Introduction
Peroxisome proliferator-activated receptors (PPARs)
are members of the nuclear receptor superfamily of ligand-inducible
transcription factors that control the expression of numerous genes
involved in adipogenesis, lipid metabolism, inflammation and
maintenance of metabolic homeostasis (1–3). Each of the 3 PPAR
isoforms, namely, PPARα, PPARβ/δ and PPARγ, organizes to form a
heterodimer with retinoid X receptor α (RXRα) and binds to the
PPAR-responsive regulatory element (PPRE) present in the target
gene promoters (4,5). Despite high structural homology, these 3
PPAR isoforms differ in their tissue distributions, ligand
specificity, and physiological roles in vivo (6,7). PPARγ is
highly expressed in white and brown adipose tissues and
overexpressed in several types of human cancer, including prostate
cancer (8,9). Due to its vital role in the regulation of insulin
sensitivity and glucose metabolism, PPARγ has been studied as the
target molecule for the development of therapeutic drugs for the
treatment of type 2 diabetes (10–12). In addition, PPARγ agonists
are being used as adjuvants in the treatment of prostate cancer
(13,14).
Although several potent endogenous ligands with
relatively low affinity for PPARγ, including free fatty acids and
eicosanoids, have been identified (15‑18), a physiologically active
endogenous ligand is yet unknown. A number of synthetic ligands
including thiazolidinediones (TZDs) exhibit high affinity for PPARγ
and exhibit robust insulin-sensitizing activities (10–12). Upon
binding to selective ligands, PPARγ undergoes a conformational
change that facilitates the dissociation of co-repressors and
recruitment of co-activators, including steroid receptor
co-activator, cAMP response element binding protein-binding
protein, and PPARγ co-activator-1α, leading to the transcriptional
activation of target genes (19–21). However, the complete
understanding of the dynamics of PPARγ necessitates the study of
the detailed mechanisms underlying the recruitment of
tissue‑specific co‑activators and co-repressors.
Protein arginine methylation is a common
post-translational modification (PTM) in various proteins and is
catalyzed by enzymes called protein arginine methyltransferases
(PRMTs) (22,23). In mammals, PRMTs that have been characterized
have been demonstrated to produce 3 types of methylarginine,
namely, mono-methylarginine, asymmetric di-methylarginine and
symmetric di-methylarginine (22,24). In epigenetic gene regulation,
PRMTs are recruited to promoters via interaction with transcription
factors as co-activators or co-repressors, followed by methylate
arginine residues in histones and other chromatin proteins (23,25).
PRMT6 is a type I PRMT enzyme located predominantly in the nucleus
that exhibits a high affinity for arginine 2 on histone H3 (H3R2)
and catalyzes H3R2 asymmetric di-methylation (H3R2me2a) (26,27). As
H3R2me2 is a repressive mark, PRMT6 activity is primarily
associated with transcriptional silencing (28). However, the
functions of PRMT6 in PPARγ regulation and adipogenesis have not
been completely identified, although it is hypothesized to regulate
numerous biological process including transcription (28,29), DNA
replication (30) and signal transduction. The present study
identified that PRMT6 co‑repressed PPARγ-dependent transcription
through H3R2me2 in PPRE and served as a key regulator of adipocyte
differentiation.
Materials and methods
Constructs, reagents and antibodies
Green fluorescent protein (GFP)-PRMT1, GFP-PRMT4,
GFP-PRMT5 and GFP-PRMT6 plasmids were obtained from Dr M T Bedford
(MD Anderson Cancer Center, Smithville, TX, USA). MS023,
pioglitazone, GW7647, GW501516, retinoic acid, dexamethasone,
methyl isobutyl xanthine (IBMX) and insulin were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Antibodies against
PRMT5 (cat. no. sc-376937), GFP (cat. no. sc-9996), GAPDH (cat. no.
sc-25778) and β-actin (cat. no. sc-47778) were procured from Santa
Cruz Biotechnology, Inc., (Dallas, TX, USA) and those against
histone H3 (cat. no. 9715) and PPARγ (cat. no. 2430) were procured
from Cell Signaling Technology, Inc., (Danvers, MA, USA).
Anti-H3R2me2a (cat. no. 07–585) and Asym24 (cat. no. 07–414) were
obtained from EMD Millipore (Billerica, MA, USA) and anti-PRMT1
(cat. no. A300-722A), anti-PRMT4 (cat. no. A300-421A) and
anti-PRMT6 (cat. no. A300–929A) were from Bethyl Laboratories
(Montgomery, TX, USA). Horseradish peroxidase (HRP)-conjugated
secondary antibodies (anti-mouse, cat. no. 315-035-003; and
anti-rabbit, cat. no. 211-035-109) were purchased from Jackson
ImmunoResearch Laboratories, Inc., (West Grove, PA, USA).
Cell culture and transfection
The human cell line 293T, human prostate cancer PC3
cell line, African green monkey kidney fibroblast CV1 cell line and
mouse embryonic fibroblast 3T3-L1 cell line were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). All
cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS; HyClone; GE Healthcare Life
Sciences) and 100 units/ml penicillin/streptomycin (HyClone
Laboratories) at 37°C and 5% CO2 in a humidified
chamber. For overexpression from mammalian expression plasmids,
TransIT-2020™ (Mirus Bio, LLC, Madison, WI, USA) was used according
to the manufacturer’s protocol. For small-interfering RNA (siRNA)
transfection, TransIT-X2™ (Mirus Bio, LLC) was used. All siRNA
duplexes were synthesized by Integrated DNA Technologies Pte. Ltd.,
(Singapore). The sequences of PRMT6-targeting siRNAs used were as
follows: Human, 5′-GAC AAG ACA CGG ACG UUU-3′ and mouse, 5′-GCU ACG
GAC UUC UGC ACG A-3′
3T3‑L1 adipocyte differentiation and Oil
Red O staining
3T3-L1 cells were differentiated into adipocytes
according to the ATCC protocol. Briefly, cells were grown for 48 h
until 100% confluence and maintained in the growth medium (DMEM
supplemented with 10% FBS and 100 units/ml
penicillin/streptomycin). Cells were incubated in a differentiation
medium (growth medium supplemented with 1 µM dexamethasone,
0.5 mM IBMX and 1 µg/ml insulin) for 48 h. Following
incubation, the medium was replaced with an adipocyte maintenance
medium (growth medium with 1 µg/ml insulin) for an
additional 48 h and cells were then fed every other day with growth
medium. For Oil Red O staining, cells were washed with PBS and
fixed with 4% paraformaldehyde for 1 h at room temperature,
followed by staining with Oil Red O (Sigma-Aldrich; Merck KGaA) for
1 h, as previously described (31)
Luciferase gene reporter assay
The transactivation assay for PPARs was measured by
reporter gene (PPRE-luciferase plasmid) analysis as described
previously (31). CV1 cells were transfected with PPRE‑Luc firefly
luciferase constructs (Addgene, Inc., Cambridge, MA, USA) and
SV40-Renilla luciferase plasmids (Addgene, Inc.) using
TransIT-2020™ (Mirus Bio, LLC). Following incubation for 24 h at
37°C, cells were treated with pioglitazone, GW7647 or GW501516 for
additional 24 h. The dual luciferase reporter assay kit (Promega
Corporation, Madison, WI, USA) was used according to the
manufacturer’s protocol and the luciferase activities were
quantified using GloMax® 20/20 (Promega Corporation).
