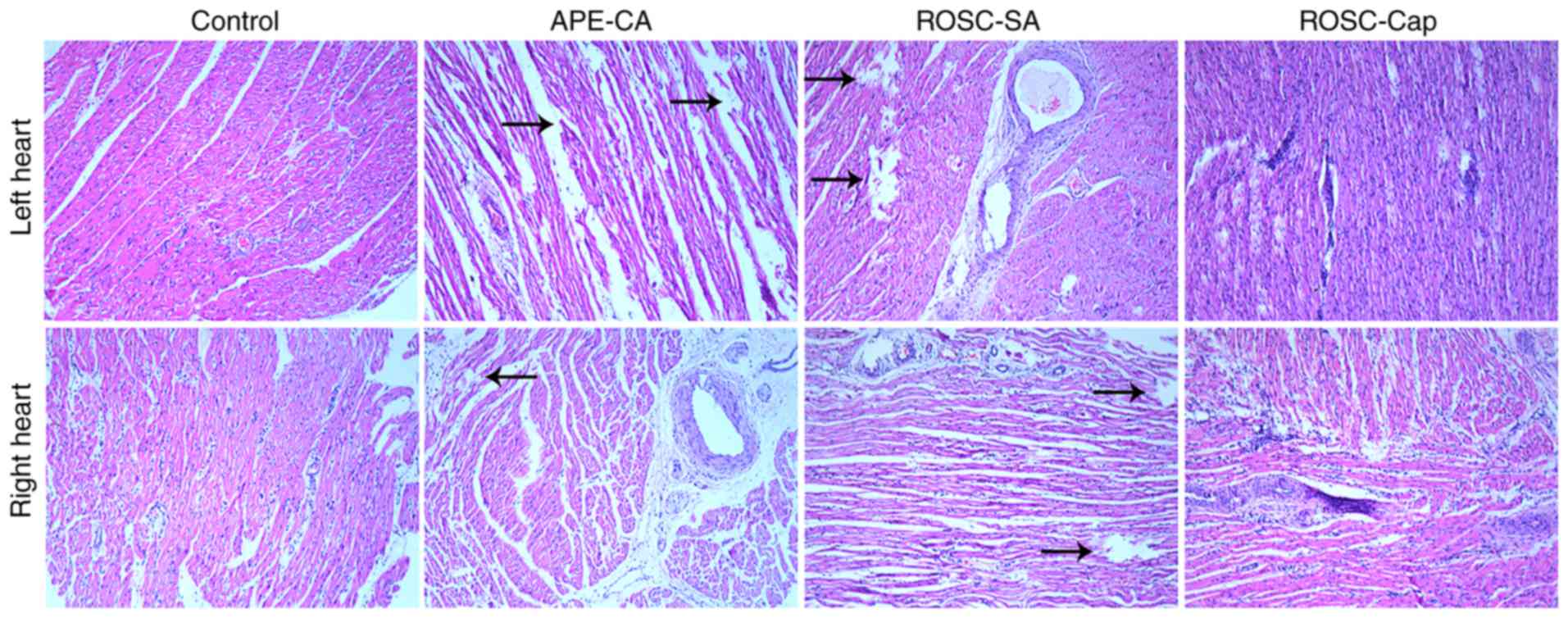
Histological analysis of the myocardium
following APE-CA
Histological analyses of the left and right
ventricular tissues isolated following APE-CA and ROSC were
performed. The control group exhibited no lesions in the myocardial
cells and the myocardial fibers were normal (Fig. 1). In contrast to the control
group, the APE-CA and ROSC groups displayed myocardial fiber
dissolution or fracture in the central zones of the left and right
ventricles, which significantly increased the pathological score
(all P<0.05). Captopril partially inhibited this effect
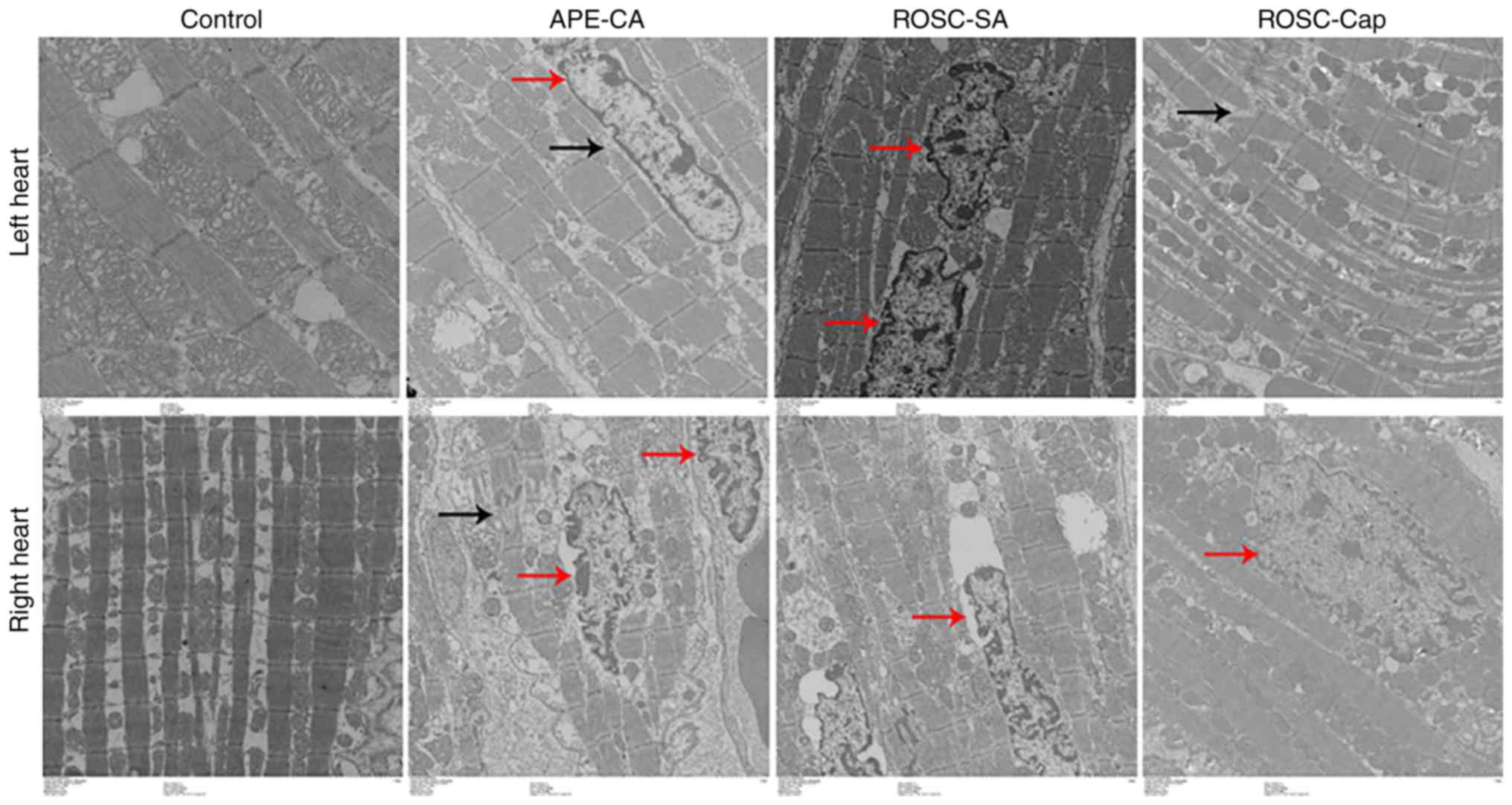
(Fig. 1). Electron microscopy
analysis of myocardial tissues in the APE-CA, ROSC-SA and ROSC-Cap
groups revealed nuclear swelling, as well as myofilament
dissolution and intercalated disks broken or dissolved (Fig. 2; black arrows), and mitochondrial
engorgement and myocardial cell apoptosis (Fig. 2; red arrows). The increase in
apoptosis in the left myocardium was less pronounced in the
captopril-treated group (Fig.
2).
Imbalance of the ACE2/ACE axis in left
and right myocardium following APE-CA
As presented in Fig.
