Introduction
Oxaliplatin (OXA), a third-generation platinum
antitumor drug, is widely used for the treatment of
gastrointestinal cancers, such as colorectal, gastric, liver and
pancreatic cancer (1). As the
first-line basic chemotherapeutic drug recommended by the National
Comprehensive Cancer Network guidelines, OXA is currently used for
the treatment of gastric and colorectal cancers (1,2).
However, despite its usefulness, OXA-based chemotherapy may cause
chemotherapy-associated liver injury. Rubbia-Brandt et al
(3) first reported that different
degrees of hepatic sinus injuries occurred in 78% of colorectal
cancer patients who received OXA-based chemotherapy. OXA-based
chemotherapy-induced hepatic injury has been reported in as many as
19-52% of patients with various types of tumors (4). This injury may manifest as hepatic
sinusoidal expansion, intrahepatic sinus platelet aggregation,
hepatocyte atrophy and necrosis, hepatic steatosis,
steatohepatitis, intrahepatic sinus hemorrhage and sinusoidal
obstruction syndrome (3,4).
At present, very little is known on the
pathophysiological mechanisms that underlie OXA-induced liver
injury. OXA has been confirmed to cause liver oxidative stress
response through certain known mechanisms. Robinson et al
(5) reported that oxidative
stress-related genes (Mt1, HO1 and SOD3) were upregulated in the
liver following OXA chemotherapy, indicating that oxidative stress
plays an important role in OXA-induced liver injury. By generating
reactive oxygen species (ROS), OXA causes a series of reactions,
such as oxidative injury of normal hepatocyte mitochondria, as well
as injury, falloff and local edema of sinusoidal endothelial cells,
thereby causing chemotherapy-related liver injury (6). Our previous study also demonstrated
that oxidative stress response plays an important role in
OXA-induced acute liver injury (7).
Non-alcoholic fatty liver disease (NAFLD), which
comprises a spectrum of liver diseases ranging from steatosis to
non-alcoholic steatohepatitis and cirrhosis, is a common hepatic
condition (8) that it is
frequently associated with visceral obesity, dyslipidemia, insulin
resistance and type-2 diabetes mellitus (8,9).
NAFLD is characterized by a low level of hepatic oxidative stress
response, chronic inflammation and fibrosis (10). It was previously reported that,
once chemotherapy-induced liver injury occurs in patients with
NAFLD, it may cause adverse consequences, such as chemotherapy
delay, dose reduction, or even cessation, as well as a higher risk
of liver failure and death compared with patients without NAFLD
(11). There is currently no
standard of care in place for OXA-induced liver injury in patients
with NAFLD. One of the hepatoprotective drugs used by clinicians is
reduced glutathione (GSH). However, whether GSH treatment exerts
protective effects against OXA-induced liver injury in NAFLD
remains unclear.
OXA itself may cause hepatic oxidative stress
response; therefore, whether OXA aggravates the already existing
hepatic oxidative stress, inflammation and fibrosis in NAFLD
remains unknown. The objectives of the present study were to
investigate whether OXA chemotherapy affects the existing hepatic
oxidative stress, inflammation and fibrosis in an NAFLD mouse
model, and to investigate the protective action of GSH against
OXA-induced liver injury in NAFLD.
Materials and methods
Ethics statement
All animal studies were performed according to the
guidelines of the Chinese Council on Animal Care and were approved
by the Affiliated Tumor Hospital of Guangxi Medical University
(Nanning, China) Committees on Animal Experimentation.
Drugs and reagents
OXA for injection (cat. no. 13092615; Jiangsu
Hengrui Medicine Co., Ltd.); GSH for injection (Chongqing Yaoyou
Medicine Co., Ltd.); alanine aminotransferase (ALT) kit (HuiLi
Biotech Co., Ltd.); aspartate aminotransferase (AST) kit, GSH kit,
superoxide dismutase (SOD) kit, glutathione peroxidase (GSH-px)
kit, malondialdehyde (MDA) kit and total protein quantification kit
[bicinchoninic acid (BCA) method] (all from Jiangcheng
Bioengineering Institute).
Animal experiments
BALB/cJ mice, aged 4-6 weeks, were purchased from
Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice
were maintained in polypropylene cages (n=6 per cage) in an
air-conditioned room (25±1°C, relative humidity 50±20%, 12-h
light/dark cycle). After 1 week of acclimatization, the NAFLD mice
were fed a high-fat diet (D12451, 45% energy from fat; Research
Diets, Inc.) for 12 weeks. At the end of the 12 weeks, the animals
were administered 8 mg/kg OXA (0.5 ml) via intraperitoneal (i.p.)
injection for 3 days; the NAFLD control group was administered
vehicle (5% glucose, 0.5 ml) for 3 days. The drug regimen was based
on previously published studies (7). Normal control mice were fed a
standard chow diet for 12 weeks, and were then administered vehicle
(5% glucose, 0.5 ml) for 3 days. The animals were anesthetized with
ketamine/xylazine (100/15 mg/kg, i.m.), and blood and liver tissue
were collected prior to sacrifice by exsanguination. Pieces of
liver tissue were snap-frozen in liquid nitrogen, or were fixed in
10% neutral-buffered formalin. To assess the impact of GSH
treatment, mice (n=10 per group) were treated with OXA (8 mg/kg,
i.p.) for 3 days, and with GSH (400 mg/kg, i.p.) 30 min prior to
the first OXA injection and once daily until 3 days after the final
OXA dose. The mice were euthanized via deep anesthesia with
isoflurane 3 days after the final dose of GSH was administered.
Blood and liver samples were collected for further analysis. The
experimental design is depicted in Fig. 1.
Histological examination
Liver tissues were fixed in 4% paraformaldehyde,
embedded in paraffin and cut into 4-µm sections.
Histological assessment of the liver tissue sections was performed
following hematoxylin and eosin (H&E) and Masson's trichrome
staining. The presence of liver injury was assessed by two
specialist liver pathologists who were blinded to the grouping.
Liver fibrosis was examined by Masson's trichrome staining. The
morphometric assessment of liver fibrosis was performed using a
fully automated Leica image processor with automated stage and
Leica Quin software 2004 (Leica Microsystems GmbH). The mean
fibrotic area was calculated from 15-18 areas per liver section
analyzed at a magnification of ×200.
Flow cytometry and intracellular cytokine
staining
Mononuclear cells (MNCs) were obtained from the
mouse livers on the indicated days. Intracellular cytokine staining
was performed as previously described (12). Briefly, MNCs were stimulated with
phorbol myristate acetate (cat. no. P1585, 50 ng/ml; Sigma-Aldrich;
Merck KGaA), Golgistop (cat. no. 554724, 2.0 µM; BD
Biosciences), and ionomycin (cat. no. 407952, 1 µM;
Calbiochem) for 5 h. The cells were stained with phycoerythrin
(PE)cy5.5-anti-CD3 (cat. no. 152312), and then fixed in 4%
paraformaldehyde (cat. no. 420801; both BioLegend). Next, the cells
were permeabilized with phosphate-buffered saline (PBS; cat. no.
SH30256.01B; HyClone) supplemented with 0.2% saponin and 0.05%
sodium azide, and then stained with PE-anti-interleukin (IL)-4
(cat. no. 504104), PE-anti-IL-10 (cat. no. 505007), PE-anti-IL-17A
(cat. no. 506903), PE-anti-interferon (IFN)-γ (cat. no. 113603),
PE-anti-tumor necrosis factor (TNF)-α (cat. no. 506305), or an
isotype-matched, irrelevant control Ab (all from BioLegend).
Stained cells were analyzed using a BD FACSAria flow cytometer (BD
Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-PCR) analysis
Total RNA isolation and cDNA synthesis were
conducted using standard methods. Total RNA was extracted using
RNeasy Mini kits (Qiagen) and reverse-transcribed into cDNA using
SuperScript III Reverse Transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.). The gene-specific primers used are listed in
Supplementary Table S1. rt-qpcr
was performed with abi prism 7500 real-time pcr system (applied
biosystems; thermo fisher scientific, inc.) with 1x sybr-green
universal pcr mastermix (takara bio, inc.). transcript levels were
calculated according to the 2−ΔΔcq method, normalized to
the expression of gapdh, and expressed as fold change compared with
the control. all samples were run in duplicate to ensure
amplification integrity.
Analysis of oxidative stress
parameters
Proteins were extracted from whole liver tissues
using RIPA buffer and quantified using the Bradford assay (Nanjing
Jiangcheng Bioengineering Institute). The SOD, MDA and GSH-Px
content of liver tissues was determined using the kits obtained
from Nanjing Jiangcheng Bioengineering Institute, according to the
manufacturer's protocols. To estimate hepatic ROS level, liver
tissues were harvested and immediately homogenized in PBS using a
Teflon homogenizer (Tissue-Tearor; BioSpec Products Inc.). Briefly,
50 µl liver homogenate was mixed with 4.85 ml of 100 mmol/l
potassium phosphate buffer (cat. no. 700621-5; Cayman Chemical) and
incubated with 2′,7′-dichlorofluorescin (DCF) diacetate
(Sigma-Aldrich; Merck KGaA) in methanol at a final concentration of
5 µmol/l for 15 min at 37°C. The hepatic ROS level was
determined as the amount of 2′,7′-dichlorofluorescein (DCF)
quantified from a DCF standard curve. The BCA protein assay kit
(Thermo Fisher Scientific, Inc.) was used to measure protein
concentration. The fluorescence of DCF was measured with a
Varioskan spectrophotometer (Thermo Fisher Scientific, Inc.) at
excitation/emission wavelengths 480/530 nm.