The data are presented as the mean ± standard deviation of three
independent experiments.
Immunoblotting and immunoprecipitation
(IP)
Whole cell extracts were obtained using a lysis
buffer [20 mM Tris-HCl (pH 8.0), 150 mM sodium chloride (NaCl), 10%
glycerol, 1% NP-40 and 2 mM ethylenediaminetetraacetic acid (EDTA)]
supplemented with protease and phosphatase inhibitor cocktails
(Roche Diagnostics, Basel, Switzerland). Following centrifugation
at 16,000 × g for 10 min at 4°C, protein concentration was
determined by Bradford assay according to the manufacturer’s
instructions (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Subsequent to boiling in SDS loading buffer for 5 min, equal
amounts (10–30 µg) of protein were resolved through 10%
SDS-PAGE and were transferred onto polyvinylidene fluoride (PVDF)
membranes. The blots were blocked with 5% skim milk/0.1% Tween
20/TBS for 1 h at room temperature and incubated overnight with
primary antibodies, followed by treatment with an HRP-linked
secondary antibody for 1 h at room temperature. All primary
antibodies were used at a dilution of 1:1,000 and secondary
antibodies at a dilution of 1:10,000. Blots were developed with the
WesternBright ECL HRP substrate (Advansta, Inc., San Jose, CA,
USA), according to the manufacturer’s protocols. For the IP assay,
equal amounts (~1 mg) of lysates were incubated with the
appropriate antibodies (1 µg of antibody to each IP
reaction) overnight at 4°C, and the antibody‑protein complex was
captured using Protein A/G Sepharose beads (Santa Cruz
Biotechnology, Inc.) for 1 h at 4°C. Following washing twice with
NP-40 lysis buffer, the complexes were eluted and analyzed by 10%
SDS-PAGE and immunoblotting, as aforementioned.
Reverse transcription‑quantitative
polymerase chain reaction (RT‑qPCR)
Total cellular RNA was extracted using the TRIsure™
RNA isolation kit (Bioline, London, UK), and cDNA was synthesized
using the SensiFAST™ cDNA synthesis kit (Bioline). qPCR
amplification was performed using 0.5 µl cDNA as the
template, 10 µl SensiFAST SYBR™ No-ROX premix (Bioline), 1
µl each of the forward and reverse primers and 7.5 µl
RNase-free water, in an Eco Real-Time PCR System (Illumina, Inc.,
San Diego, CA, USA). Reaction parameters were as follows: cDNA
synthesis at 37°C for 60 min, transcriptase inactivation at 95°C
for 5 min, then PCR cycling at 95°C for 10 sec, 58°C for 30 sec and
72°C for 20 sec for 40 cycles. Data analyses were performed using
the Eco software version 3.1 (Illumina, Inc) based on the ∆∆Cq
method (32). The primer sets for CCAAT-enhancer-binding protein α
(C/EBPα) were forward, 5′-AGG TGC TGG AGT TGA CCA GT-3′ and
reverse, 5′-CAG CCT AGA GAT CCA GCG AC-3′; the primer sets for
adipocyte Protein 2 (aP2) were forward, 5′-ATG TGT GAT GCC TTT GTG
GGA-3′ and reverse, 5′-TGC CCT TTC ATA AAC TCT TGT-3′. GAPDH was
used as control gene (forward, 5′-CTC ATG ACC ACA GTC CAT GCC
ATC-3′, and reverse, 5′-CTG CTT CAC CAC CTT CTT GAT GTC-3′).
In vitro methylation assay
GFP-PRMT6 protein was purified from the transfected
293T cells (~1 mg protein) using an anti-GFP IP method. Immobilized
GFP-PRMT6 protein was incubated with 50 µl reaction buffer
[20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM
phenylmethane sulfonyl fluoride and 1 mM dithiothreitol]
supplemented with 1 µg recombinant histone (mixture of H2A,
H2B, H3, and H4; New England Biolabs, Inc., Ipswich, MA, USA) or
recombinant human PPARγ (ProSpec-Tany TechnoGene Ltd., Rehovot,
Israel) and 1 µCi 3[H]-labeled AdoMet (specific activity:
55–85 Ci/mmol; PerkinElmer, Inc., Waltham, MA, USA) at 37°C for 1
h. The reaction was stopped by the addition of SDS loading buffer,
and the proteins were resolved on 12% SDS-PAGE gels. Proteins were
transferred onto PVDF membranes, and the tritium signal was
amplified by spraying with EN3HANCE spray (PerkinElmer, Inc.) at
room temperature. Membranes were exposed to autoradiography film
for at least 1 week at 80°C
Chromatin immunoprecipitation (ChIP)
assay
Chromatin from the differentiated 3T3-L1 cells
(1×106 cells) was used for the ChIP experiment with each
antibody (anti-PPARγ, anti-PRMT6 and anti-H3R2me2a) at 1:100
dilutions. A ChIP assay kit (EMD Millipore) was used according to
the manufacturer’s protocol. Briefly, samples were crosslinked with
1% formaldehyde and quenched with 0.125 M glycine. Samples were
then lysed in an SDS lysis buffer [50 mM Tris-HCl (pH 8.0), 1% SDS
and 10 mM EDTA] containing protease inhibitors and were sonicated
at 20% amplitude to shear DNA samples to 200‑1,000 base pair
lengths for five cycles of 30 sec each, resting for 1 min between
cycles on ice (Vibra-cell VCX750; Sonics & Materials Inc.,
Newtown, CT, USA). The lysates were diluted in an IP dilution
buffer [16.7 mM Tris-HCl (pH 8.0), 0.01% SDS, 1.1% Triton X-100,
1.2 mM EDTA and 150 mM NaCl] containing protease inhibitors.
Following agarose-bead clearing, antibodies were added to lysates
at a dilution of 1:100 overnight at 4°C, and the combined
antibody/DNA complexes were incubated with protein A/G beads for 1
h at 4°C. Beads were sequentially washed with a low salt buffer [20
mM Tris-HCl (pH 8.0), 0.1% SDS, 1% Triton X-100, 2 mM EDTA and 150
mM NaCl], high salt buffer (same buffer containing 500 mM NaCl),
and a lithium chloride (LiCl) buffer [10 mM Tris-HCl (pH 8.0), 0.25
M LiCl, 1% NP-40, 1% sodium deoxycholate and 1 mM EDTA]. The final
washing step was performed twice with Tris-EDTA buffer. Complexes
were eluted from the beads using an elution buffer [1% SDS and 0.1
M sodium bicarbonate (NaHCO3)] and reverse-crosslinked
with proteinase K for 2 h at 65°C. The samples were purified with
DNA spin columns and then analyzed by RT-qPCR as aforementioned.