3A and B, in the left myocardium, the expression levels of ACE
(P<0.001 and P<0.01, respectively), the AT1 receptor
(P<0.001 and P<0.01, respectively) and the Mas receptor
(P<0.001 for each) in the APE-CA and ROSC groups were
significantly higher compared with those in the control group.
Decreases in ACE and AT1 receptor expression were observed in the
ROSC-SA vs. those in the APE-CA group (P<0.05 and P<0.01,
respectively). However, the levels of ACE2 and Mas receptor were
not significantly different between the ROSC-SA and APE-CA groups.
Immunohistochemical analysis of the left myocardium revealed
positive staining for the Mas receptor, primarily in the cell
nuclei, of the control group, while staining was evident in the
cell nuclei and cytoplasm of the APE-CA and ROSC-SA groups
(Fig. 3C). A marked increase in
Mas receptor expression was observed in the APE-CA and ROSC groups
compared with that in the control group (P<0.05 for each;
Fig. 3C). No changes in the
proteins of the ACE2 axis and ACE axis were observed in the right
myocardium following either APE-CA or ROSC (Fig. 3D and E).
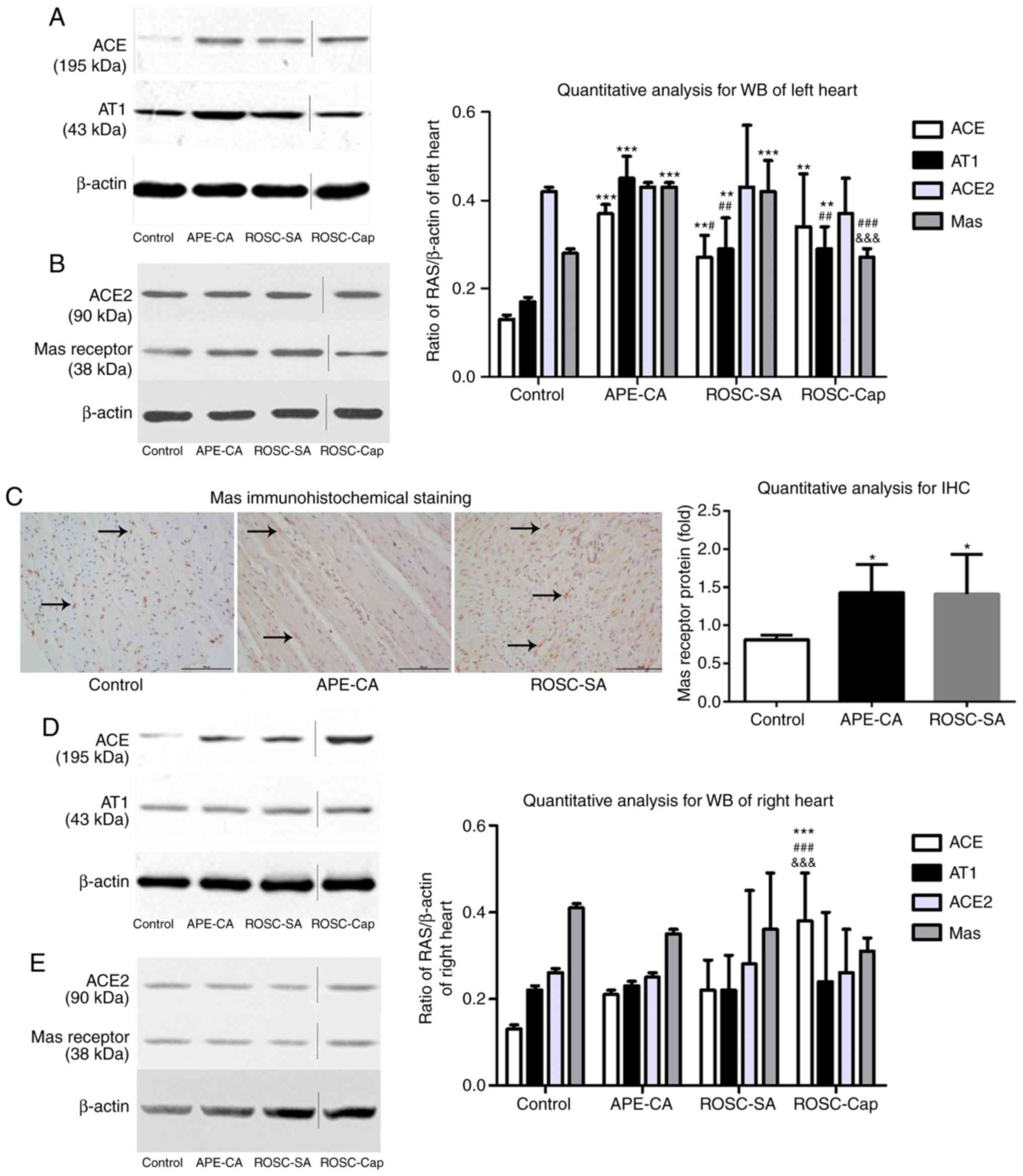 | Figure 3Expression of proteins of the RAS in