Statistical analysis
The results are expressed as means ± standard
deviation. Cumulative survival time was calculated using the
Kaplan-Meier method and was analyzed by the log-rank test. Other
datasets were evaluated by ANOVA with a post hoc Tukey's test for
multiple comparisons. All statistical analyses were performed using
SPSS 10 (SPSS Inc.) and P<0.05 was considered to indicate
statistically significant differences.
Results
OXA chemotherapy induces acute liver
injury in NAFLD mice
Balb/c mice were first fed a high-fat diet for 12
weeks to establish NAFLD and were then treated with OXA (i.p.) once
per day for 3 days (Fig. 1).
Survival curve analysis revealed that death occurred 2 days after
the administration of the final dose of OXA, and the survival rate
in this group was 50% (5/10; Fig.
2A). The weight of the OXA-treated mice was significantly lower
compared with that of the NAFLD control group (Fig. 2B; P<0.05). To evaluate
OXA-induced liver injury in NAFLD mice, the serum AST and alanine
aminotransferase (ALT) levels were assessed. Compared with the
control mice, the OXA-treated mice exhibited significantly elevated
serum ALT and AST levels following OXA treatment (Fig. 2C; P<0.05). Coinciding with the
trend of serum ALT and AST levels, more severe inflammatory cell
infiltration, hepatocyte ballooning and necrosis were observed in
the OXA-treated mice (Fig. 2D).
Moreover, the lipid vacuoles in the OXA group were more abundant
and larger compared with those observed in the control group
(Fig. 2D). Additionally, no
significant hepatic sinus expansion was observed in the liver
tissues of the OXA group mice. These results indicated that OXA
chemotherapy causes acute liver injury in NAFLD mice.
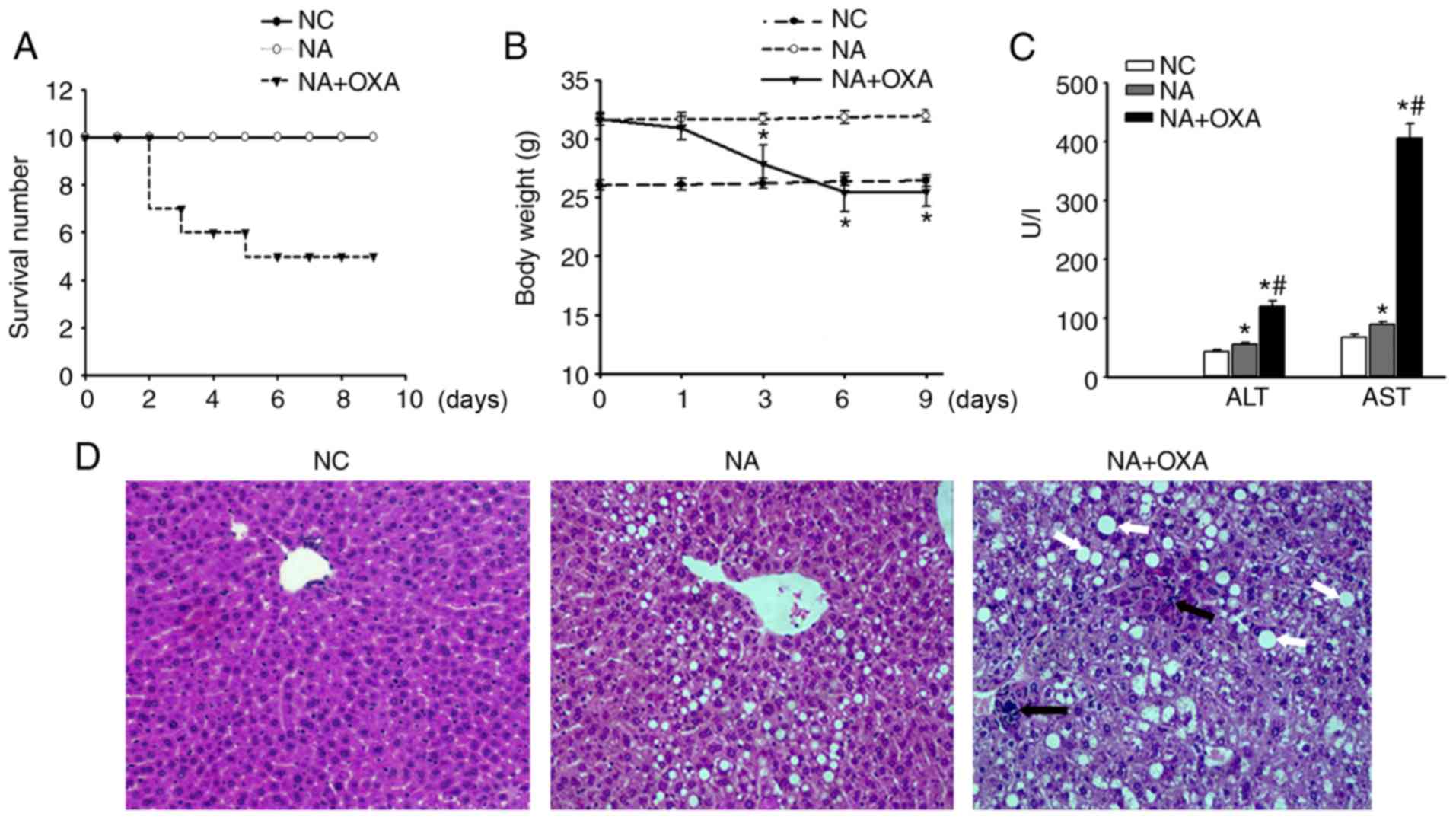 | Figure 2OXA-induced acute liver injury in
NAFLD mice. (A) Survival rates of the three groups. No mice died in
either the NC or the NA groups, and the survival rate across the 9
days was the same in both groups. (B) Changes in body weight were
observed in the three study groups. (C) ALT and AST serum levels 3
days after the administration of the final dose of OXA. The results
are presented as the mean ± standard deviation of five mice in each
group. *P<0.05 compared with the NC group;
#P<0.05 compared with the NA group. (D)
Histopathological examination of liver tissues of the control
groups and 3 days after the administration of the final dose of OXA
(H&E staining; original magnification, ×100). Black arrows
indicate inflammatory infiltration, and white arrows indicate large
lipid vacuoles. OXA, oxaliplatin; NAFLD, non-alcoholic fatty liver
disease; ALT, alanine aminotransferase; AST, aspartate
aminotransferase; NC, normal control; NA, NAFLD control; H&E,
hematoxylin and eosin. |
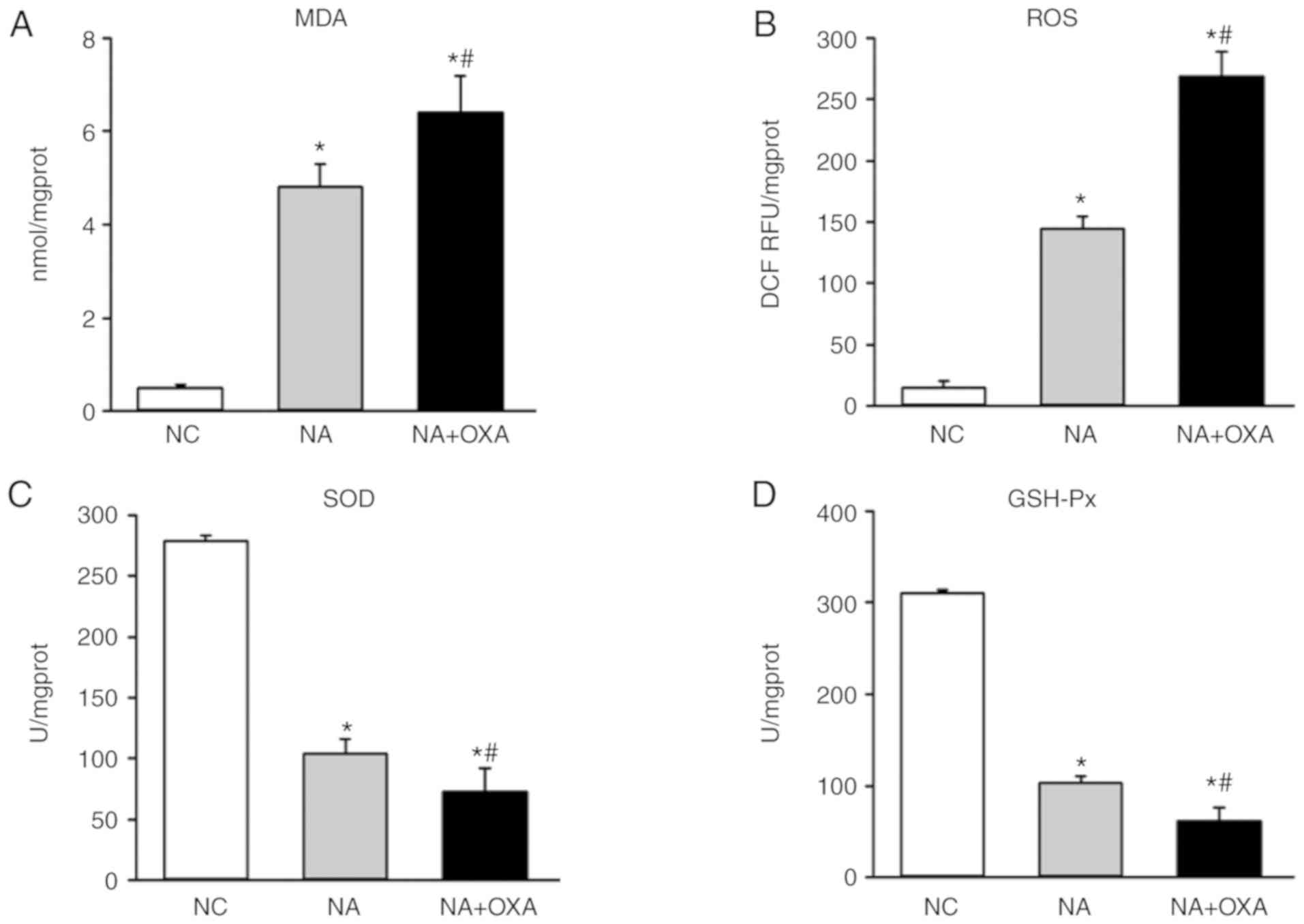
OXA chemotherapy aggravates intrahepatic
oxidative stress response in NAFLD mice
High-fat diet may induce oxidative stress in the
NAFLD liver (13). The findings
of the present study indicated that OXA-induced liver injury is
associated with oxidative stress. To observe the effects of OXA on
intra-hepatic oxidative stress in NAFLD mice, the levels of hepatic
MDA, ROS, SOD and GSH-Px were measured as an indication of the
redox status of NAFLD mice following OXA treatment. The levels of
the oxidative indicators MDA (Fig.