The primer sets used were as follows: aP2 PPRE forward, 5′-GAG CCA
TGC GGA TTC TTG-3′ and reverse, 5′-CCA GGA GCG GCT TGA TTG TTA-3′;
aP2 non-PPRE forward, 5′-CAG CCC CAC ATC CCC ACA GC-3′ and reverse,
5′-GGA TGC CCA ACA ACA GCC ACA C-3′.
Statistical analysis
Data are presented as mean ± standard deviation of
three independent experiments. Comparisons between groups were
performed using a two-tailed Student’s t-test (SigmaPlot ver. 10.0;
Systat Software, Inc., San Jose, CA, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
PRMT6 suppresses PPARγ transcriptional
activity
To investigate the regulation mechanism underlying
PPARγ transactivity by arginine methylation, a PPARγ reporter gene
assay was performed using GFP-PRMT1, GFP-PRMT4, GFP-PRMT5 or
GFP‑PRMT6 plasmids. Overexpression of PRMT6 significantly
suppressed PPARγ transactivity, but GFP-PRMT1, GFP-PRMT4 or
GFP-PRMT5 had little or no effect on PPARγ activity (Fig. 1A). In addition, the protein
expression patterns of PRMT1, PRMT4 and PRMT5 during 3T3-L1
adipogenic differentiation were not significantly altered from
visual observation; however, the PRMT6 level was increased
(Fig. 1B). To confirm the
suppression of PPARγ by PRMT6, the PPARγ transactivity in PC3 human
prostate cancer cells that exhibited stable expression of PPARγ
protein was examined. Basal and pioglitazone-induced PPARγ
activities in PC3 cells were suppressed upon PRMT6 overexpression
(Fig. 1C) but increased following
PRMT6 depletion (Fig. 1D).
However, the transactivities of PPARα, PPARβ/δ and RXRα were
unaffected by PRMT6 overexpression or depletion (Fig. 1E-L). Taken together, these results
indicate that PPARγ transactivity was negatively regulated by
PRMT6.
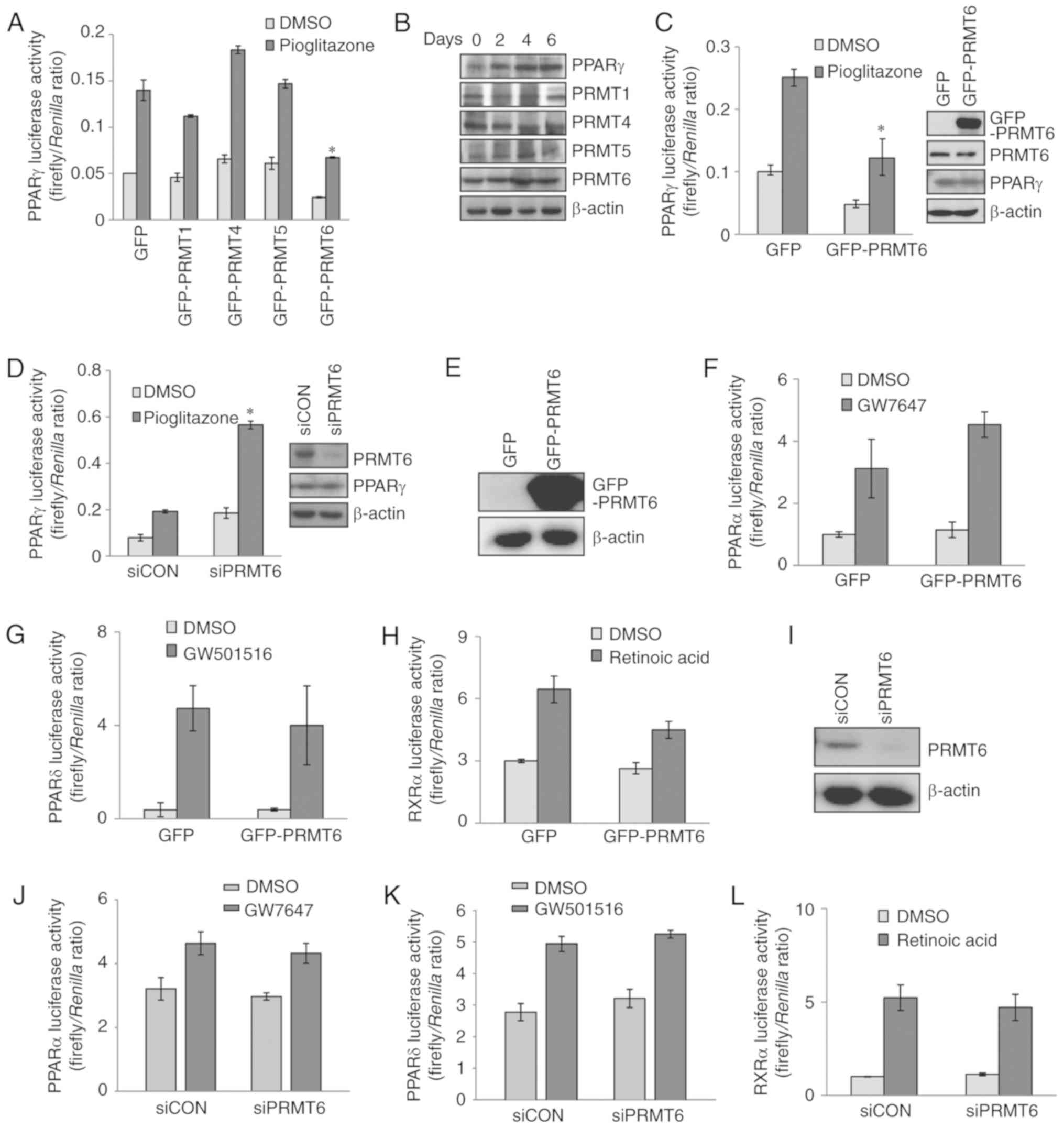 | Figure 1PRMT6 suppresses PPARγ transactivity.
(A) CV1 cells were transiently co-transfected with PPARγ,
PPRE‑firefly luciferase and SV40‑Renilla luciferase
constructs combined with GFP‑PRMTs plasmids. After 24 h, cells were
treated with 2 µM pioglitazone for an additional 24 h.