the left myocardium as determined by (A and B) WB analysis and (C)
immunohistochemistry (scale bar, 100 µm; black arrows
indicate expression of the Mas receptor). (D and E) Expression of
members of the RAS in the right myocardium as determined by WB
analysis. Vertical lines in the western blot indicate non-adjacent
bands from the same gel. Values are expressed as the mean ±
standard deviation, N=5 pigs per group. *P<0.05;
**P<0.01; ***P<0.001 vs. control group;
#P<0.05; ##P<0.01;
###P<0.001 vs. the APE-CA group;
&&&P<0.001 vs. the ROSC-SA group; APE-CA,
acute pulmonary embolism with cardiac arrest; ROSC, return of the
spontaneous circulation; SA, saline; ACE, angiotensin-converting
enzyme; RAS, renin-angiotensin system; IHC, immunohistochemistry;
AT1, angiotensin II receptor type 1; WB, western blot. |
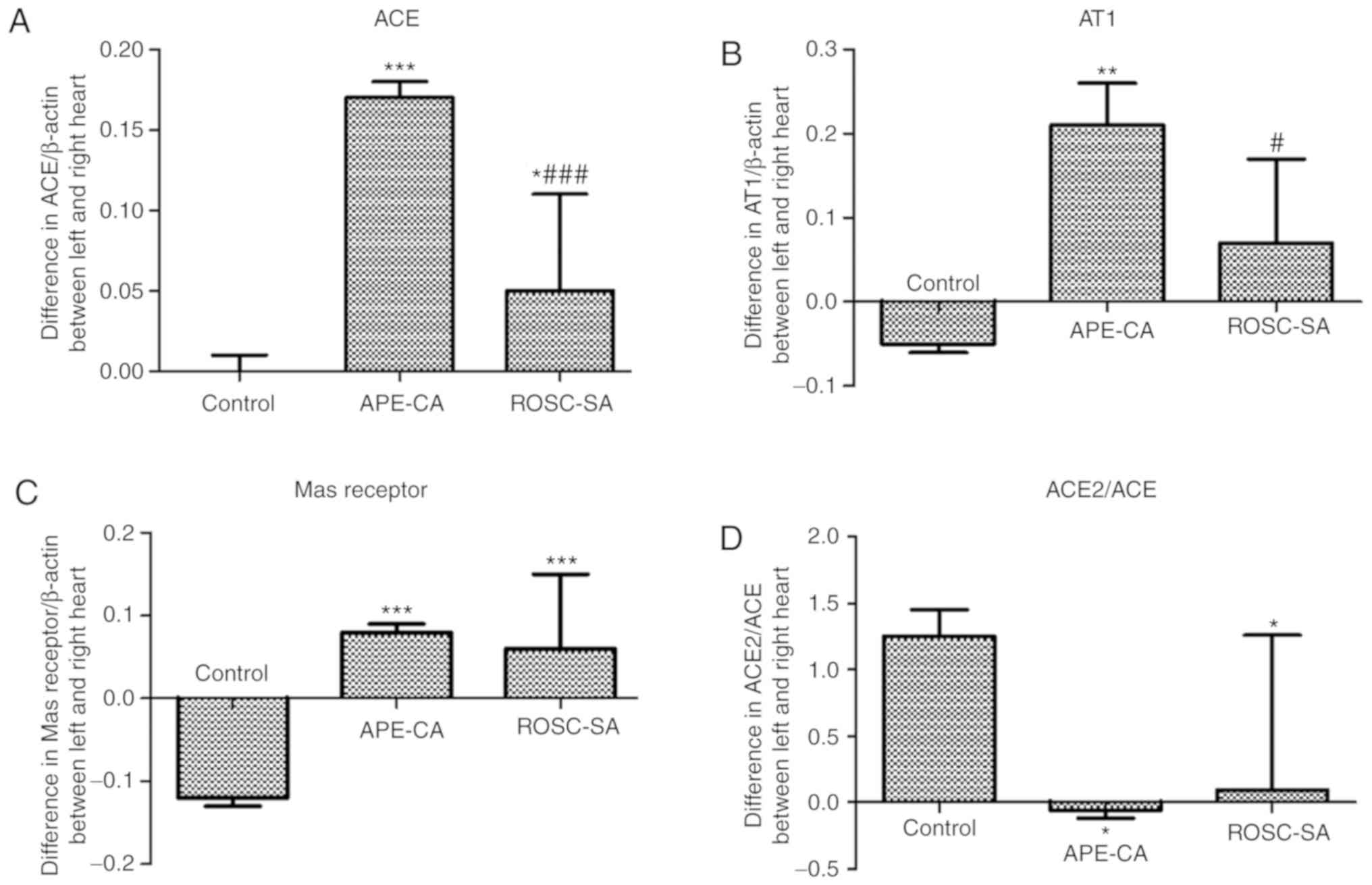
Differences in proteins of the RAS
between left and right myocardium
The present study indicated a significant increase
in the levels of ACE, AT1 and Mas in left myocardial tissues
compared with those in right myocardial tissues across the
experimental groups (Fig. 4A-C).