3A) and ROS (Fig. 3B) in the
liver tissues of the NAFLD mice were found to be significantly
increased following OXA injection (P<0.05). By contrast, the
levels of the antioxidative indices SOD (Fig. 3C) and GSH-Px (Fig. 3D) in the liver tissues of NAFLD
mice decreased following OXA treatment. The liver tissues exhibited
marked alterations in the expression of redox status indicators
following OXA injection, indicating that OXA aggravated the
intrahepatic oxidative stress response in NAFLD mice.
OXA aggravates intrahepatic inflammatory
response in NAFLD mice
Subclinical inflammation has also been reported as
one of the basic changes that occur in NAFLD (8). In order to investigate the effect of
OXA chemotherapy on intrahepatic inflammation in NAFLD mice, flow
cytometry was performed to detect the expression of inflammatory
cytokines in intrahepatic T lymphocytes in NAFLD mice following OXA
chemotherapy. The percentage of TNF-α-, IFN-γ- and IL-17-producing
T cells significantly increased 6 days after the administration of
the first dose of OXA (P<0.05), whereas no significant change in
the percentage of IL-6-producing T cells was observed (Fig. 4A and B, P>0.05). Similarly, the
TNF-α, IFN-γ and IL-17 mRNA levels in the OXA group were
significantly higher compared with those in the control group
(P<0.05; Fig. 4C). These
results indicated that OXA may aggravate intrahepatic inflammation
in NAFLD mice.
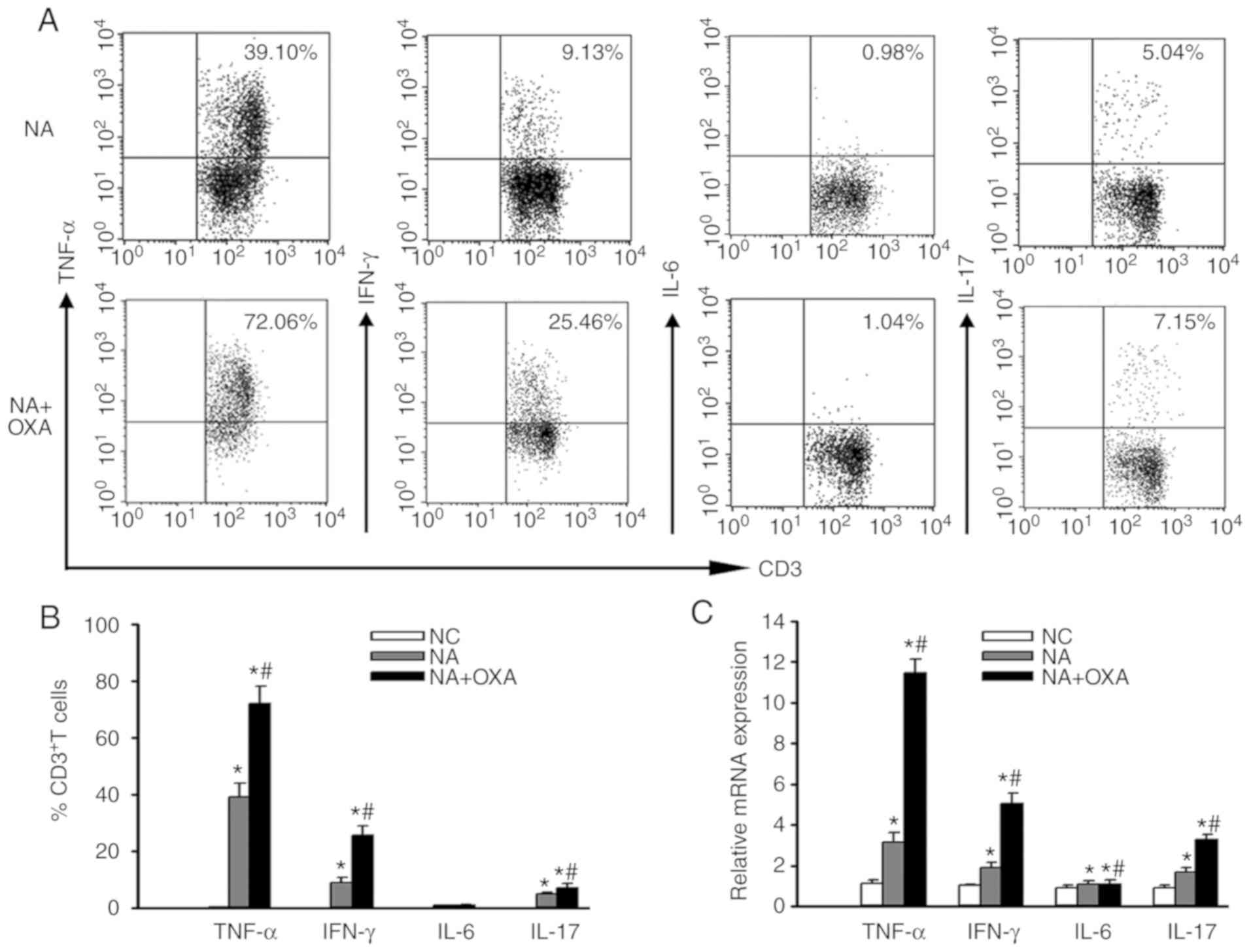 | Figure 4Effects of OXA on inflammatory
cytokine expression. (A) Representative staining of intracellular
TNF-α, IFN-γ, IL-6 and IL-17 in hepatic CD3+ T cells
from the NA + OXA group 3 days after the administration of the
final dose of OXA. (B) Percentages of CD3+ T cells
expressing TNF-α, IFN-γ, IL-6 and IL-17 from the livers of each
group of mice 3 days after the administration of the final dose of
OXA. (C) RT-PCR was employed to investigate the mRNA levels of
TNF-α, IFN-γ, IL-6 and IL-17 in the liver tissues of each group of
mice 3 days after the administration of the final dose of OXA. Data
represent the mean ± standard deviation of five mice in each group.
*P<0.05 compared with the NC group;
#P<0.05 compared with the NA group. OXA, oxaliplatin;
TNF, tumor necrosis factor; IFN, interferon; IL, interleukin;
RT-PCR, reverse transcription-polymerase chain reaction; NC, normal
control; NA, NAFLD control. |
OXA chemotherapy promotes hepatic
fibrosis in NAFLD mice
Next, the effect of OXA on hepatic fibrosis in NAFLD
mice was investigated. Hepatopathological analysis did not reveal
distinct liver fibrosis 6 days after administration of the first
dose of OXA (Fig. 2D). Masson's
trichrome staining revealed small amounts of collagen fiber
deposits in the liver tissues of NAFLD mice prior to OXA treatment,
but increased collagen fiber deposition in the liver 6 days after
administration of the first dose of OXA; these changes were more
prominent 10 days after OXA treatment, as distinct circumferential
collagen fiber deposition was observed in hepatocytes (Fig. 5A and B). Consistent with these
findings, the levels of the liver fibrosis index transforming
growth factor (TGF)-β significantly increased 6 days after
administration of the first dose of OXA (Fig. 5C). There was also a modest
increase in α-smooth muscle actin (SMA) and tissue inhibitor of
metallopeptidase (TIMP)-1 expression 6 days after administration of
the first dose of OXA, but these differences did not reach
statistical significance (Fig. 5D and
E). However, liver fibrosis was aggravated, which was confirmed
by a significant increase in the mRNA levels of TGF-β (4.85-fold;
P<0.01), α-SMA (2.23-fold; P<0.05) and TIMP-1 (2.64-fold;
P<0.01) 10 days after OXA treatment (Fig. 5C-E). These results confirmed that
OXA promotes hepatic fibrosis in NAFLD mice.