Firefly/Renilla luciferase activities were measured by a
Dual luciferase assay kit. (B) The expression levels of PRMTs
during adipogenic differentiation of 3T3-L1 cells were measured by
western blot analysis. (C and D) PC3 human prostate cancer cells
were transiently transfected with PPRE‑firefly luciferase and
SV40‑Renilla luciferase constructs combined with (C)
GFP-PRMT6 plasmids or (D) siPRMT6 duplex RNA. PRMT6 and PPARγ
protein levels were confirmed by western blot analysis. (E-H)
Luciferase reporter gene assay following transfection with
GFP-PRMT6 plasmids. (E) GFP-PRMT6 level in cells transfected with
GFP-PRMT6 plasmids was determined by western blot analysis. (F-H)
The cells were then treated with 1 µM (F) GW7647, (G)
GW501516 or (H) retinoic acid for 24 h, to examine the levels of
PPARα, PPARβ/δ and RXRα activities, respectively, in
GFP-PRMT6-overexpressing CV1 cells. (I-L) CV1 cells were
transfected with PRMT6-targeting duplex siRNA. (J-L) The cells were
then treated with 1 µM (J) GW7647, (K) GW501516 or (L)
retinoic acid for 24 h, to examine the levels of PPARα, PPARβ/δ and
RXRα activities, respectively. Error bars represent standard
deviation (n=3; *P<0.05). PRMT6, protein arginine
methyltransferase 6; PPARγ, peroxisome proliferator-activated
receptor γ; GFP, green fluorescent protein; PPRE, PPAR‑responsive
regulatory element; RXRα, retinoid X receptor α; si, small
interfering; siCON, scrambled control. |
PRMT6 regulates 3T3‑L1 adipogenic
differentiation
As PPARγ is a major regulator of adipogenic
differentiation, the present study examined whether PRMT6 regulated
adipocyte differentiation. Differentiated adipocytes were obtained
following the transfection of 3T3-L1 cells with GFP-PRMT6 plasmid
or siPRMT6 duplex RNA. In concordance with the previous results of
PPARγ transactivation (Fig. 1),
PRMT6 overexpression decreased intracellular lipid accumulation as
compared with the control (data not shown), and the knockdown of
PRMT6 expression increased the size and number of intracellular
lipid droplets (Fig. 2A). The
mRNA levels of C/EBPα and aP2, adipogenic marker genes, were
significantly increased in PRMT6-depleted cells (Fig. 2B and C). MS023 is a potent
selective inhibitor of type I PRMTs, including PRMT1, PRMT3, PRMT4,
PRMT6 and PRMT8, and has high specificity for PRMT6 (33). The
present study identified that the treatment of cells with MS023
resulted in 3T3-L1 adipocyte differentiation (Fig. 2D-F), indicating that PRMT6
controlled adipogenic differentiation processes via PPARγ
regulation.
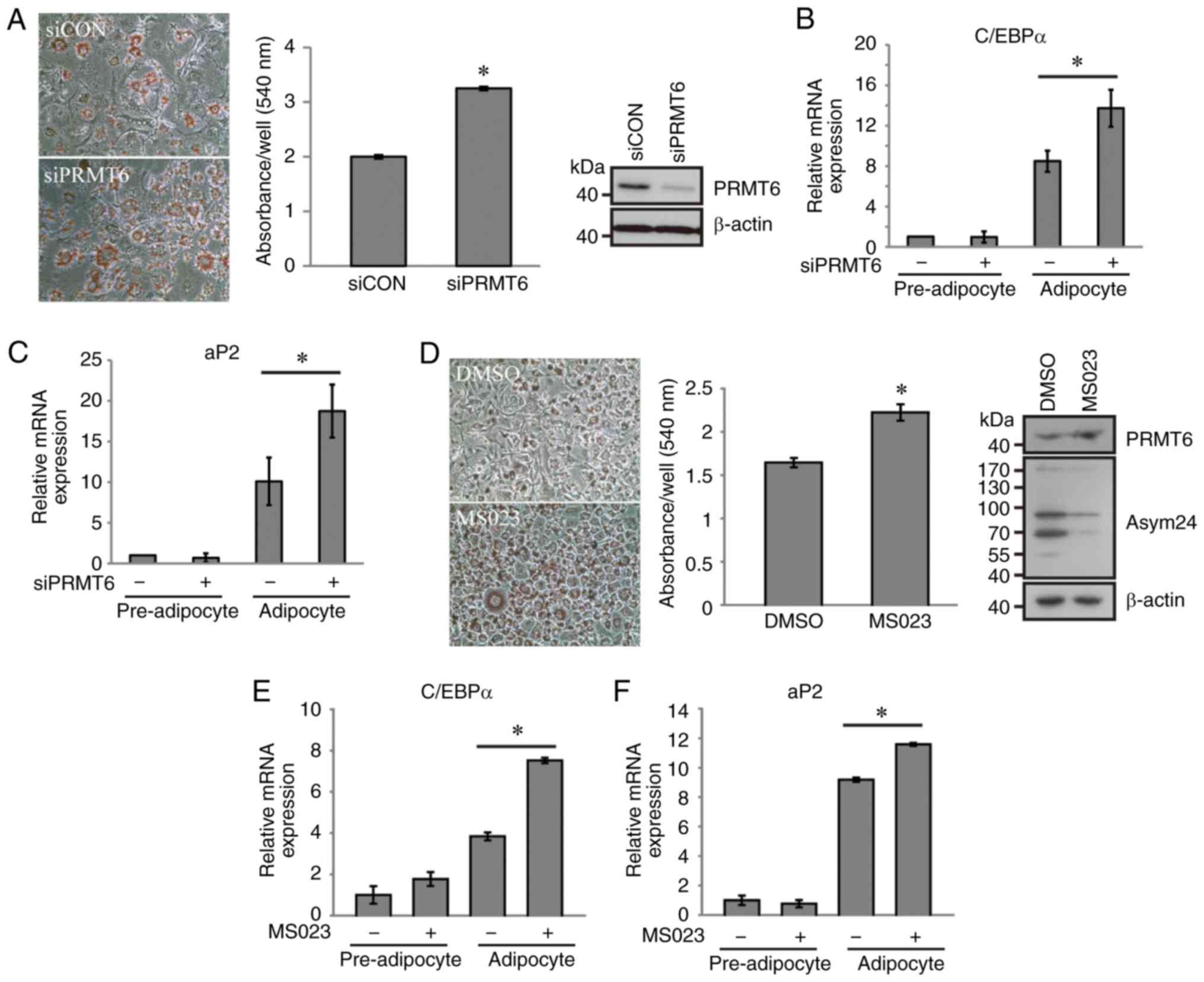 | Figure 2PRMT6 inhibits the adipogenic
differentiation of 3T3-L1. (A and B) Prior to differentiation,
3T3-L1 cells were transfected with scrambled or PRMT6-targeting
siRNA and then differentiated using a hormonal cocktail for 8 days.