In the ROSC-SA group, the differences in ACE and AT1 expression
between the left and right myocardium were significantly decreased
compared with those in the APE-CA group (P<0.001 and P<0.05;
Fig. 4A and B). The difference in
the ACE2/ACE ratio between the left and right myocardium was
reduced in the APE-CA and ROSC-SA groups compared with that in the
control group (P<0.05; Fig.
4D). This effect was likely due to the activation of ACE
expression in the left myocardium and ACE2 expression in the right
myocardium.
Effects of captopril on apoptotic
signaling following APE-CA
To elucidate the mechanism of action of captopril to
inhibit myocardial apoptosis, the levels of the
apoptosis-associated proteins Bax and Bcl-2 in the myocardium were
assessed. In the left myocardium, Bax expression was increased
(P<0.001 and P<0.05, respectively), while Bcl-2 expression
(P<0.001 and P<0.05, respectively) was decreased in the
APE-CA and ROSC-SA groups compared with those in the control group
(Fig. 5A). In the ROSC-SA group,
inhibition of myocardial apoptosis was indicated by decreased Bax
and increased Bcl-2 expression compared with that in the APE-CA
group (P<0.01 and P<0.05, respectively; Fig. 5A). Compared with the saline group,
after ROSC administration, Bax expression increased, whereas Bcl-2
expression decreased following captopril treatment in the left
myocardium (P<0.01 and P<0.05; Fig. 5A), confirming the anti-apoptotic
effect of captopril in the left heart. In the right heart, Bax
expression was significantly elevated in the APE-CA and ROSC-SA
groups (P<0.05 for each; Fig.
5B) compared with that in the control group.
Regarding the levels of caspase-3 in the left heart,
a significant increase in the APE-CA and ROSC-SA groups compared
with the control group was noted (P<0.05 and P<0.01,
respectively; Fig. 5A). In
addition, captopril treatment after ROSC decreased the levels of
caspase-3 compared with those in the saline-treated group
(0.34±0.06 vs. 0.19±0.07, P<0.001; Fig. 5A). In the right heart, the levels
of cleaved caspase-3 (17/19 KD) were significantly increased in the
ROSC-SA group compared with those in the control group (P<0.01;
Fig. 5B). However, captopril had
no inhibitory effect on cleaved caspase-3 following ROSC (Fig. 5B). These results suggest that
captopril inhibits apoptosis in the left ventricle but may be
enhanced slightly in the right myocardium.
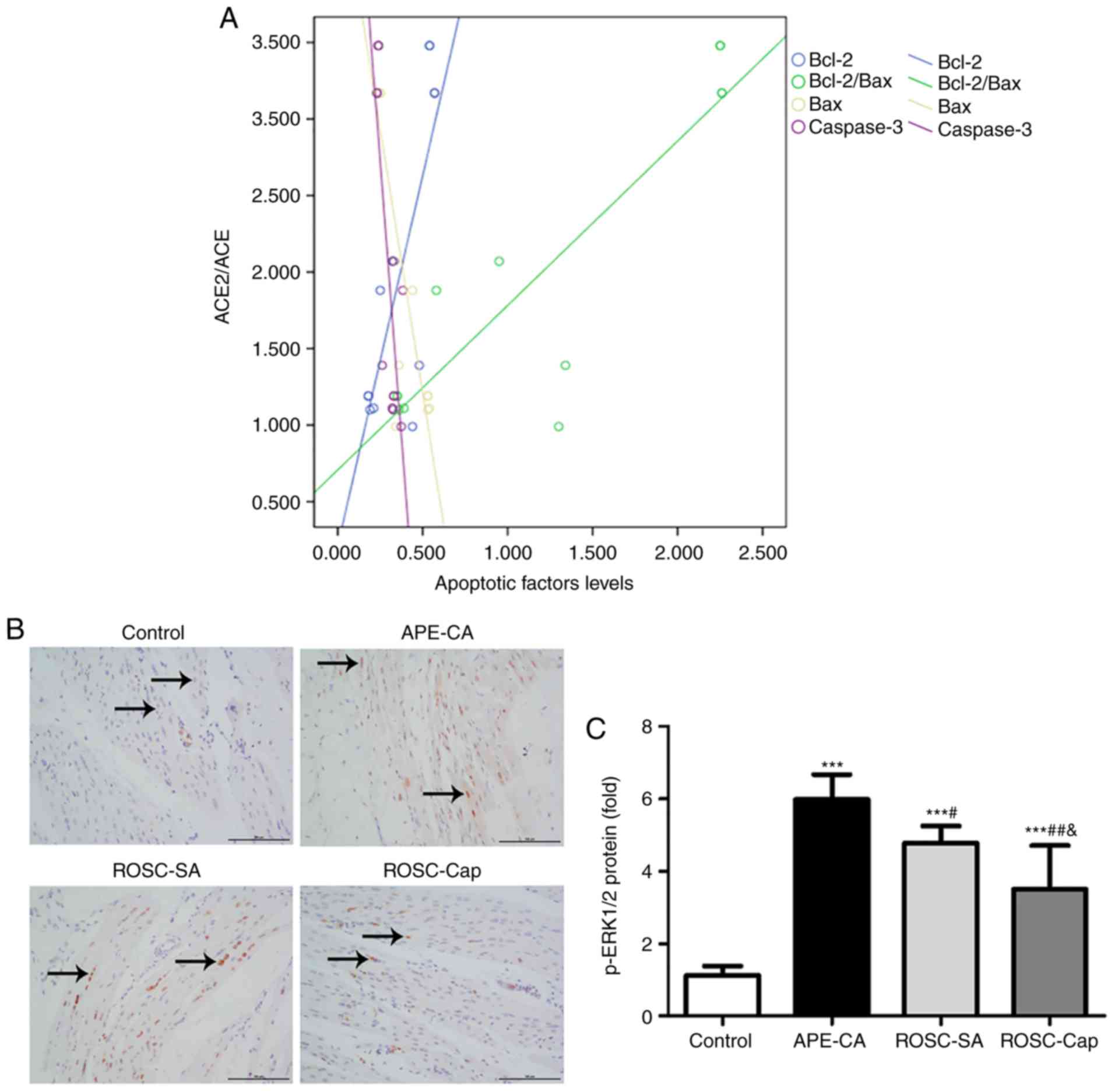
Correlation between ACE2/ACE axis
imbalance and apoptotic signaling proteins in the left
myocardium
In the present study, the ACE2/ACE ratio was
positively correlated with the levels of Bcl-2 (r=0.792, P<0.01)
and the Bcl-2/Bax ratio (r=0.884, P<0.001), and negatively
correlated with the levels Bax (r=−0.844, P<0.01) and caspase-3
(r=−0.789, P<0.01) in the control, APE-CA and ROSC-SA groups
(Fig. 6A). These results support
the hypothesis that an imbalance in the ACE2/ACE axis affects
apoptosis of the left myocardium during APE-CA.
ERK1/2 activation in the myocardium and
effects of captopril during ROSC following APE-CA