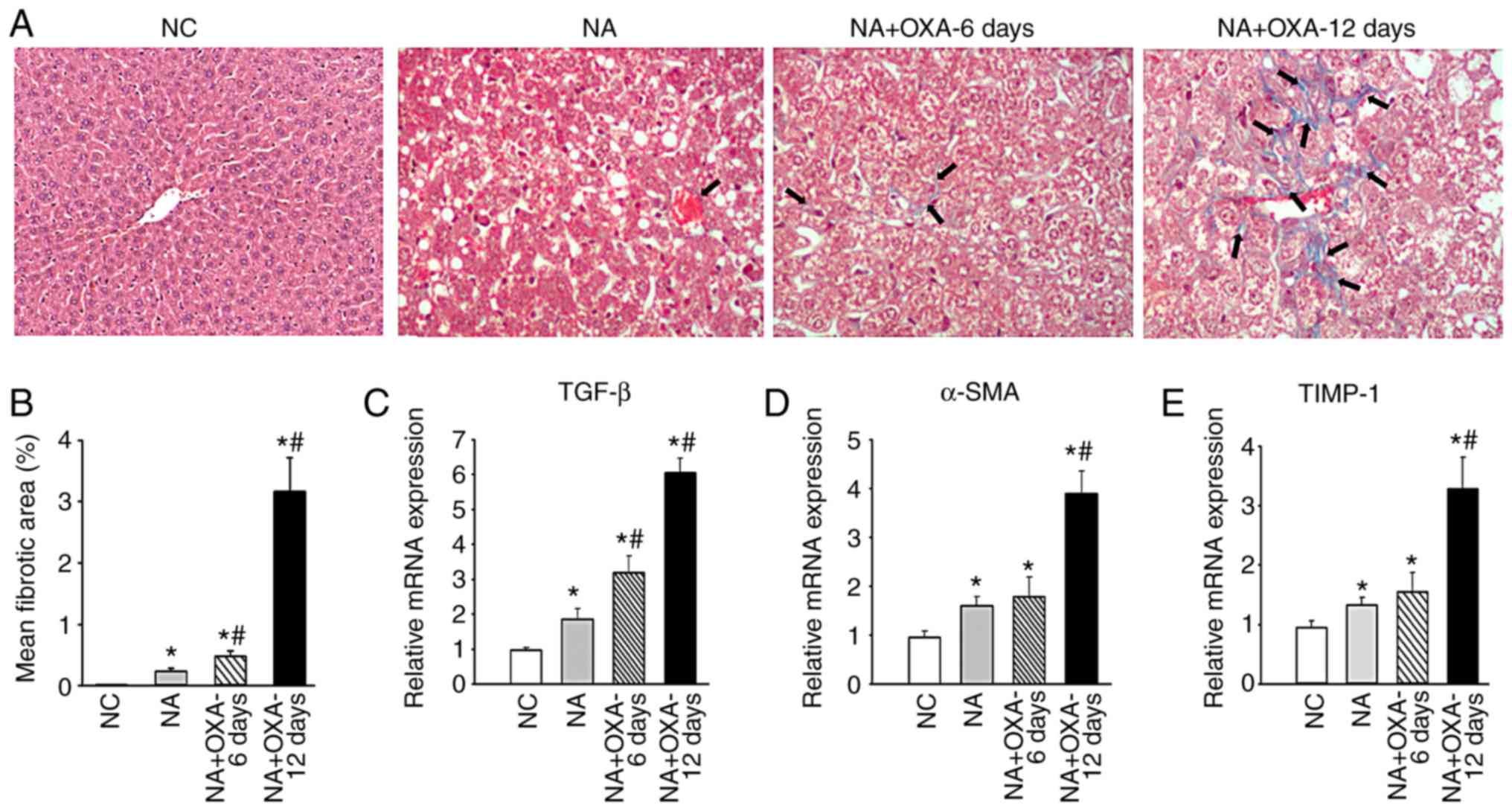 | Figure 5Effects of OXA on hepatic fibrosis in
NAFLD mice. (A) Masson's trichrome staining on the liver specimens
in groups NC, NA and NA + OXA (at 6 and 12 days after the
administration of the first dose of OXA). Black arrows indicate
areas of collagen deposition. (B) Morphometric analysis of Masson's
trichrome staining of the groups in (A). (C-E) mRNA levels of (C)
TGF-β, (D) α-SMA and (E) TIMP-1 in liver tissues analyzed by
RT-PCR. Results are presented as the means ± standard deviation
from five mice in each group. *P<0.05 compared with
the NC group; #P<0.05 compared with the NA group.
OXA, oxaliplatin; NAFLD, non-alcoholic fatty liver disease; NC,
negative control; NA, NAFLD control; TGF, transforming growth
factor; SMA, smooth muscle actin; TIMP, tissue inhibitor of
metallopeptidase; RT-PCR, reverse transcription-quantitative
polymerase chain reaction. |
GSH attenuates OXA-induced acute liver
injury in NAFLD mice
Our previous research demonstrated that treatment
with GSH attenuates OXA-induced acute liver injury in normal mice
(7). In the present study, to
test whether GSH exerts a protective effect against OXA-induced
acute liver injury in NAFLD mice, OXA-treated NAFLD mice underwent
GSH treatment 30 min prior to each OXA injection for 3 days, as
well as for 3 consecutive days after the final dose of OXA (NA +
OXA + GSH group) (Fig. 1).
Treatment with GSH did not improve the survival rate of OXA-treated
NAFLD mice (Fig. 6A). However,
hepatopathological analysis indicated severe swelling and necrosis
of hepatocytes, destruction of the hepatic architecture and
inflammatory cell infiltration in OXA-treated NAFLD mice, which was
attenuated by GSH treatment (Fig.
6B). Additionally, there were fewer and smaller lipid vacuoles
in the hepatocytes of the NA + OXA + GSH group (Fig. 6B). Consistent with histological
findings, treatment with GSH inhibited OXA-induced increases in the
serum levels of ALT (Fig. 6C;
P<0.05) and AST (Fig. 6C;
P<0.05). GSH treatment did not alter the serum levels of
albumin, coagulation times or total proteins (data not shown).
These results indicated that GSH attenuates OXA-induced acute liver
injury in NAFLD mice.