(A) The intracellular lipids were stained using Oil Red O (original
magnification, x400). (B and C) Relative mRNA expression of (B)
C/EBPα and (C) aP2 in 3T3-L1 cells was determined using
quantitative polymerase chain reaction. (D) During differentiation,
cells were treated with 5 µM MS023 and stained using Oil Red
O. (E and F) Following treatment with MS023, relative mRNA
expression of (E) C/EBPα and (F) aP2 in 3T3-L1 cells was determined
using quantitative polymerase chain reaction Error bars represent
standard deviation (n=3; *P<0.05). si, small
interfering; siCON, scrambled control; PRMT6, protein arginine
methyltransferase 6; C/EBPα, CCAAT-enhancer-binding protein α; aP2,
adipocyte Protein 2; DMSO, dimethyl sulfoxide. |
PRMT6 interacts with, but does not
methylate, PPARγ
To elucidate the mechanism underlying PRMT6-mediated
PPARγ regulation, the physical interaction between PRMT6 and PPARγ
was first examined. Ectopically‑overexpressed PPARγ protein was
co-immunoprecipitated with PRMT6 in 293T cells from visual
observation (Fig. 3A). To confirm
this result, endogenous PPARγ and PRMT6 were co-immunoprecipitated
in differentiated adipocytes. In mature adipocytes, the protein
PPARγ exhibited marked interaction with PRMT6, as evident in the
reciprocal co-IP experiments (Fig. 3B
and C; lane 2). The treatment with pioglitazone, a PPARγ
agonist, significantly decreased the interaction between PPARγ and
PRMT6 (Fig. 3B; lane 3). In
addition, the inhibition of PRMT6 with MS023 treatment led to the
disruption of this complex (Fig.
3C; lane 3). These results suggested that the complete
activation of PPARγ required the dissociation with, or inhibition
of, the negative regulator PRMT6.
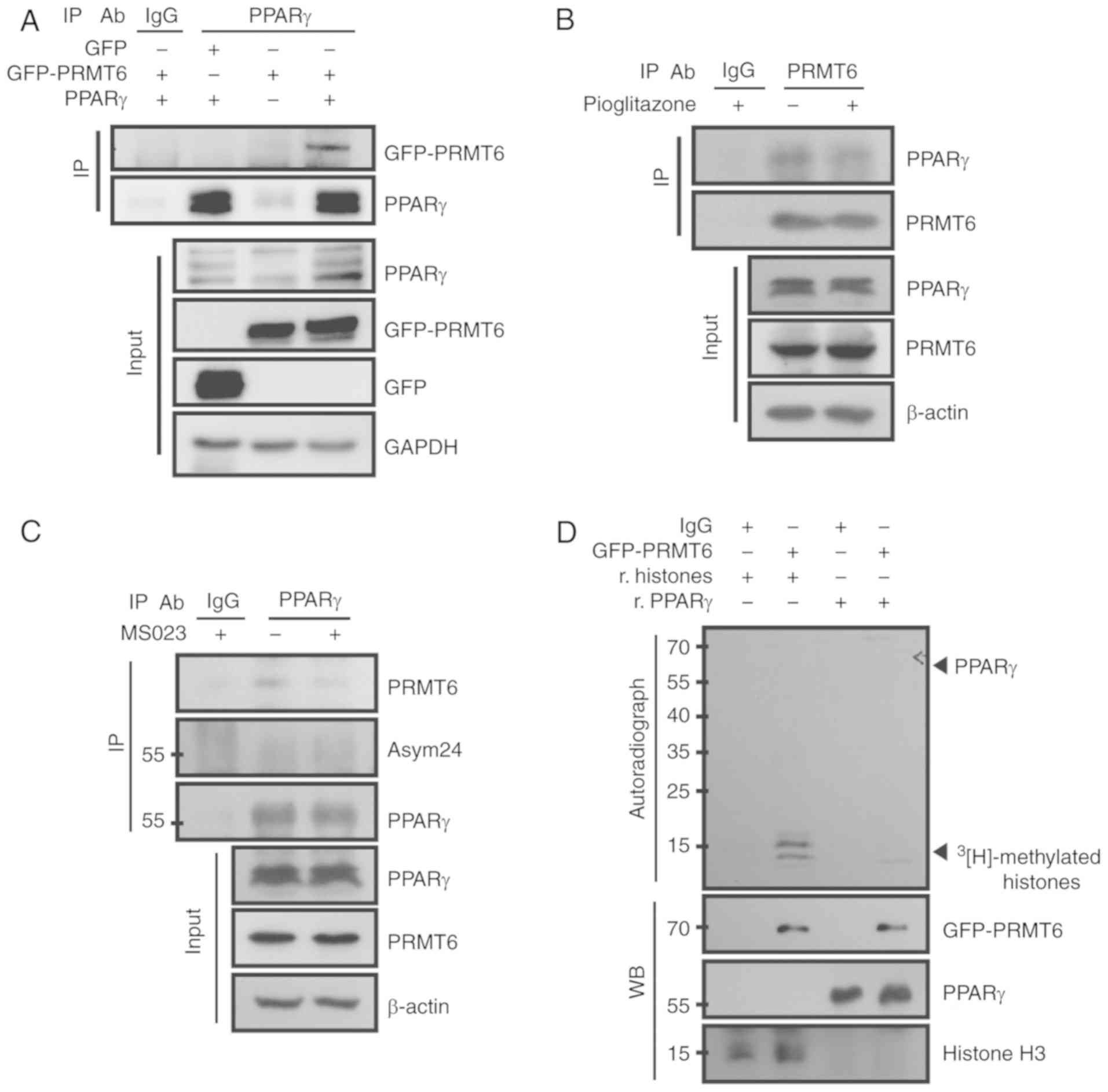 | Figure 3PRMT6 binds to, but does not
methylate, PPARγ. (A) 293T cells were co-transfected with GFP-PRMT6
and PPARγ for 24 h and then immunoprecipitated using the anti-PPARγ
antibody. (B) 3T3-L1 cells were differentiated with 2 µM
pioglitazone for 8 days, and then endogenous PR<T6 was
immunoprecipitated. (C) 3T3-L1 cells were differentiated with 5
µM MS023 for 8 days, and then endogenous PPARγ was
immunoprecipitated. (D) In vitro PRMT6 methylation assay
using recombinant PPARγ protein. PRMT6, protein arginine
methyltransferase 6; PPARγ, peroxisome proliferator-activated
receptor γ; GFP, green fluorescent protein; IP,
immunoprecipitation; IgG, immunoglobulin G; r, recombinant; WB,
western blotting; Asym24, asymmetric di-methyl arginine. |
Based on these results, we hypothesized that PPARγ
served as a substrate for PRMT6. However, no asymmetric di-methyl
arginine was detected in the immunoprecipitated PPARγ protein
(Fig. 3C). To additionally
confirm this result, in vitro methylation assays were
performed using purified PRMT6 and recombinant PPARγ protein. No
3[H]-radioactive methylation
signals were observed following treatment with recombinant PPARγ
(Fig. 3D; lane 4), while
recombinant histones, used as positive controls, were highly
methylated by PRMT6 (Fig. 3D;
lane 2). These results suggested that PPARγ protein was not a
substrate for PRMT6.