To study the molecular mechanism of action of
captopril in the myocardium, immunohistochemical analysis of
p-ERK1/2 in the left myocardium was performed. The images indicated
that p-ERK1/2-positive cells were located primarily in the nuclei
of cardiomyocytes in the control group, but in the nuclei and
cytoplasm of the APE-CA and ROSC groups (Fig. 6B). Compared to the control group,
a 5.35-fold and 4.27-fold increase in p-ERK1/2 was determined in
the APE-CA and ROSC-SA group, respectively. Of note, treatment with
captopril reduced this level by 26.57% in the ROSC-Cap group
compared with that in the ROSC-SA group (Fig. 6C). In the right myocardium, no
significant differences in the p-ERK1/2 levels were observed among
the four groups (data not shown).
Discussion
The present study provides important insight
regarding myocardial apoptosis following APE induced CA and ROSC.
First, it was demonstrated that APE-CA induces myocardial apoptosis
and myocardial fiber fracture. Furthermore, an imbalance in the
ACE2/ACE axis was revealed to be a consequence of differential
activation of the ACE axis in the left myocardium and the ACE2 axis
in the right myocardium following APE-CA and ROSC as shown in
Fig. 4. Finally, the results
revealed that captopril reduces left myocardial injury and
apoptosis, as evidenced by increased Bcl-2 expression and decreased
Bax and caspase-3 expression. However, there were some marked
increases in apoptosis in the right myocardium associated with
captopril, but they were not significant.
The major pathophysiological features of APE include
endogenous or exogenous embolus, pulmonary hypertension and acute
right ventricular dilation, ventricular interdependence, lower left
ventricular diastolic compliance, acute cardiogenic shock and even
death (19). Thus, massive APE
with CA results in an imbalance of right and left ventricular
function. Kumamaru et al (20) determined that a normal
right-to-left ventricular ratio of <0.97 was sufficient to
exclude right ventricular strain/pulmonary embolism-associated
short-term death. The present study aimed to further clarify how
pathophysiological mechanisms of APE differ between the left and
right heart.
The RAS maintains cardiovascular stability,
specifically via the classic ACE/Ang II/AT1 receptor axis and a
recently identified ACE2/Ang(1-7)/Mas receptor axis. Thus, the duality
of the RAS (its ability to stimulate apoptosis, vasoconstriction,
proliferation and fibrosis, while also being able to initiate
anti-apoptotic, vasodilatory, anti-proliferative and anti-fibrotic
pathways) is mainly driven by a balanced ACE2/ACE axis (21). The present study demonstrates that
the RAS is activated during a massive APE when the ACE2/ACE ratio
is decreased in the left, but not in the right myocardium. This
observation may be explained by the fact that, during an APE, the
blood supply from the coronary artery to the left heart is
significantly reduced and the left ventricular pressure and
coronary perfusion pressure exhibit marked decreases, leading to
activation of the RAS.