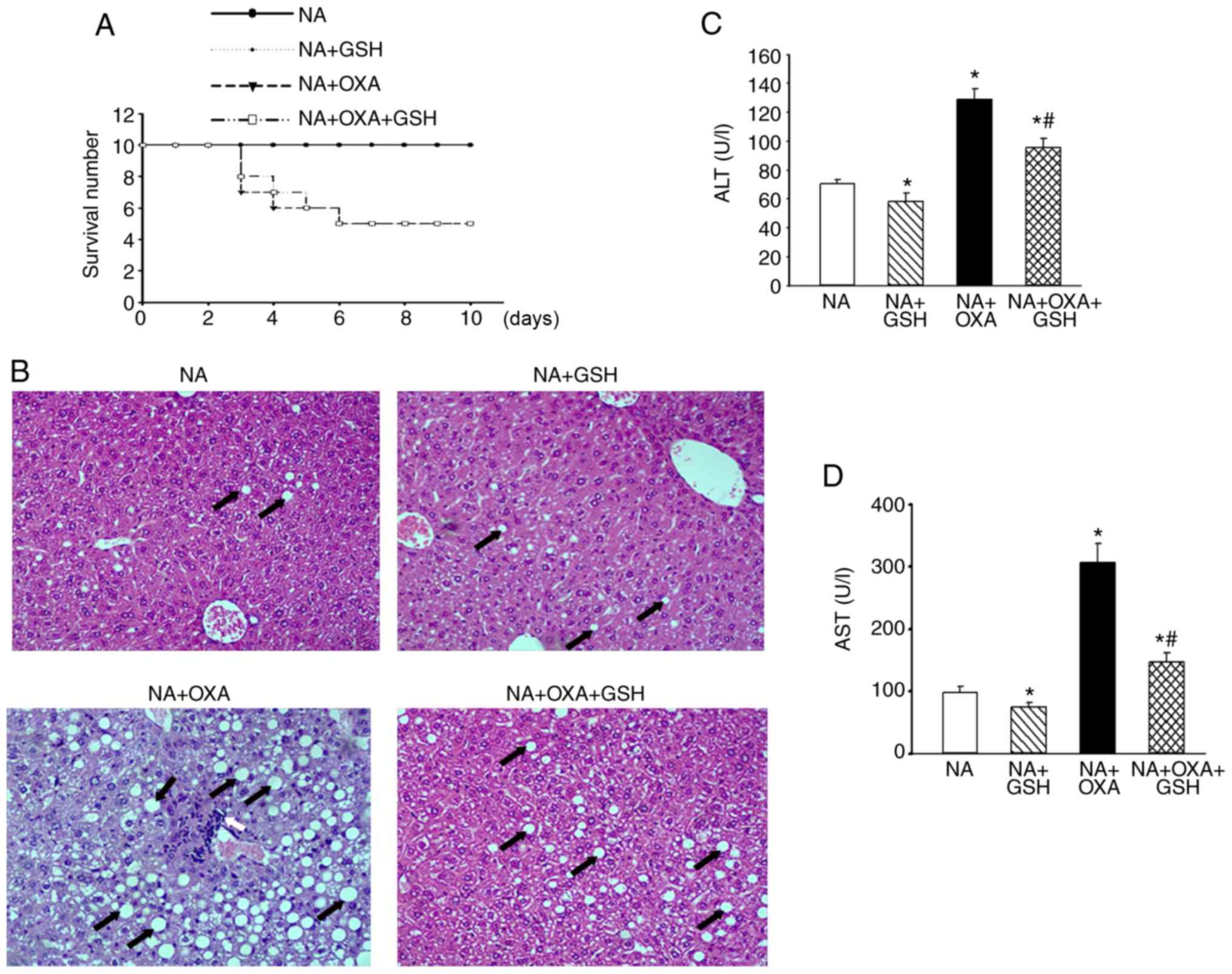 | Figure 6Treatment with GSH attenuates
OXA-induced acute liver injury in NAFLD mice. NAFLD mice were
randomly classified into four groups: i) NAFLD mice treated with
OXA for 3 days (NA + OXA); ii) NAFLD mice treated with OXA for 3
days and with GSH every day from the first day of OXA
administration until the end of the experiment (NA + OXA + GSH);
iii) NAFLD mice treated with GSH every day (NA + GSH); and iv)
NAFLD mice that received 5% glucose for 3 days, serving as control
(NA). (A) The survival rates of the four groups were assessed. No
mice died in either the NA or NA + GSH groups, and the survival
rate across the 10 days was the same in both group. (B) Liver
histopathology was examined in each group 6 days after the final
dose of OXA (H&E staining, original magnification, ×100). White
arrow indicates inflammatory infiltration, and black arrows
indicate lipid vacuoles. The serum (C) ALT and (D) AST levels of
each group were evaluated 6 days after the final dose of OXA. The
results are presented as the mean ± standard deviation from five
mice in each group. *P<0.05 compared with the NA
group; #P<0.05 compared with the NA + OXA group. GSH,
reduced glutathione; OXA, oxaliplatin; NAFLD, non-alcoholic fatty
liver disease; H&E, hematoxylin and eosin; ALT, alanine
aminotransferase; AST, aspartate aminotransferase; NA, NAFLD
control. |
GSH attenuates OXA-aggravated oxidative
stress and hepatic inflammation in NAFLD mice
To test whether GSH exerts a protective effect
against the oxidative stress aggravated by OXA in NAFLD mice,
OXA-administered NAFLD mice underwent GSH treatment. The analysis
of oxidative stress indices indicated that the MDA (Fig. 7A; P<0.05) and ROS (Fig. 7B; P<0.05) levels in the liver
tissues of the NA + OXA + GSH group were significantly lower
compared with those in non-GSH-treated mice (NA + OXA group)
following OXA injection. The levels of SOD (Fig. 7C; P<0.05) and GSH-Px (Fig. 7D; P<0.05) in the liver tissues
of the NA + OXA + GSH group were significantly higher compared with
those in the NA + OXA group. These results indicated that GSH
treatment may mitigate the OXA-aggravated intra-hepatic oxidative
stress response in NAFLD mice following OXA chemotherapy.
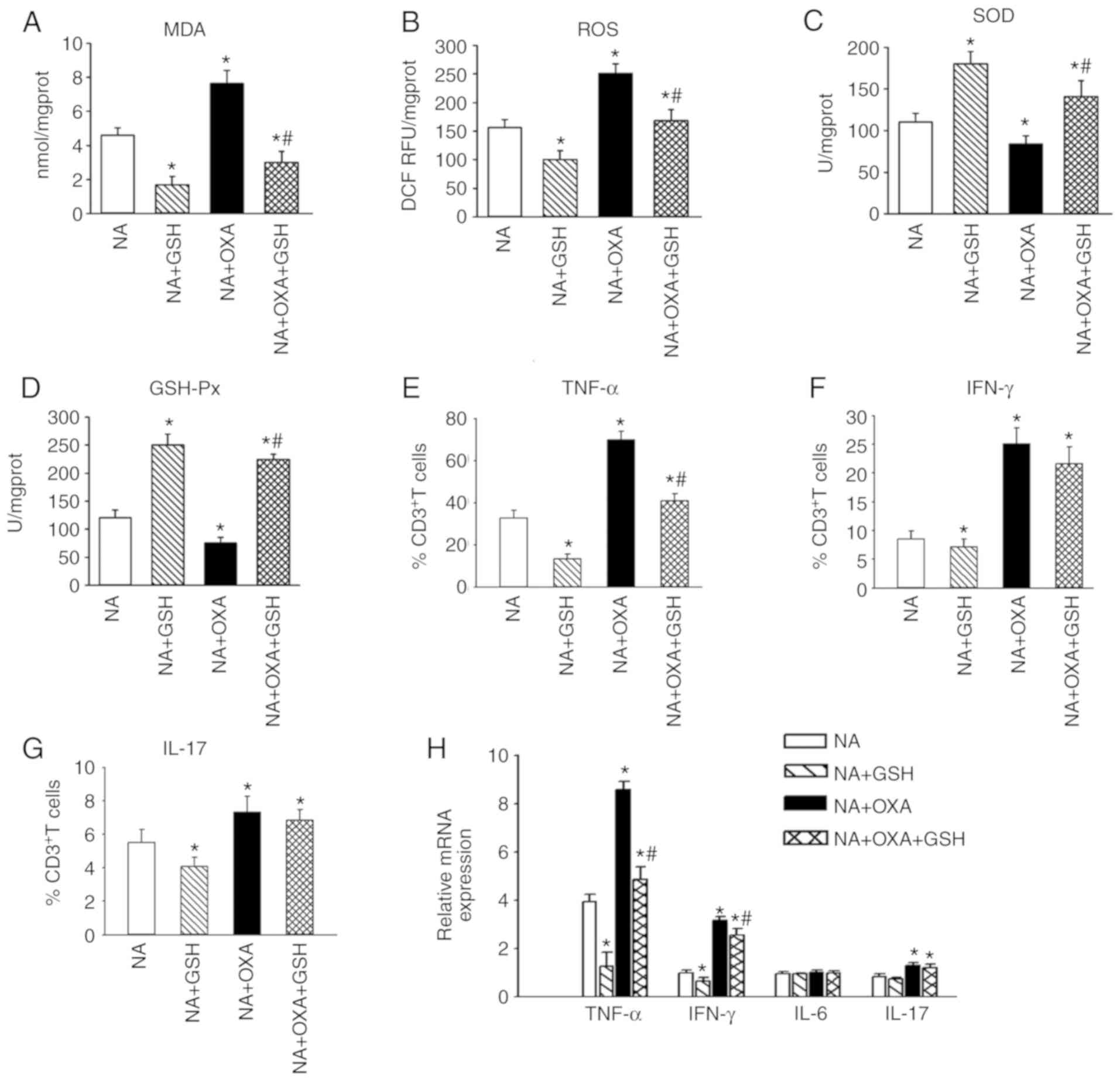 | Figure 7GSH attenuates OXA-aggravated hepatic
oxidative stress and inflammation in NAFLD mice. (A-D) RT-PCR was
employed to investigate the mRNA levels of (A) MDA, (B) ROS, (C)
SOD and (D) GSH-Px in the liver tissues of each group 9 days after
the first dose of OXA. (E-G) Flow cytometry detection of the
intracellular cytokines (E) TNF-α, (F) IFN-γ and (G) IL-17 was
performed on CD3+ T cells from each group 9 days after
the first dose of OXA. (H) mRNA levels of TNF-α, IFN-γ and IL-17 in
the liver tissues of each group 3 days after the final dose of OXA.
The results are presented as the mean ± standard deviation from
five mice in each group. *P<0.05 compared with the NA
group; #P<0.05 compared with the NA + OXA group. GSH,
reduced glutathione; OXA, oxaliplatin; NAFLD, non-alcoholic fatty
liver disease; RT-PCR, reverse transcription-polymerase chain
reaction; MDA, malondialdehyde; ROS, reactive oxygen species; SOD,
superoxide dismutase; GSH-Px, glutathione peroxidase; TNF, tumor
necrosis factor; IFN, interferon; IL, interleukin; NA, NAFLD
control. |
The findings of the present study confirmed that OXA
can increase the expression of the inflammatory cytokines TNF-α,
IFN-γ and IL-17 in the livers of NAFLD mice. Subsequently, the
expression of these cytokines was detected in liver tissues
following GSH treatment. It was observed that treatment with GSH
reduced the percentage of hepatic TNF-α+ T cells in the
NA + OXA + GSH group (Fig. 7E;
P<0.05), whereas no significant difference in the proportion of
hepatic IFN-γ+ and IL-17+ T cells was
observed compared with the NA + OXA group (Fig. 7F and G; P>0.05). Similarly,
treatment with GSH resulted in lower TNF-α mRNA levels in the liver
tissues of the NA + OXA + GSH group, whereas no significant
differences in the IFN-γ, IL-6 and IL-17 mRNA levels were observed
compared with those in the NA + OXA group (Fig. 7H). These results indicated that
GSH treatment suppressed the OXA-aggravated inflammation by
reducing the expression of the inflammatory cytokine TNF-α, but did
not affect the expression of IFN-γ, IL-6 or IL-17.