PRMT6 represses PPARγ target gene
expression by generating repressive mark H3R2me2a
As PRMT6 serves the role of an epigenetic regulator
through the methylation of the histone H3R2me2a (28,29), the
present study examined whether PRMT6 was recruited to the promoter
region of PPARγ target genes in 3T3‑L1 cells. Using a ChIP assay,
it was verified that PPARγ and PRMT6 were recruited to the PPRE,
but not the non-PPRE region, of the aP2 gene upon complete
differentiation of the cells (Fig.
4A). In addition, the H3R2me2a level was increased in the PPRE
region in response to PRMT6 recruitment, which was suggestive of
the mechanism that inhibits the PRMT6-mediated transcriptional
activity of PPARγ. It was confirmed that treatment with MS023
markedly decreased the level of H3R2me2a in the PPRE region
(Fig. 4B), presumably leading to
an increase in adipogenesis (Fig.
2D-F). Taken together, these data demonstrated that PRMT6
served as a co-repressor that generated the H3R2me2a repressive
mark in the PPRE region, resulting in the suppression of PPARγ
functions during adipogenic differentiation (Fig. 4C).
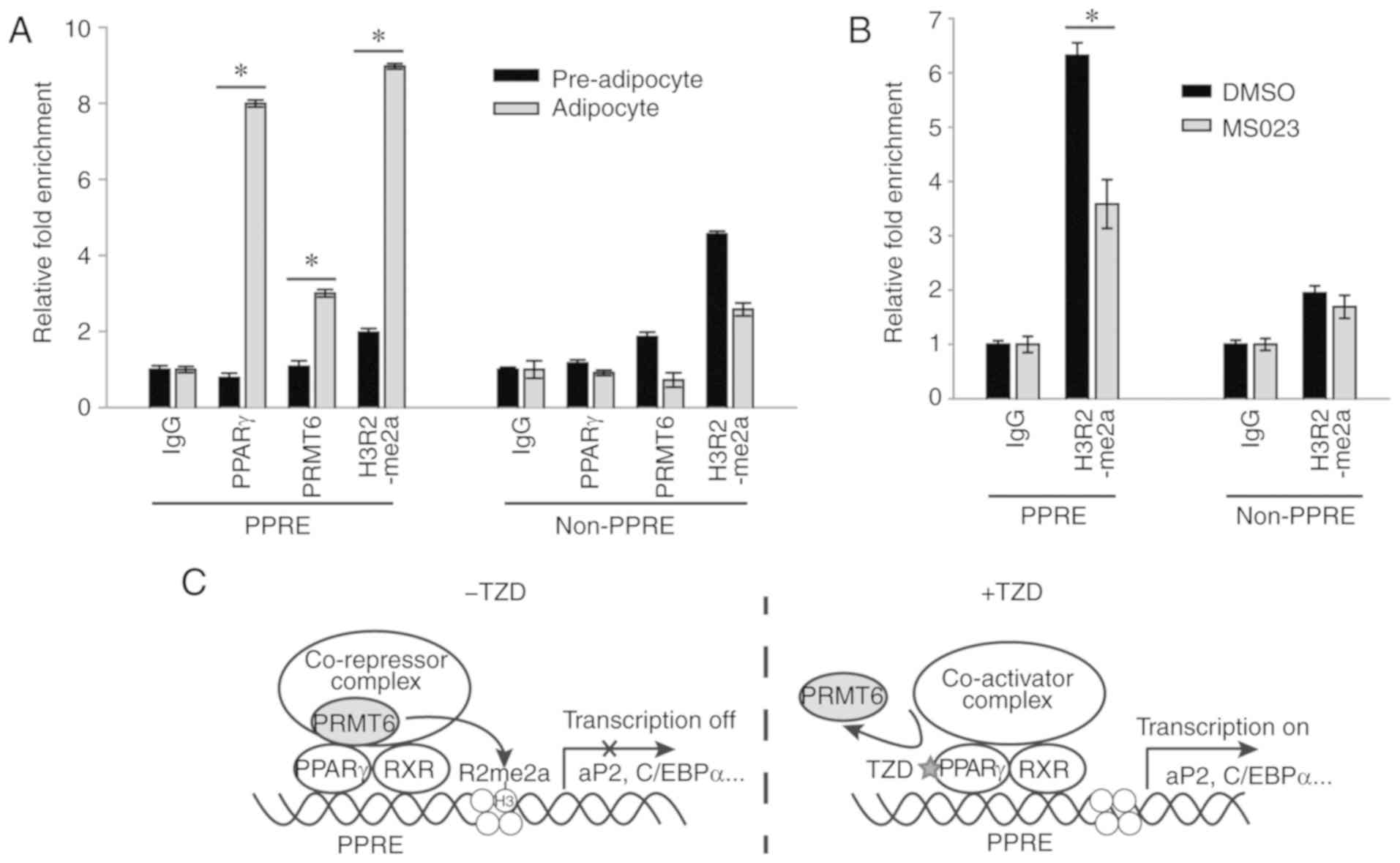 | Figure 4PRMT6 serves as a co-repressor of
PPARγ by generating the repressive mark H3R2me2a. (A) ChIP analysis
for PPARγ, PRMT6, or H3R2me2a in PPRE or adjacent non‑PPRE of aP2
gene. Pre‑adipocyte and differentiated (6 days) 3T3‑L1 cells were
fixed and subjected to ChIP assay. (B) During differentiation,
3T3-L1 cells were treated with MS023 (5 µM) for 6 days, and
the presence of H3R2me2a at the aP2 promoter site was measured by
ChIP assay. Error bars represent standard deviation (n=3;
*P<0.05). (C) A graphic model for the regulation of
PPARγ-mediated adipogenic gene expression by PRMT6. PRMT6 binds to
PPARγ in the absence of a ligand, which generates an H3R2me2a
repressive mark. When ligands, including TZD, bind to PPARγ, the
interaction between PRMT6 and PPARγ is weakened and the repressive
mark is decreased, resulting in the activation of adipogenic gene
transcription. PRMT6, protein arginine methyltransferase 6; PPARγ,
peroxisome proliferator-activated receptor γ; ChIP, chromatin
immunoprecipitation assay; H3R2me2a, arginine 2 on histone H3
asymmetric di-methylation; PPRE, PPAR-responsive regulatory
element; aP2, adipocyte Protein 2; C/EBPα, CCAAT-enhancer-binding
protein α; TZD, thiazolidinediones. |
Discussion
PRMT6 is one of the type I PRMTs present in the
nucleus, and the diverse physiological functions of PRMT6 are
suggestive of its importance (23,34). Epigenetically, it serves the
role of a transcriptional repressor by methylating the R2 residue
of histone H3 within chromatin (28,29). However, to the best of our
knowledge, the role of PRMT6 in the regulation of PPARγ, one of the
nuclear receptors, has not been identified previously. The present
study demonstrated that PRMT6 negatively regulated PPARγ
transactivity without affecting the activity of other isoforms
(PPARα and PPARβ/δ) and RXRα. Using a 3T3-L1 adipocyte cell
differentiation model, the inhibitory role of PRMT6 in adipogenesis
and differentiation that was predominantly controlled by PPARγ was
confirmed. The siRNA-mediated depletion of PRMT6 or the inhibition
of PRMT6 by MS023 promoted adipogenic differentiation, indicating
that the enzymatic activity of PRMT6 is essential for the
regulation of PPARγ function.