Zagorski et al (22) reported that the mortality rate due
to severe pulmonary embolism in rats was 37.8%, which is similar to
the mortality rate of 47.4% in the model of the present study.
Schultz et al (23)
employed an in vivo APE model in pigs and mentioned that ‘a
stable model of high-risk PE in pigs with an acceptable variance
and mortality rate may therefore be difficult to develop.’ This
suggests that the mortality rates in this model are expected to be
high. This would justify the high percentage of mortality observed
in the present study. Of note, a large proportion of APE patients
die of heart failure following ROSC (4). A study including 778 comatose
survivors of CA reported that 65% of patients died within 1week
after resuscitation, and >50% of the deaths were attributed to
heart failure (24,25). Previous studies confirm that
myocardial apoptosis has a major role in cardiac dysfunction
(5,26). When a myocardial cell is
stimulated by certain pathological or physiological factors, the
intracellular mitochondrial channel releases macromolecules,
including cytochrome c (Cyt-C), pro-caspase-2 and
pro-caspase-9. Cleavage of these released macromolecules by
proteases activates apoptotic peptidase activating factor-1 and
apoptosis factor-1 to form a complex that subsequently activates
caspase-9 and caspase-3. Bcl-2 regulates the mitochondrial membrane
permeability, the release of the abovementioned pro-apoptotic
factors from the mitochondria and mitochondrial apoptosis.
Constitutive expression of Bcl-2 attenuates Bax-mediated Cyt-C
release, and inhibits caspase activation and transfer of
apoptosis-inducing proteins from the mitochondria to the nucleus
(27). Recently, it was
demonstrated that certain ribosomal proteins (rp), including rpL3,
are released from the nucleolus and activate mitochondrial
apoptotic pathways by increasing the Bax/Bcl-2 ratio (28).
In the present APE-CA model, an increased Bax
expression in the left and right myocardium, increased caspase-3
activation and downregulation of Bcl-2 in the left myocardium were
observed. Furthermore, in captopril-treated pigs, the expression of
Bcl-2 in the left myocardium was increased and that of Bax and
caspase-3 was decreased compared with that in SA-treated pigs,
confirming the anti-apoptotic effect of captopril only on the left
heart following ROSC.
The present results are consistent with those of
previous studies indicating that the classic ACE/AngII/AT1 receptor
axis promotes myocardial apoptosis, while the novel
ACE2/Ang(1-7)/Mas receptor axis inhibits apoptosis.
Liu et al (7) demonstrated
that in H9c2 cells, AngII increases the apoptotic index four-fold
of that in untreated control cells (P<0.01). Fabris et al
(29), reported that the ACE
inhibitor ramipril is effective in preventing ovariectomy-induced
cardiac apoptosis associated with reduced cardiac ACE and AT1
receptor gene expression. Another previous study demonstrated that
in endothelial cells isolated from the coronary arteries of dogs
following cardiopulmonary bypass, captopril markedly inhibited
hemoglobin-based oxygen carrier-induced apoptosis by reducing
associated decreases in the Bcl-2/Bax ratio and increases in
caspase-3 cleavage (30). Qi
et al (8) reported that in
a model of myocardial ischemia induced by coronary artery ligation
surgery, ACE2 overexpression significantly preserved cardiac
function and reduced the infarct size, presumably by inhibiting
cell apoptosis. Furthermore, Ang-(1-7)
treatment significantly protected H9C2 rat cardiomyocytes from
hypoxia/reoxygenation-induced oxidative injury by reducing cell
apoptosis (31). Wan et al
(32) indicated that genetic
silencing of intracellular ACE markedly stimulated ACE2 expression,
caused a downregulation of caspase-3 and Bax expression, and
promoted Bcl-2 expression in H9c2 cardiomyocytes in response to
anoxia/reoxygenation.
It has been proposed that ERK activation is
associated with the progression of heart failure. Increased
expression of p-ERK has been reported in pressure overload-induced
heart failure (33). The
association of ERK1/2 with lipopolysaccharide-induced H9c2 cell
apoptosis has also been reported (34). Lei et al (11) recently demonstrated that the
increase of p-ERK1/2 in high glucose-induced H9c2 cells may be
significantly suppressed by treatment with Ang-(1-7).
In addition, irbesartan or ramipril treatment prevented high salt
diet-induced left ventricular hypertrophy and lowered ERK
phosphorylation (12). It was
also confirmed that a loss of ACE2 leads to increased p-ERK1/2 in
myocardial infarct and peri-infarct regions (35). The observations of the present
study support the collective results of the aforementioned studies,
in terms of inhibition of left cardiac ERK1/2 activation by
captopril treatment corresponding to a lower incidence of
myocardial apoptosis.
Of note, the present study has several limitations.
First, the sample size was relatively small and the statistical
power may have been insufficient to detect additional inter-group
differences. The present study focused on the roles of imbalances
in the ACE2/ACE axis, not on the ACE2 axis alone, in myocardial
apoptosis in massive APE. Thus, future studies addressing the
direct involvement of the ACE2 axis in massive APE should be
designed using recombinant ACE2 or ACE2 gene knockout models.
In conclusion, the present study indicated that an
imbalance in the ACE2/ACE axis is associated with apoptosis
following massive APE and demonstrated the protective effects of
post-resuscitation captopril against myocardial apoptosis.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81372025), the 2015
Annual Special Cultivation and Development Project for Technology
Innovation Base of Beijing Key Laboratory of Cardiopulmonary
Cerebral Resuscitation (grant no. Z151100001615056), the Natural
Science Foundation of Beijing Municipality (grant no. 7173253) and
the Beijing Municipal Administration of Hospitals’ Youth Programme
(grant no. QML20170105). The funders had no role in the study
design, data collection and analysis, writing of the manuscript or
decision to publish.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
CSL, MRX and HLX designed the study; HLX, LXZ, JY,
NT and LA performed the experiments and analyzed the data; QTL
provided chest CT scans; HLX wrote the manuscript. All authors
approved the final version for publication.