GSH does not attenuate OXA-aggravated
liver fibrosis in NAFLD mice
Whether GSH treatment attenuates OXA-aggravated
liver fibrosis in NAFLD mice was next investigated. GSH treatment
did not decrease the mean fibrotic area in the liver tissues of the
NA + OXA + GSH group compared with the NA + OXA group (Fig. 8A and B; P>0.05). Additionally,
treatment with GSH resulted in a mild decrease in the levels of
α-SMA, TGF-β and TIMP-1 compared with the NA + OXA group, but the
differences did not reach statistical significance (Fig. 8C-E; P>0.05). These results
indicated that GSH treatment did not mitigate hepatic fibrosis in
NAFLD mice after OXA chemotherapy.
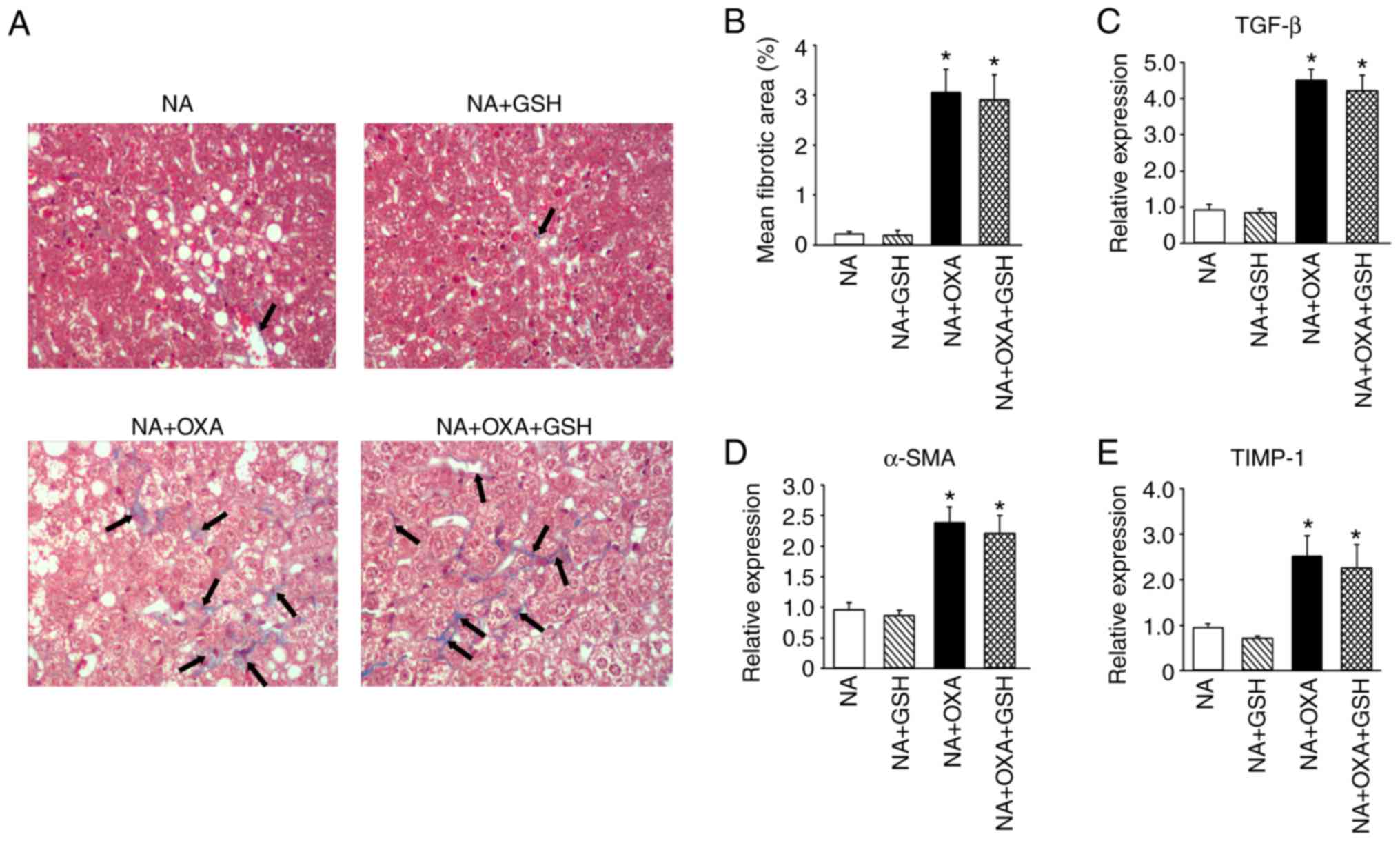 | Figure 8GSH does not attenuate OXA-aggravated
liver fibrosis in NAFLD mice. (A) Masson's trichrome staining was
performed on liver specimens of NAFLD mice from each group
(original magnification, ×100). Black arrows indicate areas of
collagen deposition. (B) Morphometric analysis of Masson's
trichrome staining of the groups in (A). (C-E) mRNA levels of (C)
TGF-β, (D) α-SMA and (E) TIMP-1 in liver tissues analyzed by
RT-PCR. The results are presented as the means ± standard deviation
from five mice in each group. *P<0.05 compared with
the NA group. OXA, oxaliplatin; NAFLD, non-alcoholic fatty liver
disease; GSH, reduced glutathione; NA, NAFLD control; TGF,
transforming growth factor; SMA, smooth muscle actin; TIMP, tissue
inhibitor of metallopeptidase; RT-PCR, reverse
transcription-quantitative polymerase chain reaction. |
Discussion
There are currently several challenges associated
with the clinical studies attempting to investigate the effect of
OXA chemotherapy on the progression of NAFLD. First, non-invasive
methods, such as ultrasound and magnetic resonance elastography,
may achieve an accurate diagnosis of moderate-to-severe steatosis
(defined as histological degree ≥30%) (14); however, they have limited
sensitivity in mild steatosis (defined as histological degree
5-30%) and may result in the misdiagnosis of patients as healthy
controls (14). Liver biopsy, the
current gold standard for diagnosing NAFLD, is costly, invasive and
potentially risky (14).
Therefore, liver biopsy or non-invasive methods may be unsuitable
for screening patients to assess the progression of NAFLD following
OXA chemotherapy, or for patient follow-up after therapeutic
intervention. Animal models can overcome the shortcomings of the
abovementioned clinical studies for investigating the effect of OXA
chemotherapy on the progression of NAFLD. We previously established
an animal model with OXA-induced acute liver injury in normal
healthy mice (7). In the present
study, OXA was used to induce acute liver injury in mice with
high-fat diet-induced NAFLD. More severe lipid accumulation,
inflammatory cell infiltration and hepatocyte necrosis were
observed in the NAFLD mice following OXA chemotherapy. Moreover,
the levels of serum ALT and AST were consistent with the
histological findings. These results demonstrated that OXA may
induce acute liver injury in NAFLD mice. To the best of our
knowledge, this is the first report of a reproducible experimental
NAFLD model with OXA-induced acute liver injury, which may be used
to explore in detail the pathogenesis of this condition.
It was previously indicated that long-term high-fat
diet can significantly increase free radical production and alter
the body's redox status, ultimately resulting in chronic oxidative
stress in liver tissues (13).
The present study demonstrated that high-fat diet also induced
chronic oxidative stress in the liver tissues of NAFLD mice. This
oxidative stress was further exacerbated by OXA chemotherapy, as
confirmed by a significant increase in the levels of MDA and ROS in
the liver tissues of NAFLD mice, as well as a significant decrease
in the activity of the antioxidant enzymes SOD and GSH-Px.
Oxidative stress may lead to hepatocellular injury through several
mechanisms, including lipid peroxidation, which can directly
promote cell necrosis and activate the apoptotic Fas-ligand pathway
(15). Oxidative stress occurs
when ROS are produced at levels exceeding those capable of being
sequestered by normal cellular antioxidant defenses (13,15). Excessive amounts of ROS may exert
direct deleterious effects on cells through lipid peroxidation,
protein degradation and DNA damage (15,16). In line with the generation of high
amounts of ROS, the livers of NAFLD mice treated with OXA exhibited
severe pathological changes, as demonstrated by a higher degree of
inflammatory cell infiltration, hepatocyte necrosis and increased
serum transferase activity. This indicates that OXA can amplify the
existing oxidative stress in NAFLD, and induce oxidative liver
injury.