In the absence of the ligand, PPARγ is associated
with several corepressor molecules, including nuclear receptor
corepressor, silencing mediator of retinoid and thyroid hormone
receptor, paired amphipathic helix protein Sin3 and
receptor-interacting protein 140 (5,19,20,35). Subsequent to
binding with the ligand, PPARγ undergoes conformational change,
resulting in a decrease in its binding affinity to the
co-repressors. In the subsequent steps, a series of co-activators
combine together to form an active complex (19,21). The present
study demonstrated that PPARγ stably associated with PRMT6 and that
this binding was decreased in the presence of the PPARγ agonist
pioglitazone, suggesting that PRMT6 may serve as a typical
co-repressor to regulate PPARγ functions. This interaction was also
disrupted upon PRMT6 inhibition, indicative of the importance of
PRMT6 activity for the binding between these two proteins.
Several previous studies have supported the
regulation of PPARγ mediated by various post‑translational
modifications, including phosphorylation, SUMOylation,
ubiquitination and acetylation (36,37). The present study failed to
detect any evidence of the direct arginine methylation of PPARγ by
PRMT6. Instead, it was confirmed that PRMT6 performed H3R2me2a
methylation in the promoter region of PPARγ target genes during the
adipocyte differentiation process. Taken together, the data from
the present study suggested that there are two methods through
which PRMT6 represses PPARγ: i) PRMT6 directly interacts PPARγ; and
ii) PRMT6 generates the H3R2me2a repressive mark. Additional
studies are warranted to evaluate the formation of
PPARγ-PRMT6-H3R2me2a and other co-repressor complexes. At present,
2 previous studies have suggested that PRMT(s) may be involved in
the regulation of PPARγ and adipogenesis. Brunmeir and Xu (37)
suggested that coactivator-associated arginine methyltransferase 1
(CARM1)/PRMT4 served as a coactivator of PPARγ and promoted
adipocyte differentiation. Quinn et al (38) demonstrated
that PRMT5 promoted the expression of PPARγ target genes by binding
to, and subsequently methylating, chromatin histones at adipogenic
promoters. Therefore, PRMT6 as a co-repressor and CARM1/PRMT4 and
PRMT5 as co-activators of PPARγ may suggest that a series of
arginine methylations by PRMTs optimize the activation process of
PPARγ.
The activation of PPARγ by TZDs results in the
improvement in insulin sensitivity through the promotion of fatty
acid uptake into the adipose tissue, leading to an increase in the
production of adiponectin and decrease in the levels of
inflammatory mediators including tumor necrosis factor‑α,
plasminogen activator inhibitor-1 and interleukin-6 (38,39).
However, chronic activation PPARγ by TZD causes severe
side‑effects, including weight gain, fluid retention and
osteoporosis, thereby increasing the risk of congestive heart
failure, myocardial infarction, cardiovascular diseases and
all-cause mortality in patients (40,41). The present study
demonstrated that the functional effects of PPARγ were enhanced by
the PRMT6 selective inhibitor MS023, suggesting that PRMT6
inhibitors may serve potential roles to synergize the effects of
TZD in PPARγ-associated metabolic diseases.
Funding
The present study was supported by the Sookmyung
Women’s University Research Grant (grant no. 1-1503-0044) and a
grant from the National Research Foundation of Korea and the Korean
government Ministry of Science and ICT (grant no.
NRF-2015R1D1A1A01059600).
Availability of data and materials
All data used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. All materials used are included in Materials and
methods.
Authors’ contributions
JWH, YSS, and YKK conceived and designed the
experiments. JWH and YSS performed experiments. GUB, and SNK
provided experimental assistance and conceptual advice. JWH and YKK
supervised the experiments and wrote the manuscript. All authors
read and commented on the manuscript and agreed to the publication
of the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
PRMT
|
protein arginine methyltransferase
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
PPRE
|
PPAR-responsive regulatory element
|
|
RXRα
|
retinoid X receptor α
|
|
TZD
|
thiazolidinedione
|
|
PTM
|
post-translational modification
|
|
ChIP
|
chromatin immunoprecipitation
|
|
CARM1
|
coactivator-associated arginine
methyltransferase 1
|
Acknowledgments
Not applicable.
References
|
1
|
Ahmadian M, Suh JM, Hah N, Liddle C,
Atkins AR, Downes M and Evans RM: PPARγ signaling and metabolism:
The good, the bad and the future. Nat Med. 19:557–566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Willson TM, Lambert MH and Kliewer SA:
Peroxisome proliferator-activated receptor gamma and metabolic
disease. Annu Rev Biochem. 70:341–367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grygiel-Górniak B: Peroxisome
proliferator-activated receptors and their ligands: Nutritional and
clinical implications-a review. Nutr J. 13:172014. View Article : Google Scholar
|
|
4
|
Tyagi S, Gupta P, Saini AS, Kaushal C and
Sharma S: The peroxisome proliferator-activated receptor: A family
of nuclear receptors role in various diseases. J Adv Pharm Technol
Res. 2:236–240. 2011. View Article : Google Scholar
|
|
5
|
Janani C and Ranjitha Kumari BD: PPAR
gamma gene-a review. Diabetes Metab Syndr. 9:46–50. 2015.
View Article : Google Scholar
|
|
6
|
Braissant O, Foufelle F, Scotto C, Dauça M
and Wahli W: Differential expression of peroxisome
proliferator-activated receptors (PPARs): Tissue distribution of
PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology.
137:354–366. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Auboeuf D, Rieusset J, Fajas L, Vallier P,
Frering V, Riou JP, Staels B, Auwerx J, Laville M and Vidal H:
Tissue distribution and quantification of the expression of mRNAs
of peroxisome proliferator-activated receptors and liver X
receptor-alpha in humans: No alteration in adipose tissue of obese
and NIDDM patients. Diabetes. 46:1319–1327. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tachibana K, Yamasaki D, Ishimoto K and
Doi T: The role of PPARs in cancer. PPAR Res. 2008.102737:2008.
|
|
9
|
Koeffler HP: Peroxisome
proliferator‑activated receptor gamma and cancers. Clin Cancer Res.