Ethics approval and consent to
participate
The Committee on the Ethics of Animal Experiments of
Capital Medical University approved the study procedures (permit
no. 2010-D-013). Experimental protocols were designed in strict
compliance with the Animal Care Guidelines of the Institutional
Animal Care and Use Committee of Capital Medical University
(Beijing, China) as well as the Utstein-style guidelines (13).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests regarding this study.
Acknowledgments
The authors would like to thank Miss Nathalie in
Medjaden Bioscience, Ltd. (Hong Kong, China) for assisting with the
preparation of this manuscript
References
|
1
|
Dahhan T, Alenezi F, Samad Z and Rajagopal
S: Echocardiography in the risk assessment of acute pulmonary
embolism. Semin Respir Crit Care Med. 38:18–28. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiménez D, Lobo JL, Barrios D, Prandoni P
and Yusen RD: Risk stratification of patients with acute
symptomatic pulmonary embolism. Intern Emerg Med. 11:11–18. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White RH: The epidemiology of venous
thromboembolism. Circulation. 107(23 Suppl 1): I4–I8. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yin Q, Li X and Li C: Thrombolysis after
initially unsuccessful cardiopulmonary resuscitation in presumed
pulmonary embolism. Am J Emerg Med. 33:132.e1-e22015. View Article : Google Scholar
|
|
5
|
Sabbah HN: Apoptotic cell death in heart
failure. Cardiovasc Res. 45:704–712. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Mello WC: Local renin angiotensin
aldosterone systems and cardiovascular diseases. Med Clin North Am.
101:117–127. 2017. View Article : Google Scholar
|
|
7
|
Liu JJ, Li DL, Zhou J, Sun L, Zhao M, Kong
SS, Wang YH, Yu XJ, Zhou J and Zang WJ: Acetylcholine prevents
angiotensin II-induced oxidative stress and apoptosis in H9c2
cells. Apoptosis. 16:94–103. 2011. View Article : Google Scholar
|
|
8
|
Qi YF, Zhang J, Wang L, Shenoy V, Krause
E, Oh SP, Pepine CJ, Katovich MJ and Raizada MK:
Angiotensin-converting enzyme 2 inhibits high-mobility group box 1
and attenuates cardiac dysfunction post-myocardial ischemia. J Mol
Med (Berl). 94:37–49. 2016. View Article : Google Scholar
|
|
9
|
Xiao HL, Li CS, Zhao LX, Yang J, Tong N,
An L and Liu QT: Captopril improves postresuscitation hemodynamics
protective against pulmonary embolism by activating the
ACE2/Ang-(1–7)/Mas axis. Naunyn Schmiedebergs Arch Pharmacol.
389:1159–1169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian Y, Li H, Liu P, Xu JM, Irwin MG, Xia
Z and Tian G: Captopril pretreatment produces an additive
cardioprotection to isoflurane preconditioning in attenuating
myocardial ischemia reperfusion injury in rabbits and in humans.
Mediators Inflamm. 2015.819232:2015.
|
|
11
|
Lei Y, Xu Q, Zeng B, Zhang W, Zhen Y, Zhai
Y, Cheng F, Mei W, Zheng D, Feng J, et al: Angiotensin-(1–7)
protects cardiomyocytes against high glucose-induced injuries
through inhibiting reactive oxygen species-activated leptin-p38
mitogen-activated protein kinase/extracellular signal-regulated
protein kinase 1/2 pathways, but not the leptin-c-Jun N-terminal
kinase pathway in vitro. J Diabetes Investig. 8:434–445. 2017.
View Article : Google Scholar :
|
|
12
|
Le Corvoisier P, Adamy C, Sambin L,
Crozatier B, Berdeaux A, Michel JB, Hittinger L and Su J: The
cardiac renin-angiotensin system is responsible for high-salt
diet-induced left ventricular hypertrophy in mice. Eur J Heart
Fail. 12:1171–1178. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Idris AH, Becker LB, Ornato JP, Hedges JR,
Bircher NG, Chandra NC, Cummins RO, Dick W, Ebmeyer U, Halperin HR,
et al: Utstein-style guidelines for uniform reporting of laboratory
CPR research A statement for healthcare professionals from a task
force of the American Heart Association the American College of
Emergency Physicians, the American College of Cardiology, the
European Resuscitation Council, the Heart and Stroke Foundation of
Canada, the Institute of Critical Care Medicine, the Safar Center
for Resuscitation Research, and the Society for Academic Emergency
Medicine. Writing Group. Circulation. 94:2324–2336. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pantazopoulos IN, Xanthos TT, Vlachos I,
Troupis G, Kotsiomitis E, Johnson E, Papalois A and Skandalakis P:
Use of the impedance threshold device improves survival rate and
neurological outcome in a swine model of asphyxial cardiac
arrest*. Crit Care Med. 40:861–868. 2012. View Article : Google Scholar
|
|
15
|
Travers AH, Rea TD, Bobrow BJ, Edelson DP,
Berg RA, Sayre MR, Berg MD, Chameides L, O’Connor RE and Swor RA:
Part 4: CPR overview: 2010 American Heart Association Guidelines
for Cardiopulmonary Resuscitation and Emergency Cardiovascular
Care. Circulation. 122(18 Suppl 3): pp. S676–S684. 2010, View Article : Google Scholar
|
|
16
|
Stadlbauer KH, Rheinberger K, Wenzel V,
Raedler C, Krismer AC, Strohmenger HU, Augenstein S, Wagner-Berger
HG, Voelckel WG, Lindner KH and Amann A: The effects of nifedipine
on ventricular fibrillation mean frequency in a porcine model of
prolonged cardiopulmonary resuscitation. Anesth Analg. 97:226–230.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang G, Zhang Q, Yuan W, Wu J and Li C:
Enalapril protects against myocardial ischemia/reperfusion injury
in a swine model of cardiacarrest and resuscitation. Int J Mol Med.