Hepatic inflammation is considered to be the main
driver of hepatic tissue damage, triggering the progression from
NAFLD to severe fibrogenesis and, ultimately, hepatocellular
carcinoma (17). In the present
study, we observed that the levels of the hepatic inflammatory
cytokines TNF-α, IFN-γ and IL-17 were increased at baseline, and
increased markedly after OXA chemotherapy in NAFLD mice. The
inflammatory cytokines TNF-α, IFN-γ and IL-17 have been shown to
cause hepatocyte injury by triggering a potent cytotoxic immune
response and cell death (17-19). Among these factors, TNF-α acts as
a pivotal mediator in the progression of acute liver injury. The
overproduction of TNF-α through its receptor activates caspase-3, a
member of the family of cysteine proteases, which, in turn,
triggers hepatocellular necrosis and the apoptotic pathway
(18). TNF-α-induced ROS
generation sustains c-Jun N-terminal kinase (JNK) activation and
leads to hepatocyte apoptosis (20). In addition to the rapid increase
in the expression of inflammatory cytokines, the NAFLD mice
exhibited an aggravation of liver injury and lower survival rate
following OXA chemotherapy. Therefore, the findings of the present
study demonstrated that the hepatic inflammation already present in
NAFLD is further exacerbated by OXA chemotherapy, resulting in more
severe liver injury.
In addition to the observed direct damage to
cellular components, recent evidence indicates that oxidative
stress plays an important role in the progression of inflammatory
disorders. Oxidative stress activates various inflammatory
pathways, such as nuclear factor (NF)-κB and NLRP3, which
subsequently induce the expression of the inflammatory cytokines
TNF-α, IL-1 and TGF-β1 (21,22). Excessive ROS generation activates
the JNK and caspase pathways, ultimately leading to TNF-α-induced
cell death (23). Oxidative
stress also promotes the migration of inflammatory cells across the
endothelial barrier, leading to tissue injury (24). Therefore, it is reasonable to
hypothesize that oxidative stress, which is exacerbated by OXA, may
contribute to the rapid increase in the production of inflammatory
cytokines in NAFLD mice after OXA chemotherapy, further aggravating
liver injury. Moreover, the aggravation of hepatic oxidative stress
and inflammation after OXA chemotherapy may explain why the risk of
liver failure, or even death, in patients with NAFLD is
significantly higher compared with that in patients without NAFLD.
However, the exact mechanism underlying the interaction between
oxidative stress and inflammation after OXA chemotherapy in NAFLD
requires further investigation.
The present study also demonstrated that OXA
chemotherapy promotes hepatic fibrosis in NAFLD mice, with
increased collagen fiber deposition in the liver. OXA has been
shown to upregulate the expression of collagen I and TGF-β
(5). Consistent with this result,
we found that TGF-β levels increased in both the early and late
stages of OXA-induced acute liver injury in NAFLD mice. TGF-β is
the most effective cytokine in promoting liver fibrosis, as it can
activate hepatic stellate cells (HSCs), promote mass collagen
synthesis, and induce gradual deposition of extracellular matrix
(ECM) (25). TGF-β activates
receptor-activated Smads (R-Smads), leading to transcriptional
induction of α-SMA, the main marker of transdifferentiation of HSCs
(26). By upregulating TIMPs to
reduce ECM degradation, TGF-β further promotes the occurrence and
progression of liver fibrosis (25). In the present study, α-SMA and
TIMP-1 were found to be upregulated during the late stages of
OXA-induced acute liver injury. These findings indicated that OXA
promotes liver fibrosis in NAFLD mice. Furthermore, ROS are known
to promote signaling pathway protein phosphorylation, activation of
HSCs, and marked upregulation of TGF-β expression (27). Additionally, the inflammatory
cytokine IL-17 has been shown to be a profibrotic factor through
HSC activation (28). In the
present study, a marked increase in the ROS and IL-17A levels in
liver tissues was observed following OXA chemotherapy. Therefore,
it was hypothesized that the aggravation of OXA
chemotherapy-induced oxidative stress and inflammation further
promoted the development of liver fibrosis in NAFLD mice.
GSH is the most abundant cellular thiol antioxidant,
attaining concentrations in the high millimolar range in the liver
(29). GSH can alleviate
oxidative stress by serving as a substrate for antioxidative
enzymes, including GSH-Px, which converts hydroperoxide into less
harmful fatty acids, water and GSH disulfide (29). Increased ROS production causes
extensive damage to hepatocytes, and ROS can be neutralized by GSH
(30). In addition, GSH affects
the function of the immune system and regulates cytokine production
(29,30). As it is essential to normal liver
function, GSH has been extensively used in the clinical setting to
suppress liver inflammation (16,29). We previously reported that GSH
treatment can significantly inhibit oxidative stress response and
acute liver injury induced by OXA in normal mice (7). Accordingly, in the present study, we
observed a significant improvement in the cellular redox status,
including marked decreases in the levels of ROS, MDA and TNF-l, as
well as significantly elevated SOD and GSH-Px activities in the
NAFLD mouse liver after OXA chemotherapy, which was accompanied by
decreased liver tissue injury. Taken together, these findings
suggest that GSH attenuates OXA-induced liver injury in NAFLD mice
by inhibiting hepatic oxidative stress and inflammatory cytokine
production.
Passive GSH uptake is limited, due to an unfavorable
concentration gradient between the plasma and the cytosol (31). Therefore, non-toxic compounds,
such as γ-glutamylcysteine (31)
and L-oxothiazolidine carboxylic acid (32), which can increase endogenous
intracellular GSH levels and protect against several types of
insults, may be more effective in reducing OXA-induced
hepatotoxicity, which warrants further investigation.
However, GSH treatment did not completely inhibit
OXA chemotherapy-induced liver injury in NAFLD mice, as the ALT and
AST levels remained higher compared with those in the NAFLD control
group. Additionally, GSH treatment did not increase the survival
rate of the NAFLD mice following OXA chemotherapy. These results
indicated that GSH treatment does not fully inhibit OXA-induced
liver injury. GSH is the most important and ubiquitous antioxidant
molecule produced in the liver, where it serves as the principal
non-protein thiol involved in antioxidant cell defense (29). Consistently, GSH prevented
oxidative liver damage induced by OXA in NAFLD mice, which was
confirmed by the significant decrease in the levels of hepatic ROS
and MDA following GSH treatment. GSH also reduced the inflammatory
cytokine TNF-α level in liver tissues, but did not reduce the
levels of IFN-γ and IL-17. Therefore, GSH treatment did not
completely suppress the OXA-induced hepatic inflammation in NAFLD
mice. Furthermore, GSH treatment did not reverse the OXA-induced
increase in collagen fiber deposition or the upregulated expression
of TGF-β, α-SMA and TIMP-1 in NAFLD mice, indicating that GSH
treatment did not attenuate OXA-aggravated liver fibrosis in NAFLD
mice. The pathogenesis of OXA-induced liver injury involves various
factors and interactions. It is difficult to completely alleviate
liver injury and suppress the development of liver fibrosis using a
single hepatoprotective drug, as oxidative stress response,
inflammation, fibrosis, thrombosissis and coagulation abnormalities
must also be taken into consideration (5,33).
Thus, it is crucial to employ a comprehensive therapy scheme to
treat OXA-induced liver injury in a background of NAFLD.
Diagnosis of OXA-induced liver injury is mainly
based on liver histopathology (3,5).
In the present study, the clotting time and serum albumin of NAFLD
mice did not differ significantly before and after OXA chemotherapy
(data not shown). Therefore, the liver function tests of
biosynthetic capacity may not accurately reflect the
hepatoprotective effect of GSH. In this study, the hepatoprotective
effect of GSH against OXA-induced liver injury in NAFLD mice was
investigated only by liver histopathology and serum ALT and AST
levels, without liver function tests of biosynthetic capacity.
In conclusion, the present study demonstrated that
OXA induces acute liver injury in NAFLD mice. Moreover, OXA
aggravates hepatic oxidative stress in NAFLD mice, as shown by an
increase in the ROS and MDA levels and a decrease in the SOD and
GSH-Px levels. OXA aggravates hepatic inflammation in NAFLD mice by
increasing the expression of the inflammatory cytokines TNF-α,
IFN-γ and IL-17. Furthermore, OXA exacerbates hepatic fibrosis in
NAFLD mice, as confirmed by an increase in collagen fiber
deposition and TGF-β, α-SMA and TIMP-1 levels. Treatment with
exogenous GSH can mitigate OXA-induced hepatocyte injury in NAFLD
mice by inhibiting hepatic oxidative stress and the production of
the pro-inflammatory cytokine TNF-α. However, GSH does not
attenuate OXA-aggravated liver fibrosis in NAFLD mice. Therefore,
these results suggest that GSH may be utilized as a therapeutic
agent for the prevention of liver injury caused by OXA treatment in
NAFLD; however, GSH does not completely inhibit the OXA-induced
liver injury. Therefore, to minimize OXA-induced liver injury in
NAFLD, it may be possible to use a concomitant hepatoprotective
treatment, whereas all factors that may cause liver damage after
OXA chemotherapy should be taken into consideration.
Supplementary Materials
Funding
The present study was partially supported by the
National Natural Science Foundation of China (grant no. 81460418),
the Self-Raised Funds of Guangxi Health Department (grant no.
Z2016483), the Guangxi Natural Science Foundation (grant no.