9:1–9. 2003.PubMed/NCBI
|
|
10
|
Bermúdez V, Finol F, Parra N, Parra M,
Pérez A, Peñaranda L, Vílchez D, Rojas J, Arráiz N and Velasco M:
PPAR-gamma agonists and their role in type 2 diabetes mellitus
management. Am J Ther. 17:274–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Larsen TM, Toubro S and Astrup A:
PPARgamma agonists in the treatment of type II diabetes: Is
increased fatness commensurate with long‑term efficacy? Int J Obes
Relat Metab Disord. 27:147–161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HI and Ahn YH: Role of peroxisome
proliferator-activated receptor-gamma in the glucose-sensing
apparatus of liver and beta-cells. Diabetes. 53(Suppl 1): S60–S65.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blanquicett C, Roman J and Hart CM:
Thiazolidinediones as anti-cancer agents. Cancer Ther. 6:25–34.
2008.PubMed/NCBI
|
|
14
|
Joshi H, Pal T and Ramaa CS: A new dawn
for the use of thiazolidinediones in cancer therapy. Expert Opin
Investig Drugs. 23:501–510. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moore KJ, Rosen ED, Fitzgerald ML, Randow
F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman
BM and Freeman MW: The role of PPAR-gamma in macrophage
differentiation and cholesterol uptake. Nat Med. 7:41–47. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Villacorta L, Schopfer FJ, Zhang J,
Freeman BA and Chen YE: PPARgamma and its ligands: Therapeutic
implications in cardiovascular disease. Clin Sci (Lond).
116:205–218. 2009. View Article : Google Scholar
|
|
17
|
Schopfer FJ, Lin Y, Baker PR, Cui T,
Garcia-Barrio M, Zhang J, Chen K, Chen YE and Freeman BA:
Nitrolinoleic acid: An endogenous peroxisome proliferator-activated
receptor gamma ligand. Proc Natl Acad Sci USA. 102:2340–2345. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Behl T, Kaur I, Goel H and Kotwani A:
Implications of the endogenous PPAR-gamma ligand,
15-deoxy-delta-12, 14-pros-taglandin J2, in diabetic retinopathy.
Life Sci. 153:93–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Viswakarma N, Jia Y, Bai L, Vluggens A,
Borensztajn J, Xu J and Reddy JK: Coactivators in PPAR-regulated
gene expression. PPAR Res. 2010:2010. View Article : Google Scholar
|
|
20
|
Berger J and Moller DE: The mechanisms of
action of PPARs. Annu Rev Med. 53:409–435. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qi C, Zhu Y and Reddy JK: Peroxisome
proliferator-activated receptors, coactivators, and downstream
targets. Cell Biochem Biophys. 32:Spring;187–204. 2000. View Article : Google Scholar
|
|
22
|
Bedford MT and Clarke SG: Protein arginine
methylation in mammals: Who, what, and why. Mol Cell. 33:1–13.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blanc RS and Richard S: Arginine
methylation: The coming of age. Mol Cell. 65:8–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bedford MT: Arginine methylation at a
glance. J Cell Sci. 120:4243–4246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jain K, Jin CY and Clarke SG: Epigenetic
control via allosteric regulation of mammalian protein arginine
methyltransferases. Proc Natl Acad Sci USA. 114:10101–10106. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boulanger MC, Liang C, Russell RS, Lin R,
Bedford MT, Wainberg MA and Richard S: Methylation of Tat by PRMT6
regulates human immunodeficiency virus type 1 gene expression. J
Virol. 79:124–131. 2005. View Article : Google Scholar :
|
|
27
|
Frankel A, Yadav N, Lee J, Branscombe TL,
Clarke S and Bedford MT: The novel human protein arginine
N-methyltransferase PRMT6 is a nuclear enzyme displaying unique
substrate specificity. J Biol Chem. 277:3537–3543. 2002. View Article : Google Scholar
|
|
28
|
Guccione E, Bassi C, Casadio F, Martinato
F, Cesaroni M, Schuchlautz H, Lüscher B and Amati B: Methylation of
histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually
exclusive. Nature. 449:933–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hyllus D, Stein C, Schnabel K, Schiltz E,
Imhof A, Dou Y, Hsieh J and Bauer UM: PRMT6-mediated methylation of
R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev.
21:3369–3380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
El-Andaloussi N, Valovka T, Toueille M,
Steinacher R, Focke F, Gehrig P, Covic M, Hassa PO, Schär P,
Hübscher U and Hottiger MO: Arginine methylation regulates DNA
polymerase beta. Mol Cell. 22:51–62. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim SN, Choi HY, Lee W, Park GM, Shin WS
and Kim YK: Sargaquinoic acid and sargahydroquinoic acid from
Sargassum yezoense stimulate adipocyte differentiation through
PPARalpha/gamma activation in 3T3-L1 cells. FEBS Lett.
582:3465–3472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Eram MS, Shen Y, Szewczyk M, Wu H,
Senisterra G, Li F, Butler KV, Kaniskan HÜ, Speed BA, Dela Seña C,
et al: A potent, selective, and cell-active inhibitor of human type
I protein arginine methyltransferases. ACS Chem Biol. 11:772–781.
2016. View Article : Google Scholar
|
|
34
|
Yang Y and Bedford MT: Protein arginine
methyltransferases and cancer. Nat Rev Cancer. 13:37–50. 2013.
View Article : Google Scholar
|
|
35
|
Cohen RN: Nuclear receptor corepressors
and PPARgamma. Nucl Recept Signal. 4:e0032006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Beekum O, Fleskens V and Kalkhoven E:
Posttranslational modifications of PPAR-gamma: Fine-tuning the
metabolic master regulator. Obesity (Silver Spring). 17:213–219.
2009. View Article : Google Scholar
|
|
37
|
Brunmeir R and Xu F: Functional regulation
of PPARs through post‑translational modifications. Int J Mol Sci.
19:2018. View Article : Google Scholar
|
|
38
|
Quinn CE, Hamilton PK, Lockhart CJ and
McVeigh GE: Thiazolidinediones: Effects on insulin resistance and
the cardiovascular system. Br J Pharmacol. 153:636–645. 2008.
View Article : Google Scholar
|
|
39
|
Hauner H: The mode of action of
thiazolidinediones. Diabetes Metab Res Rev. 18(Suppl 2): S10–S15.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rizos CV, Elisaf MS, Mikhailidis DP and
Liberopoulos EN: How safe is the use of thiazolidinediones in
clinical practice? Expert Opin Drug Saf. 8:15–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nesto RW, Bell D, Bonow RO, Fonseca V,
Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith
S, et al: Thiazolidinedione use, fluid retention, and congestive
heart failure: A consensus statement from the American Heart
Association and American Diabetes Association. Diabetes Care.
27:256–263. 2004. View Article : Google Scholar
|