38:1463–1473. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rezkalla S, Kloner RA, Khatib G and Khatib
R: Beneficial effects of captopril in acute coxsakievirus B3 murine
myocarditis. Circulation. 81:1039–1046. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pinsky MR: The right ventricle:
Interaction with the pulmonary circulation. Crit Care. 20:2662016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumamaru KK, George E, Ghosh N, Quesada
CG, Wake N, Gerhard-Herman M and Rybicki FJ: Normal ventricular
diameter ratio on CT provides adequate assessment for critical
right ventricular strain among patients with acute pulmonary
embolism. Int J Cardiovasc Imaging. 32:1153–1161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwai M and Horiuchi M: Role of the
ACE2/angiotensin1-7/Mas axis in the cardiovascular system.
Hypertens Res. 33:1108–1109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zagorski J, Debelak J, Gellar M, Watts JA
and Kline JA: Chemokines accumulate in the lungs of rats with
severe pulmonary embolism induced by polystyrene microspheres. J
Immunol. 171:5529–5536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schultz J, Andersen A, Gade IL, Ringgaard
S, Kjaergaard B and Nielsen-Kudsk JE: A porcine in-vivo model of
acute pulmonary embolism. Pulm Circ. 8:20458932177382172018.
View Article : Google Scholar :
|
|
24
|
Brain Resuscitation Clinical Trial I Study
Group: A randomized clinica1 study of thiopental loading in
comatose survivors of cardiac arrest. N Engl J Med. 314:397–403.
1986. View Article : Google Scholar
|
|
25
|
Brain Resuscitation Clinical Trial II
Study Group: A randomized clinical study of a calcium-entry
blocker(lidoflazine)in the treatment of comatose survivors of
cardiac arrest. N Engl J Med. 324:I225–1231. 1991.
|
|
26
|
Sun XQ, Zhang R, Zhang HD, Yuan P, Wang
XJ, Zhao QH, Wang L, Jiang R, Jan Bogaard H and Jing ZC: Reversal
of right ventricular remodeling by dichloroacetate is related to
inhibition of mitochondria-dependent apoptosis. Hypertens Res.
39:302–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pagliara V, Saide A, Mitidieri E,
d’Emmanuele di Villa Bianca R, Sorrentino R, Russo G and Russo A:
5-FU targets rpL3 to induce mitochondrial apoptosis via
cystathionine-β-synthase in colon-cancer cells lacking p53.
Oncotarget. 7:50333–50348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fabris B, Candido R, Bortoletto M, Toffoli
B, Bernardi S, Stebel M, Bardelli M, Zentilin L, Giacca M and
Carretta R: Stimulation of cardiac apoptosis in ovariectomized
hypertensive rats: Potential role of the renin-angiotensin system.
J Hypertens. 29:273–281. 2011. View Article : Google Scholar
|
|
30
|
Zhou Li T, Yao R, Yang Y, Zhou Q, Wu C, Li
W, You Q, Zhao Z, Yang XL, et al: Angiotensin-converting enzyme
inhibitor captopril reverses the adverse cardiovascular effects of
polymer-ized hemoglobin. Antioxid Redox Signal. 21:2095–2108. 2014.
View Article : Google Scholar
|
|
31
|
Zhao P, Li F, Gao W, Wang J, Fu L, Chen Y
and Huang M: Angiotensin1-7 protects cardiomyocytes from
hypoxia/reoxygenation-induced oxidative stress by preventing
ROS-associated mitochondrial dysfunction and activating the Akt
signaling pathway. Acta Histochem. 117:803–810. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wan W, Jiang X, Li X, Zhang C and Yi X:
Silencing of angiotensin-converting enzyme by RNA interference
prevents H9c2 cardiomyocytes from apoptosis induced by
anoxia/reoxygenation through regulation of the intracellular
renin-angiotensin system. Int J Mol Med. 32:1380–1386. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi J, Yu J, Tan Y, Chen R, Xu W, Chen Y,
Lu J, Liu Q, Wu J, Gu W and Zhang M: Mechanisms of Chinese Medicine
Xinmailong’s protection against heart failure in
pressure-overloaded mice and cultured cardiomyocytes. Sci Rep.
7:428432017. View Article : Google Scholar
|
|
34
|
Li FF, Yuan Y, Liu Y, Wu QQ, Jiao R, Yang
Z, Zhou MQ and Tang QZ: Pachymic acid protects H9c2 cardiomyocytes
from lipopolysaccharide-induced inflammation and apoptosisby
inhibiting the extracellular signal-regulated kinase 1/2 and p38
pathways. Mol Med Rep. 12:2807–2813. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kassiri Z, Zhong J, Guo D, Basu R, Wang X,
Liu PP, Scholey JW, Penninger JM and Oudit GY: Loss of
angiotensin-converting enzyme 2 accelerates maladaptive left
ventricular remodeling in response to myocardial infarction. Circ
Heart Fail. 2:446–455. 2009. View Article : Google Scholar : PubMed/NCBI
|