2016GXNSFBA380218), the Guangxi Basic Ability Promotion Project of
Middle-aged and Young Teachers in Colleges and Universities (grant
no. 2017KY0121), and the Guangxi Key Laboratory of Molecular
Medicine in Liver Injury and Repair (grant no. 16-140-46-18).
Availability of data and materials
The data generated and analyzed during the present
study are available from the corresponding authors on reasonable
request.
Authors' contributions
YLu is the first author and performed all steps of
the experiments. YLi, XH, SW, JW assisted YLu in conducting the
experiments. YLu wrote the manuscript. CY helped in literature
survey and study design. All authors contributed toward data
analysis, drafting and critically revising the paper, provided
final approval of the version to be published, and agree to be
accountable for all aspects of the study.
Ethics approval and consent to
participate
All animal studies were performed according to the
guidelines of the Chinese Council on Animal Care and were approved
by the Affiliated Tumor Hospital of Guangxi Medical University
Committees on Animal Experimentation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Riddell IA: Cisplatin and oxaliplatin: Our
current understanding of their actions. Met Ions Life Sci.
18:2018.PubMed/NCBI
|
|
2
|
Formica V, Zaniboni A, Loupakis F and
Roselli M: Noninferiority of three months versus six months of
oxaliplatin-based adjuvant chemotherapy for resected colon cancer.
How should IDEA findings affect clinical practice. Int J Cancer.
143:2342–2350. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rubbia-Brandt L, Audard V, Sartoretti P,
Roth AD, Brezault C, Le Charpentier M, Dousset B, Morel P, Soubrane
O, Chaussade S, et al: Severe hepatic sinusoidal obstruction
associated with oxaliplatin-based chemotherapy in patients with
metastatic colorectal cancer. Ann Oncol. 15:460–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duwe G, Knitter S, Pesthy S, Beierle AS,
Bahra M, Schmelzle M, Schmuck RB, Lohneis P, Raschzok N, Öllinger
R, et al: Hepatotoxicity following systemic therapy for colorectal
liver metastases and the impact of chemotherapy-associated liver
injury on outcomes after curative liver resection. Eur J Surg
Oncol. 43:1668–1681. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Robinson SM, Mann J, Vasilaki A, Mathers
J, Burt AD, Oakley F, White SA and Mann DA: Pathogenesis of FOLFOX
induced sinusoidal obstruction syndrome in a murine chemotherapy
model. J Hepatol. 59:318–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chun YS, Laurent A, Maru D and Vauthey JN:
Management of chemotherapy-associated hepatotoxicity in colorectal
liver metastases. Lancet Oncol. 10:278–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin Y, Li Y, Hu X, Liu Z, Chen J, Lu Y,
Liu J, Liao S, Zhang Y, Liang R, et al: The hepatoprotective role
of reduced glutathione and its underlying mechanism in
oxaliplatin-induced acute liver injury. Oncol Lett. 15:2266–2272.
2018.PubMed/NCBI
|
|
8
|
Issa D, Patel V and Sanyal AJ: Future
therapy for non-alcoholic fatty liver disease. Liver Int. 38(Suppl
1): S56–S63. 2018. View Article : Google Scholar
|
|
9
|
Marchesini G, Bugianesi E, Forlani G,
Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N,
Melchionda N and Rizzetto M: Nonalcoholic fatty liver,
steatohepatitis, and the metabolic syndrome. Hepatology.
37:917–923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fiorucci S, Biagioli M and Distrutti E:
Future trends in the treatment of non-alcoholic steatohepatitis.
Pharmacol Res. 134:289–298. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinez MA, Vuppalanchi R, Fontana RJ,
Stolz A, Kleiner DE, Hayashi PH, Gu J, Hoofnagle JH and Chalasani
N: Clinical and histologic features of azithromycin-induced liver
injury. Clin Gastroenterol Hepatol. 13:369–376e3. 2015. View Article : Google Scholar
|
|
12
|
Lu Y, Wang X, Yan W, Wang H, Wang M, Wu D,
Zhu L, Luo X and Ning Q: Liver TCRγδ(+) CD3(+) CD4(−) CD8(−) T
cells contribute to murine hepatitis virus strain 3-induced hepatic
injury through a TNF-α-dependent pathway. Mol Immunol. 52:229–236.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Masarone M, Rosato V, Dallio M, Gravina
AG, Aglitti A, Loguercio C, Federico A and Persico M: Role of
oxidative stress in pathophysiology of nonalcoholic fatty liver
disease. Oxid Med Cell Longev. 2018:95476132018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Green CJ, Parry SA, Gunn PJ, Ceresa CDL,
Rosqvist F, Piché ME and Hodson L: Studying non-alcoholic fatty
liver disease: The ins and outs of in vivo, ex vivo and in vitro
human models. Horm Mol Biol Clin Investig. Aug 11–2018.Epub ahead
of print. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koek GH, Liedorp PR and Bast A: The role
of oxidative stress in non-alcoholic steatohepatitis. Clin Chim
Acta. 412:1297–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Y, Dong H, Thompson DC, Shertzer HG,
Nebert DW and Vasiliou V: Glutathione defense mechanism in liver
injury: Insights from animal models. Food Chem Toxicol. 60:38–44.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Del Campo JA, Gallego P and Grande L: Role
of inflammatory response in liver diseases: Therapeutic strategies.
World J Hepatol. 10:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao B: Hepatoprotective and
anti-inflammatory cytokines in alcoholic liver disease. J
Gastroenterol Hepatol. 27(Suppl 2): S89–S93. 2012. View Article : Google Scholar
|
|
19
|
Yano A, Higuchi S, Tsuneyama K, Fukami T,
Nakajima M and Yokoi T: Involvement of immune-related factors in
diclofenac-induced acute liver injury in mice. Toxicology.
293:107–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rutherford A and Chung RT: Acute liver
failure: Mechanisms of hepatocyte injury and regeneration. Semin
Liver Dis. 28:167–174. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mittal M, Siddiqui MR, Tran K, Reddy SP
and Malik AB: Reactive oxygen species in inflammation and tissue
injury. Antioxid Redox Signal. 20:1126–1167. 2014. View Article : Google Scholar :
|
|
22
|
Zhang X, Zhang JH, Chen XY, Hu QH, Wang
MX, Jin R, Zhang QY, Wang W, Wang R, Kang LL, et al: Reactive
oxygen species-induced TXNIP drives fructose-mediated hepatic
inflammation and lipid accumulation through NLRP3 inflammasome
activation. Antioxid Redox Signal. 22:848–870. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng Y, Ren X, Yang L, Lin Y and Wu X: A
JNK-dependent pathway is required for TNFalpha-induced apoptosis.
Cell. 115:61–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Wetering S, van Buul JD, Quik S, Mul
FP, Anthony EC, ten Klooster JP, Collard JG and Hordijk PL:
Reactive oxygen species mediate Rac-induced loss of cell-cell
adhesion in primary human endothelial cells. J Cell Sci.
115:1837–1846. 2002.PubMed/NCBI
|
|
25
|
Fabregat I and Caballero-Diaz D:
Transforming growth factor-β-induced cell plasticity in liver
fibrosis and hepatocarcinogenesis. Front Oncol. 8:3572018.
View Article : Google Scholar
|
|
26
|
Kisseleva T and Brenner DA: Role of
hepatic stellate cells in fibrogenesis and the reversal of
fibrosis. J Gastroenterol Hepatol. 22(Suppl 1): S73–S78. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin W, Tsai WL, Shao RX, Wu G, Peng LF,
Barlow LL, Chung WJ, Zhang L, Zhao H, Jang JY and Chung RT:
Hepatitis C virus regulates transforming growth factor beta1
production through the generation of reactive oxygen species in a
nuclear factor kappaB-dependent manner. Gastroenterology.
138:2509–2518. 2518.e12010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan Z, Qian X, Jiang R, Liu Q, Wang Y,
Chen C, Wang X, Ryffel B and Sun B: IL-17A plays a critical role in
the pathogenesis of liver fibrosis through hepatic stellate cell
activation. J Immunol. 191:1835–1844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yuan L and Kaplowitz N: Glutathione in
liver diseases and hepatotoxicity. Mol Aspects Med. 30:29–41. 2009.
View Article : Google Scholar
|
|
30
|
Wang J, Chen Y, Gao N, Wang Y, Tian Y, Wu
J, Zhang J, Zhu J, Fan D and An J: Inhibitory effect of glutathione
on oxidative liver injury induced by dengue virus serotype 2
infections in mice. PLoS One. 8:e554072013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peter C, Braidy N, Zarka M, Welch J and
Bridge W: Therapeutic approaches to modulating glutathione levels
as a pharmacological strategy in Alzheimer's disease. Curr
Alzheimer Res. 12:298–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gross CL, Giles KC and Smith WJ:
L-oxothiazolidine 4-carboxylate pretreatment of isolated human
peripheral blood lymphocytes reduces sulfur mustard cytotoxicity.
Cell Biol Toxicol. 13:167–173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zou X, Wang Y, Peng C, Wang B, Niu Z, Li Z
and Niu J: Magnesium isoglycyrrhizinate has hepatoprotective
effects in an oxaliplatin-induced model of liver injury. Int J Mol
Med. 42:2020–2030. 2018.PubMed/NCBI
|